
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient enantioselective liquid–liquid extraction of 1,2-amino-alcohols using SPINOL based phosphoric acid hosts†
Erik B.
Pinxterhuis‡
a,
Jean-Baptiste
Gualtierotti‡
a,
Hero J.
Heeres
 b,
Johannes G.
de Vries
*ac and
Ben L.
Feringa
*a
b,
Johannes G.
de Vries
*ac and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, Faculty of Mathematics and Natural Sciences, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bDepartment of Chemical Engineering, ENTEG, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands
cLeibniz Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany. E-mail: johannes.devries@catalysis.de
First published on 21st July 2017
Abstract
Access to enantiopure compounds on large scale in an environmentally friendly and cost-efficient manner remains one of the greatest challenges in chemistry. Resolution of racemates using enantioselective liquid–liquid extraction has great potential to meet that challenge. However, a relatively feeble understanding of the chemical principles and physical properties behind this technique has hampered the development of hosts possessing sufficient resolving power for their application to large scale processes. Herein we present, employing the previously untested SPINOL based phosphoric acids host family, an in depths study of the parameters affecting the efficiency of the resolution of amino-alcohols in the optic of further understanding the core principles behind ELLE. We have systematically investigated the dependencies of the enantioselection by parameters such as the choice of solvent, the temperature, as well as the pH and bring to light many previously unsuspected and highly intriguing interactions. Furthermore, utilizing these new insights to our advantage, we developed novel, highly efficient, extraction and resolving protocols which provide remarkable levels of enantioselectivity. It was shown that the extraction is catalytic in host by demonstrating transport in a U-tube and finally it was demonstrated how the solvent dependency could be exploited in an unprecedented triphasic resolution system.
Introduction
There is a large demand for enantiopure compounds, not only for the pharmaceutical and fine chemical industries,1,2 but also for the fragrance & flavour, agrochemical and food & feed industries.3 Some chiral molecules can be obtained from agriculture or fermentation, in the past referred to as the “chiral pool”, however, the number of chiral compounds obtained in this way is rather limited.3–6 Therefore the development of efficient methods to obtain chiral compounds in enantiopure form has been the centre of much attention over the past decades. This occurred via two distinctive pathways. The synthetic approach relies on the asymmetric transformation to create enantiopure compounds via such methods as the reaction of prochiral compounds to single enantiomers using chiral catalysts or the differentiation of one of the enantiomers via kinetic resolution.7 The separatory approach relies on selectively removing one enantiomer from a racemate via crystallisation or chromatography.8,9 While asymmetric synthesis has clearly shown to be incredibly powerful in answering the need for chiral molecular diversity, it needs longer development times and thus struggles to achieve the requirements defined by time to market pressure, although this problem can largely be abated by the use of high throughput experimentation.10,11 In addition, many reported catalytic procedures are too slow to be cost-effective and require extensive development to increase the turnover frequency. In contrast, resolution of racemates is still a preferred method as often the cost of the racemate is rather low and the processes are easy to develop. One example of this is the production of enantiopure BINOL via entrainment crystallisation.12 Although these crystallisation methods fit better with the time to market demands, there is only a limited number of low-cost resolving agents available. In addition, these processes often suffer from solids handling issues in the plant.13–17 Other resolution techniques such as chromatography, electrophoresis, or the use of membranes are either too slow or require high cost systems hindering large sale applications.18–34Enantioselective Liquid–Liquid Extraction (ELLE) has been well studied as an answer to these issues and many reports as to the potential efficiency, scalability and costs have been published.25,35–43 Since this method relies on the catalytic use of a chiral host and since mixing and separation of the two phases are the two major unit operations used it has a much reduced operational cost. In addition, the method can be scaled up quite easily as was shown earlier by us, for instance through the use of existing centrifugal contactor-separators.44,45 This means that it has the potential to be even more viable, both economically and environmentally, than crystallisation. However, despite all the potential advantages of ELLE, it has, to the best of our knowledge, never been applied in large-scale production due to the fact that few developed chiral hosts are sufficiently selective (αop > 3; for an explanation of how this value is obtained see below),46 to allow a reduction of the number of extractions (stages).2,43 Most of the reported host structures,43 such as crown ethers, BINOL derivatives, metal complexes (Pd, Cu, Ln, Zn, Co, Ru), tartrates, Cinchona alkaloids or guanidinium derivatives or our own BINOL based phosphoric acid system, though promising,47 function only at a proof of concept level with extraction values falling just short of applicability. Another underlying issue within these systems is that the fundamental chemical principles (kinetics, interactions, active complexes…) that drive them are poorly understood making further optimisation and developments difficult. As a direct result the developments in the field of ELLE has slowed significantly with only a few recent results appearing such as the work of Schuur48 and Tang.49 Therefore a more detailed understanding of these systems is of paramount importance for the development of new, highly selective, hosts.
With the recent advancements in the field of organocatalysis, numerous new selector candidates have emerged. Their identification and study may provide the required insight into the core principles behind ELLE to bridge the gap between concept and application and explain the dependencies of the enantioselection on different parameters. Herein, we describe the results of an in depths study, from a fundamental point of view, of the interactions and parameters determining the efficiency of the resolution of amino-alcohols by several new selector candidates. These hosts are capable of separating aminoalcohols with sufficient operational selectivity (vide infra) to reach the required level of efficiency to become industrially viable.
