
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom imine to amine: an unexpected left turn. Cis-β iron(II) PNNP′ precatalysts for the asymmetric transfer hydrogenation of acetophenone†
Karl Z.
Demmans
 ,
Chris S. G.
Seo
,
Alan J.
Lough
and
Robert H.
Morris
*
,
Chris S. G.
Seo
,
Alan J.
Lough
and
Robert H.
Morris
*
Department of Chemistry, University of Toronto, 80 Saint George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: rmorris@chem.utoronto.ca
First published on 14th July 2017
Abstract
A novel PNN ligand bearing an orthophenylene group and a primary amine was synthesized with the aid of a palladium-catalyzed amination and reacted with phosphonium dimers [–PR2CH2CH(OH)–]2[Br]2 R = Et, iPr, Cy, Ph, xylyl, and o-Tol, and [Fe(OH2)6]2+ to produce a new series of cis-β iron(II) PNNP′ precatalysts cis-β-[Fe(Br)(CO)(PNNP′)]BPh4 as a pair of diastereomers. The more stable orthophenylene amido group was chosen to imitate and replace the enamido moiety of a highly active iron precatalyst for the asymmetric transfer hydrogenation (ATH) of ketones in an attempt to prevent its deactivation caused by reduction of the enamido group. This objective was partially achieved using the complex with a PEt2 group which catalyzed the transfer hydrogenation in isopropanol of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equivalents of acetophenone to racemic 1-phenylethanol. With a low acetophenone to catalyst ratio of 500 to 1, the catalytic activity was moderate and the enantiomeric excess (ee) of the product 1-phenylethanol ranged surprisingly from 94% (R) to 95% (S) depending on the nature of PR2 and whether the precatalyst contained an imine or amine donor. The amine precatalyst with a PEt2-group produced a more stable hydride species when activated, allowing the reaction mixture to be heated to 75 °C to obtain a TON of 8821 for acetophenone while retaining the high ee of 95% (S). The activation pathway in basic isopropanol (iPrOH) was studied for three precatalysts to elucidate that the cis-β precatalysts rearrange to form trans hydride complexes. The study suggests that the enantioselectivity of these complexes is determined by from which side of the PNNP′ plane the hydride transfer occurs.
000 equivalents of acetophenone to racemic 1-phenylethanol. With a low acetophenone to catalyst ratio of 500 to 1, the catalytic activity was moderate and the enantiomeric excess (ee) of the product 1-phenylethanol ranged surprisingly from 94% (R) to 95% (S) depending on the nature of PR2 and whether the precatalyst contained an imine or amine donor. The amine precatalyst with a PEt2-group produced a more stable hydride species when activated, allowing the reaction mixture to be heated to 75 °C to obtain a TON of 8821 for acetophenone while retaining the high ee of 95% (S). The activation pathway in basic isopropanol (iPrOH) was studied for three precatalysts to elucidate that the cis-β precatalysts rearrange to form trans hydride complexes. The study suggests that the enantioselectivity of these complexes is determined by from which side of the PNNP′ plane the hydride transfer occurs.
Introduction
The asymmetric hydrogenation of ketones to produce enantiopure alcohols is a widely studied reaction with significant importance in the flavour, fragrance, agricultural, and pharmaceutical industries.1–5 Due to the fact that industrial catalysts typically use less earth-abundant metals such as ruthenium4,6,7 for this reaction, the use of base metals such as iron for the reduction of polar bonds has recently become a focus of research.8–12In the field of ATH, our group reported iron(II) PNNP′ precatalysts based on (S,S)-DPEN (Fig. 1) that when activated in basic iPrOH produced an iron hydride capable of reducing aromatic ketones enantioselectively at a turnover number (TON) of approx. 5000 and a turnover frequency (TOF) as high as 200 s−1 at 28 °C with ee up to 98% for the case of 1b.13,14
 | ||
| Fig. 1 Select iron(II) PNNP complexes developed in the Morris group.13,15,16 | ||
An investigation into the mechanism of action of these catalyst systems showed that the active hydride in the system, trans-FeH(CO)(PNNP′) (2 in Fig. 1) has a ligand with a unique structure containing a secondary amine and an enamido donor: PNNP′ = (S,S)-PPh2CH2CH2NHCHPh-CHPhNCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPPh2.13,17 Precatalyst 1a is deprotonated at the methylene next to the imino donor to produce a neutral enamido species, which forms 2 under basic iPrOH solution.
CHPPh2.13,17 Precatalyst 1a is deprotonated at the methylene next to the imino donor to produce a neutral enamido species, which forms 2 under basic iPrOH solution.
A pathway to deactivation of the catalyst was found to be the reduction of the CC bond. The fully saturated precatalyst, [FeCl(CO)(PNNP)][BF4] (3 in Fig. 1), was isolated and shown to be a very poor catalyst for both direct and transfer hydrogenation.17 The objective of this investigation was to replace the CC bond with an orthophenylene group to prevent reduction and shut down the deactivation pathway and allow the recycling of the catalyst. Recent developments in palladium-catalyzed amination18–21 allowed us to afford the key PNN building blocks needed to prepare the precatalysts 5a–f and 6a–e, shown in Fig. 2 in their major diastereomeric form.
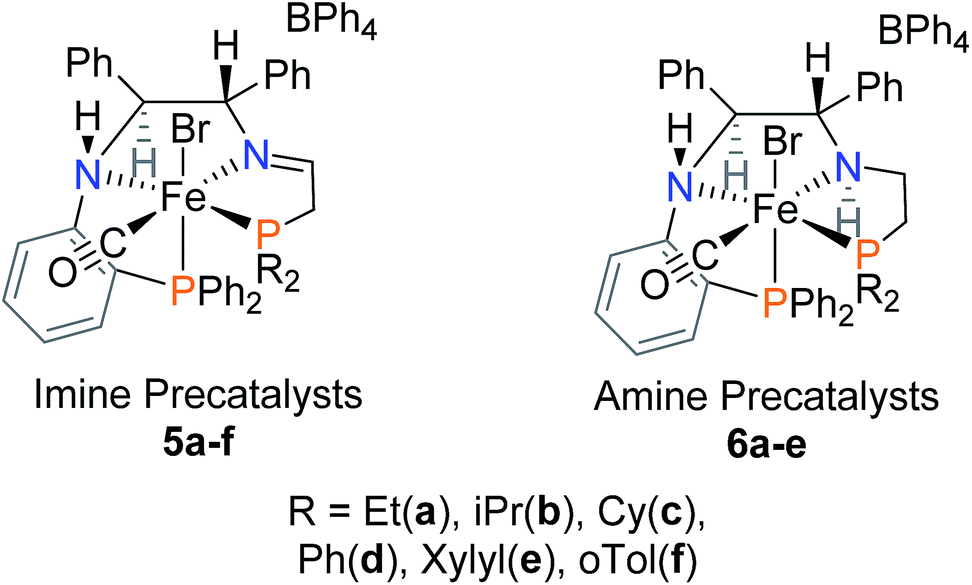 | ||
| Fig. 2 Imine and amine precatalysts bearing the orthophenylene linker. | ||
This article describes their preparation, catalytic testing, and a study of their activation in basic iPrOH solution.
At first glance, the cis-β ligand structures of 5a–f and 6a–e are quite different from those of the trans structures of 1–3 due to the steric requirements of our new ligands with the orthophenylene group. A further goal of this project is to understand how this change in structure influences catalyst activity and selectivity.
Examples of iron catalysts with cis-β-PNNP ligands have been reported by Mezzetti's and our own group. Mezzetti et al. have also crystallized cis-β iron(II) PNNP precatalysts (Fig. 3), wherein the phosphine donors are linked via an ethylene or propylene bridge to create a macrocycle.8,22–26
 | ||
| Fig. 3 Selected cis-β iron(II) PNNP complexes developed in the Mezzetti group. | ||
Our first iron catalyst to achieve an ee of 99% for ketone reduction was iron precatalyst 4 (Fig. 1) bearing a dicyclohexylphosphine group.16 Interestingly, this precatalyst crystallizes in the cis-β configuration, placing the more flexible ethyldiphenylphosphine group in the apical position.27 While more enantioselective, this catalyst system was much less active than that from 1a,b. The current study probes the influence of the cis-β structures of complexes 5a–f and 6a–e on the activity and enantioselectivity of transfer hydrogenation catalyst systems derived from them.
