
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic [2 + 2 + 2] cycloaddition with indium(III)-activated formaldimines: a practical and selective access to hexahydropyrimidines and 1,3-diamines from alkenes†
Hui
Zhou
,
Hetti Handi
Chaminda Lakmal
,
Jonathan M.
Baine
,
Henry U.
Valle
,
Xue
Xu
and
Xin
Cui
 *
*
Department of Chemistry, Mississippi State University, Mississippi State, MS 39762, USA. E-mail: xcui@chemistry.msstate.edu
First published on 24th July 2017
Abstract
Catalytic [2 + 2 + 2] cycloaddition with imines has, for the first time, been developed as a practical and selective approach for direct construction of hexahydropyrimidine derivatives from various alkenes. With formaldimines as reagents and simple InCl3 as the catalyst, this ionic [2 + 2 + 2] approach is applicable for a wide scope of alkenes and allenes with various electronic and steric properties, as well as substitution patterns. Through facile hydrolysis of the resulting hexahydropyrimidines, this catalytic process also provides a new synthetic strategy for the aminomethylamination of alkenes and allenes to practically access 1,3-diamine derivatives.
Introduction
Catalytic [2 + 2 + 2] cycloaddition reaction represents one of the most efficient approaches for constructing six-membered rings from simple and readily available building blocks.1 A number of catalytic systems have been developed to produce aromatic rings from alkynes, and partially saturated cyclic compounds from at least one triple bond system. Cycloaddition that produces saturated cyclohexanes or their heterocyclic analogues from all double bonds, especially simple alkenes, remains as the most challenging process and largely underdeveloped.1a,1b Among different combinations, a well-organized catalytic [2 + 2 + 2] cycloaddition with one molecule of alkene and two molecules of imine would enable direct assembly of hexahydropyrimidines (HHPs) (Scheme 1). As a particularly attractive class of N-hetereocycles,2 HHPs commonly present as key structural units in numerous natural products,3 drugs,4 and biologically active molecules.5 Besides rich medicinal and biological applications, HHPs have also been developed as polymer stabilizers6 and chelating ligands for functional metal complexes.7 Synthetically, hydrolysis of the aminal moiety of HHPs would readily produce 1,3-diamine derivatives, another class of molecules that are of synthetic and biological importance.8 However, this strategy has never been practically utilized because traditional syntheses of HHPs mainly rely on condensation reactions with presynthesized 1,3-diamines.2,9 Development of the aforesaid [2 + 2 + 2] system would provide a direct synthesis of HHPs from alkenes, yet synthetically constitutes a two-step aminoalkyl amination approach for preparing 1,3-diamines from alkenes, a highly applicable but challenging new process that awaits extensive development (Scheme 1).8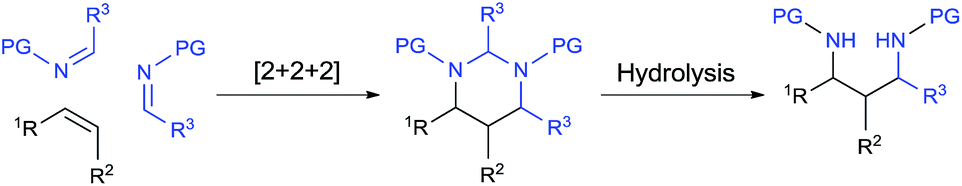 | ||
| Scheme 1 Catalytic [2 + 2 + 2] pathway for transforming alkenes to hexahydropyrimidines and 1,3-diamines. | ||
Low-valent transition metal-catalyzed [2 + 2 + 2] cycloaddition has emerged as a major tool for the synthesis of carbo- and heterocycles beyond aromatic systems.1b–f New catalytic systems involving RhI,1b,10 Ni0,1b,11 and CoI1b resulted in effective usage of alkenes as cycloaddition partners. While isocyanates have been proven to cyclize with 1,3-dienes or allenes to form dihydropyrimidine-2,4-diones (Scheme 2A),11c–e imines, which bear less electrophilic carbon centers, have not been reported to undergo cyclization with alkenes to form saturated N-heterocycles, including HHPs.
