
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient stabilisation of a dihydrogenphosphate tetramer and a dihydrogenpyrophosphate dimer by a cyclic pseudopeptide containing 1,4-disubstituted 1,2,3-triazole moieties†‡
Disha
Mungalpara
a,
Arto
Valkonen
 b,
Kari
Rissanen
b and
Stefan
Kubik
*a
b,
Kari
Rissanen
b and
Stefan
Kubik
*a
aTechnische Universität Kaiserslautern, Fachbereich Chemie – Organische Chemie, Erwin-Schrödinger-Straße, 67663 Kaiserslautern, Germany. E-mail: kubik@chemie.uni-kl.de
bUniversity of Jyvaskyla, Department of Chemistry, Nanoscience Center, P.O. Box 35, Jyväskylä FI-40014, Finland
First published on 12th July 2017
Abstract
A cyclic pseudooctapeptide 2 is described containing 1,4-disubstituted 1,2,3-triazole moieties. This compound features eight converging hydrogen bond donors along the ring, namely four amide NH and four triazole CH groups, which enable 2 to engage in interactions with anions. While fully deprotonated sulfate anions exhibit only moderate affinity for 2, protonated anions such as dihydrogenpyrophosphate and dihydrogenphosphate anions are strongly bound. Complexation of the phosphate-derived anions involves sandwiching of a dihydrogenpyrophosphate dimer or a dihydrogenphosphate tetramer between two pseudopeptide rings. X-ray crystallography provided structural information, while 1H NMR spectroscopy, mass spectrometry, and isothermal titration calorimetry demonstrated that these complexes are stable in solution (2.5 vol% water/DMSO) and can even be transferred without decomposition into the gas phase. The observed high thermodynamic stabilities are attributed to the mutual reinforcement of the interactions between the individual complex components, namely, hydrogen-bonding between the anions, multiple hydrogen bonding interactions between the anion aggregates and the triazole CH and NH hydrogen bond donors of 2, and potential dispersive interactions between the closely arranged pseudopeptide rings. Pseudopeptide 2 thus represents a promising lead for the construction of phosphate receptors, whose binding selectivity makes use of the unique ability of certain anions to assemble into higher aggregates.
Introduction
Coulomb's law states that particles with a like electrical charge repel each other.1 As a consequence, two isolated cations or anions are expected to maximise their distance thus minimising the electrostatic repulsion. Repulsion can be avoided if the charges of the ions are screened by an appropriate ligand as in complexes of larger crown ethers folding around two cations,2 or complexes of certain anion receptors binding to two halides.3 A special situation arises in anion coordination chemistry4 if the anion is protonated. In this case, Coulomb repulsion of the anions can be compensated by hydrogen-bonding interactions, allowing protonated anions to form dimers or larger aggregates in which every individual component is negatively charged. While there recently has been controversy in the literature about the exact nature of the underlying interactions,5 such aggregates are generally believed to be stabilised by attractive electrostatic interactions in the hydrogen bond that balance electrostatic repulsion.6Aggregates of anions have been experimentally detected in the solid state,7–10 and in solution.11 They can be stabilised by organic ligands as the examples of complexes demonstrate containing hydrogensulfate anions,7a–f sulfate anions,7g sulfate anions bridged by water molecules,7h or dihydrogenphosphate anions.8–10 Phosphates, in particular, have been shown to assemble into a wide variety of different structures, ranging from dimers8 to larger linear9 or cyclic oligomers.10 There is ample evidence for the supramolecular stabilisation of anion aggregates in the solid state, but reports that demonstrate the survival of these complexes in solution are scarce.11 A notable exception came from the Flood group who showed that their cyanostar receptor stabilises the hydrogensulfate dimer.12 NMR spectroscopy clearly revealed the existence of the respective complex in chloroform. The authors were even able to detect the signal of the bridging protons in the anion dimer by NMR spectroscopy and they demonstrated the stability of the complex in the gas phase by mass spectrometry. In addition, a bis-calix[4]pyrrole receptor has recently been described by the Sessler group, which was shown to bind two dihydrogenphosphate anions, two sulfate anions bridged by water molecules, and two pyrophosphate anions.13
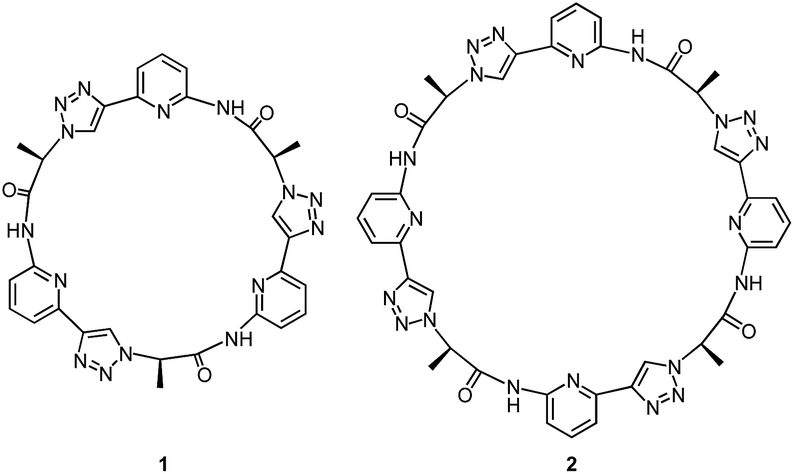
Our group recently introduced the cyclic pseudopeptide 1 containing 1,4-disubstituted 1,2,3-triazole moieties.14 This compound prefers conformations in solution with the protons on the NH groups and those on the triazole moiety converging to the centre of the macrocycle, well preorganised for anion binding. Binding of oxoanions such as sulfate and dihydrogenphosphate (DHP) was detected in 2.5 vol% water/DMSO, but the binding equilibria were rather complex, likely because of the inability of 1 to saturate all hydrogen bond acceptors of the investigated anions. In the case of sulfate, we detected the formation of complexes with two receptor molecules binding to one anion. DHP recognition, on the other hand, involved binding of two DHP anions to one receptor in solution, a complex stoichiometry consistent with the dimer of the DHP anion acting as guest. The crystal structure of the corresponding complex revealed that two molecules of 1 bind to a linear DHP trimer in the solid state, confirming the ability of 1 to stabilise DHP aggregates.