Results and discussion
Synthesis of the (S,S) isomer of benzoPPh2DPEN and the iron imine precatalysts
Following the work of Liang et al., wherein achiral PNN ligands based on N,N-dimethylethylenediamine were synthesized,21 a new chiral PNN ligand precursor bearing a primary amine was prepared (Scheme 1). A total of four steps were required: a monotosylation of (S,S)-DPEN as described by Tietze et al.,28 a Buchwald–Hartwig amination, a nucleophilic aromatic substitution to install diphenylphosphine, and a tosyl deprotection as shown in Scheme 1.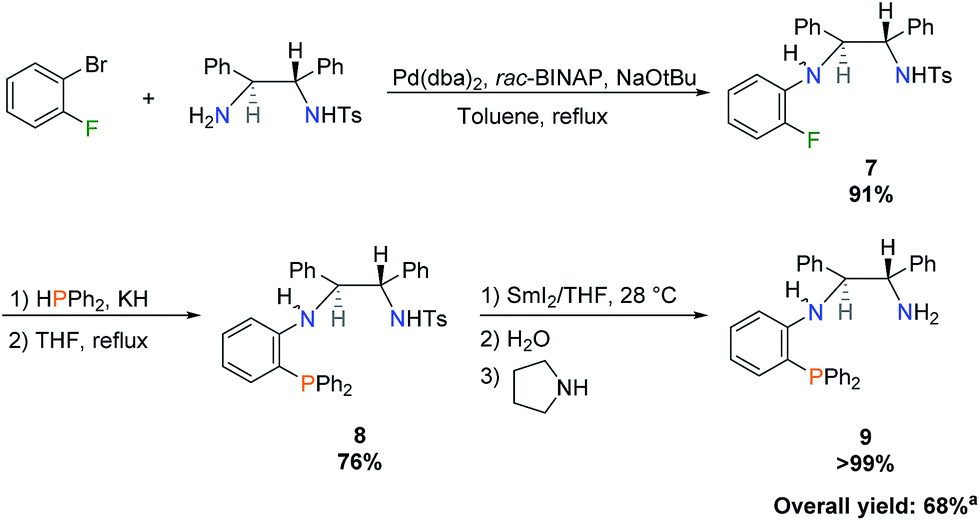 | ||
| Scheme 1 Synthesis of benzoPPh2DPEN (S,S) isomer 9. aPercent overall yield was calculated based on molar amount of monotosylated-(S,S)-DPEN. | ||
In greater detail, 1-bromo-2-fluorobenzene was chosen as the coupling reagent to monotosylated-(S,S)-DPEN in order to selectively perform Buchwald–Hartwig amination on the bromo substituent, leaving the fluoro group unaltered for subsequent phosphination.18–21 This reaction was scaled up to produce 3 g of benzoFTsDPEN (7) at the cost of refluxing in toluene for several days. Nucleophilic aromatic substitution using bulky potassium diphenylphosphide proceeded slowly in refluxing THF due to the kinetic barrier imposed by the sterically-encumbered ortho position. This reaction was also attempted in refluxing 1,4-dioxane to increase the reaction rate, but low conversions were obtained due to the lower dielectric constant of 1,4-dioxane and poor solubility of the starting material. The product, benzoPPh2TsDPEN (8), was isolated by simple filtration after stirring the crude residue in diethyl ether. Attempts were also made to perform Buchwald–Hartwig amination with monotosylated-(S,S)-DPEN and (2-bromophenyl)diphenylphosphine, but as the PNN ligand formed, it substituted BINAP from the palladium catalyst, halting the reaction immediately. The tosyl deprotection step required extremely reducing conditions and could not be achieved via other reductants such as sonication with magnesium powder in anhydrous methanol,29 or sodium naphthalenide in cold THF.30 A solution of samarium diiodide in THF was prepared and the deprotection methodology was adapted from the literature procedure31 to isolate benzoPPh2DPEN (9) as an air-stable colourless oil.
This novel, chiral PNN molecule with a primary amine condensed readily with aldehydes in the presence of an iron salt following protocols previously established to produce the new iron(II) PNNP′ precatalysts as shown in Scheme 2.32,33
 | ||
| Scheme 2 Synthesis of iron PNNP′ imine precatalysts 5a–f. Percent values represent overall yields based on the molar amount of 9. | ||
In short, various air- and moisture-stable phosphonium dimers (10a–f) were deprotonated in basic condition to release phosphine aldehydes. Through the iron-templated condensation reaction with 9, dicationic bis(acetonitrile) iron complexes (11a–f) were presumably formed as signalled by the emergence of a typical pink colour for such complexes.16,34 Ligand exchange was performed with potassium bromide under carbon monoxide gas, followed by salt metathesis in methanol to isolate the imine precatalysts 5a–f as tan powders. Chirality was set at the amino nitrogen upon coordination to iron, creating a pair of diastereomers labelled as (S)-NH and (R)-NH. In most cases, the phosphorus and proton NMR of the isolated powders suggested an S:R ratio of 95:5. The diastereomers 5d co-crystallized in a 1:1 mixture as confirmed by single crystal X-ray diffraction (Fig. 4). The altered ratio of diastereomers (from 95:5 to 50:50) after co-crystallization was reflected in the NMR data as shown in the ESI (see ESI, Fig. S7 and 8).† Thus there is slow interconversion of the diastereomers.
