
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the role of boroxines in metal-free carbon–carbon homologations using diazo compounds and boronic acids†
Claudio
Bomio
 ,
Mikhail A.
Kabeshov
,
Arthur R.
Lit
,
Shing-Hing
Lau
,
Janna
Ehlert
,
Claudio
Battilocchio
and
Steven V.
Ley
*
,
Mikhail A.
Kabeshov
,
Arthur R.
Lit
,
Shing-Hing
Lau
,
Janna
Ehlert
,
Claudio
Battilocchio
and
Steven V.
Ley
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB21EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk
First published on 15th June 2017
Abstract
By means of computational and experimental mechanistic studies the fundamental role of boroxines in the reaction between diazo compounds and boronic acids was elucidated. Consequently, a selective metal-free carbon–carbon homologation of aryl and vinyl boroxines using TMSCHN2, giving access to TMS-pinacol boronic ester products, was developed.
Introduction
Carbon–carbon bond forming reactions have been at the centre of organic synthesis for decades allowing efficient and quick assembly of complex molecular structures.1–6 Among many others, organoboron compounds are often the reagents of choice as they promote C–C bond formation reactions with high chemo- and stereoselectivity without the need for expensive, sometimes unstable and toxic transition metal catalysts.7,8 A large number of highly efficient methods have been described where a boron atom plays initially a role of the Lewis acid followed by a σ-bond migration thus allowing a metal-free coupling reaction between an electrophile (organoborane, organoboronic ester, dihaloborane)9–13 and a nucleophile (organolithium intermediates, sulfur ylides, diazo compounds).9,14–23Coupling between boronic acids and diazo compounds has attracted considerable interest in the synthetic community as boronic acids are readily available, usually stable and more atom efficient reagents when compared to their ester analogues.16,24–26 Additionally, the homologation using TMS-diazomethane (TMSCHN2) and organoboron compounds is useful to install a trimethylsilyl and boron functionality in a metal-free fashion.27–32 These doubly functionalised carbon substrates are of interest, as they may be used to generate sequential or orthogonal functionalization at the same carbon atom.28,29 To date, the homologation with TMSCHN2 has only been performed in combination with boronic acids to access the TMS-free homologation products.26,33 While several proposed mechanisms have been reported, the role of a boronic acid anhydride (boroxine) intermediate in these reactions remains unclear.16,26,33
In this work, we report a detailed computational and experimental study unveiling the crucial role of the boronic acid anhydrides–boroxines in these coupling reactions with diazo compounds and demonstrate how this knowledge can be applied to improve the scope of the method.
Results and discussion
Initially, the intriguing difference between the behaviour of the p-methoxyphenyl boronic acid (1) and the respective p-methoxyphenylboroxine (4) in their reactions with TMS diazomethane (TMSCHN2, 2) was noteworthy (Scheme 1).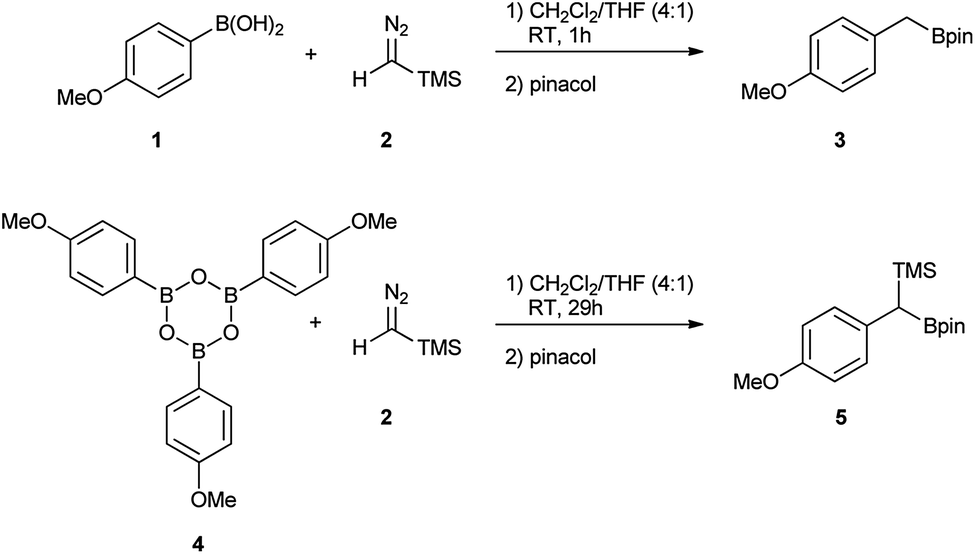 | ||
| Scheme 1 Reactions of the boronic acid 1 and boroxine 4 with TMS diazomethane 2. | ||
When mixing the boronic acid 1 with TMSCHN22 at ambient temperature, nearly instant decolourisation of the reaction mixture (originating from TMSCHN2) and strong gas evolution was observed. As a result, the formation of a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of homologation product 3 and Bpin ester of the starting material 6 was isolated. By contrast, when using boroxine 4 instead of boronic acid 1 under the same conditions, no gas evolution and only very slow decolourisation was observed. As expected, the TMS-homologated product 5 was isolated as the main product in 75% yield after 29 h of stirring at RT (Scheme 1).