Here, we show that the higher cyclic oligomer of 1, cyclic pseudooctapeptide 2, has an even more pronounced propensity to stabilise such anion aggregates. Compound 2 was originally synthesised in the hope that it would fold around oxoanions as some of the large macrocyclic receptors introduced by Katayev and Sessler,15 thus yielding simple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes. We found, however, that 2 prefers to bind a DHP tetramer and a dimer of dihydrogenpyrophosphate (DHPP) anions by sandwiching these aggregates between two macrocyclic rings. The complexes persist in solution (2.5 vol% water/DMSO) and can also be detected in the gas phase. Considering that the DHP complex comprises six individual components this stability is remarkable, clearly illustrating the substantial stabilising effect of receptor 2. Pseudopeptide 2 thus represents a promising lead compound for the construction of phosphate receptors, whose binding selectivity is not governed by size or shape complementarity between the host and individual anions but makes use of the unique ability of certain protonated anions to assemble into higher aggregates.
:1 complexes. We found, however, that 2 prefers to bind a DHP tetramer and a dimer of dihydrogenpyrophosphate (DHPP) anions by sandwiching these aggregates between two macrocyclic rings. The complexes persist in solution (2.5 vol% water/DMSO) and can also be detected in the gas phase. Considering that the DHP complex comprises six individual components this stability is remarkable, clearly illustrating the substantial stabilising effect of receptor 2. Pseudopeptide 2 thus represents a promising lead compound for the construction of phosphate receptors, whose binding selectivity is not governed by size or shape complementarity between the host and individual anions but makes use of the unique ability of certain protonated anions to assemble into higher aggregates.
Results and discussion
Synthesis and structural characterisation
Synthesis of 2 was based on the strategy developed for 1 (Scheme 1). It started with building block 3 of which one portion was TMS deprotected and an equivalent portion converted into the azide by substitution of the mesylate group. The products thus obtained were then coupled under copper(I)-catalysis to afford dimer 4. This dimer, which was also used for the synthesis of 1,14 was now chain elongated to the corresponding linear tetramer 5 by using a related three step sequence. The terminal groups of 5 were prepared for cyclisation and the subsequent cyclisation was then performed under dilution conditions to suppress oligomerisation. After chromatographic purification, 2 was obtained in analytically pure form.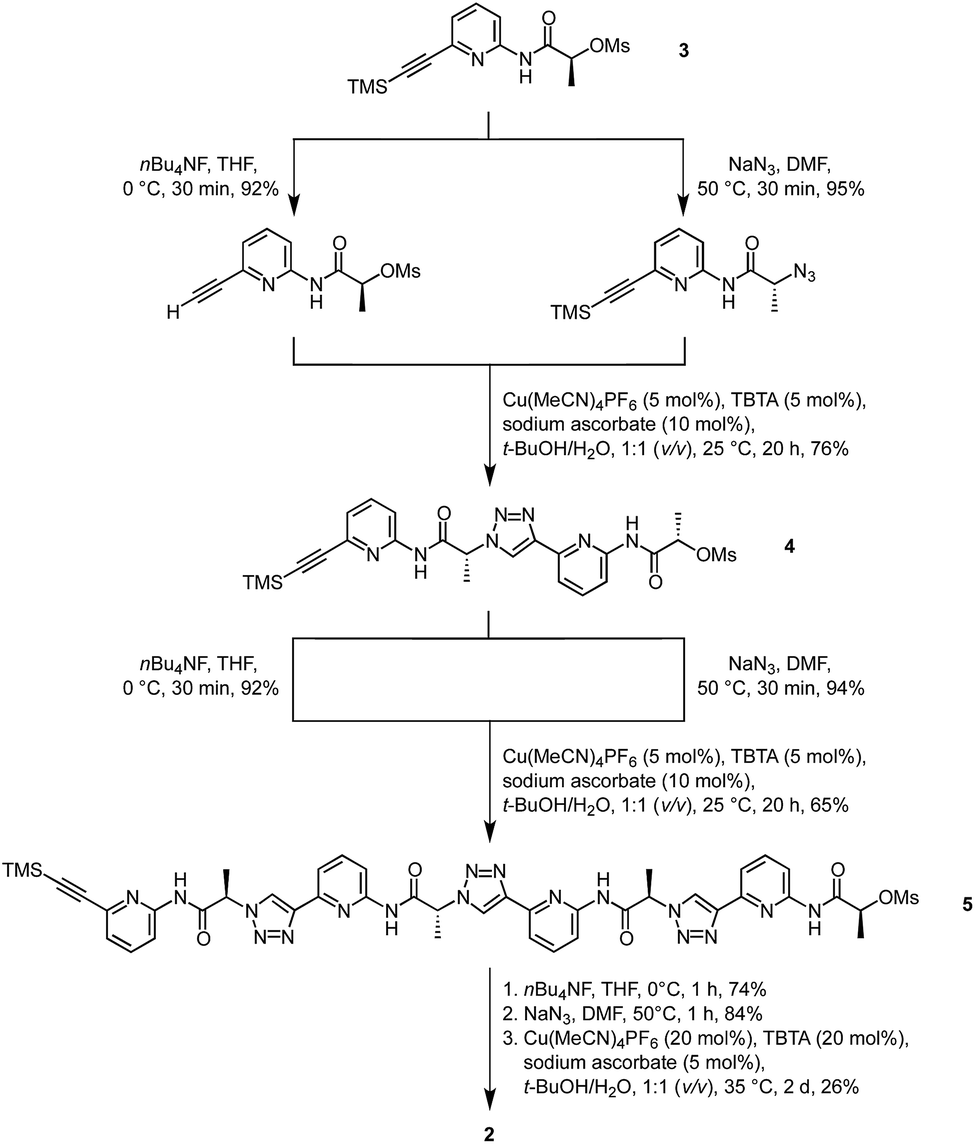 | ||
| Scheme 1 Synthesis of cyclic pseudopeptide 2. | ||
It should be noted that a side product was isolated during purification that did not exhibit the simple 1H NMR spectrum of the C4 symmetric 2 but a more complex one. This side product also represents a cyclic tetramer according to MS whose reduced symmetry is presumably caused by partial epimerisation. Epimerisation likely only occurs during or after cyclisation since no evidence for the presence of stereoisomers could be found in the 1H NMR spectra of the linear precursors.