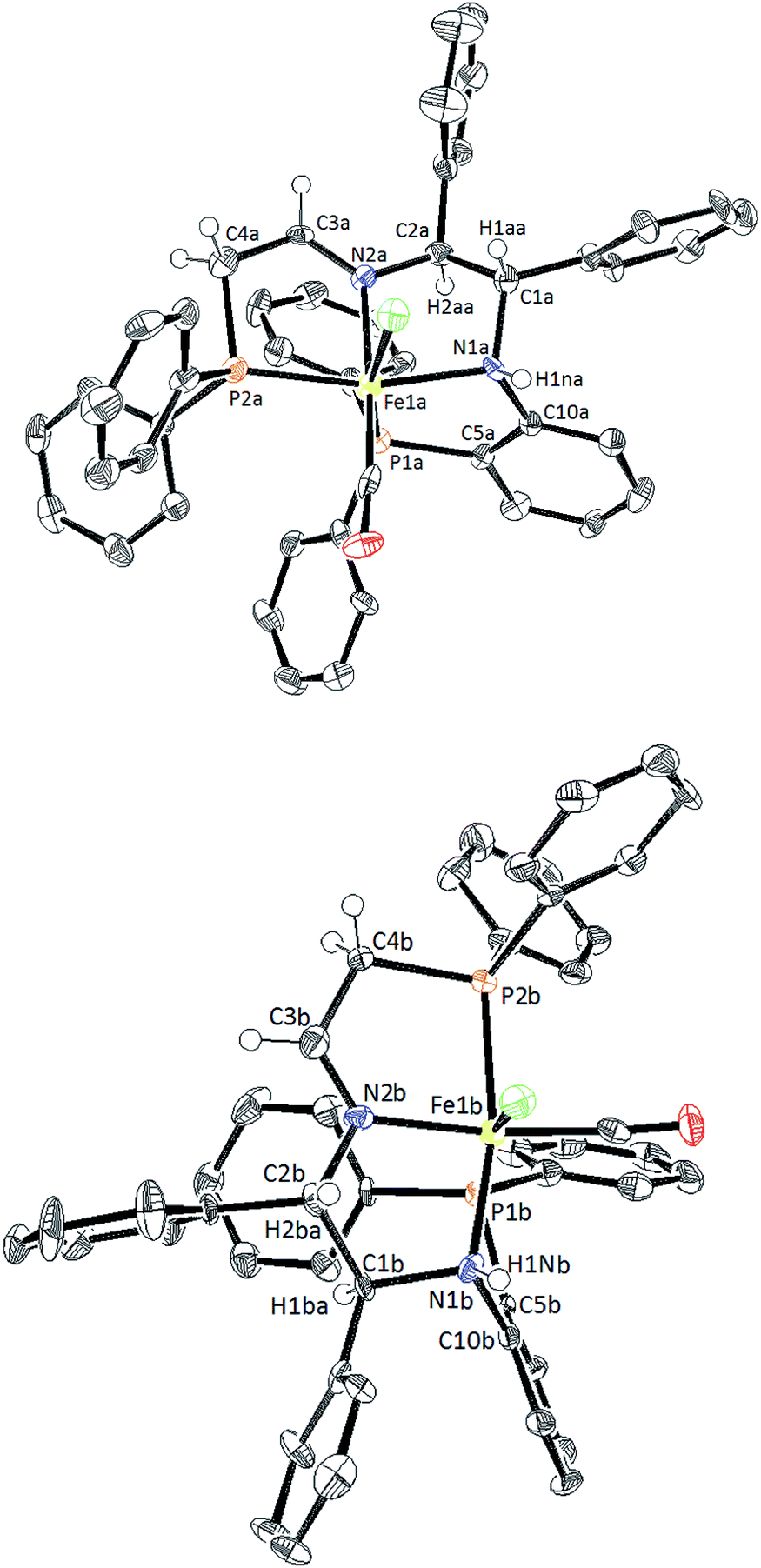 | ||
| Fig. 4 ORTEP plot of 5d containing the pair of diastereomers, with (R)-NH on the top and (S)-NH on the bottom. The molecules have been rotated to display the difference in the gauche and anti-conformation of the (R)-NH and (S)-NH, respectively with respect to the CH of the adjacent CHPh group. Thermal ellipsoids are drawn at 50% probability level. Aromatic ring protons and the BPh4 counteranions were omitted for clarity. Selected interatomic distances (Å) and angles (deg): for (R)-NH: Fe1a–N1a 2.077(8), Fe1a–N2a 1.976(8), F1a–P1a 2.247(3), Fe1a–P2a 2.233(3), N1a–C10a 1.440(12), P1a–C5a 1.819(10), N2a–C3a 1.275(11), C3a–C4a 1.485(14), N1a–C1a 1.521(11), N2a–C2a 1.485(10); P1a–Fe1a–P2a 103.80(13). For (S)-NH: Fe1b–N1b 2.047(9), Fe1b–N2b 1.982(8), Fe1b–P1b 2.260(3), Fe1b–P2b 2.248(3), N1b–C10b 1.472(12), P1b–C5b 1.823(10), N2b–C3b 1.263(11), C3b–C4b 1.476(14), N1b–C1b 1.545(10), N2b–C2b 1.497(10); P1b–Fe1b–P2b 102.00(13). | ||
Upon further investigation it was found that the major isomer (S)-NH contained a more sterically-favoured orientation by placing the orthophenylene and the adjacent phenyl group from the DPEN backbone in the anti-conformation. The torsion angle of the PNNP′ ligand for the major isomer is −56.8° (P2b–N2b–N1b–P1b Fig. 4), while the torsion angle for the minor isomer is +64.9° (P2a–N2a–N1a–P1a).
Synthesis of the iron amine precatalysts
The precatalysts bearing two amino ligands were also synthesized to provide the secondary amine in the structure of an analogue to the active hydride catalyst (2 in Fig. 1). Amine precatalysts 6a–e were synthesized as outlined in Scheme 3. | ||
| Scheme 3 Synthesis of iron PNNP′ amine precatalysts 6a–e. Percent values represent overall yields based on the molar amount of 9. | ||
The desired PNNP′ ligands (12a–e) were prepared as shown in Scheme 3 by reductive amination of the phosphine aldehyde, released in basic conditions from 10a–e (as in Scheme 2), with 9. These ligands as a crude residue were placed on iron without further purification, presumably forming the dicationic bis(acetonitrile) iron complexes 13a–e. Ligand exchange followed by salt metathesis allowed for the isolation of the cis-β-[Fe(CO)(Br)(PNNP′)][BPh4] amine precatalysts 6a–e in moderate yields. Four isomers could possibly arise due to having two chiral nitrogen centres, but the NMR data showed that only one pair of diastereomers was isolated in the powdered product. The major diastereomer of 6a crystallized as a single isomer as confirmed by single crystal X-ray diffraction as shown in Fig. 5.
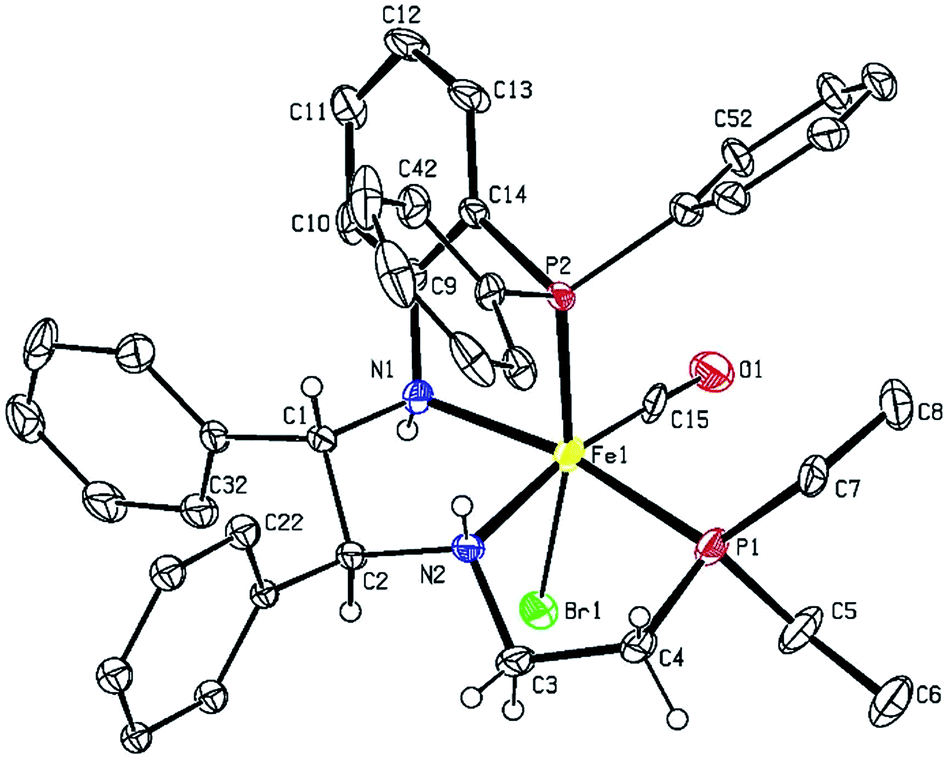 | ||
| Fig. 5 Crystal structure of the major diastereomer of 6a. Thermal ellipsoids are drawn at the 50% probability level. Aromatic ring protons and the BPh4 counteranion were omitted for clarity. Selected interatomic distances (Å) and angles (deg): Fe1–N1 2.050(4), Fe1–N2 2.055(5), Fe1–P1 2.2282(14), Fe1–P2 2.2294(14), N1–C9 1.457(6), P2–C14 1.818(5), N2–C3 1.495(7), C3–C4 1.536(7), N1–C1 1.513(6), N2–C2 1.502(6); P1–Fe1–P2 103.04(5). | ||
The major isomer (S,S)-NH,NH contained a more sterically-favoured alternating anti-disposition of the orthophenylene N–H, the C–H of the adjacent CHPh group, and ethylene linker N–H. The torsion angle of the PNNP′ ligand for the major isomer is −58.8° (P1–N2–N1–P2).