:1 mixture of homologation product 3 and Bpin ester of the starting material 6 was isolated. By contrast, when using boroxine 4 instead of boronic acid 1 under the same conditions, no gas evolution and only very slow decolourisation was observed. As expected, the TMS-homologated product 5 was isolated as the main product in 75% yield after 29 h of stirring at RT (Scheme 1).
The decomposition of TMSCHN2 (2) to diazomethane is known to occur under acidic conditions and is due to the formation of H3O+ through an equilibrium between boronic acids or boroxines and their anionic tetrahedral species in the presence of water. Thereby this decomposition can rationalise the difference in the products obtained (Scheme 1).34,35 In order to further understand the role of boroxines in the coupling of diazo compounds with boronic acids, quantum chemistry calculations at density functional theory (ωB97xD/cc-pVTZ//ωB97xD/cc-pVDZ with SMD solvation model (solvent = dichloromethane, ε = 8.93) as implemented in Gaussian 09) level were therefore performed (Fig. 1).36–42
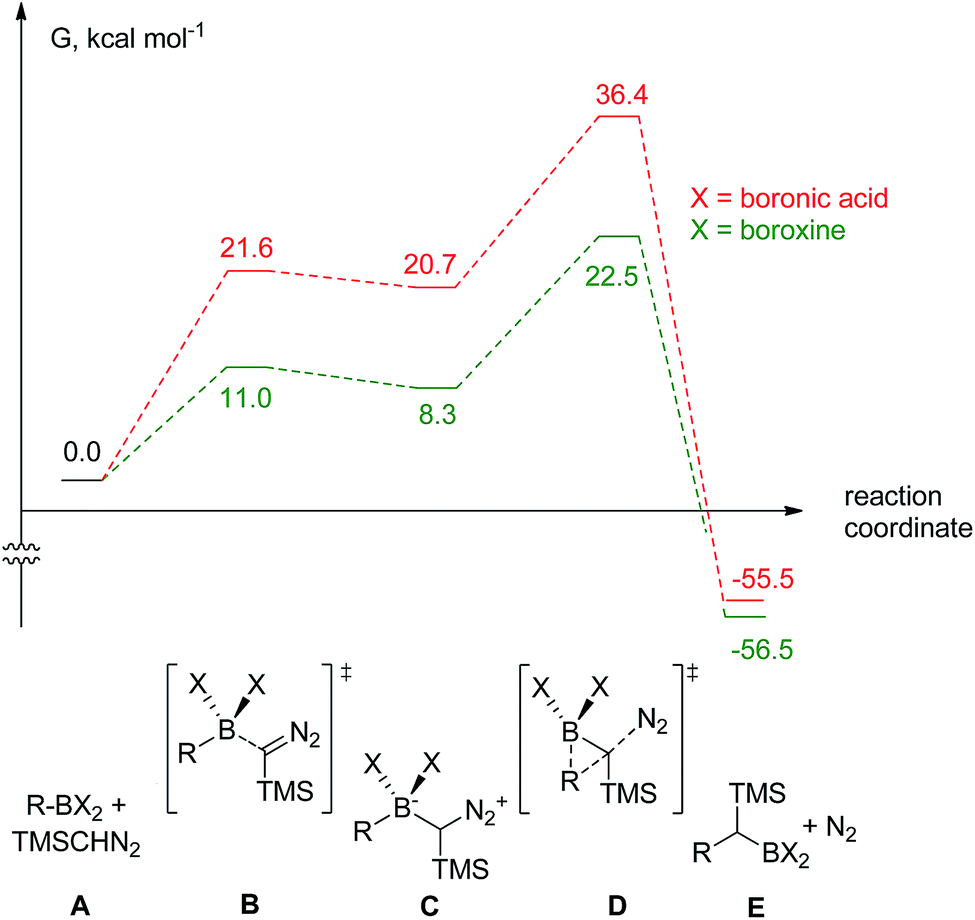 | ||
| Fig. 1 The computed reaction pathway for the coupling between boronic species and TMS diazomethane (R = Ph). | ||
It was found that both reactions, using boronic acid or boroxine in combination with TMSCHN2 (2), proceed via a two-step mechanism involving a sequence of a coordination transition state (point B, Fig. 1), unstable intermediate (point C, Fig. 1) and the highest energy point on the coordinate for both examples, which corresponds to the transition state for migratory insertion (point D, Fig. 1). In contrast to other reactions involving boronic acids and boroxines, where similar reactivity for both is usually observed,34 the striking difference of 13.9 kcal mol−1 (36.4 vs. 22.5, respectively; Fig. 1) for the activation Gibbs energies was computed. According to these computational results, only the boroxine, not the boronic acid, is expected to be reactive towards TMSCHN2 (2) (the barrier of 36.4 kcal mol−1 cannot be achieved under conventional conditions of organic reactions in solution).43,44
In order to check whether similar differences in reactivity are expected for reactions of diazomethane with boroxines and boronic acids, additional calculations using the same computational model, were performed. It can be concluded from the analysis that diazomethane should behave similarly to TMSCHN2 (2) and react smoothly with boroxines (Gact = 18.5 kcal mol−1). However, the reaction between diazomethane and boronic acids might be possible, but would require significantly higher activation energy (Gact = 29.2 kcal mol−1). Additionally, only mono addition of TMSCHN2 (2) to boroxine is expected due to the significantly higher activation barrier (27.2 kcal mol−1, lower diastereomeric transition state) for the second insertion into the benzylic boroxine intermediate (Fig. 1E).