Results and discussion
Principles of enantioselective liquid liquid extraction
For a better understanding of the results we here give a short description of the methodology used and the meaning of the units. Enantioselective Liquid Liquid Extraction (ELLE) is based on the principles of host–guest chemistry and extraction. An ELLE system is, composed of two immiscible (or partially immiscible) liquid phases with the racemate soluble only in one of the phases and the chiral host only in the other. Enantioselective extraction occurs via chiral recognition in the form of preferred binding of one of the two enantiomers to the host in the other phase, as described by Lehn50 and Cram,51 followed by back extraction in a third phase (usually the same solvent as the feeding phase). The efficiency of this process is expressed as its operational selectivity (αop) which represents the ratio of the distributions of the enantiomers between the two phases as shown in eqn (1a). The distribution is defined as the ratio between the concentration of one enantiomer in the organic phase and the concentration of the same enantiomer in the aqueous phase (eqn (1b)). To achieve full resolution of racemates a complete selectivity is not required since, once a minimum amount of selectivity is reached, multistage extraction processes can be used to achieve complete enantioseparation.43–45 The threshold at which a process has a sufficient operational selectivity for application depends on the amount of process steps required to achieve full resolution of the racemate (Nmin) which has to remain as low as possible. The relationship between the operational selectivity and the minimal number of fractional extraction steps to achieve this is given by the Fenske equation (eqn (1c)) and is displayed graphically in Fig. 1 for ee = 99%. It is commonly accepted that an αop of 1.5, which represents 25 steps is the minimum to remain within feasibility. However, due to the exponential drop of Nmin as αop increases, processes with an αop significantly above this value are much preferred. At an αop > 3 (Nmin < 10 steps) the decrease in number of steps verses rise of αop diminishes notably with an optimum at 7 where the Nmin no longer shows a significant decrease upon a further rise of αop and therefore can be considered ideal.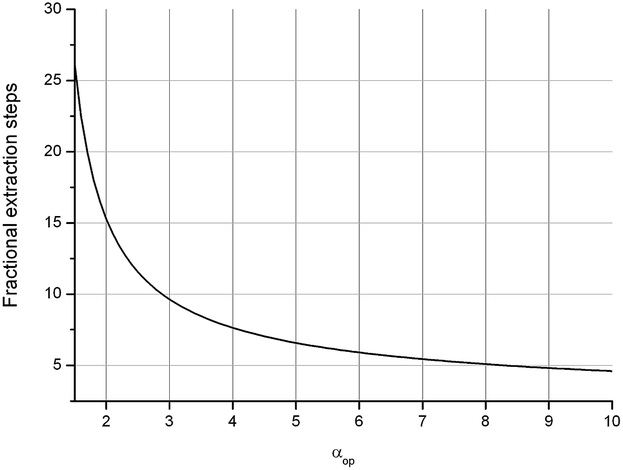 | ||
| Fig. 1 Relationship between number of stages and αop according to eqn (1c) for ee = 99%. | ||
In addition to the operational selectivity another parameter is important as to the scalability and application of the ELLE process. The ability to recover the guest from the host is a key factor which has to be measured via back-extraction experiments. This is tested on laboratory levels via the use of a U-tube extractor where a release phase is added to the system. This system, based on the developments of Cram, allows a quantative measurement of the release of the guest by the host and its applicability to large scale processes.
Host family studied
SPINOL derived ligands have been used for highly enantioselective metal-catalyzed catalytic transformations, outclassing in many cases the more traditional BINOL and H8-BINOL derived alternatives, in terms of enantio-discrimination.52,53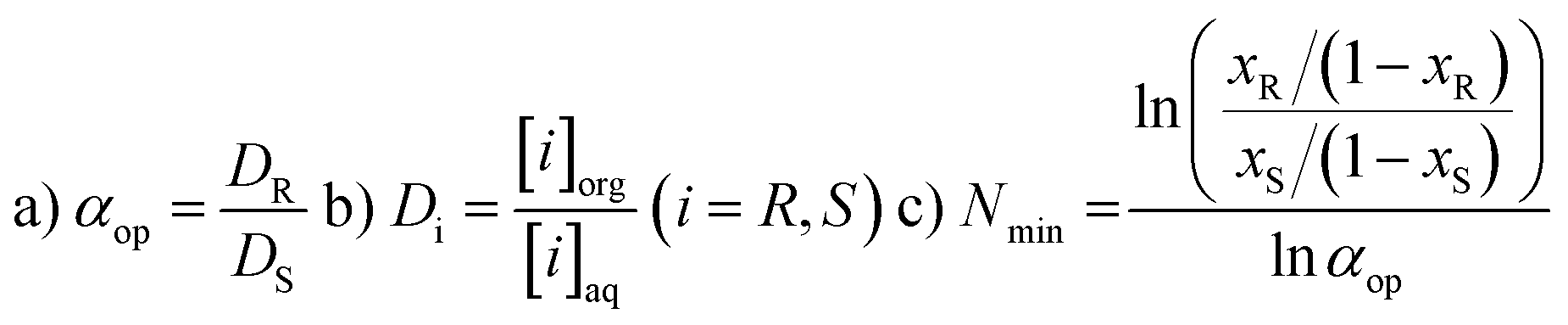 | (1) |
However, these ligands require a significantly greater synthetic effort to obtain compared to the other backbones and are therefore underused in fields outside asymmetric catalysis. Nevertheless, SPINOL-based hosts could be highly suited for ELLE as long as they are stable under the conditions of the extraction and if sufficient turnovers can be obtained. For this study we have selected four well-described SPINOL based phosphoric acids as potential hosts, (Fig. 2) which we obtained via known synthetic routes (see ESI† for more information).
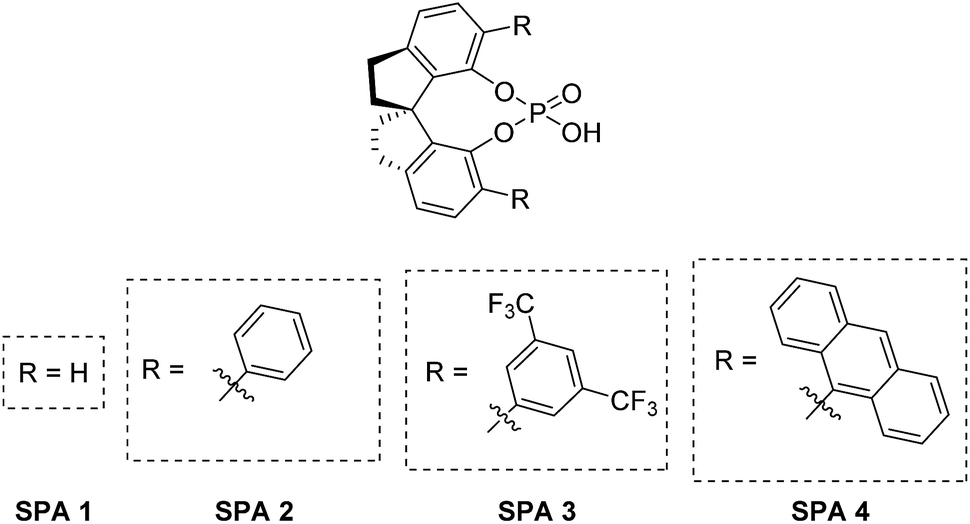 | ||
| Fig. 2 Chiral host for ELLE. | ||
Evaluation of the capacity towards the enantioselective liquid liquid extraction of various racemates by hosts