Factors contributing to the preferential formation of cis-β isomers
While we had postulated that a cis-β structure was formed as an intermediate during the activation of 1a,17 the crystallization of 4 (Fig. 1)27 raises the question of the importance of cis-β structures in catalysis. By comparing the crystal structures of 1a, 4, 5d, and 6a, two trends dictating the formation of a trans or cis-β PNNP′ iron precatalyst could be discerned. First, the steric bulk of the two phosphine donors play a significant role as they are forced in close contact upon coordination to the iron centre. Precatalysts 1a and 4 differ in steric bulk of one phosphine moiety, increasing from diphenylphosphino to dicyclohexylphosphino, respectively. This increase for 4 causes the diphenylphosphino moiety on the flexible ethylene linker to move out of the plane to decrease the steric strain. Precatalyst 1a and 5d both contain two diphenylphosphino moieties, yet 5d also folds into a cis-β structure. The second factor is the flexibility of the linker connecting the amino donor to the phosphine donor. The tetrahedral amino donor would always force the phosphine donor out of the plane, but precatalysts with a flexible ethylene linker may shift to negate this effect. The rigid orthophenylene linker cannot bend; therefore, it acts to extend the direction forced by the tetrahedral amino, placing the phosphine donor either above or below the plane, thus creating diastereomers. Reducing the steric bulk from diphenylphosphine in 6d to diethylphosphine in 6a does not produce a trans PNNP′ precatalyst; thus, the cis-β structure is likely due to the tetrahedral nitrogen and the rigidity of the orthophenylene linker.Catalytic testing
Precatalysts 5a–f and 6a–e were preliminarily screened for the ATH of acetophenone in iPrOH for the set period of 1 h at room temperature. The activity of these systems was much lower than that of 1a,b and 4 which reached completion in seconds under these conditions.13,16In summary, the most active catalysts were 5c,d and 6a,d. The most intriguing result was that the ee of 1-phenylethanol ranged from 94% (R) to 90% (S). All of the activated imine precatalysts produced (R)-1-phenylethanol with a high ee, whereas the activated amine precatalysts 6d,e produced (R)-1-phenylethanol in slight excess and 6a–c produced (S)-1-phenylethanol with a high ee. Given the varying activities and enantioselectivities of all of the precatalysts synthesized, we decided to focus further studies on 5d and 6d for a direct comparison of the imine and amine precatalysts as well as 6a to elucidate the origin of the enantioselectivity.
The reaction progress of these selected catalysts (and related systems) under more optimized conditions are shown in Fig. 6 and the yields and ee are listed in Table 2. As mentioned, samples of the imine-containing complex 5d with PPh2 groups were available as a powder containing 95:5 (S)-NH:(R)-NH and as crystals containing a 50:50 ratio. Crystals of the amine-containing complexes 6a and 6d were used in the following sections instead of the powdered samples as the crystals contain solely the major isomer. This led to a slight discrepancy in the ee for complex 6d between Tables 1 and 2. The ATH of acetophenone with the crystals occurred more rapidly. The amine-containing complex with the diethylphosphine group 6a was the most active and enantioselective, producing 1-phenylethanol in 95% ee but, remarkably, in the (S) configuration. Explanations for these observations are provided in the mechanism section.
 | ||
| Fig. 6 ATH of acetophenone with crystals of the precatalysts in optimized conditions (top – conversion, bottom – ee, positive is (R), negative is (S)). Only the first 1000 minutes of the catalytic profile was shown to compare the rate of catalysis for these precatalysts. | ||
| Precatalyst | Label, R group | Conversion (%) | ee (%), 1-phenylethanol |
|---|---|---|---|
|
a Reactions performed under argon at 28 °C. Time = 1 h. Acetophenone:KOtBu:precatalyst ratio was 500:8:1. Volume of iPrOH = 1.5 mL. Final concentrations: [acetophenone] = 0.742 M; [KOtBu] = 0.0119 M; [Fe] = 0.00148 M; [iPrOH] = 12.0 M. Conversions and ee were determined using a gas chromatograph containing a chiral column. Di-tert-butylbenzene was used as an external standard.
b Acetophenone:KOtBu:precatalyst ratio was 6121:8:1. Volume of iPrOH = 9.0 mL. Final concentrations: [acetophenone] = 0.441 M; [KOtBu] = 5.74 × 10−4 M; [Fe] = 7.2 × 10−5 M; [iPrOH] = 12.4 M.
c Time = 3 min. Ref. 13.
d Time = 2 h. Ref. 16.
e This ee has a large error (±10) due to the low conversion.
|
|||
| trans-[FeCl(CO)(PNNP′)]-[BF4] | 1a, Ph | 82b,c | 78 (R) |
| 1b, xylyl | 82b,c | 90 (R) | |
| cis-β-[FeBr(CO)(PNNP′)]-[BPh4] | 4, Cy | 71b,d | 98 (R) |
| Powered imine | 5a, Et | 0.7 | 70 (R)e |
| 5b, iPr | 1.1 | 80 (R)e | |
| 5c, Cy | 22.9 | 76 (R) | |
| 5d, Ph | 22.8 | 79 (R) | |
| 5e, xylyl | 3.2 | 90 (R)e | |
| 5f, oTol | 3.4 | 60 (R)e | |
| Powdered amine | 6a, Et | 12.3 | 90 (S) |
| 6b, iPr | 0.6 | 60 (S)e | |
| 6c, Cy | 2.4 | 80 (S)e | |
| 6d, Ph | 10.5 | 15 (R) | |
| 6e, xylyl | 3.8 | 50 (R)e | |
| Precatalyst, R | Label | Conversion (%) | ee (%), 1-phenylethanol |
|---|---|---|---|
|
a Reactions performed under argon at 28 °C. Time = 180/500 min. Conversion and ee at 180/500 min. Acetophenone:KOtBu:precatalyst ratio was 500:8:1. Volume of iPrOH = 9.0 mL. Final concentrations: [acetophenone] = 0.037 M; [KOtBu] = 5.74 × 10−4 M; [Fe] = 7.2 × 10−5 M; [iPrOH] = 13.1 M. Conversions and ee were determined as in Table 1.
|
|||
| Imine, Ph | 5d powder | 66/91 | 78/78 (R) |
| Imine, Ph | 5d | 84/97 | 80/80 (R) |
| Amine, Et | 6a | 93/97 | 95/95 (S) |
| Amine, Ph | 6d | 25/38 | 20/8 (S) |
Catalysis with 6d displayed a steady decrease in the ee of (S)-1-phenylethanol (an increase in the relative amount of (R)-1-phenylethanol), while 5d and 6a maintained a constant ee within the allotted reaction time. An intriguing result was obtained during the ATH of 3′,5′-bis(trifluoromethyl)acetophenone, wherein the ee steadily increased from 0% to +24% (Fig. 7).
 | ||
| Fig. 7 ATH of 3′,5′-bis(trifluoromethyl)acetophenone with crystals of precatalyst 6d in optimized conditions. ee, positive is (R), negative is (S). Values of ee at low conversions at early times have large errors. | ||
The closest analogue to 1a is the amine complex with the PPh2 groups, 6d, which has disappointing activity and enantioselectivity. The minor change of adding an orthophenylene to precatalyst 1a led to a dramatic decrease in the activity. The triphenylphosphine-like moiety of 5d and 6d is too sterically demanding and this leads to slow catalyst activation, ligand lability, and catalyst instability. Studies into the catalyst activation as described in the next section support these assertions.
Decreasing the bulk of one of the donors to PEt2CH2– creates a more stable catalyst. When the substrate:precatalyst ratio and temperature are increased to 6121:1 and 75 °C, 5d deactivated after 4% conversion but 6a displayed no signs of deactivation. With multiple additions of 6121 equivalents of acetophenone to a heated solution of 6a in a closed vial, a TON of 8821 could be obtained in 8.5 hours (see ESI, page S12†). In this case the 90% ee for (S)-1-phenylethanol was retained. When precatalyst 6a was heated at 85 °C with an argon flow over the solution to remove some acetone and with an initial substrate:precatalyst ratio of 54000:1 an equilibrium was reached after three days. Then another 54000:1 equivalents of acetophenone was added to the reaction mixture. This process was repeated to reach a TON of 150000 over 11 days. Due to the equilibrium process, the ee of the product alcohol degraded to zero over the course of the reaction.