To verify the computed results, control experiments were performed (Table 1). As already previously described, when using boronic acid 1, a fast decomposition of TMSCHN2 (2) occurred with formation of the homologated product 3 and only traces of the TMS homologation product 5 being observed. The conversion of the starting material 1 into 3 using 1.03 eq. of TMSCHN2 (2) gave only a 43% yield (entry 1). In contrast, when using boroxine (4) and 3.10 eq. of TMSCHN2 (2) (1.03 eq. per boron) the exclusive formation of the TMS homologation product 5 was observed (75% yield, entry 2). Addition of DIPEA to stabilise TMSCHN2 (2) led to much slower generation of diazomethane but had no effect on the final product distribution (entries 3 and 5). When water was added to the reaction (in order to shift the equilibrium towards the boronic acid 1) lower conversion towards homologation products 3 and 5 was observed, regardless of the presence of DIPEA. Moreover, it can be observed that water has the effect to increase the rate of decomposition of TMSCHN2 (entries 4, 6 and 7). Finally, the addition of large excess of base using boroxine 5 significantly decreases the reaction rate (entry 8). This can also explain why only small amount of TMS homologation product 5 is observed when boronic acid 1 is used in presence of DIPEA at room temperature (entries 3 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R-BX2 | TMSCHN2 | Timeb | DIPEA | H2O | Yielda (5:3:6) |
| a NMR yields. R = 4-MeOPh. b Time required for full disappearance of TMSCHN2. c TMSCHN2 not fully consumed. | ||||||
| 1 | RB(OH)2 | 1.03 eq. | 1 h | — | — | 1%:43%:48% |
| 2 | (RBO)3 | 3.10 eq. | 29 h | — | — | 75%:0%:10% |
| 3 | RB(OH)2 | 1.03 eq. | 15 h | 2.1 eq. | — | 2%:40%:51% |
| 4 | RB(OH)2 | 1.03 eq. | 10 h | 2.1 eq. | 2.0 eq. | 0%:14%:83% |
| 5 | RB(OH)2 | 1.03 eq. | 65 h | 6.0 eq. | — | 2%:42%:47% |
| 6 | RB(OH)2 | 1.03 eq. | 23 h | 6.0 eq. | 2.0 eq. | 0%:16%:80% |
| 7 | RB(OH)2 | 1.03 eq. | 1 h | — | 2.0 eq. | 0%:19%:78% |
| 8 | (RBO)3 | 3.10 eq. | 67c h | 6.0 eq. | — | 80%:0%:17% |
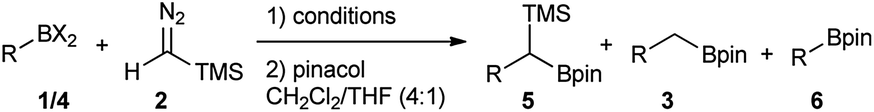
To rank organoboron reagents by their reactivity towards TMSCHN2 (2), further computational studies were performed and the results were subsequently compared with reactivity trends previously reported in the literature (Fig. 2). From all the computed examples, organoborane 14 showed the highest reactivity towards TMSCHN2, followed by difluoroorganoborane 13, boroxine 12 and catecholborane 8. Organoboranes are known to react fast with TMSCHN2 (2) at −78 °C while catecholboranes need heating (60 °C) over an extended time period (12 h).27,45 Both the reported reactivities as well as the experimentally observed behaviour of boroxines are in good correlation with our calculations. To prove that both the MIDA 7 and pinacol boranes 8 are not reactive, the substrates were treated with TMSCHN2 (2) at reflux in toluene for 3 d. In all cases, no homologation products were observed. Again, this is in agreement with our calculations.