These chiral hosts were tested in the two-phase enantioselective extraction of a range of chiral compounds that are often used in enantiopure form as building blocks in organic synthesis (Fig. 3) and highly promising initial results were obtained. 6,6′-Unsubstituted SPINOL phosphoric acid (SPA 1) gave racemic extraction for all families whereas 6,6′-phenyl substituted SPA 2 showed modest enantio-discrimination for phenylglycine (7, 6% ee) and 1,2-diphenylethan-1-amine (15, 10% ee) and good to excellent selectivites in the cases of 1-amino-2,3-dihydro-1H-inden-2-ol (10, 18% ee) and phenylglycinol (11, 40% ee). 6,6′-(3,5-diCF3) Phenyl substituted SPA 3 showed a similar trend though, overall, with slightly lower selectivities, while 6,6′-(5-anthracyl) substituted SPA 4 proved selective only for phenylglycinol (11, 37%). Highly encouraged by these results we next measured the αop for the most selective guest–host pairs. We obtained in the case of trans-inden-2-ol (10) an αop of 3.8 when using SPA 2, while for phenylglycinol (11) we obtained, respectively, 5.1 and 3.6 for SPA 2, SPA 3 as well as and an exceptional αop of 34.8 for SPA 4. These results are significantly better than anything previously reported47 and open the way towards industrial application. The result obtained with SPA 4 in the resolution of 11 is particularly striking as its efficiency is, to the best of our knowledge, higher than any previously reported system including the pioneering work of Cram who obtained with his best system an αop of 31 for phenylglycine methyl ester.54 In all cases the (R)-enantiomer of the SPINOL based hosts preferentially extracts the (R)-enantiomer of the corresponding amino acid or amino alcohol.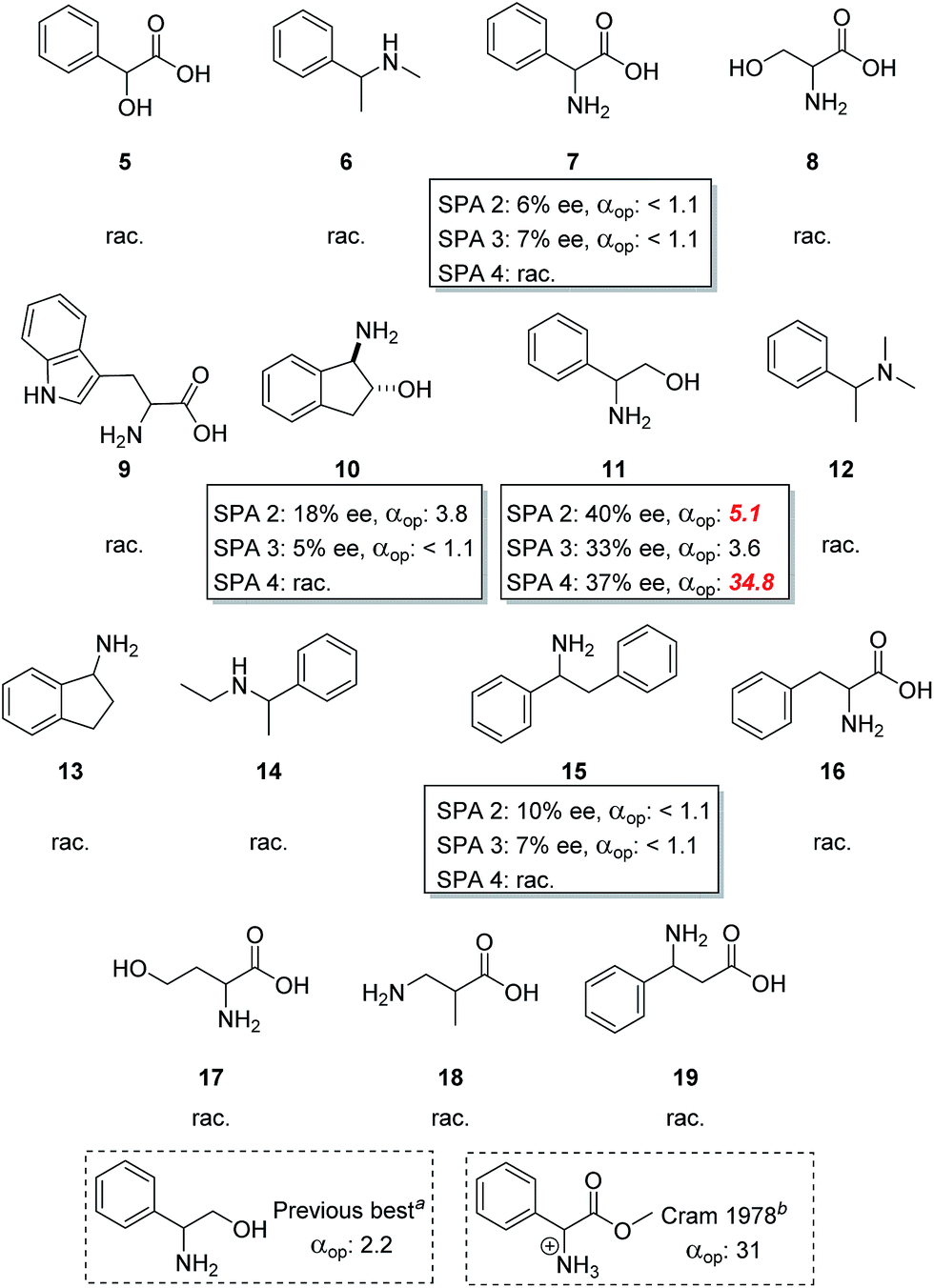 | ||
| Fig. 3 Screening of compound classes. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stirrbars. A solution of the racemic guest (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH 5.0) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was then cooled to 6 °C and stirred at 900 rpm for 16 h. The phases were then allowed to separate over a period of 2 min. The aqueous phase was then removed and an aliquot injected into a reverse phase HPLC for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). aSee ref. 43. bSee ref. 50. | ||
Study and optimisation of the ELLE of amino-alcohols by hosts
In view of these exceptional results we further examined the parameters affecting the ELLE of phenylglycinol (11) using SPA 2 and SPA 4 to gain insight into the mode of action of these hosts.The temperature of the extraction usually strongly affects the operational selectivity; prior experience has taught us that a delicate balance between solubility and selectivity exists with an optimum often at around 6 °C and that small changes can strongly impact the process. We therefore studied the variation of ELLE over a 2–40 °C range using the two best hosts (Fig. 4a). Interestingly, both revealed unexpected differences in behaviour. With SPA 2 the highest ee and αop were obtained at 2 °C (48% ee, αop = 15.3) with diminishing results at increasing temperatures although it retained much of its exceptional selectivity even at 40 °C (33% ee, αop = 3.5). At 6 °C, a maximum in the amount extracted was observed, however at lower ee's. SPA 4 showed a far greater sensitivity towards temperature. The highest ee's and αop's are obtained in a very small window around 6 °C (37% ee, αop = 34.8). Much lower enantioselection was observed at both lower and higher temperatures even though these results remain exceptionally high for ELLE (αop = 3.6–4.3). Interestingly, this exceptionally high αop is due to a reduction in the overall amounts extracted at 6 °C which results in near perfect selectivity for one enantiomer rather than any enhancement of extraction of the desired enantiomer, this therefore results in fewer steps needed to achieve high ee's and therefore an overall increase in efficiency. A similar behaviour is observed with SPA 2 at 2 °C. Initially all measurements were performed in a two to one guest to host (G/H) ratio for there to be an equivalent of each enantiomer to the catalyst. However, we were also interested in the effect the G/H ratio had on our system. To put this in perspective: in a continuous extractive separation, the host is used in catalytic amounts with respect to the total amount of guest. Nevertheless, in each separatory stage the concentrations of host and racemate can be varied to achieve the optimum result. This could even include a situation where the concentration of host is higher than that of the racemate. We thus measured the effectiveness of the ELLE process with SPA 2 and SPA 4 over a G/H range of 0.25 to 3.0 (Fig. 4b). We observe that when an excess of guest over host is used (G/H > 2) a progressive erosion of ee and alpha is observed as expected due to a decreasing proportion of guest which can be extracted which mathematically leads to lower distribution values and therefore ee's and αop. When a sub-stoichiometric amount of total guest to host is used (G/H < 1) the host extracts more guest regardless of configuration and an erosion of ee and alpha can be observed.