1H NMR spectroscopy indicates that 2 adopts an averaged C4 symmetric conformation in DMSO-d6. The NOESY NMR spectrum in the same solvent features crosspeaks between the NH, triazole CH, and the C*H signals, the latter of which correspond to the protons on the stereogenic centres. These crosspeaks account for the spatial proximity of the respective sets of protons (see ESI‡). Since no crosspeak is visible between the NH signal and the signal of the aromatic C3H, the preferred conformation of 1, characterised by a converging arrangement of the NH, triazole CH, and C*H protons, seems to be retained upon ring enlargement.
Anion binding studies
Binding studies were restricted to 2.5 vol% water/DMSO as solvent and to the oxoanions sulfate, dihydrogenphosphate (DHP), and (di)hydrogenpyrophosphate [(D)HPP] as guests to allow comparison with the previous work on receptor 1, whose interactions with the same anions was studied under analogous conditions.14 Anions such as halides and nitrate were excluded because they did not exhibit detectable affinity to 1 in DMSO.The ESI mass spectrum of a solution of 2 in dichloromethane containing 1 equiv. of TBA sulfate, recorded in the negative mode, exhibits two major signals (see ESI‡). Besides the signal of the deprotonated receptor also a strong signal is visible whose m/z ratio can be assigned to the 1:1 complex 2·SO42− with one TBA cation partially balancing the two negative charges. No evidence for the presence of higher complexes could be detected. The preferential formation of a 1:1 complex is also consistent with the shape of the ITC isotherm obtained by titrating 2 with TBA sulfate in 2.5 vol% H2O/DMSO. The resulting stability constant logKa amounts to 3.1 (ΔH = +6.1 kJ mol−1, TΔS = +23.6 kJ mol−1), rendering the sulfate complex of 2 more than one order of magnitude less stable than the corresponding 1:1 complex of 1.14 Moreover, ring enlargement causes sulfate binding to go from exothermic for 1 to endothermic for 2 in 2.5 vol% water/DMSO.
The stoichiometry of the sulfate complex suggests that 2 can indeed fold around an anion as predicted, but binding seems to be not very efficient. While the exact mode of complex formation unfortunately could not be elucidated, it is very likely that it differs profoundly from the one observed for protonated phosphate-derived anions (vide infra).
 | ||
| Fig. 1 Molecular structure of 22·DHPP2 showing the 2:2 association of the pseudopeptide and two DHPP anions with the thermal ellipsoids shown at the 50% probability level (a). The TBA cations and the hydrogen atoms except those on the NH and triazole CH groups are omitted for clarity. (b) shows the same structure as a stick model from the top (left) and the side (right) to further illustrate the arrangement of the binding partners. | ||
The pseudopeptide rings in 22·DHPP2 adopt a slightly distorted conformation with a lower than the ideal C4 symmetry. Each vacant oxygen atom of the anion dimer is hydrogen-bonded to two NH groups of 2, one from the upper and one from the lower ring in the sandwich. The N⋯O distances range between 2.76 and 2.84 Å. Four opposing triazole CH bonds face the oxygen atoms of the anion dimer that are involved in the hydrogen-bonding interactions. The C⋯O distances of 3.65–3.85 Å indicate that this interaction is also weakly stabilising. The other four triazole CH bonds face the bridging oxygen atom of the anions at distances of 3.65–3.80 Å. Two TBA counterions fill the bowl-shaped cavities of the pseudopeptide rings, one on each side forming the sandwiched 2 × 2 × 2 complex (see ESI‡). The remaining two TBA cations could not be located due to the severe disorder in the crystal.
The space filling model shown in Fig. S1a‡ illustrates that the overall structure of the complex is very compact and that the DHPP dimer perfectly fills the space between the two pseudopeptide rings. Both rings are structurally nicely complementary, allowing them to closely approach each other in the sandwich as visible in the respective side view in Fig. S1b.‡ The arrangement of NH and CH hydrogen bond donors from both rings above one another causes the methyl groups in the side chains of one ring to be arranged above the aromatic rings of the other pseudopeptide (Fig. 1b), indicating that dispersive interactions between the rings could contribute to stabilise the whole aggregate.
NMR spectroscopy demonstrates that the binding mode seen in the crystal structure of the DHPP complex of 2 persists in solution. The effects of TBA DHPP on the 1H NMR spectrum of 2 in 2.5 vol% D2O/DMSO-d6 are shown in Fig. 2.
 | ||
| Fig. 2 1H NMR spectrum of 2 (0.5 mM) in 2.5 vol% D2O/DMSO-d6 in the absence (a) and the presence of 0.25 equiv. (b), 0.5 equiv. (c), 0.75 equiv. (d), 1.0 equiv. (e), and 2.0 equiv. (f) of TBA DHPP. The signals of the triazole and C*H protons are marked in green and blue, respectively. | ||
According to these spectra, the addition of TBA DHPP to a solution of 2 causes the appearance of a new set of signals. This signal set becomes progressively larger as the amount of the salt increases while the signals of the free receptor simultaneously decrease, showing that complex formation is slow on the NMR time-scale. The complex is practically fully formed after addition of 1 equiv. of the salt.
The observed downfield shift of most signals is consistent with anion complexation, while the new signal set associated with the complex furthermore illustrates that complex formation involves a symmetry reduction from C4 of the free receptor to C2 in the complex. This overall symmetry is consistent with the structure of the complex found in the crystal structure. NMR spectroscopy thus indicates that the DHPP complex of 2 possesses equivalent structures in solution and the solid state.
To obtain structural information about this complex in solution, a ROESY NMR spectrum of a 1:1 mixture of 2 and TBA DHPP in 2.5 vol% D2O/DMSO-d6 was recorded. NOE crosspeaks are visible in this spectrum between individual pairs of triazole CH and C*H signals, indicating the spatial proximity of the corresponding protons. These crosspeaks also show that two types of symmetry equivalent pairs of triazole CH and C*H protons exist in the complex, which is consistent with the crystal structure. Additional exchange peaks appear between the two triazole CH and between the two C*H signals, accounting for the dynamics of the complex in solution. These exchange signals can either be caused by the exchange of individual pseudopeptide rings or the conversion of their mutual arrangement in the complex. Unfortunately, no intermolecular crosspeaks are visible between, for example, methyl protons of 2 and aromatic protons that would confirm the sandwich-type arrangement of the two rings. Nevertheless, the ROESY NMR spectrum provides evidence for a symmetry relationship of protons of 2 that mirrors the one in the crystal structure. The ROESY NMR spectrum thus support the conclusion derived from the 1D NMR spectra.