Base activation and hydride studies of 5d, 6a, and 6d
A study of the activation of the precatalysts and the production of the iron hydride species serves to shed light on the catalytic cycle and the origin of the enantioselectivity. Here we examine the stepwise deprotonation of the complexes with varying equivalents of base in THF to give mixtures of amido complexes followed by the formation of hydrides by treatment with iPrOH. A summary of the NMR chemical shifts and 2JPP coupling constants of all of the precatalysts and activated complexes can be found in the ESI (see ESI, page S10).† All of the structures proposed herein are based on the data collected from crystal structures. The relative amounts of the isomers and free ligand in each reaction are estimated based on the integration of their 31P NMR signals.From previous studies of the reaction of KOtBu with 1a in THF, it is known that the precatalyst is deprotonated once at the amino group and once at the methylene group adjacent to the imino group thus producing one amido and one enamido nitrogen donor in a five-coordinate neutral iron complex.13
When isopropanol is added to this solution, it protonates the nitrogen of the amido donor; the enamido nitrogen is less basic and is not protonated. The isopropoxide then transfers a hydride to iron to form the active neutral iron hydride amine complex with the release of acetone. We designed the current catalytic system so that a weakly basic but unreducible orthophenylene amido would replace the enamido group of 1a; a free orthophenylene amine is expected to be acidic like aniline and become even more acidic upon coordination to the iron(II). Consequently, the orthophenylene amido would remain deprotonated under the basic catalytic conditions in iPrOH.
The reaction of the powder of 5d with one equivalent of potassium tert-butoxide (KOtBu) is shown in Scheme 4. The acidic aniline-like amine is deprotonated to form 14d as the major species as well as cis-beta-15d and 16d and two other very minor species. In Scheme 4, 15d is shown to form directly from 5d by deprotonation. The proposed cis-β structure of 15d is based on the large 2JPP coupling constant of 59 Hz in the 31P NMR spectrum. The absence of the bromide ligand in 15d is based on the large Δδ of the two 31P doublets, also observed for the crystallographically characterized complex 16d. As drawn, the CO and C6H4PPh2 groups have switched positions, possibly by dissociation of this bulky phosphine moiety. 14d has the NMR characteristics of a trans complex.
 | ||
| Scheme 4 Reaction of 5d with one equivalent of KOtBu. | ||
The five-coordinate imine–amido complex 16d crystallized from the crude mixture produced as in Scheme 4 (Fig. 8). It is square pyramidal with a calculated degree of trigonality τ of 0.38.35 The trigonal planar amido nitrogen N1, the imine nitrogen N2 and the rigid orthophenylene group hold Fe1, N2, N1 and P1 in a plane. The CH2CH2PPh2 arm is bent slightly out of the plane, as shown in the top of Fig. 8, with a P1–N1–N2–P2 torsion angle of −21.3°. Overlap between the filled p orbital on N1 and an empty d orbital on Fe(II) results in pi-bonding and a shortening in the nitrogen–iron bond length from 2.047(9) Å in 5d-(S)-NH to 1.819(9) Å in 16d. A bisamido iron complex formed from orthodiaminobenzene has similar Fe–N distances (1.896(2) Å, 1.915(2) Å).36 The CO and P2 donor atoms form a Y shape across from the Fe1–N1 bond with a P2–Fe1–C17 angle of 87.3(3).
 | ||
| Fig. 8 ORTEP plot of the cation of 16d. A torsion angle view of the PNNP ligand (top) and a view of the open site (bottom). Thermal ellipsoids are drawn at 50% probability level. Most aromatic ring protons and the BPh4 counteranion were omitted for clarity. Selected interatomic distances (Å), angles (deg), and torsion angles (deg): Fe1–N1 1.818(9), Fe1–N2 1.946(8), Fe1–P1 2.230(3), Fe1–P2 2.228(3), N1–C5 1.420(12), P1–C6 1.776(11), N2–C3 1.296(12), C3–C4 1.49(1), N1–C1 1.49(1), N2–C2 1.49(1); P1–Fe1–P2 104.4(1); P1–N1–N2–P2 −21.3. | ||
In order to observe hydride complexes, the powder of 5d was treated with base and then iPrOH as in ATH catalysis conditions, resulting in the formation of three products (Scheme 5). After 1 day the hydride complex 17d and the doubly deprotonated complex 18d became the major species in the mixture, and the alkoxide complex 19d disappeared. The 13C–1H HSQC spectrum revealed that the imine of 5d had been reduced since none of the products have imine resonances. Complex 17d formed slowly upon addition of iPrOH, requiring at least five minutes to obtain an appreciable amount for NMR analysis. This was likely due to the hindered approach of iPrOH to the sterically encumbered iron centre of amido complexes like 18d. The structure of 16d shows that there is likely to be an ortho-CH from phenyl groups blocking the open site of the iron.
 | ||
| Scheme 5 Products of the activation of 5d. | ||
Iron complex 17d contains the hydride and the amine proton on opposite sides of the PNNP′ ligand. This hydride species is expected to be catalytically less active in ATH than 2 (Fig. 1) which has Fe–H and N–H aligned on the same side of PNNP ligand to take advantage of the rate enhancing “N–H effect”.37,38
In order to verify the identity of complexes 17d–19d, crystals of 6d were reacted with one equivalent of KOtBu (Scheme 6). This is a simpler reaction than that of Scheme 5 which required the reduction of the imine of 5d as one of the steps. Deprotonation of the acidic orthophenylene amine led to the formation of the singly deprotonated complex 20d with a small amount of 18d. Addition of iPrOH led to the formation of the same mixture of complexes 17d–19d, as in Scheme 5, confirming that the imine of 5d was indeed reduced. Activation of 6d led to a much lower concentration of the hydride species 17d, affording a decreased activity compared to the imine precatalyst 5d. The major species of the activation step was the alkoxide complex 19d which shifted to the doubly deprotonated complex 18d slowly over time (see ESI, Fig. S17†).
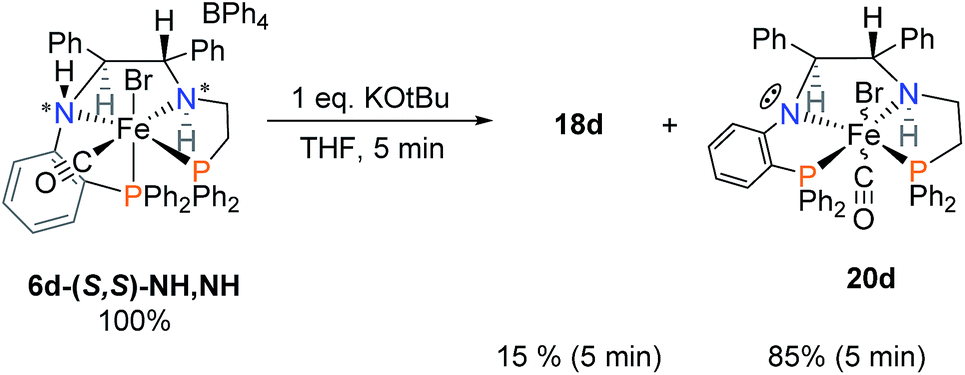 | ||
| Scheme 6 Reaction of 6d with one equivalent of KOtBu. | ||
The activation of 6a bearing the diethylphosphine group was accomplished in a similar manner. First, the species resulting from reacting 6a with one equivalent of KOtBu in THF are shown in Scheme 7. The major product 20a remained pyramidal at the orthophenylene amido nitrogen, allowing the bromide to remain fixed on the iron centre. The doubly deprotonated complex 18a was present in small amount alongside the six-coordinate iron complex with the cis-β structure cis-beta-20a which would logically result from direct deprotonation of 6a.
 | ||
| Scheme 7 Reaction of 6a-(S,S)-NH,NH with one equivalent of KOtBu. | ||
After the reaction of 6a with excess base, the addition of iPrOH immediately produced the hydride complex 21a as shown in Scheme 8. The amine proton of 21a is in the thermodynamically favoured location, below the plane of the PNNP′ ligand, whereas the carbonyl ligand now can be found above the plane. This places the Fe–H and N–H bonds in alignment to participate in the rate enhancing “N–H effect” on hydride transfer to the ketone in the outersphere. The proposed alkoxide complex 19a was formed as a minor species, disappearing after 3 days.