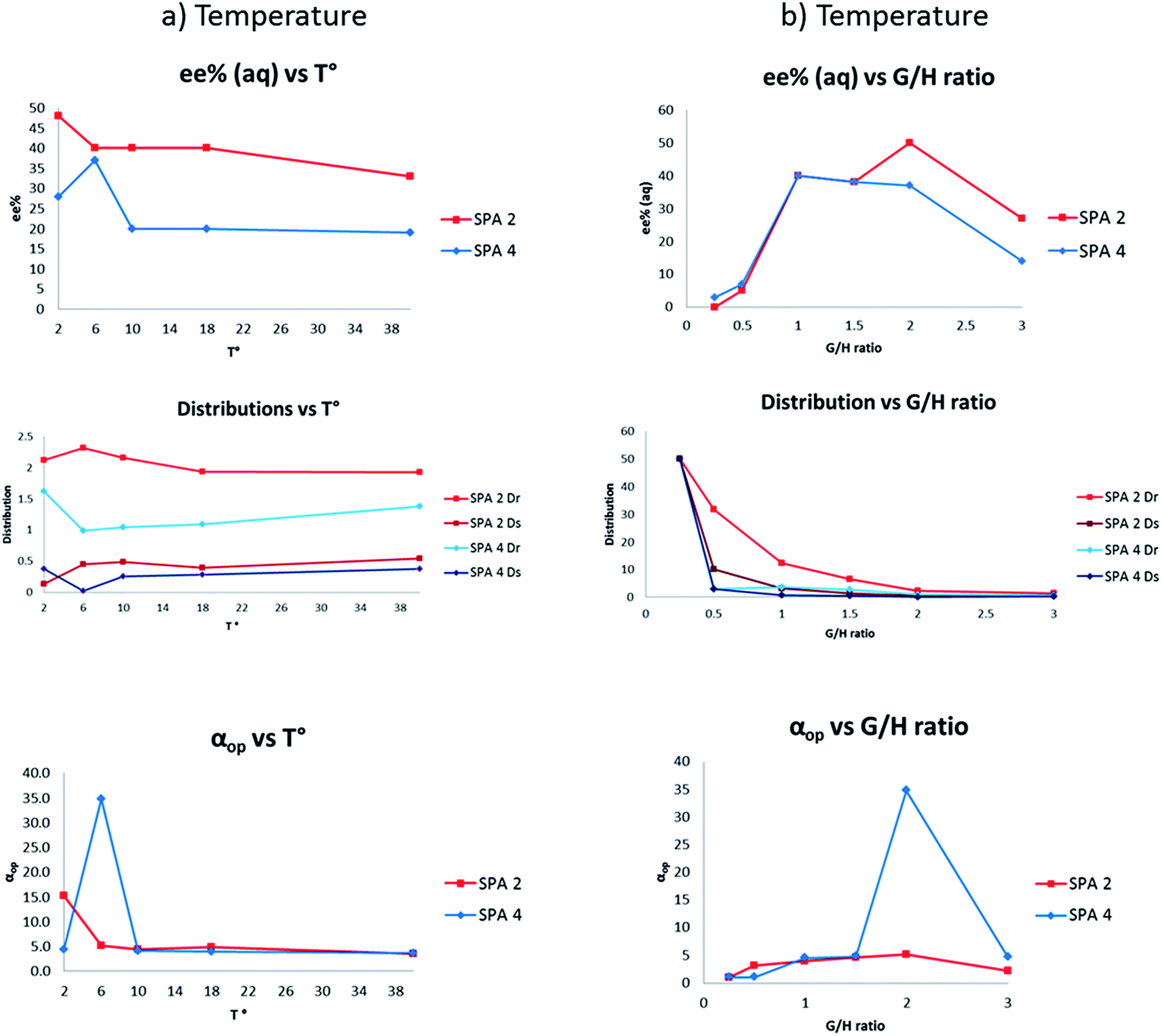 | ||
| Fig. 4 T° and G/H screenings. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stir bars. (a) A solution of racemic 11 (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH 5.0) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was immediately cooled to the indicated temperature and stirred at 900 rpm for 16 h. (b) A solution of racemic 11 (0.4 ml, indicated concentration) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH 5.0) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was then immediately cooled to 6 °C and stirred at 900 rpm for 16 h. In all cases, the phases were then allowed to separate over a period of 2 min. The aqueous phase was removed and an aliquot injected into a reverse phase HPLC equipped with chiral columns for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). For numerical values and entire pH distribution graphs see ESI.† | ||
The more interesting behaviour of these systems appears in the 1–2 G/H range (excess of total guest, substoichiometric amount of each enantiomer) were loss in selectivity is less than expected. This may indicate that the extraction is not solely determined by a competition between enantiomers for the host which, one would expect, would give a linear erosion of selectivity as observed when G/H < 1 but appears to also be determined by the two association constants between each enantiomer and host.
We next determined the effect of the pH on the enantioselectivity of the extraction. As the recognition between host and guest relies on acid–base interactions, we reasoned that pH could prove a crucial parameter for enantio-discrimination. While surprisingly little pH dependency was noted when BINOL derived phosphoric acids were used47 (estimated pKa 3.5–3.9),55 SPINOL based phosphoric acids have predicted pKa's (around 4.9)55 far closer to the pKb of phenylglycinol (around 5.5)56 which could have a positive effect as to the selectivity. We therefore studied the effect of the pH over a range of pH = 2–12 (Fig. 5). Both SPA 2 and SPA 4 showed similar behaviours in terms of ee of the extraction with two significant maxima for each, a first one at pH 5 (40 and 37% ee respectively) and a second one at respectively pH 9.6 (44% ee) and pH 8.4 (52% ee), respectively. From the measured distributions it is clear that above pH 7 the amounts extracted are too high for good selectivity, this is, however, due to some solubility of the neutral guest into the organic phase,57 though the effect of the catalyst remains visible. Below pH = 7, where the amounts extracted is determined by the catalyst, both SPA 2 and SPA 4 show interesting behaviours. SPA 2 has a maximum in extraction at pH 5 while SPA 4 is in a local minimum with the amount of undesired enantiomer extracted dropping to almost zero. In both cases these local variations result in optimal αop values despite their opposite trends, making any attempts at determining structure–activity relationships difficult.