 | ||
| Scheme 2 Potential catalytic [2 + 2 + 2] pathways for synthesizing HHPs from alkenes. | ||
Alternatively, acid-catalyzed [2 + 2 + 2] cycloaddition provides an ionic approach for the synthesis of HHPs, although being rarely developed. Recently Sun and coworkers have successfully demonstrated a catalytic [2 + 2 + 2] system with triazines and allenes.12 While the AuI-catalyst was designed to employ amidoallenes and allenoates for the cycloaddition, a general reactivity toward various alkenes, as well as allenes without activating substituents, remain to be developed. The expected reactivity should require new mechanistic pathways that do not rely on specific electronic properties of the alkene substrates. Among different strategies, a proposed [2 + 2 + 2] process starting with an intermolecular aza-Prins reaction with an acid-complexed imine is particularly attractive as it would be generally applicable for various olefin compounds (Scheme 2). However, although the intramolecular aza-Prins reactions are well studied13 and widely utilized in target syntheses,14 catalytic intermolecular aza-Prins processes are highly challenging due to the insufficient nucleophilicity of the iminium species.13d,15 On the other hand, for a [2 + 2 + 2] cycloaddition toward HHPs, the iminium intermediates are expected to react with one alkene and one imine sequentially. Therefore, selectivity issues brought by alkene and imine as competitive nucleophiles, such as potential imine trimerization or oligomerization,16 and iminium-initiated styrene oligomerization, must be addressed, especially for a totally intermolecular version (Scheme 2B).
Herein we wish to report the first catalytic [2 + 2 + 2] system that enables direct construction of HHPs from alkenes and allenes with a wide range of electronic properties and substitution patterns (Scheme 2C). Environmentally benign and inexpensive InCl3 (ref. 17) was demonstrated to be a practical and selective catalyst for the three-component cyclization via an intermolecular aza-Prins reaction of N-sulfonyl formaldimines. Furthermore, the formed HHP derivatives could be readily hydrolyzed to afford various sulfonyl-protected 1,3-diamine derivatives.
Results and discussion
Initial experiments were performed to identify an effective catalyst for the proposed [2 + 2 + 2] reaction of styrene (1a) (Table 1). Compared to many other imines, formaldimines tend to feature thermodynamically and kinetically benefited reactivity as a result of their weaker imine π bond and less steric hindrance. While N-aryl and N-alkyl formaldimine equivalents have recently been explored as effective aminomethylation reagents,12,18 we envisioned N-sulfonyl-protected formaldimine would form a more electrophilic iminium with acids to facilitate an intermolecular aza-Prins reaction. Furthermore, the electron-withdrawing sulfonyl group would turn the imine less nucleophilic to suppress its oligomerization.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (%)b | Entry | Catalyst | Yield (%)b |
| a Carried out with 1a (0.1 mmol), 2a (0.3 mmol), and InCl3 (0.02 mmol) in 1.5 mL anhydrous 1,2-dichloroethane (DCE). b Isolation yields. c Not detected. d 10 mol% InCl3, 60 hours. e Reaction carried at room temperature. | |||||
| 1 | — | n.d.c | 13 | Cul | n.d. |
| 2 | CF3CO2H | n.d. | 14 | Cu(OTf)2·C6H6 | n.d. |
| 3 |

|
n.d. | 15 | Zn(OTf)2 | n.d. |
| 16 | Pd(OAc)2 | n.d. | |||
| 17 | RhCl2 | n.d. | |||
| 4 | BF3·Et2O | trace | 18 | RuCl3 | n.d. |
| 5 | B(C6F5)3 | trace | 19 | La(OTf)3 | 13 |