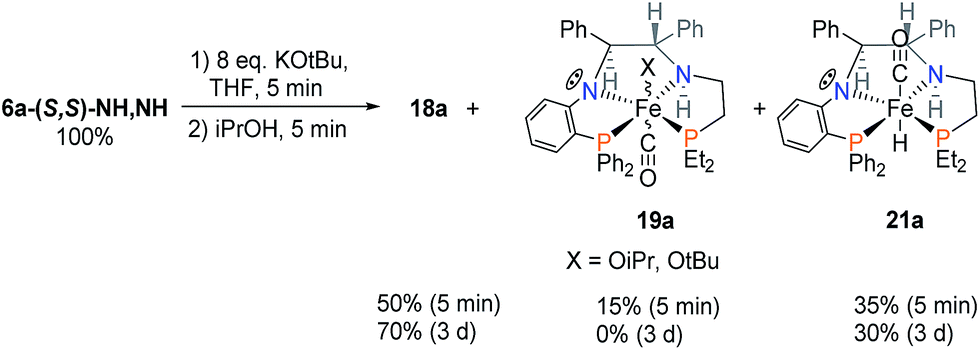 | ||
| Scheme 8 Products of the activation of 6a. | ||
Proposed catalytic cycle and explanation of activity and enantioselectivity of iron precatalysts
Key evidence such as the reversal of enantioselectivity of precatalysts 5d and 6a, the askew catalytic profile of 3′,5′-bis(trifluoromethyl)acetophenone with precatalyst 6d, and base studies was used to conclude that there is more than one catalytic cycle at work. The nature of the precatalyst determines which catalytic cycle dominates. We propose that the activated imine precatalysts form an unobserved kinetic hydride (23 in Scheme 9) that has the Fe–H and N–H aligned above the plane of the PNNP′ ligand, while activated amine precatalysts 6a–e produce the hydride species (21 in Scheme 9) that have aligned Fe–H and N–H below the plane. The reversal of the enantioselectivity is then caused by altering from which side of the PNNP′ plane transfer of the hydride to the substrate takes place. In both cases, the phenyl group of the approaching acetophenone points away from the (S,S)-CHPhCHPh backbone due to the steric bulk around the active site in 21 and 23. See the ESI† for the three-dimensional space-filling models of 1-phenylethoxide hydrogen bonded to the NH in the active site from above or below the plane of the PNNP′ ligand (see ESI, pages S66–S68†). | ||
| Scheme 9 Proposed mechanism for the reduction of acetophenone with precatalysts 5 and 6. | ||
In more detail, the reaction of 5 with one equivalent of KOtBu leads to the formation of 16 (Scheme 9) wherein the carbonyl ligand is below the plane of the PNNP ligand (as exemplified for 5d in Scheme 4). The reduction of the imine via iPrOH in TS16,22 selectively occurs above the plane of the PNNP ligand to form the unobservable complex 22 in which the lone pair rests above the plane (Scheme 9). Subsequent formation of the hydride complex 23 occurs relatively quickly given the very low concentration of precatalyst in iPrOH. In a step-wise outer-sphere pericyclic transition state TS23,22 the prochiral acetophenone is reduced to produce (R)-1-phenylethanol and reform 22. Over time, intermediate 23d rearranges to the observed hydride complex 17d, wherein the Fe–H and N–H are on opposite sides of the PNNP′ ligand and therefore exhibit the lowest catalytic activity. Although slow, 17 may also hydrogenate acetophenone via a step-wise hydride transfer to acetophenone to form a bound (R)-1-phenylethoxide iron complex, which can be protonated by iPrOH to release (R)-1-phenylethanol.
Activation of the amine precatalysts 6 is proposed to occur via deprotonation of both amines and subsequent rearrangement of a cis-β structure to form the square pyramidal complexes 18. As complex CO-Up-18 is formed in the equilibrium, iPrOH may quickly donate a proton and hydride through an outer-sphere mechanism to produce the active hydride complex 21. Prochiral acetophenone is preferentially reduced to (S)-1-phenylethanol, reforming CO-Up-18 in the process.
The activity of the imine precatalyst 5d is an interesting case as the precatalyst is available as a powder containing 95:5 (S)-NH:(R)-NH and crystals containing a 50:50 ratio. The activity of the powder is significantly lower than that of the crystals. We postulate that the minor diastereomer would rearrange more easily to place the carbonyl ligand below the plane of the PNNP′ ligand. The diastereomeric structures shown in Scheme 2 demonstrate that the (S)-NH isomer requires loss of the bromide ligand and for the CO and the triphenylphosphine-like part of the ligand to swap coordination sites. The (R)-NH isomer, upon loss of the bromide ligand, would simply need to shift the carbonyl and the triphenylphosphine moiety to produce 16d. The crystals of 16d were also tested in the ATH of acetophenone to obtain a comparable activity to the crystals of 5d (see ESI, page S11†). These precatalysts all obtain an ee of 79–80% (R), demonstrating that the precatalysts likely produce the same active hydride, but in varying quantities.
For precatalyst 5d, the more active hydride 23d initially forms then rearranges to 17d as the reaction proceeds. This effect on activity was not apparent at low substrate:precatalyst ratios and temperatures, but when the substrate:precatalyst ratio was increased to 6121:1 and the reaction temperature to 75 °C, the catalyst's activity ceased after 4% conversion. The increased temperature likely enhanced the rate of rearrangement from hydride 23d to 17d. For precatalyst 6a, complex 21a is the most stable hydride allowing for the ATH of acetophenone to produce (S)-1-phenylethanol enantioselectively with the highest activity. Since 21a is the hydride that is favored by thermodynamics when R = Et, and 6a is more stable than 5d, 6a directly produces more of an active hydride than 5d at increased substrate:precatalyst ratio and temperature. With multiple additions of substrate, a TON of 8821 was obtained in 8.5 h while retaining the ee of 90% for (S)-1-phenylethanol.
Precatalyst 6d presented an unusual result for investigation. Using the crystals of 6d, a large amount of 21d was produced, allowing for the production of (S)-1-phenylethanol in excess. The relative amount of (R)-1-phenylethanol increased as the reaction proceeded, which suggests that the active hydride was slowly converted to 23d. This was mirrored in the ATH of 3′,5′-bis(trifluoromethyl)acetophenone as the ee was initially 0%, and increased to +20% over the course of catalysis. The change in ee coincided perfectly with the rate of ATH, rapidly increasing during the first 150 minutes, then subsiding until equilibrium is reached (see Fig. 7). These tests suggest that a solution of activated 6d contains two competing hydride species.
The discrepancy in ee for 6d between Tables 1 and 2 is due to the precatalyst being available either as a powder containing a mixture of the (R,S) and the (S,S) diastereomers or crystals containing solely the (S,S) diastereomer. In basic conditions, the bromide of the (S,S) diastereomer decoordinates to allow for a facile shift of the carbonyl ligand above the plane of the PNNP. This produces an excess of CO-Up-18 and thus more of the (S)-1-phenylethanol when using crystals of 6d. Using the (R,S) diastereomer, the bromide would decoordinate and the carbonyl would shift down to fill the empty coordination site below the plane of the PNNP ligand. This produces more of CO-Down-18 which would rearrange more quickly to 22d to produce the (R)-1-phenylethanol. Therefore the powdered precatalyst 6d, containing the (R,S) diastereomer would yield a slight excess of the (R)-1-phenylethanol as shown in Table 1.