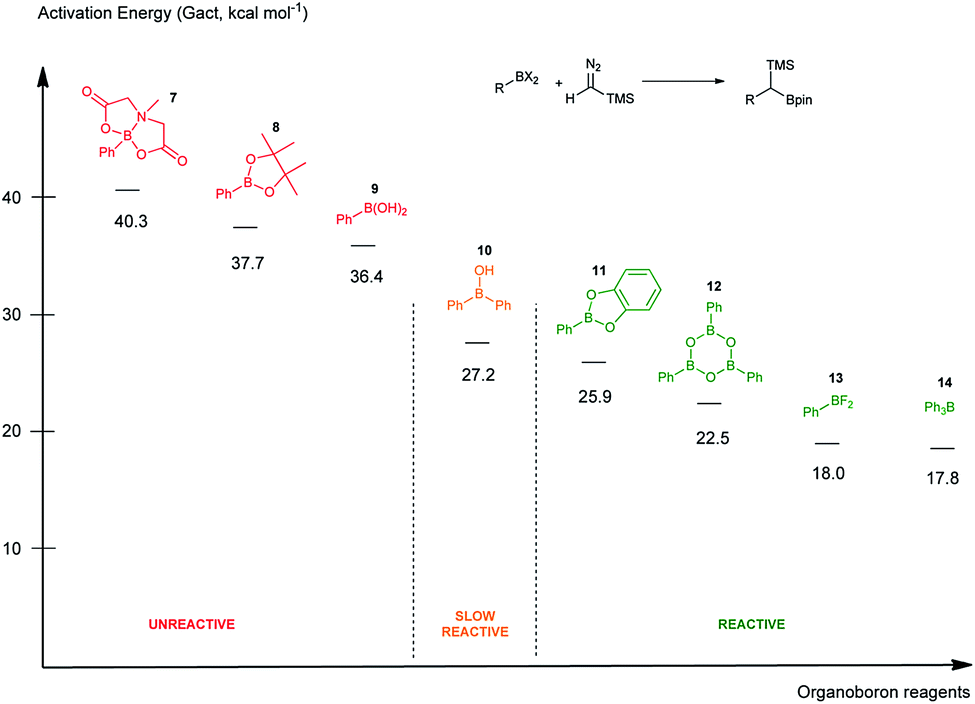 | ||
| Fig. 2 Computed reactivities of organoboron reagents towards TMS diazomethane. | ||
To conclude therefore, it was determined that boroxines are ideal reagents for reactions with diazo compounds, as they allow excellent reactivity and reasonable atom economy. Additionally, these results corroborate the fact that boroxines are most likely the reactive species in the coupling between diazo compounds and boronic acids.26
Our studies continued with the optimisation of reaction conditions by using boroxines, obtained via dehydration of boronic acids using a Dean–Stark apparatus (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T | Time | DIPEA | Yield |
| a 1.03 eq. TMSCHN2 per boron atom. b NMR yield. c Aldehyde formation observed. d Reaction performed with boronic acid 1. | |||||
| 1 | CH2Cl2/THF (4:1) |
24 °C | 29 h | — | 75b,c% |
| 2 | Toluene | 60 °C | 3 h | — | 78c% |
| 3 | Toluene | 60 °C | 3 h | 3.0 eq. | 88% |
| 4 | Toluene | 85 °C | 1 h | 3.0 eq. | 87% |
| 5 | Toluene | 85 °C | 1 h | 3.6 eq. | 93% |
| 6 | Toluene | 85 °C | 1 h | 3.6 eq. | 50b,d% |
As already described in Scheme 1, the desired product 5 could be obtained in 75% yield after stirring at rt for 29 h. To our delight, it was found that, unlike in other reported homologation reactions with TMSCHN2 (2), using just a slight excess of 0.033 equiv. of TMSCHN2 (2) per boron atom was sufficient to reach full conversion.26,33 To speed up the process, the reaction was performed at 60 °C in toluene, to give 78% of the desired product (5) after just 3 h (Table 2, entry 2). As a side product, the corresponding aldehyde was observed (approximately 10%), which is likely to arise from the oxidation of the TMS-boroxine intermediate. It was possible to suppress the formation of this byproduct by adding DIPEA to the reaction mixture, which resulted in an improved yield of 88% for compound 5 (Table 2, entry 3). Increasing the temperature