| 6 | MgCl2 | n.d. | 20 | BiCl3 | 49 |
| 7 | AlCl3 | n.d. | 21 | ln(OTf)3 | 37 |
| 8 | Sc(OTf)3 | 23 | 22 | lnCl 3 | 98 |
| 9 | Mn(acac)3 | n.d. | 23d | lnCl3 | 92 |
| 10 | FeCl3 | n.d. | 24e | ln(OTf)3 | <20 |
| 11 | CoCl3 | n.d. | 25e | lnCl3 | <20 |
| 12 | NiBr2 | n.d. | 26 | lnBr3 | 75 |
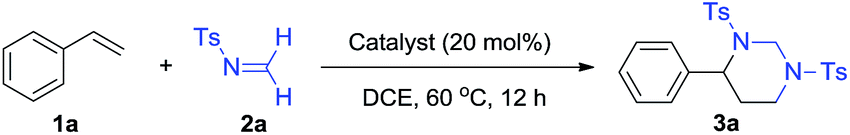
Although sulfonyl formaldimine has been shown to react with various strong nucleophiles,19 there was no desired reaction observed with styrene under catalyst-free condition at 60 °C (entry 1). Two representative Brønsted acids, trifluoroacetic acid and BINOL-derived phosphoric acid, were tested as catalysts under the same conditions and did not form any desired product (entries 2 and 3). Various Lewis acids, including main group elements, as well as both early and late transition metals with different valences, were then screened with 20 mol% loading at 60 °C (entries 4–21). Moderate to good yields of the desired [2 + 2 + 2] product were observed with several catalysts, including Sc3+, Fe3+, La3+, Bi3+ and In3+.20 While In(OTf)3 gave a poorly selective reaction with several side products observed, InCl3 was found to catalyze a clean reaction to produce the desired 4-phenyl-1,3-ditosylhexahydropyrimidine (3a) in 98% yield (entry 22). Decreasing the catalyst loading to 10 mol% could still afford 3a in 92% yield albeit with prolonged time (entry 23). Reactions at room temperature were less effective with In3+ catalysts (entries 24 and 25). Moreover, InBr3 was employed as an alternative catalyst and afforded 3a in 75% yield (entry 26).
Under the optimized conditions, the InCl3-catalyzed [2 + 2 + 2] cycloaddition was evaluated by employing various alkenes (Table 2). Using N-tosyl formaldimine 2a,21 electron-rich styrene derivatives bearing para- and meta-alkyl groups could all be cyclized to form the corresponding HHP products (3b–3d). Electron-rich styrene derivatives, such as 3b–3d, all displayed high reactivity, although slight decrease in selectivity toward HHPs was observed. Considering their increased nucleophilicity that would raise competitive side reactions, such as polymerization, slow addition of these olefins was used and indeed effectively ensured good yields. The structure of 3c was further confirmed by X-ray analysis on the single crystal. Remarkably, electron-poor 4-trifluoromethylstyrene worked well, affording HHP 3e in 89% yield at 90 °C. Halogenated styrenes with F, Cl, and Br atoms at different positions, including sterically hindered ortho-chlorostyrene and ortho-bromostyrene, all served as excellent substrates for the cycloaddition in up to 99% yield (3f–3j). In addition to expanded aromatic olefins such as 2-vinylnaphthalene (3k), a conjugated diene was shown to produce bicyclic vinyl HHP 3l, although in decreased yield. Other internal alkenes with both cis- and trans-configurations are also suitable substrates for synthesizing 2,3-disubstituted HHPs. For example, tricyclic HHP 3m was synthesized from indene in 60% yield as a single cis-diastereomer. Using cis-β-methylstyrene and trans-stilbene, 2,3-disubstuted HHPs 3n and 3o were produced respectively in satisfying yields with excellent trans-diastereoselectivity. Finally, N-protecting groups beyond tosyl were examined (2b and 2c). HHP 3p was synthesized in 69% yield with a 2-naphthalenesulfonyl group, which can be deprotected under milder conditions.22 Moreover, sterically hindered 2-mesitylenesulfonyl was also well tolerated to form HHP 3q.