In summary, the enantioselectivity and activity of the activated catalysts depend on two factors: the equilibrium between CO-Up-18, CO-Down-18, and 22, as well as the equilibrium between 23 and 17. Initially imine precatalysts form an excess of 22, while amine precatalysts form an excess of CO-Down-18. As the reaction proceeds precatalysts 5d and 6d create an excess of 17, whereas precatalyst 6a creates an excess of 21.
Conclusion
A highly versatile synthetic protocol via a Buchwald–Hartwig amination was performed to synthesize the new air-stable, chiral PNN ligand 9 bearing an orthophenylene and a primary amine with an overall yield of 68%. A series of PNNP′ ligands were also synthesized via reductive amination, but were not isolated. Eleven new cis-β iron(II) PNNP′ precatalysts 5a–f and 6a–e were isolated as a pair of diastereomers and preliminarily tested in the ATH of acetophenone. Two factors contributed to the formation of cis-β precatalysts: the bulk of the phosphine donors as well as the flexibility of the linker connecting the amino donor to the phosphine donor. Imine precatalyst 5d crystallized as a pair of diastereomers while amine precatalyst 6a crystallized as a single diastereomer. This permitted an interesting comparison of the activity of powdered 5d containing a (S)-NH:(R)-NH ratio of 95:5 with the activity of the isolated crystals with a 50:50 ratio. The data suggested that the minor isomer may lead to a higher concentration of the active hydride. While the activity of these precatalysts were low compared to previously published iron(II) PNNP′ complexes 1a,b and 4, the enantioselectivity varied dramatically ranging from +94% with 5e to −95% with 6a. The mechanism of activation was studied with 5d and 6d, for a direct comparison of an imine and an amine precatalyst, as well as with 6a to elucidate the reversal of the ee. The studies revealed that the imine precatalyst 5d and amine precatalyst 6d form identical thermodynamic products when activated; it is the formation of different kinetic hydrides that lead to the significant difference in ee between these two precatalysts. The reversal of the enantioselectivity, from 5d to 6a, is proposed to be caused by the formation of hydrides on the opposite sides of the plane of the PNNP′ ligand (Fig. 9). In systems employing a C2-symmetric catalyst, the ee would not change regardless of which side the substrate approached from,39 but in the current case a reversal of the ee is observed due to the formation of a C1-symmetric catalyst. This proposal is supported by the structure determinations of the hydride species produced from 5d and 6a which contained hydrides on opposite sides of the PNNP′ ligand.
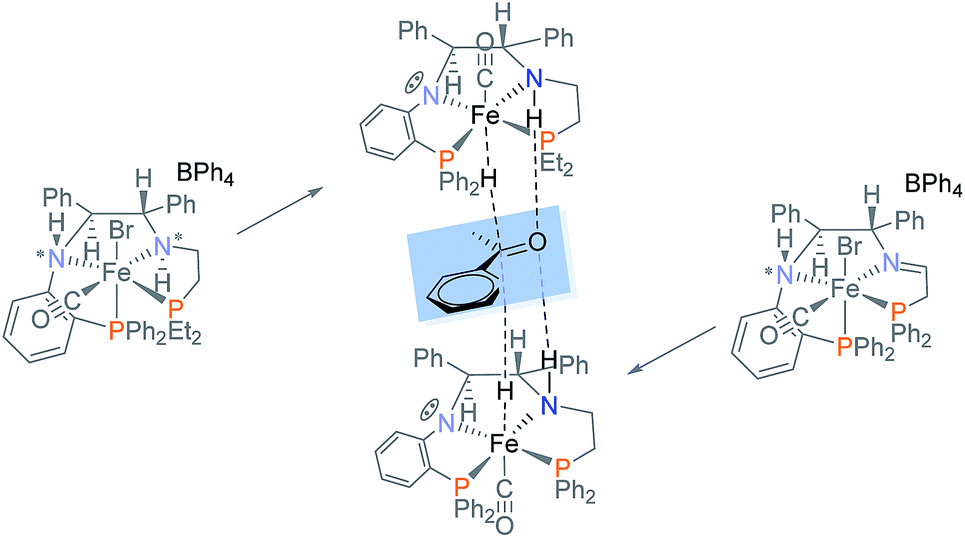 | ||
| Fig. 9 Amine complex 5d and imine complex 6a lead to hydrides that produce opposite enantiomers. | ||
When the ATH of acetophenone was heated to 75 °C and the substrate:precatalyst ratio was increased from 500:1 to 6121:1, 5d deactivated readily while 6a reacted cleanly. With multiple additions of 6121 equivalents of acetophenone to a heated solution of 6a, a TON of 8821 was obtained in 8.5 hours while retaining an ee of 90% for (S)-1-phenylethanol. The objective of this study was to increase the stability of active hydride 2 by replacing the enamido with an orthophenylene group. This was partially achieved using the complex with a PEt2 group which catalyzed the transfer hydrogenation of 150000 equivalents of acetophenone to racemic 1-phenylethanol.
The methodology described here may prove quite useful for groups studying asymmetric catalysis. Upon simple condensation with aldehydes, a plethora of new unsymmetrical, chiral tetradentate PNNX (where X = N, P, O, etc.) ligands may be synthesized. Ligand 9 may also be used as a building block to study the effect of shortening the metallocycle ring size by one atom in catalyst systems which currently use the PNN-fragment 24 and its tetradentate variants (Scheme 10).22,26,40–46
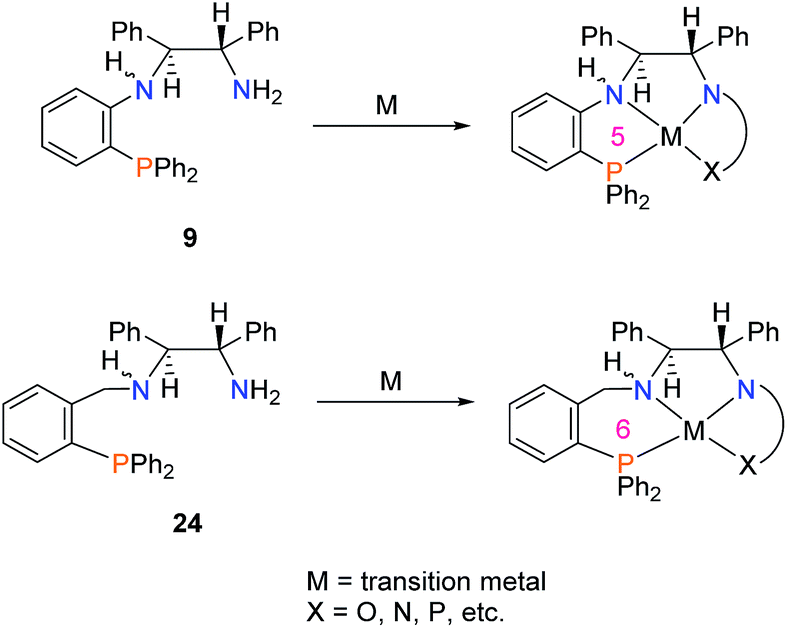 | ||
| Scheme 10 Employing 9 as a substitute for the commonly used ligand 24 to shorten the metallocycle ring size by one. | ||
Acknowledgements
Robert H. Morris thanks NSERC for a Discovery Grant and the Canada Council for the Arts for a Killam Research Fellowship. NSERC is thanked for a PGS D awarded to Karl Z. Demmans. The authors wish to acknowledge the Canadian Foundation for Innovation, project number 19119, and the Ontario Research Fund for funding of the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers. Digital Specialty Chemicals is thanked for supplying chemicals. We thank Samantha S. A. M. Smith for her assistance.Notes and references
- S. P. Hameed, M. Chinnapattu, G. Shanbag, P. Manjrekar, K. Koushik, A. Raichurkar, V. Patil, S. Jatheendranath, S. S. Rudrapatna, S. P. Barde, N. Rautela, D. Awasthy, S. Morayya, C. Narayan, S. Kavanagh, R. Saralaya, S. Bharath, P. Viswanath, K. Mukherjee, B. Bandodkar, A. Srivastava, V. Panduga, J. Reddy, K. R. Prabhakar, A. Sinha, M. B. Jimenez-Diaz, M. S. Martinez, I. Angulo-Barturen, S. Ferrer, L. M. Sanz, F. J. Gamo, S. Duffy, V. M. Avery, P. A. Magistrado, A. K. Lukens, D. F. Wirth, D. Waterson, V. Balasubramanian, P. S. Iyer, S. Narayanan, V. Hosagrahara, V. K. Sambandamurthy and S. Ramachandran, J. Med. Chem., 2014, 57, 5702–5713 CrossRef PubMed.