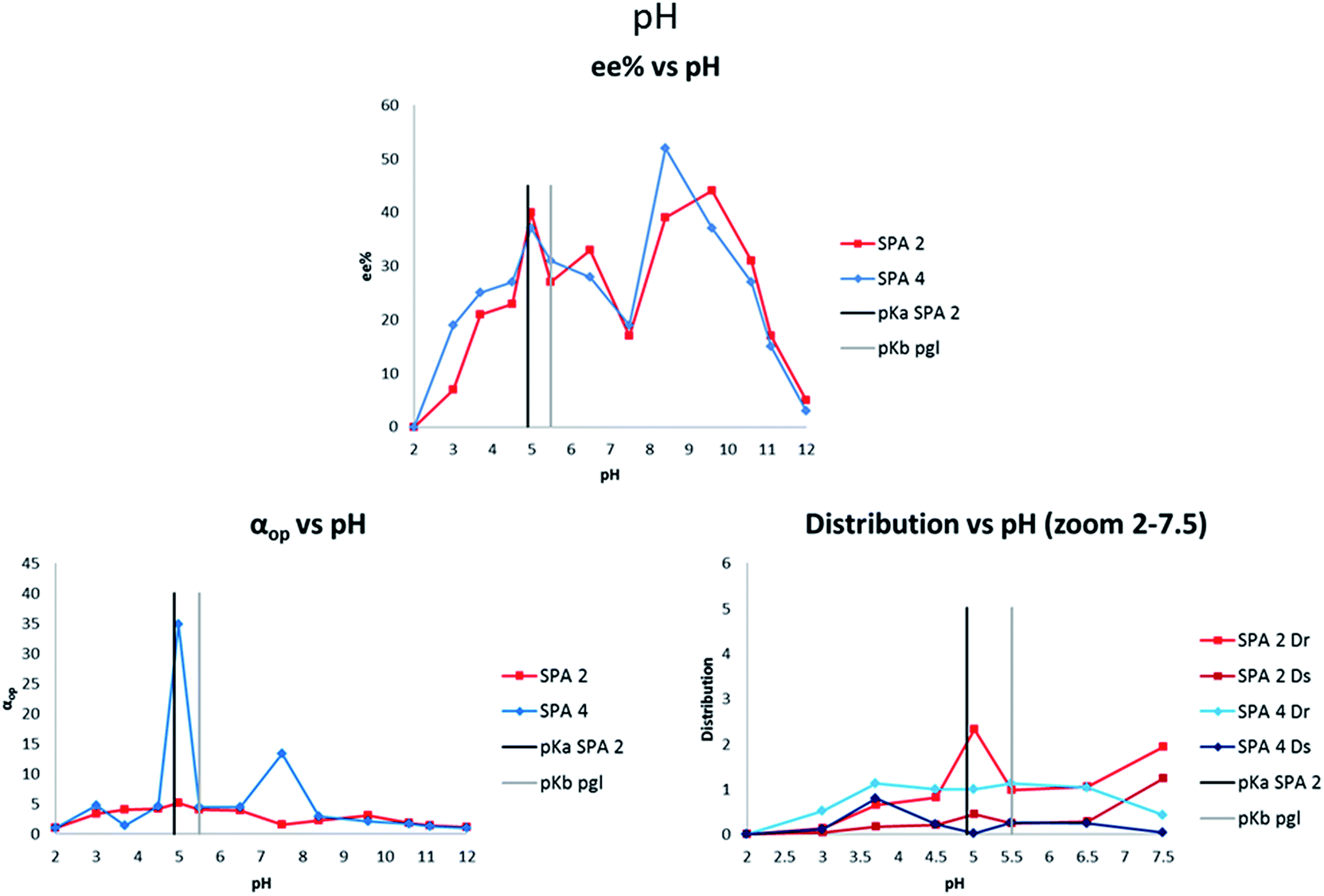 | ||
| Fig. 5 pH screenings. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stir bars. A solution of racemic 11 (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, indicated pH) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was then immediately cooled to 6 °C and stirred at 900 rpm for 16 h. In all cases, the phases were then allowed to separate over a period of 2 min. The aqueous phase was removed and an aliquot injected into a reverse phase HPLC equipped with chiral columns for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). For numerical values and entire pH distribution graphs see ESI.† | ||
One hypothesis towards explaining this high pH dependency would be that the changing ionic strength of the media affects the ion-pairing distance of the G/H complex, altering the chiral recognition between guest and host. At a pH close to both the pKa of the host and pKb of the guest, the complex might tighter ion pairs with stronger interactions which would explain the ee optimum at pH 5. As a control to check the influence of the buffer on the system, an extraction without buffer was performed and we obtained an ee of 37% and an αop of 5.6 which showed that the buffer was not key to the extraction mechanism but important for maintaining an optimal pH as it varied strongly over the course of the extraction.
Finally we turned our attention to the solvent effect on the ELLE with these hosts. Due to limitations in solvent miscibility and the requirement that the guest must only be soluble in one phase, we limited our study to various halogenated and aromatic solvents (Fig. 6). Both SPA 2 and SPA 4 show similar trends with chloroform, which was the optimal solvent in both cases (40% ee, αop = 5.1; 37% ee, αop = 34.8, respectively). When switching to DCM or tetrachloromethane, a loss of ee and lower extraction efficacy is observed. Most astonishingly, when switching from chlorinated solvents to toluene with both hosts we observed a reversal in selectivity, with near perfect retention of αop in the case of SPA 4 (−8% ee, αop = 3.8; −39% ee, αop = 35.0, respectively).