The fact that the addition of the exact amount of TBA DHPP required for the formation of the 2:2 complex (1 equiv.) is sufficient to convert 2 into the respective complex suggests that it is thermodynamically very stable. Stability is in fact so high that 22·DHPP2 can also be transferred into the gas phase without decomposition as demonstrated by ESI mass spectrometry. In the mass spectrum recorded in the negative mode of an equimolar solution of 2 and TBA DHPP in dichloromethane only two major signals are visible at 771.89 and 1279.02 (Fig. 3). The peak with the lower m/z ratio can be assigned to the trianion [(2·DHPP)2·TBA]3− while the other corresponds to the same ion with an additional TBA cation. The isotope patterns of the two peaks confirm their respective charge states. The mutual stabilisation of the DHPP dimer and two rings of pseudopeptide 2 therefore seem to survive transfer of the complex from solution to the gas phase.
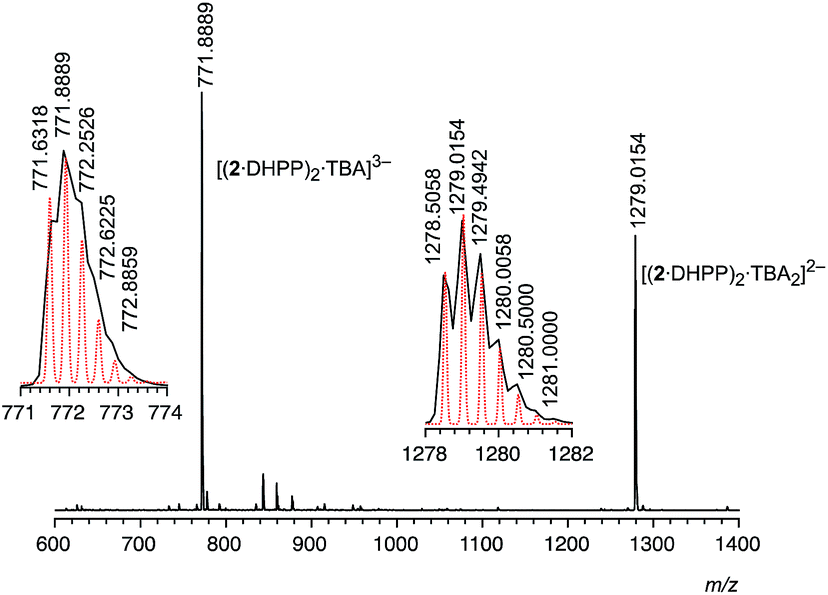 | ||
| Fig. 3 ESI mass spectrum (negative mode) of a solution of 2 (0.5 mM) in dichloromethane containing 1 equiv. of TBA DHPP. The dotted red lines in the insets show the calculated isotopic pattern of the respective peak. | ||
We also investigated the complex between 2 and DHPP by isothermal titration calorimetry (ITC). This titration showed that complex formation is strongly exothermic in 2.5 vol% water/DMSO. The obtained binding isotherm features a single sharp transition at a 1:1 molar ratio of the binding partners (Fig. 4), which is consistent with the stoichiometry derived from the crystal structure and from MS. The steep step moreover suggests cooperativity because even small amounts of the salt lead to the final 2:2 complex. The presence of other potential complex species, also absent in the NMR spectroscopic binding study (Fig. 2), is not evident.
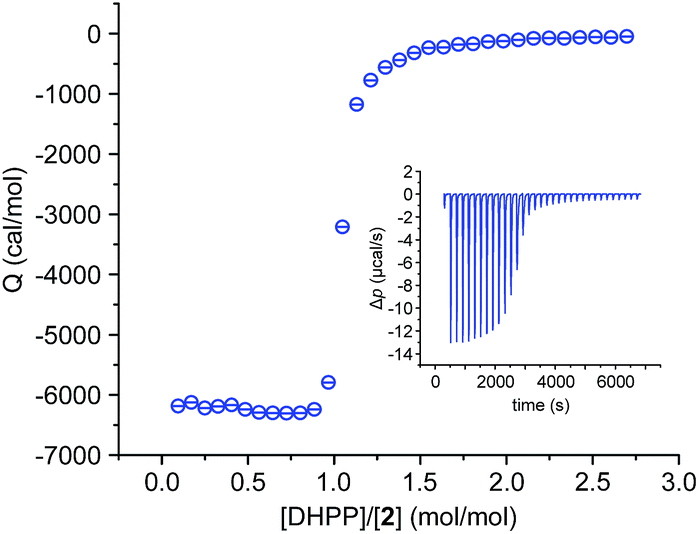 | ||
| Fig. 4 ITC binding isotherm obtained by titrating a solution of TBA DHPP (6.1 mM) into a solution of 2 (0.4 mM) in 2.5 vol% H2O/DMSO. The inset shows the heat pulses of the measurement that were used to obtain the isotherm. | ||
In spite of its higher stoichiometry, formation of the DHPP complex of 2 thus seems to proceed in a relatively simple fashion, which allowed us to fit the ITC data to a simplified binding model by neglecting the dimerisation equilibrium of the anion and assuming that the DHPP dimer binds as a single entity to two molecules of 2 in a stepwise fashion. We thus obtained two binding constants, the first one describing binding of the DHPP dimer to one pseudopeptide ring and the second one the subsequent formation of the final complex (for details, see ESI‡). With a logK11 of 6.3 and a logK21 of 6.4 both binding constants are practically equal in size, leading to a logβ of 12.7 for the overall stability of the complex. This logβ, although not representing the actual stability constant as the dimerisation equilibrium of the anion is missing, clearly shows that DHPP binding proceeds very efficiently. The fact that the second stability constant is larger than log(K11/4) is consistent with cooperative binding,17 and the thermodynamic parameters associated with the individual stability constants indicate that DHPP binding is not only enthalpically (negative ΔH) but also entropically favourable (positive TΔS).
ITC did not allow us, unfortunately, to estimate the dimerisation constant of the DHPP dimer directly because titrating a solution of TBA DHPP into 2.5 vol% H2O/DMSO produced only small exothermic heat effects. As the association constant of the DHP dimer was previously determined to amount to a logKa of 1.71 in DMSO,11g it is likely that also the DHPP dimer (or even the cyclic DHP tetramer, vide infra) is not present in 2.5 vol% H2O/DMSO to a significant extent at the concentrations used for the binding studies. Its stabilisation by 2 can therefore be attributed to the presence of the pseudopeptide rings that hold the anions together.