- C. J. Smith, A. Ali, M. L. Hammond, H. Li, Z. Lu, J. Napolitano, G. E. Taylor, C. F. Thompson, M. S. Anderson, Y. Chen, S. S. Eveland, Q. Guo, S. A. Hyland, D. P. Milot, C. P. Sparrow, S. D. Wright, A.-M. Cumiskey, M. Latham, L. B. Peterson, R. Rosa, J. V. Pivnichny, X. Tong, S. S. Xu and P. J. Sinclair, J. Med. Chem., 2011, 54, 4880–4895 CrossRef CAS PubMed.
- D. De Vita, F. Pandolfi, R. Cirilli, L. Scipione, R. Di Santo, L. Friggeri, M. Mori, D. Fiorucci, G. Maccari, R. S. Arul Christopher, C. Zamperini, V. Pau, A. De Logu, S. Tortorella and M. Botta, Eur. J. Med. Chem., 2016, 121, 169–180 CrossRef CAS PubMed.
- W. S. Knowles and R. Noyori, Acc. Chem. Res., 2007, 40, 1238–1239 CrossRef CAS PubMed.
- P. Dupau, Top. Organomet. Chem., 2012, 42, 47–64 CrossRef CAS.
- R. Noyori and T. Okhuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- A. Lee, S. M. So, A. J. Lough, H. Kim and J. Chin, Asian J. Org. Chem., 2014, 3, 1102–1107 CrossRef CAS.
- R. Bigler, R. Huber, M. Stockli and A. Mezzetti, ACS Catal., 2016, 6, 6455–6464 CrossRef CAS.
- Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed.
- K. Z. Demmans, O. W. K. Ko and R. H. Morris, RSC Adv., 2016, 6, 88580–88587 RSC.
- J. F. Sonnenberg, K. Y. Wan, P. E. Sues and R. H. Morris, ACS Catal., 2017, 7, 316–326 CrossRef CAS.
- R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed.
- W. Zuo, S. Tauer, D. E. Prokopchuk and R. H. Morris, Organometallics, 2014, 33, 5791–5801 CrossRef CAS.
- A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed.
- S. A. M. Smith and R. H. Morris, Synthesis, 2015, 47, 1775–1779 CrossRef CAS.
- W. Zuo, D. E. Prokopchuk, A. J. Lough and R. H. Morris, ACS Catal., 2016, 6, 301–314 CrossRef CAS.
- A. Dumrath, C. Luebbe and M. Beller in Palladium-catalyzed cross-coupling reactions, ed. Á. Molnár, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 445–489 Search PubMed.
- A. Dumrath, C. Luebbe, H. Neumann, R. Jackstell and M. Beller, Chem.–Eur. J., 2011, 17, 9599–9604 CrossRef CAS PubMed.
- F. Kügler, J. Ermert, P. Kaufholz and H. H. Coenen, Molecules, 2015, 20, 470–486 CrossRef PubMed.
- W.-Y. Lee and L.-C. Liang, Dalton Trans., 2005, 1952–1956 RSC.
- R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., Int. Ed., 2015, 54, 5171–5174 CrossRef CAS PubMed.
- R. Bigler, R. Huber and A. Mezzetti, Synlett, 2016, 27, 831–847 CrossRef CAS.
- R. Bigler and A. Mezzetti, Org. Lett., 2014, 16, 6460–6463 CrossRef CAS PubMed.
- R. Bigler and A. Mezzetti, Org. Process Res. Dev., 2016, 20, 253–261 CrossRef CAS.
- R. Bigler, E. Otth and A. Mezzetti, Organometallics, 2014, 33, 4086–4099 CrossRef CAS.
- S. A. M. Smith, A. J. Lough and R. H. Morris, IUCrData, 2017, 2, x170452 Search PubMed.
- L. F. Tietze, Y. Zhou and E. Topken, Eur. J. Org. Chem., 2000, 2247–2252 CrossRef CAS.
- B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017–1018 RSC.
- S. C. Bergmeier and P. P. Seth, Tetrahedron Lett., 1999, 40, 6181–6184 CrossRef CAS.
- T. Ankner and G. Hilmersson, Org. Lett., 2009, 11, 503–506 CrossRef CAS PubMed.
- P. E. Sues, A. J. Lough and R. H. Morris, Organometallics, 2011, 30, 4418–4431 CrossRef CAS.
- A. A. Mikhailine, E. Kim, C. Dingels, A. J. Lough and R. H. Morris, Inorg. Chem., 2008, 47, 6587–6589 CrossRef CAS PubMed.
- W. Zuo and R. H. Morris, Nat. Protoc., 2015, 10, 241–257 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- P. O. Lagaditis, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2011, 133, 9662–9665 CrossRef CAS PubMed.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- K. Abdur-Rashid, M. Faatz, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2001, 123, 7473–7474 CrossRef CAS PubMed.
- J.-X. Gao, H. Zhang, X.-D. Yi, P.-P. Xu, C.-L. Tang, H.-L. Wan, K.-R. Tsai and T. Ikariya, Chirality, 2000, 12, 383–388 CrossRef CAS PubMed.
- Y.-y. Li, J.-s. Chen, C.-b. Yang, Z.-r. Dong, B.-z. Li, H. Zhang, J.-x. Gao and I. Takao, Chem. Res. Chin. Univ., 2004, 20, 180–184 CAS.
- C. Sui-Seng, F. N. Haque, A. Hadzovic, A.-M. Pütz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De Iuliis and R. H. Morris, Inorg. Chem., 2009, 48, 735–743 CrossRef CAS PubMed.
- N. Meyer, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5605–5610 CrossRef CAS PubMed.
- J. F. Sonnenberg, N. Coombs, P. A. Dube and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 5893–5899 CrossRef CAS PubMed.
- J. x. Gao, P. p. Xu, X. d. Yi, C. b. Yang, H. Zhang, S. h. Cheng, H. l. Wan, K. r. Tsai and T. Ikariya, J. Mol. Catal. A: Chem., 1999, 147, 105–111 CrossRef CAS.
- Z.-b. Cheng, S.-l. Yu, Y.-y. Li, Z.-r. Dong, G.-s. Sun, K.-l. Huang and J.-x. Gao, Chem. Res. Chin. Univ., 2011, 27, 170–173 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental considerations, methods for syntheses of the PNN ligand, methods for syntheses of precatalysts 5 and 6, experimental protocols for ATH, base and hydride studies, and crystallization of 16d, summary of 31P chemical shifts, 2JPP coupling, IR CO stretch, and suggested structures, reaction profiles for the ATH of acetophenone in optimized conditions, reaction profiles for the ATH of acetophenone for higher TON reactions, NMR spectra of newly synthesized chemicals, NMR spectra of base activation studies, 3D space-filling (van der Waals) models, crystal data summary, gas chromatograph for the ATH of acetophenone and 3′,5′-bis(trifluoromethyl)acetophenone. CCDC 1551973–1551975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02558k |
| This journal is © The Royal Society of Chemistry 2017 |