| a Carried out with 1 (0.1 mmol), 2 (0.3 mmol), and InCl3 (0.02 mmol) in 1.5 mL anhydrous DCE. Isolation yields are shown. b Slow addition of 1 in 3 h followed by stirring for 9 h. c The structure was determined by X-ray crystal analysis. d 90 °C. e Starting with cis-β-methylstyrene. Diastereomeric ratio was determined by crude HNMR. f Starting with trans-stilbene. |
|---|

|
With the success on alkenes, research efforts were then extended to examine allenes as substrates for the synthesis of vinylidenehexahydropyrimidines, a class of HHP derivatives bearing alkenyl sp2 carbon on the ring (Table 3). Interestingly, terminal arylallenes exclusively cyclize with formaldimine 2a with their terminal double bonds, resulting in the formation of 5-arylidenehexahydropyrimidines (5-AHHPs, 5). Under the standard conditions, phenylallene and its derivatives with methyl groups at para-, meta-, and ortho-positions all selectively formed 5-AHHPs 5a–5d, respectively. Halogenated arylallenes with different substitution patterns were all ideal substrates for the [2 + 2 + 2] cycloaddition with 62–96% yield (5e–5g). The structure of 5e was further confirmed by X-ray analysis on the single crystal. Moreover, 1,1-disubstituted allene 4h was able to form 5-AHHP 5h bearing a tetrasubstituted olefin unit, albeit in a lower yield. More broadly, an internal allene, 1-methyl-3-phenylallene, was shown to form 5-AHHPs 5i, with exclusive regioselectivity and 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio.
:1 E/Z ratio.
 | (1) |
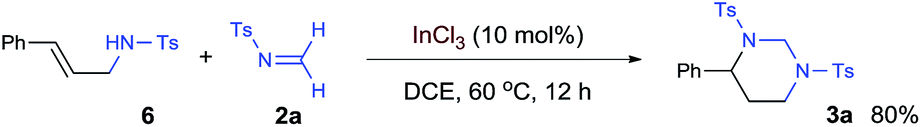 | (2) |

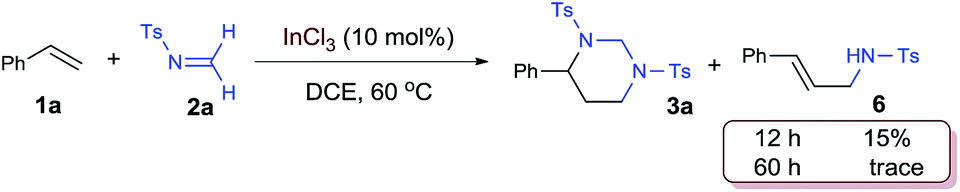
The [2 + 2 + 2] cycloaddition is believed to start with catalytic activation of formaldimine to form an In-complexed iminium species, which was attacked by the alkene and then another imine. The proposed “iminium–alkene–imine” sequence is supported by the observation of an allylamide compound 6 when the reaction was interrupted at an earlier time [eqn (1)]. The styrenyl and N-tosyl aminomethyl units in this compound clearly indicate the coupling of one styrene and one imine. While a 15% yield of 6 was observed during a reaction with 10 mol% catalyst, this compound disappeared at the end of the reaction. Furthermore, isolated compound 6 was proven to react with imine 2a and formed HHP 3a in a high yield under the standard reaction conditions [eqn (2)].
To shed more light on the proposed aza-Prins step, the diastereoselection of the [2 + 2 + 2] process with internal olefin 1n was studied as a mechanistic probe [eqn (3)]. Both cis- and trans-1n led to the formation of trans-3n with the same diastereomeric ratio of 8:1, which is consistent with a stepwise mechanism and indicates that aza-Prins step may form a relatively long-lived carbocation before it was quenched by the second formaldimine.