X-ray crystallography showed that crystals of 2 grown from acetone in the presence of TBA HPP contain analogous 2 × 2 × 2 sandwich-type arrangements of two pseudopeptide rings and DHPP dimers as in the structure 22·DHPP2 shown in Fig. 1. The crystals obtained were, however, centrosymmetric (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and thus contain both enantiomers of 2 (see Fig. S2‡). The pseudopeptide rings in 2(rac)2·DHPP2 adopt similar conformations, again with a lower than the ideal C4 symmetry, independent of whether the DHPP dimer is complexed by the all-R or the all-S-enantiomer of 2. The binding modes detected in the structure in Fig. 1 are retained in 2(rac)2·DHPP2. Specifically, the vacant oxygen atoms of the DHPP dimer hydrogen-bond to NH groups of 2 with N⋯O distances ranging between 2.74 and 2.84 Å for all-R-2 and 2.71 and 2.86 Å for all-S-2. Moreover, pairs of opposing triazole CH bonds in the pseudopeptide sandwiches are oriented towards the oxygen atoms of the anion dimer that are involved in the hydrogen-bonding interactions (C⋯O distances for all-R-2: 3.62–4.09 Å and all-S-2: 3.61–4.36 Å), while the other four triazole CH bonds face the bridging oxygen atom of the anions (C⋯O distances for all-R-2: 3.65–3.72 Å and all-S-2: 3.71–3.80 Å). The O⋯O distances in the DHPP dimers amount to 2.51–2.53 Å for the complex with all-R-2 and 2.49–2.54 Å for the one with all-S-2. Overall, 2(rac)2·DHPP2 is thus structurally closely related to the complex shown in Fig. 1 with only small differences in the distances, conformations of the pseudopeptide rings, and arrangement of the binding partners.
) and thus contain both enantiomers of 2 (see Fig. S2‡). The pseudopeptide rings in 2(rac)2·DHPP2 adopt similar conformations, again with a lower than the ideal C4 symmetry, independent of whether the DHPP dimer is complexed by the all-R or the all-S-enantiomer of 2. The binding modes detected in the structure in Fig. 1 are retained in 2(rac)2·DHPP2. Specifically, the vacant oxygen atoms of the DHPP dimer hydrogen-bond to NH groups of 2 with N⋯O distances ranging between 2.74 and 2.84 Å for all-R-2 and 2.71 and 2.86 Å for all-S-2. Moreover, pairs of opposing triazole CH bonds in the pseudopeptide sandwiches are oriented towards the oxygen atoms of the anion dimer that are involved in the hydrogen-bonding interactions (C⋯O distances for all-R-2: 3.62–4.09 Å and all-S-2: 3.61–4.36 Å), while the other four triazole CH bonds face the bridging oxygen atom of the anions (C⋯O distances for all-R-2: 3.65–3.72 Å and all-S-2: 3.71–3.80 Å). The O⋯O distances in the DHPP dimers amount to 2.51–2.53 Å for the complex with all-R-2 and 2.49–2.54 Å for the one with all-S-2. Overall, 2(rac)2·DHPP2 is thus structurally closely related to the complex shown in Fig. 1 with only small differences in the distances, conformations of the pseudopeptide rings, and arrangement of the binding partners.
We attribute the presence of both enantiomers of 2 in 2(rac)2·DHPP2 to the basic conditions arising upon protonation of the HPP anion. The fact that the crystals contain only the two homochiral forms of 2 can either be explained by assuming a dynamic shift of the racemisation equilibrium to the stereoisomers interacting best with the anion and/or by crystal packing effects. An additional low resolution racemic crystal structure (2(rac)2·DHPP2-2) was obtained from crystals grown in DMSO/DCM and using HPP TBA as guest (see ESI and Fig. S3‡).
Further evidence for the basic conditions of the crystallisation conditions came from the presence of several 4-hydroxy-4-methylpentan-2-one molecules, the aldol adduct of acetone, in the crystals grown from this solvent. NMR spectroscopy showed that formation of this aldol adduct is induced by TBA HPP alone and does not require the presence of 2 (see ESI‡). HPP thus seems to be sufficiently basic in acetone to mediate the aldol reaction and potentially also the racemisation of 2. In water at pH 7, HPP dominates in the HPP/DHPP protonation equilibrium by a factor of ca. 2,18 but this situation may differ in organic solvents and the extent of HPP protonation can be further shifted by 2. No aldol adduct forms in acetone when adding TBA DHPP (see ESI‡), explaining why 2 does not racemise in the presence of this anion.
These results thus demonstrate that 2 very efficiently interacts with DHPP in 2.5 vol% water/DMSO. The cavity between two appropriately arranged pseudopeptide rings is obviously perfectly suited to host the dimer of the anion. The respective 22·DHPP2 complex is stabilised by multiple hydrogen bonding interactions between the substrate and triazole CH and NH hydrogen bond donors of 2, which likely cause a mutual reinforcement of the anion dimer and the pseudopeptide sandwich. Additional dispersive interactions between the closely arranged pseudopeptide rings potentially further stabilise the complex. High overall stability is thus achieved, in spite of the entropic disadvantage associated with the 2:2 stoichiometry. Indications for stability are the slow complexation equilibrium on the NMR time-scale, the near complete complex formation when all components are present in the required 1:1 ratio, and the survival of the complex even upon transfer into the gas phase.