to 85 °C led to full conversion after 1 h without affecting the yield. By increasing the amount of DIPEA from 3.00 equiv. to 3.60 equiv. the yield of the homologation could be further improved to 93%. Further attempts to increase the yield by adding more DIPEA did not lead to any improvements. Finally, applying the optimised conditions to the boronic acid 1 led to the formation of the desired TMS homologated product 5 in moderate yield (50%) which can be attributed to the very poor solubility of boronic acid and water in toluene and easier formation of the boroxine at elevated temperature (entry 6).
Having the optimal conditions in hand, the scope was expanded to other boronic acids (see Scheme 2). The reaction showed broad functional group tolerance (–NO2, halogen, ester, amide, thiophene). The conditions could also be applied to vinyl boronic acid precursors (31 to 34). Significant difference in reactivity could be observed with substrates bearing strongly electron withdrawing groups in para position (29 and 30), where the use of DIPEA led to the protodeborated byproducts. In order to suppress protodeboration, the reaction with electron poor substrates had to be performed without the presence of base, with unavoidable 10–15% aldehyde formation as a side product.
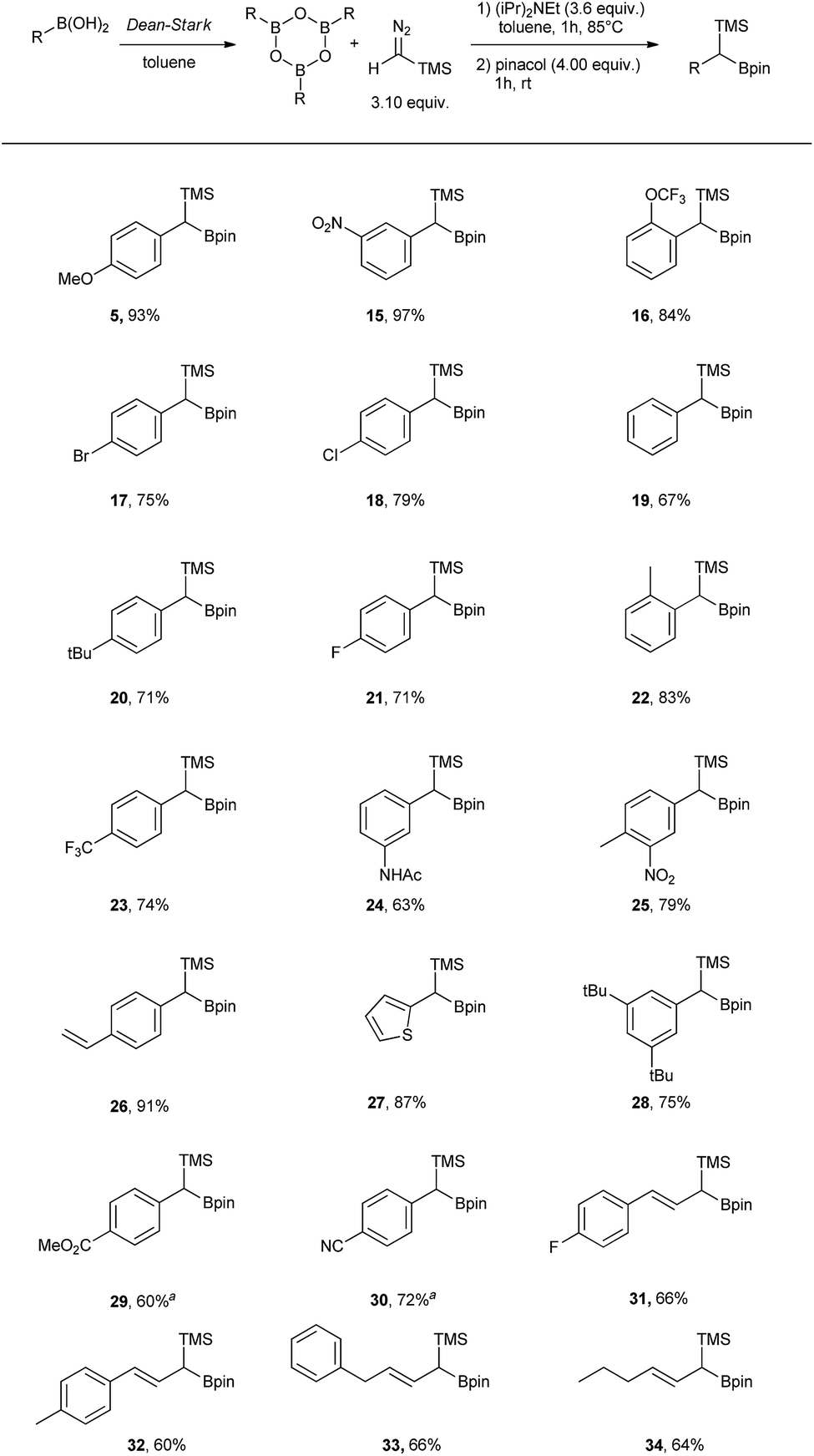 | ||
| Scheme 2 Scope table of the homologation reaction using TMSCHN2 and boroxines. a Reaction performed without DIPEA. General comment: TMS-boroxine intermediates are prone to protodeboration and oxidation and can therefore not be isolated. | ||
In addition to the scope, the homologation was performed on a gram-scale with three boronic acids (5, 18, 30) in order to demonstrate the practical usefulness of this reaction. As shown in Fig. 3 the homologation proceeded smoothly on scale by applying identical reaction conditions.
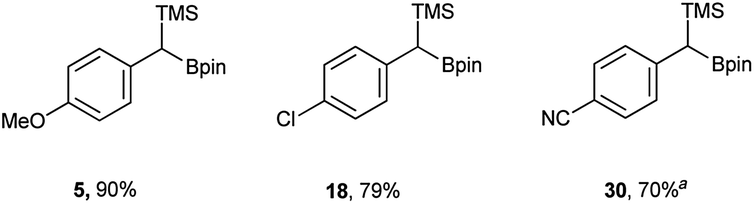 | ||
| Fig. 3 Yields of scaled up reactions (0.1 mol). a Reaction performed without DIPEA. | ||