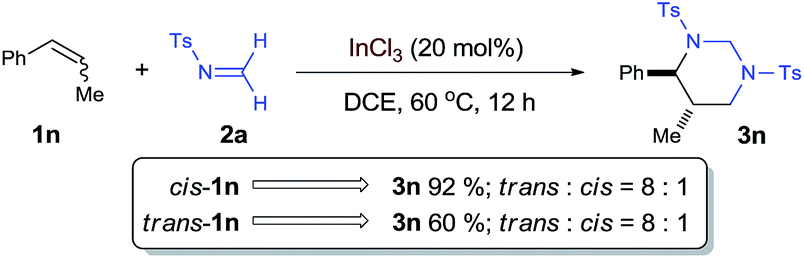 | (3) |
A plausible mechanism of this new catalytic [2 + 2 + 2] process is illustrated (Scheme 3). As a key step, highly electrophilic N-tosyl formaldiminium A initiated an intermolecular aza-Prins reaction to an alkene (1) and generate carbocation B, which selectively attacked on another imine 2a, forming formaldiminium D. Alternatively, B could experience an elimination reaction followed by protonation on the nitrogen center to form allylammonium C. The resulting allylamide 8, which supports the “iminium–alkene–imine” pathway, could reform carbocation B through the reversible reactions. Finally a ring closure of intermediate D formed the HHP and regenerated the catalyst. The regioselectivity of the cycloaddition with allenes can also be well understood. When iminium A selectively attacked the central carbon of the allene unit in 6, an allyl cation species, which is presented by the two resonance structures E and E′, was formed. Subsequent electrophilic attack on an imine 2 occurred selectively on the less hindered allyl carbon to yield iminium F, which exclusively produced 5-AHHPs (7).
 | ||
| Scheme 3 A plausible mechanism for In-catalyzed [2 + 2 + 2] cycloaddition. | ||
As a synthetic application, both HHP 3d and 5-AHHP 5e were readily hydrolyzed with catalytic amount of hydrochloric acid (Scheme 4). Tosyl-protected 1,3-diamines 7d and 7e were afforded in 88% and 92% yield, respectively. The simple removal of the aminal units in HHPs offers a practical strategy for the aminomethylamination of both alkenes and allenes.
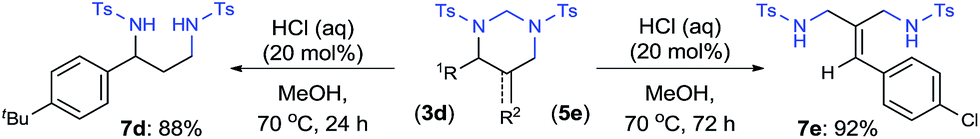 | ||
| Scheme 4 Synthesis of 1,3-diamine derivatives through Hydrolysis of HHPs and 5-AHHPs. | ||
Conclusions
In summary, catalytic [2 + 2 + 2] cycloaddition has, for the first time, been proven to cyclize both alkenes and allenes with imines, affording saturated hexahydropyrimidine derivatives. Using environmentally benign catalyst InCl3, along with N-sulfonyl formaldimines as highly reactive nitrogen source, this process features broad alkene scope, operational simplicity, and exclusive regioselectivity. Mechanistic probing experiments showed consistency with the expected “iminium–alkene–imine” addition pathway, which also result in the exclusive regioselectivity of the cycloaddition with allenes. As a result, a general and practical synthesis of hexahydropyrimidines, as well as 1,3-diamines, from various alkenes and allenes has been established. This catalytic system would encourage further development of catalytic ionic cycloaddition as effective strategy for [2 + 2 + 2] reactions that produce saturated cyclic structures. More broadly, the new catalytic pathway would stimulate further exploration of various tandem reactions that rapidly transform alkenes to highly functionalized molecules.Acknowledgements
We are grateful for financial support from the Mississippi State University Office of Research and Economic Development and Department of Chemistry.Notes and references
- (a) S. Ma, Handbook of cyclization reactions, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) G. Domínguez and J. Pérez-Castells, Chem.–Eur. J., 2016, 22, 6720 CrossRef PubMed; (c) M. Amatore and C. Aubert, Eur. J. Org. Chem., 2015, 265 CrossRef CAS; (d) G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2011, 40, 3430 RSC; (e) B. R. Galan and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2830 CrossRef CAS PubMed; (f) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307 CrossRef CAS; (g) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525 RSC.