While pyrophosphate binding has also been observed for 1, the information that could be derived about the complex of the smaller pseudopeptide was less clear.14 ITC indicated that 1 binds one HPP anion with a stability constants logKa of 6.6. Higher complexes involving, for example, two pseudopeptides and one anion were also detected, however, whose composition and stabilities could not be fully characterised. In contrast, DHPP binding by 2 leads to a structurally well-defined and stable complex, demonstrating that ring enlargement significantly improves the structural complementarity between receptor and the DHPP anion in its dimeric form.
 | ||
| Fig. 5 Molecular structure of 22·DHP4 showing the 2:4 association of the pseudopeptide and four cyclically arranged DHP anions with the thermal ellipsoids shown at the 50% probability level (a). The TBA cations and the hydrogen atoms except those on the NH and triazole CH groups are omitted for clarity. (b) Shows the same structure as a stick model from the top (left) and the side (right) to further illustrate the arrangement of the binding partners. | ||
The four phosphorus atoms are arranged in the DHP tetramer of 22·DHP4 at practically equal distances with two opposing phosphorus atoms located above and two below the plane of a square, rendering the whole arrangement overall C2v symmetric (see Fig. S4‡). Each DHP anion interacts with two neighbouring ones via hydrogen-bonding interactions between three of the four oxygen atoms. One of these oxygen atoms bind to oxygen atoms of both neighbours while each of the other two oxygen atoms binds to only one of the two neighbouring DHP anions. The corresponding O⋯O distances range between 2.57 and 2.61 Å. As a result of the arrangement of the four anions, the oxygen atoms not involved in the stabilisation of the aggregate diverge and are available for the interactions with the pseudopeptide.
Because of the square-like structure of the DHP tetramer, the conformations of the two pseudopeptide rings in 22·DHP4 are less distorted than in 22·DHPP2 albeit not fully C4 symmetric. As in 22·DHPP2, each of the four diverging hydrogen bond acceptors of the DHP tetramer binds to two NH groups of 2, one from each ring of the sandwich (N⋯O distances: 2.70–2.92 Å). Each of the triazole CH protons further engages in hydrogen-bonding interactions with an oxygen atom of the DHP tetramer that is involved in stabilisation of the anion aggregate (C⋯O distances: 3.18–3.38 Å). These attractive interactions presumably contribute to the stability of the overall complex. The arrangement of the two pseudopeptide rings resembles that in 22·DHPP2 in that the side chain methyl groups of one ring are arranged close to the aromatic planes of the other one (see Fig. S5‡). The TBA cations that could be located in the structure occupy space between the 22·DHP4 complexes rather than the bowl-shaped cavities of the pseudopeptides as in 22·DHPP2. Overall, the crystal structure solution contains 4 22·DHP4 complexes, 11 TBA cations and one DMSO molecule. Due to high disorder the locations of the 5 remaining TBA cations remain unknown.
1H NMR spectroscopy and mass spectrometry demonstrated that the 2:4 stoichiometry found in the solid-state structure of 22·DHP4 is also present in solution and can furthermore be transferred to the gas phase. In Fig. 6, 1H NMR spectra of 2 in the presence of various amounts of TBA DHP in 2.5 vol% D2O/DMSO-d6 are shown.
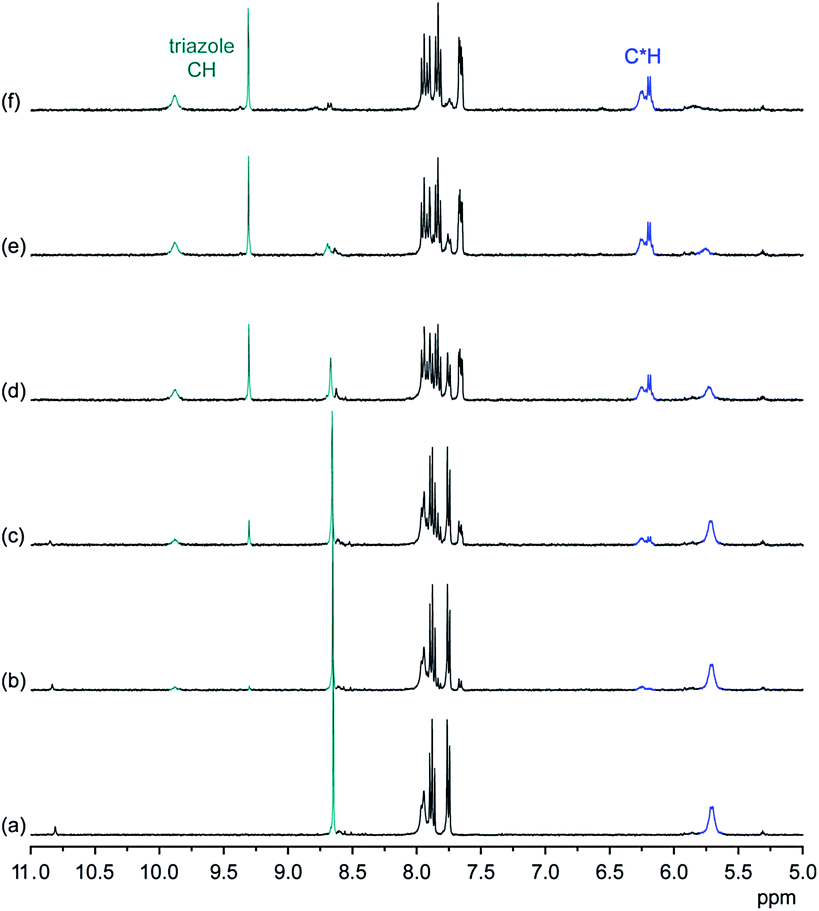 | ||
| Fig. 6 1H NMR spectrum of 2 (0.5 mM) in 2.5 vol% D2O/DMSO-d6 in the absence (a) and the presence of 0.5 equiv. (b), 1.0 equiv. (c), 2.0 equiv. (d), 3.0 equiv. (e), and 5.0 equiv. (f) of TBA DHP. The signals of the triazole and C*H protons are marked in green and blue, respectively. | ||
These spectra illustrate that formation of the DHP complex of 2 is again slow on the NMR time-scale and associated with a symmetry reduction, consistent with the C2v symmetry of the bound DHP tetramer seen in the crystal structure of 22·DHP4. In contrast to DHPP binding, where one triazole CH signal moves upfield, all four triazole CH and C*H signals are shifted downfield upon DHP binding. Moreover, the splitting of the C*H signals is smaller than in the case of the DHPP complex and one signal in the pairs of triazole CH and C*H signals is consistently broader than the corresponding other one. While all of these features account for slight structural differences between the DHPP and the DHP complexes of 2, 1H NMR spectroscopy supports the assumption that the binding modes found in the crystal structures of the respective complexes are preserved in solution. DHP binding to 2 seems to be somewhat less efficient because 5 equiv. of TBA DHP are required for the almost complete disappearance of the signals of the free receptor in the 1H NMR spectrum.