Conclusions
We have shown experimentally and by means of DFT computations that boroxines of the corresponding boronic acids are likely to be the reactive intermediates in the homologation reaction with diazo compounds. Consequently, we have developed a metal-free, robust and scalable approach towards bench stable benzyl and allyl TMS-Bpin products with high functional group tolerance using TMSCHN2 and boroxines. Current investigations are directed towards the selective functionalisation of the TMS-Bpin products.Experimental
General procedure for preparation of TMS-Bpin products
The reaction was carried out in dry conditions under an atmosphere of argon. To a mixture of boroxine (0.15 mmol, 1.0 equiv.) and N,N-diisopropylethylamine (0.094 mL, 0.54 mmol, 3.6 equiv.) in toluene (0.75 mL) was added (trimethylsilyl)diazomethane (0.23 mL, 0.465 mmol, 2 M in hexanes, 3.1 equiv.). The reaction mixture was stirred at 85 °C for 1 h and allowed to cool down to room temperature. Pinacol (70.9 mg, 0.60 mmol, 4.0 equiv.) was added and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with a saturated aqueous solution of NH4Cl and the aqueous phase was extracted with EtOAc. The combined organic extracts were washed with brine, dried (MgSO4) and concentrated in vacuo. The crude residue was purified by silica gel flash column chromatography to afford the desired TMS-Bpin product.Acknowledgements
We are grateful to the Swiss National Science Foundation (C. Bo.), the Croucher Foundation (S. H. L), Erasmus+ (J. E.), and the Engineering and Physical Sciences Research Council (M. K., C. Ba., and S. V. L. grant no. EP/K009494/1, EP/M004120/1 and EP/K039520/1) for financial support.Notes and references
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723–6733 Search PubMed.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613–1616 CrossRef CAS PubMed.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS PubMed.