- A. R. Katritzky, Comprehensive heterocyclic chemistry III, Elsevier, Amsterdam, New York, 1st edn, 2008 Search PubMed.
- (a) B. M. Williams and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 2191 CrossRef CAS PubMed; (b) Y. Ochi, S. Yokoshima and T. Fukuyama, Org. Lett., 2016, 18, 1494 CrossRef CAS PubMed; (c) I. Bosque, J. C. Gonzalez-Gomez, M. I. Loza and J. Brea, J. Org. Chem., 2014, 79, 3982 CrossRef CAS PubMed; (d) M. M. Cao, Y. Zhang, S. D. Huang, Y. T. Di, Z. G. Peng, J. D. Jiang, C. M. Yuan, D. Z. Chen, S. L. Li, H. P. He and X. J. Hao, J. Nat. Prod., 2015, 78, 2609 CrossRef CAS PubMed; (e) K. Drandarov, A. Guggisberg and M. Hesse, Helv. Chim. Acta, 1999, 82, 229 CrossRef CAS.
- G. Satzinger, W. Herrmann and F. Zimmermann, in Analytical Profiles of Drug Substances and Excipients, 1978, vol. 7, pp. 277–295 Search PubMed.
- (a) D. Horvath, J. Med. Chem., 1997, 40, 2412 CrossRef CAS PubMed; (b) M. S. Y. Khan and M. Gupta, Pharmazie, 2002, 57, 377 CAS.
- C. E. Ramey and C. J. Rostek, Hexahydropyrimidines as Hindered Amine Light Stabilizers, ACS Symp. Ser., 1985, 149–155 CrossRef CAS.
- M. Mayr and M. R. Buchmeiser, Macromol. Rapid Commun., 2004, 25, 231 CrossRef CAS.
- (a) X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557 RSC; (b) Y. Liu, Y. Xie, H. Wang and H. Huang, J. Am. Chem. Soc., 2016, 138, 4314 CrossRef CAS PubMed; (c) J. Hu, Y. Xie and H. Huang, Angew. Chem., Int. Ed., 2014, 53, 7272 CrossRef CAS PubMed.
- (a) N. Ahmed, S. Tarannum and Z. N. Siddiqui, RSC Adv., 2015, 5, 50691 RSC; (b) A. R. Katritzky, S. K. Singh and H. Y. He, J. Org. Chem., 2002, 67, 3115 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. Yoshida, Y. Tajima, M. Kobayashi, K. Masutomi, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2015, 54, 8241 CrossRef CAS PubMed; (b) T. J. Martin and T. Rovis, Angew. Chem., Int. Ed., 2013, 52, 5368 CrossRef CAS PubMed; (c) D. M. Dalton and T. Rovis, Org. Lett., 2013, 15, 2346 CrossRef CAS PubMed; (d) A. T. Brusoe, R. V. Edwankar and E. J. Alexanian, Org. Lett., 2012, 14, 6096 CrossRef CAS PubMed; (e) A. T. Brusoe and E. J. Alexanian, Angew. Chem., Int. Ed., 2011, 50, 6596 CrossRef CAS PubMed; (f) K. Zhang and J. Louie, J. Org. Chem., 2011, 76, 4686 CAS; (g) R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2782 CrossRef CAS PubMed.
- For selected recent examples, see: (a) P. Kumar, D. M. Troast, R. Cella and J. Louie, J. Am. Chem. Soc., 2011, 133, 7719 CrossRef CAS PubMed; (b) Z. Qiu and Z. Xie, Angew. Chem., Int. Ed., 2009, 48, 5729 CrossRef CAS PubMed; (c) M. Morimoto, Y. Nishida, T. Miura and M. Murakami, Chem. Lett., 2013, 42, 550 CrossRef CAS; (d) T. Miura, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2010, 132, 15836 CrossRef CAS PubMed; (e) H. Hoberg, D. Bärhausen, R. Mynott and G. Schroth, J. Organomet. Chem., 1991, 410, 117 CrossRef CAS.