The signals observed in the ESI mass spectrum of a solution of 2 (0.5 mM) containing 2 equiv. of TBA DHP confirm the 2:4 composition of the complex and thus support the complex stoichiometry assigned crystallographically. The major signals in this spectrum at 783.89 and at 1297.00 can be assigned to a triply negatively charged ion [(2·DHP2)2·TBA]3− and to a doubly negatively charged anion [(2·DHP2)2·TBA2]2−, respectively (see ESI‡). A minor third signal is visible that corresponds to the 1:1 complex between 2 and a DHP anion. Mass spectrometry thus again provides evidence that the complex, which in this case comprises an aggregate of four negatively charged ions hold together by hydrogen-bonding and further stabilised by two pseudopeptide rings, is rather stable.
Complex formation is exothermic according to ITC and the observed binding isotherm supports formation of a complex with a higher stoichiometry (see ESI‡). Part of this isotherm could be fitted by using the model also employed for the DHPP complex and the results suggested that the DHP complex is indeed less stable than the DHPP complex. Unfortunately, the complexity of the equilibria underlying formation of the 2:4 DHP complex of 2 did not allow deriving more detailed quantitative information from this titration.
The binding studies with DHP thus show that efficient anion recognition by 2 is not restricted to DHPP but extends to DHP. Both anions are able to form aggregates with the DHPP dimer and the cyclic tetramer of DHP being structurally closely related in terms of size and arrangement of hydrogen bond acceptors. Both anion aggregates therefore perfectly fit into the space between two suitably arranged pseudopeptide rings. Individual anions are connected by hydrogen bonds, while the whole aggregates are further stabilised by interactions with hydrogen bond donors of the two pseudopeptide rings that protrude into the cavity between them. The DHP complex of 2 comprises six individual components, which should render its formation entropically unfavourable. The binding studies nevertheless indicate that thermodynamic stability is substantial. The comparison of DHP binding of 2 with that of 1, which binds two DHP anions in solution while a linear DHP trimer is bound to two pseudopeptide rings in the solid state,14 again shows that the larger pseudopeptide forms the structurally better defined complex.
Conclusions
Ring enlargement of the cyclic pseudopeptide 1, affording the corresponding cyclic pseudooctapeptide 2, turned out to have profound consequences on oxoanion binding in 2.5 vol% water/DMSO. In spite of the higher number of hydrogen bond donors along the ring, the larger receptor 2 binds fully deprotonated sulfate anions with a lower affinity than 1. Protonated anions, on the other hand, which have the intrinsic ability to overcome charge repulsion by inter-anion hydrogen bond formation, can form aggregates that are strongly bound to 2. The preferred substrates are the structurally closely related DHPP dimer and the cyclic DHP tetramer. Their incorporation into the space between two pseudopeptide rings causes the mutual reinforcement of the interactions between the individual complex components. As a consequence, the complexes persist in solution and can be transferred into the gas phase without decomposition in spite of the fact that are made up of up to six components.Interestingly, the sandwich-type binding mode observed for the DHPP and DHP complexes of 2 is somewhat reminiscent of the binding mode we previously observed for an anion-binding cyclopeptide.19 In this case, shielding of the bound anion from the surrounding solvent combined with hydrophobic interactions between the cyclopeptide rings in the sandwich complex cause anion binding to even occur in competitive aqueous media. Should similar principles govern stability of the phosphate complexes of 2, water-soluble analogues of the pseudopeptide might be able to form such complexes even under aqueous conditions. Another attractive feature of 2 is the possibility to target large anionic aggregates that can only be formed by certain anions. Anion recognition is thus associated with characteristic properties of the anions, which could render the selectivity potentially larger than if the typically relatively small differences in shape and coordination strength of individual anions are targeted. Both aspects render 2 a highly interesting lead structure for the development of phosphate selective receptors acting in water. Work in this context is currently underway.
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Johannes Lang and Sebastian Kruppa, Physical Chemistry at the Department of Chemistry in Kaiserslautern, for help with the ESI MS measurements, the Academy of Finland (KR grants no. 263256, 265328 and 292746), and the Universities of Kaiserslautern and Jyvaskyla for financial support.Notes and references
- C. A. Coulomb, Mémoires de l'Académie Royale des Sciences, 1788, pp. 569–577 Search PubMed.
- (a) M. Mercer and M. R. Truter, J. Chem. Soc., Dalton Trans., 1973, 2469–2473 RSC; (b) J. D. Owen and M. R. Truter, J. Chem. Soc., Dalton Trans., 1979, 1831–1835 RSC; (c) J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- (a) A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed. Engl., 1997, 36, 2340–2342 CrossRef CAS; (b) K. Chellappan, J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899–2903 CrossRef CAS PubMed; (c) D. Meshcheryakov, V. Böhmer, M. Bolte, V. Hubscher-Bruder, F. Arnaud-Neu, H. Herschbach, A. Van Dorsselaer, I. Thondorf and W. Mögelin, Angew. Chem., Int. Ed., 2006, 45, 1648–1652 CrossRef CAS PubMed; (d) O. B. Berryman, C. A. Johnson II, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2008, 47, 117–120 CrossRef CAS PubMed; (e) C. A. Johnson II, O. B. Berryman, A. C. Sather, L. N. Zakharov, M. M. Haley and D. W. Johnson, Cryst. Growth Des., 2009, 9, 4247–4249 CrossRef.
- (a) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC, Cambridge, 2006 Search PubMed; (b) K. Bowman-James, A. Bianchi and E. García-España, Anion Coordination Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) P. A. Gale, E. N. W. Howe and X. Wu, Chem, 2016, 1, 351–422 CrossRef CAS; (d) for an excellent review about receptors for phosphate-derived anions, see: A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- (a) F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS PubMed; (b) G. Frenking and G. F. Caramori, Angew. Chem., Int. Ed., 2015, 54, 2596–2599 CrossRef CAS PubMed; (c) F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2015, 54, 2600–2602 CrossRef CAS PubMed.
- (a) I. Mata, I. Alkorta, E. Molins and E. Espinosa, ChemPhysChem, 2012, 13, 1421–1424 CrossRef CAS PubMed; (b) I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. Phys. Lett., 2013, 555, 106–109 CrossRef CAS; (c) I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2015, 119, 183–194 CrossRef CAS PubMed.