- N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC.
- R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- M. L. Crawley and B. M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, Wiley, Hoboken, NJ, USA, 2012 Search PubMed.
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10606 CrossRef CAS.
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- H. C. Brown, Organic Synthesis via Boranes, Wiley-Interscience, New York, 1975 Search PubMed.
- O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed.
- G. A. Molander and D. W. Ryu, Angew. Chem., Int. Ed., 2014, 53, 14181–14185 CrossRef CAS PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 4–9 Search PubMed.
- A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed.
- V. K. Aggarwal, Y. F. Guang and A. T. Schmidt, J. Am. Chem. Soc., 2005, 127, 1642–1643 CrossRef CAS PubMed.
- J. Hooz and S. Linke, J. Am. Chem. Soc., 1968, 90, 6891–6892 CrossRef CAS.
- J. Hooz and S. Linke, J. Am. Chem. Soc., 1968, 90, 5936–5937 CrossRef CAS.
- D. J. Pasto and P. W. Wojkowski, J. Org. Chem., 1971, 36, 1790–1792 CrossRef CAS.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- C. Peng, W. Zhang, G. Yan and J. Wang, Org. Lett., 2009, 11, 1667–1670 CrossRef CAS PubMed.
- H. Li, Y. Zhang and J. Wang, Synthesis, 2013, 45, 3090–3098 CrossRef CAS.
- D. N. Tran, C. Battilocchio, S.-B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC.
- C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed.
- J.-S. Poh, S.-H. Lau, I. G. Dykes, D. N. Tran, C. Battilocchio and S. V. Ley, Chem. Sci., 2016, 7, 6803–6807 RSC.
- J.-P. Goddard, T. Le Gall and C. Mioskowski, Org. Lett., 2000, 2, 1455–1456 CrossRef CAS PubMed.
- M. G. Civit, J. Royes, C. M. Vogels, S. A. Westcott, A. B. Cuenca and E. Fernández, Org. Lett., 2016, 18, 3830–3833 CrossRef CAS PubMed.
- E. La Cascia, A. B. Cuenca and E. Fernández, Chem.–Eur. J., 2016, 22, 18737–18741 CrossRef CAS PubMed.
- D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 230–236 CrossRef CAS.
- D. J. S. Tsai and D. S. Matteson, Organometallics, 1983, 2, 236–241 CrossRef CAS.
- C. H. Burgos, E. Canales, K. Matos and J. A. Soderquist, J. Am. Chem. Soc., 2005, 127, 8044–8049 CrossRef CAS PubMed.
- C. Wu, G. Wu, Y. Zhang and J. Wang, Org. Chem. Front., 2016, 3, 817–822 RSC.
- Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- E. Kühnel, D. D. P. Laffan, G. C. Lloyd-Jones, T. Martínez Del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef PubMed.
- Y. S. Lin, G. De Li, S. P. Mao and J. Da Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. A. Kabeshov, O. Kysilka, L. Rulíšek, Y. V. Suleimanov, M. Bella, A. V. Malkov and P. Kočovský, Chem.–Eur. J., 2015, 21, 12026–12033 CrossRef CAS PubMed.
- S. Schenker, C. Schneider, S. B. Tsogoeva and T. Clark, J. Chem. Theory Comput., 2011, 7, 3586–3595 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmami, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jamarmillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowski, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Xia, A. S. Dudnik, Y. Li and V. Gevorgyan, Org. Lett., 2010, 12, 5538–5541 CrossRef CAS PubMed.
- J. K. Lam, H. V. Pham, K. N. Houk and C. D. Vanderwal, J. Am. Chem. Soc., 2013, 135, 17585–17594 CrossRef CAS PubMed.
- R. C. Neu and D. W. Stephan, Organometallics, 2012, 31, 46–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02264f, additional data is available from the University of Cambridge Data Repository Website: https://doi.org/10.17863/CAM.10823. |
| This journal is © The Royal Society of Chemistry 2017 |