- S. Peng, S. Cao and J. Sun, Org. Lett., 2017, 19, 524 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. Katamura, T. Shimizu, Y. Mutoh and S. Saito, Org. Lett., 2017, 19, 266 CrossRef CAS PubMed; (b) B. E. Daniels, J. Ni and S. E. Reisman, Angew. Chem., Int. Ed., 2016, 55, 3398 CrossRef CAS PubMed; (c) D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 9202 CrossRef CAS PubMed; (d) I. M. Pastor and M. Yus, Curr. Org. Chem., 2012, 16, 1277 CrossRef CAS; (e) A. P. Dobbs, S. J. J. Guesné, R. J. Parker, J. Skidmore, R. A. Stephenson and M. B. Hursthouse, Org. Biomol. Chem., 2010, 8, 1064 RSC; (f) G. Q. Liu, B. Cui, R. Xu and Y. M. Li, J. Org. Chem., 2016, 81, 5144 CrossRef CAS PubMed; (g) V. Durel, C. Lalli, T. Roisnel and P. V. D. Weghe, J. Org. Chem., 2016, 81, 849 CrossRef CAS PubMed.
- B. V. Subba Reddy, P. N. Nair, A. Antony, C. Lalli and R. Grée, Eur. J. Org. Chem., 2017, 1805 CrossRef CAS.
- (a) C. Olier, M. Kaafarani, S. Gastaldi and M. P. Bertrand, Tetrahedron, 2010, 66, 413 CrossRef CAS; (b) H. Kinoshita, O. J. Ingham, W. W. Ong, A. B. Beeler and J. A. Porco, J. Am. Chem. Soc., 2010, 132, 6412 CrossRef CAS PubMed; (c) W. W. Ong, A. B. Beeler, S. Kesavan, J. S. Panek and J. A. Porco Jr, Angew. Chem., Int. Ed., 2007, 46, 7470 CrossRef CAS PubMed.
- S. M. Weinreb, Top. Curr. Chem., 1997, 190, 131 CrossRef CAS.
- J. S. Yadav, A. Antony, J. George and B. V. Subba Reddy, Eur. J. Org. Chem., 2010, 591 CrossRef CAS.
- For selected recent examples, see: (a) Y. Zheng, Y. Chi, M. Bao, L. Qiu and X. Xu, J. Org. Chem., 2017, 82, 2129 CrossRef CAS PubMed; (b) S. Liu, P. Yang, S. Peng, C. Zhu, S. Cao, J. Li and J. Sun, Chem. Commun., 2017, 53, 1152 RSC; (c) X. Lian, L. Lin, K. Fu, B. Ma, X. Liu and X. Feng, Chem. Sci., 2017, 8, 1238 RSC; (d) C. Zhu, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 11867 CrossRef CAS PubMed; (e) S. Oda, B. Sam and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 8525 CrossRef CAS PubMed; (f) S. Oda, J. Franke and M. J. Krische, Chem. Sci., 2016, 7, 136 RSC.
- (a) Y. Numajiri, B. P. Pritchett, K. Chiyoda and B. M. Stoltz, J. Am. Chem. Soc., 2015, 137, 1040 CrossRef CAS PubMed; (b) A. Sloan Devlin and J. Du Bois, Chem. Sci., 2013, 4, 1059 RSC; (c) H. Kinoshita, K. Inomata, M. Hayashi, T. Kondoh and H. Kotake, Chem. Lett., 1986, 15, 1033 CrossRef.
- S. Kobayashi, T. Busujima and S. Nagayama, Chem.–Eur. J., 2000, 6, 3491 CAS.
- N-tosylaldimines with a phenyl or n-butyl group on the α-carbon are not effective substrates in the current catalytic system.
- B. Nyasse, L. Grehn, H. L. S. Maia, L. S. Monteiro and U. Ragnarsson, J. Org. Chem., 1999, 64, 7135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1538722 and 1554489. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02576a |
| This journal is © The Royal Society of Chemistry 2017 |