- (a) P. Colomban, M. Pham-Thi and A. Novak, Solid State Ionics, 1987, 24, 193–203 CrossRef CAS; (b) M. Malchus and M. Jansen, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 54, 494–502 CrossRef; (c) P. H. Toma, M. P. Kelley, T. B. Borchardt, S. R. Byrn and B. Kahr, Chem. Mater., 1994, 6, 1317–1324 CrossRef CAS; (d) M. E. Light, P. A. Gale and M. B. Hursthouse, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o705–o706 CAS; (e) R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525–10529 CrossRef CAS PubMed; (f) M. N. Hoque, U. Manna and G. Das, Supramol. Chem., 2016, 28, 284–292 CrossRef CAS; (g) K. Pandurangan, J. A. Kitchen, S. Blasco, E. M. Boyle, B. Fitzpatrick, M. Feeney, P. E. Kruger and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2015, 54, 4566–4570 CrossRef CAS PubMed; (h) D. A. Jose, K. Kumar, B. Ganguly and A. Das, Inorg. Chem., 2007, 46, 5817–5819 CrossRef CAS PubMed.
- (a) D. M. Rudkevich, W. Verboom, Z. Brzozka, M. J. Palys, W. P. R. V. Stauthamer, G. J. van Hummel, S. M. Franken, S. Harkema, J. F. J. Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1994, 116, 4341–4351 CrossRef CAS; (b) P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Chem. Commun., 2007, 5214–5216 RSC; (c) J. Ju, M. Park, J.m. Suk, M. S. Lah and K.-S. Jeong, Chem. Commun., 2008, 3546–3548 RSC; (d) P. Dydio, T. Zieliński and J. Jurczak, Org. Lett., 2010, 12, 1076–1078 CrossRef CAS PubMed.
- (a) F. A. Cotton, B. A. Frenz and D. L. Hunter, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 302–304 CrossRef; (b) N. Ohama, M. Machida, T. Nakamura and Y. Kunifuji, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 962–964 CrossRef; (c) J. M. Karle and I. L. Karle, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1605–1608 CrossRef; (d) M. E. Light, S. Camiolo, P. A. Gale and M. B. Hursthouse, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o727–o729 CAS; (e) V. Amendola, M. Boiocchi, D. Esteban-Gómez, L. Fabbrizzi and E. Monzani, Org. Biomol. Chem., 2005, 3, 2632–2639 RSC; (f) B. Lou, X. Guo and Q. Lin, J. Chem. Crystallogr., 2009, 39, 469–473 CrossRef CAS; (g) A. Rajbanshi, S. Wan and R. Custelcean, Cryst. Growth Des., 2013, 13, 2233–2237 CrossRef CAS; (h) B. Wu, C. Huo, S. Li, Y. Zhao and X.-J. Yang, Z. Anorg. Allg. Chem., 2015, 641, 1786–1791 CrossRef CAS.
- (a) M. A. Hossain, M. Işıklan, A. Pramanik, M. A. Saeed and F. R. Fronczek, Cryst. Growth Des., 2012, 12, 567–571 CrossRef CAS PubMed; (b) V. Blažek, K. Molčanov, K. Mlinarić-Majerski, B. Kojić-Prodić and N. Basarić, Tetrahedron, 2013, 69, 517–526 CrossRef.
- (a) R. H. Wood and D. F. Platford, J. Solution Chem., 1975, 4, 977–982 CrossRef CAS; (b) F. Rull, A. D. Vallel, F. Sobron and S. Veintemillas, J. Raman Spectrosc., 1989, 20, 625–631 CrossRef CAS; (c) S. Valiyaveettil, J. F. J. Engbersen, W. Verboom and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1993, 32, 900–901 CrossRef; (d) E. A. Katayev, J. L. Sessler, V. N. Khrustalev and Y. A. Ustynyuk, J. Org. Chem., 2007, 72, 7244–7252 CrossRef CAS PubMed; (e) V. Blažek, N. Bregović, K. Mlinarić-Majerski and N. Basarić, Tetrahedron, 2011, 67, 3846–3857 CrossRef; (f) G. Baggi, M. Boiocchi, L. Fabbrizzi and L. Mosca, Chem.–Eur. J., 2011, 17, 9423–9439 CrossRef CAS PubMed; (g) N. Bregović, N. Cindro, L. Frkanec, K. Užarević and V. Tomišić, Chem.–Eur. J., 2014, 20, 15863–15871 CrossRef PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- Q. He, M. Kelliher, S. Bähring, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 7140–7143 CrossRef CAS PubMed.
- D. Mungalpara, H. Kelm, A. Valkonen, K. Rissanen, S. Keller and S. Kubik, Org. Biomol. Chem., 2017, 15, 102–113 CAS.
- (a) E. A. Katayev, G. D. Pantos, M. D. Reshetova, V. N. Khrustalev, V. M. Lynch, Y. A. Ustynyuk and J. L. Sessler, Angew. Chem., Int. Ed., 2005, 44, 7386–7390 CrossRef CAS PubMed; (b) J. L. Sessler, E. Katayev, G. D. Pantos, P. Scherbakov, M. D. Reshetova, V. N. Khrustalev, V. M. Lynch and Y. A. Ustynyuk, J. Am. Chem. Soc., 2005, 127, 11442–11446 CrossRef CAS PubMed; (c) E. A. Katayev, N. V. Boev, V. N. Khrustalev, Y. A. Ustynyuk, I. G. Tananaev and J. L. Sessler, J. Org. Chem., 2007, 72, 2886–2896 CrossRef CAS PubMed; (d) J. Cai, B. P. Bay, N. J. Young, X. Yang and J. L. Sessler, Chem. Sci., 2013, 4, 1560–1567 RSC.
- M. K. Deliomeroglu, V. M. Lynch and J. L. Sessler, Chem. Sci., 2016, 7, 3843–3850 RSC.
- D. D. Perrin, Pure Appl. Chem., 1969, 20, 133–236 CrossRef.
- G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097–16103 CrossRef CAS PubMed.
- S. Kubik, R. Goddard, R. Kirchner, D. Nolting and J. Seidel, Angew. Chem., Int. Ed., 2001, 40, 2648–2651 CrossRef CAS PubMed.
Footnotes |
| † In memoriam Fritz Vögtle (1939–2017). |
| ‡ Electronic supplementary information (ESI) available: Synthetic details, NMR spectroscopic and MS spectrometric characterisation of 2, NMR spectroscopic and mass spectrometric binding studies, ITC titrations, and crystal structures. CCDC 1555955–1555958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02700a |
| This journal is © The Royal Society of Chemistry 2017 |