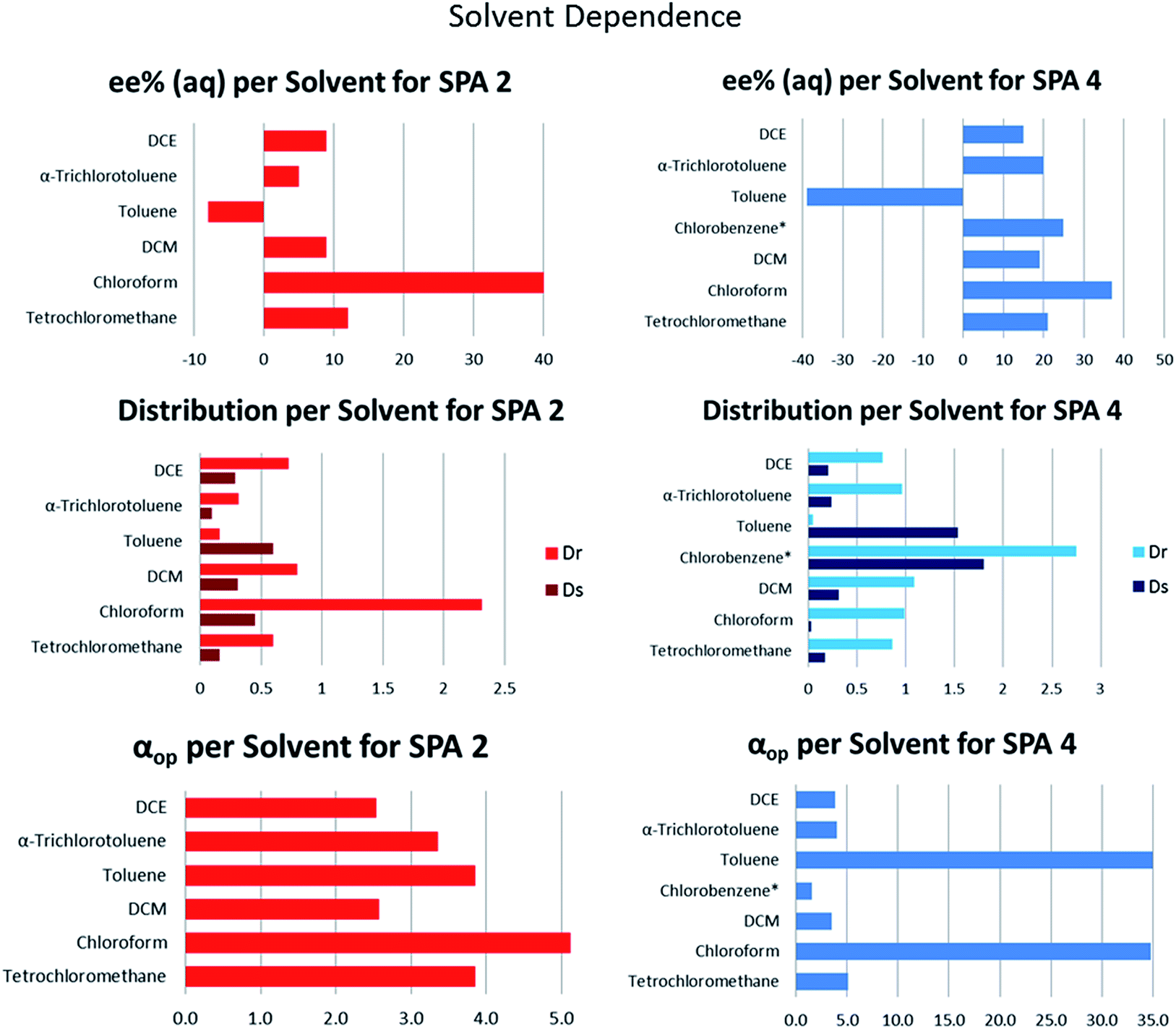 | ||
| Fig. 6 Solvent screenings. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stir bars. A solution racemic 11 (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH 5.0) was carefully added to a solution of the corresponding host in the indicated solvent (0.4 ml, 1.0 mM). The two-phasic system was then immediately cooled to 6 °C and stirred at 900 rpm for 16 h. In all cases, the phases were then allowed to separate over a period of 2 min. The aqueous phase was removed and an aliquot injected into a reverse phase HPLC equipped with chiral columns for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). For numerical values and entire pH distribution graphs see ESI.† | ||
In order to further investigate this behaviour we tested halogenated aromatic solvents such as chlorobenzene or trichlorotoluene but did not see any significant effect, with αop values remaining in the same range as with DCM. Mechanistic investigations were attempted to better understand the peculiarities observed with this system. However, attempts at DFT modelling of the system, crystal growth of the host–guest complex or its NMR characterisation proved unsuccessful. Nevertheless, observation of a basic physical model leads us to believe that a key π–π interaction exits between the guest and the host backbone. In the absence of toluene, one enantiomer can interact favourably with the backbone whereas the other cannot, determining the selectivity of the extraction. When toluene is present it can act as a π–π interaction relay allowing for the second enantiomer to also interact with the backbone. This, in addition to being the less sterically hindered of the pair results in an inversion of selectivity. Further investigations into these host–guest complexes are underway and will be reported at a later date.
It has often been observed in ELLE that the interactions between host and guest are so specific that even slight changes in guest structure significantly alter the outcome of the extraction making generalisation of a method over a family of compounds highly difficult. However, in view of the efficiency of our system we were curious as to the behaviour of these hosts towards other amino alcohols. We therefore selected a family of similar amino-alcohols and tested their resolution via ELLE using SPA 2 and SPA 4 under the optimized conditions (Fig. 7).
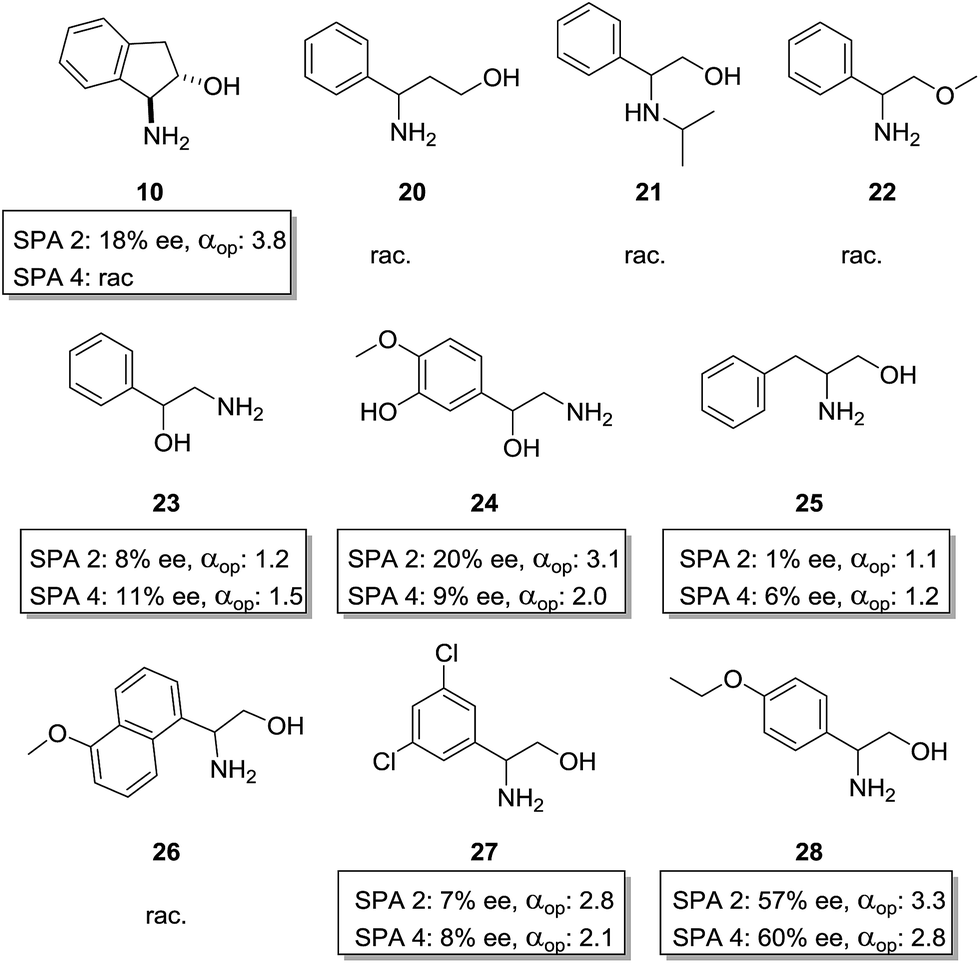 | ||
| Fig. 7 Amino-alcohol screening. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stir bars. In a standard experiment, a solution of the racemic guest (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH = 5.0) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was then immediately cooled to 6 °C and stirred at 900 rpm for 16 h. The phases were then allowed to separate over a period of 2 min. The aqueous phase was removed and an aliquot injected into a reverse phase HPLC equipped with chiral columns for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). | ||
We first studied the importance of the 1,2-amino-alcohol moiety. When cyclic trans-1-hydroxy-2-amino compound 10 was used only SPA 2 retained some selectivity (αop = 3.8), 1,3-aminoalcohol 20, N-isopropyl phenylglycinol 21 and O-methyl phenylglycinol 22 were extracted as a racemate by both hosts. 1-Amino-2-hydroxy compound 23 was extracted with moderate selectivity (SPA 4, αop = 1.5). We also tested a similar norepinephrine metabolite, normetanephrine (24), which is cheaply available in the racemic version but only available in the enantiopure form by total synthesis. With this compound we obtained an αop of above 3 with SPA 2, which is sufficient for efficient large scale resolution of this biologically relevant molecule.58 Overall this shows how important the 1,2-amino-alcohol motif is and how specific its interactions with the phosphoric acid via H-bonding are. Altering the chain length between moieties, increasing the steric bulk around these positions or removing H-bond donor or acceptor positions results in a total loss of selectivity.
We then studied the importance of the aromatic ring of the guest in terms of selectivity. When phenylalaninol (25) was used only minor selectivity could be observed (SPA 4, αop = 1.24) in its extraction, naphthyl substituted aminoalcohol 26 was too insoluble in water for ELLE whereas substituted phenylglycinol derivatives 27 and 28 could be extracted with lower but still significant selectivities (SPA 2, αop = 2.8 and SPA 2, αop = 3.3). Overall this confirmed that π-stacking was significantly involved in determining the guest–host affinity.
In view of the extraordinary sensitivity of the extraction of 11 towards the pH of the aqueous phase, we decided to further optimise the pH for the extraction of 28 (Fig. 8). This resulted in a pH dependency which had a similar profile to that of 11 with local maxima and minima arranged around the estimated pKb/pKa ranges of the host–guest system with the highest ee at pH 5 (57% and 60% ee for SPA2 and SPA 4, respectively). However, the distribution of the preferred enantiomer showed a drop in extraction at that pH whereas extraction of the other enantiomer remained at the same level unlike in the case of 11. This caused the operational selectivity to be at a minimum at pH 5. The optimum pH for the extraction of 28 was 3.5 with αop's nearly doubling, reaching 6.4 for SPA 2 and 5.3 for SPA 4. While this shows how hard it is to predict what the optimal extraction pH will be for each individual compound as no trend can be observed, it also demonstrates that these hosts can be efficiently applied to a number of amino alcohols.
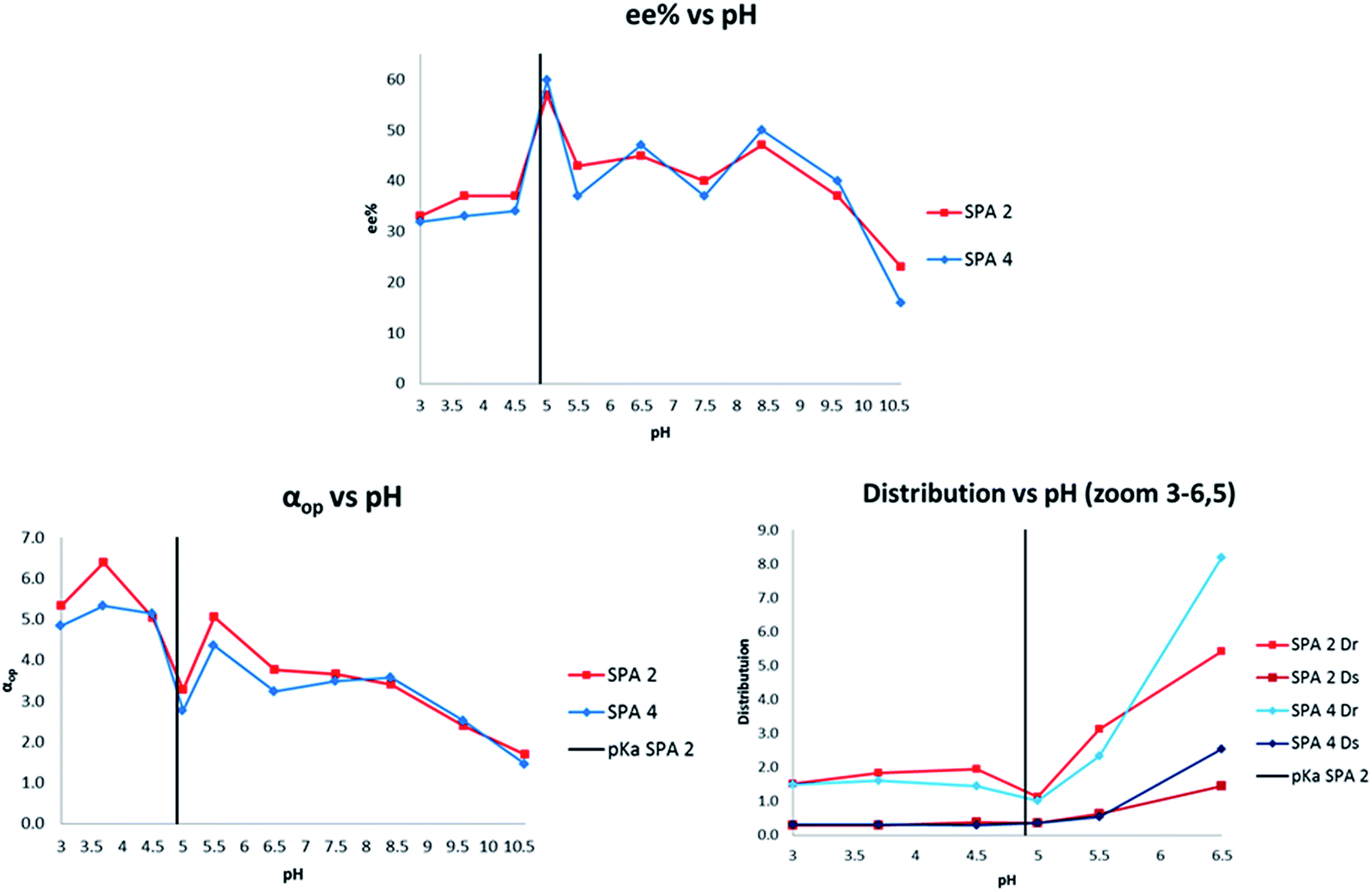 | ||
| Fig. 8 pH dependency of 28. Conditions: all extraction experiments were conducted in 2 ml auto-sampler vials with crimp cap seals equipped with stir bars. In a standard experiment, a solution of racemic 28 (0.4 ml, 2 mM) dissolved in a phosphate buffer solution (buffer strength 0.1 M, pH = 5.0) was carefully added to a solution of the corresponding host in chloroform (0.4 ml, 1.0 mM). The two-phasic system was then immediately cooled to 6 °C and stirred at 900 rpm for 16 h. The phases were then allowed to separate over a period of 2 min. The aqueous phase was removed and an aliquot injected into a reverse phase HPLC equipped with chiral columns for determination of the ee, distribution and αop. All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). For numerical values see ESI.† | ||
Applicability and scalability
With this highly efficient, optimized system in hand we turned our attention to the scalability of the process toward multistage reactors, running a U-tube experiment which serves as a useful test to judge the capacity of a host to deliver a guest from a feeding to a release phase in a catalytic manner (Fig. 9a).44 A blank run showed that at pH 5 there was no leaching of guest into the second aqueous phase over 24 h so any transfer observed can solely be attributed to the host.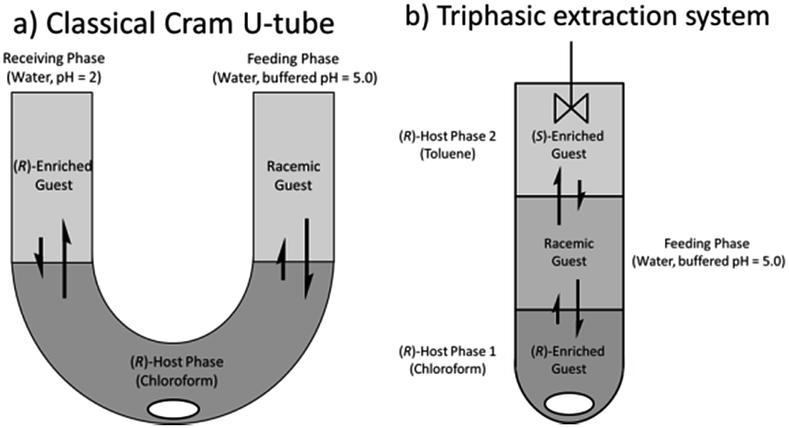 | ||
| Fig. 9 U-tube and tri-phasic reactor. Internal diameters: 1.0 cm, 0.9 cm, respectively. Stirring: for both 900 rpm, temperature: for both: 6 °C initial set-up: (a) (R)-host phase: SPA 4 in chloroform (10 ml, 0.5 mM). Feeding phase: 5 ml of a 2 or 20.0 mM solution of racemic phenylglycinol dissolved in a phosphate buffer solution (buffer strength 0.1 M) pH = 5.0. Receiving phase: 5 ml of a HCl solution in doubly distilled water such as pH = 2.0. At regular intervals an aliquot of the receiving phase was injected into a reverse phase HPLC equipped with a chiral column for determination of the ee, distribution and αop. (b) (R)-host phase 1: SPA 4 in chloroform (3 ml, 1.0 mM) feeding phase: 5 ml of a 2.0 mM solution of racemic phenylglycinol dissolved in a phosphate buffer solution (buffer strength 0.1 M) pH = 5.0. (R)-Host phase 2: SPA 4 in toluene (3 ml, 1.0 mM). All extraction experiments were carried out in triplo and with a simultaneous blank reaction (concentration of host = 0.0 mM). For numerical values see ESI.† | ||
When a 2 mM guest solution was used as feeding phase an ee of 41% could be measured in the receiving phase after an hour. However, further aliquots after two and three hours showed a slow erosion of ee occured over time. This is due to a depletion of the desired enantiomer of the guest in the feeding phase and subsequent transport of the other enantiomer as was observed earlier by us,56 indicating as well that the host was undergoing multiple transport release cycles. When a 20 mM feeding phase was used to overcome this issue, a 50% ee could be observed in the receiving phase after just 10 minutes with the ee staying globally constant over several hours while the amount of guest accumulated in the receiving phase rose steadily. Only after 24 hours was a notable loss in ee observed (29%) due, again, to depletion of the feeding phase. This showed that this system could be adapted to multistage reactors with high efficiency, potentially reaching 99% ee in under five steps if a cascade of centrifugal contactor separators is employed as was shown by our group in 2009 for 3,5-dinitrobenzoyl-(R),(S)-leucine45 and that a catalytic process was achieved with multiple turnovers, the first turnover being achieved in approximatively 40 min. In the centrifugal contactor separators these extractions are achieved in a matter of seconds.
In addition, the unique solvent dependency of this system can also be exploited in a novel fashion via simultaneous extraction of both enantiomers with a single catalyst in a vertical variation of the W-tube first suggested by Cram.59 When a triphasic system is set-up as shown below (Fig. 9b) with the bottom phase containing the host in chloroform ((R)-host phase 1), the middle phase containing the aqueous racemic guest solution (2 mM, feeding phase) and the top phase containing the host in toluene ((R)-host phase 2) extraction of the guest occurs from the feeding phase into both host phases which respectively yield, after back-extraction,60 an ee of 41% for the chloroform phase and an ee of 48% of the opposite enantiomer for the toluene phase.
The triphasic system therefore allows for the simultaneous extraction of both enantiomers with a single host is, to the best of our knowledge, unique and opens many new possibilities for ELLE extraction.
Conclusions
In summary, we have studied the efficiency of a previously unexplored family of chiral hosts towards the enantioselective liquid–liquid extraction of a range of 1,2-aminoalcohols including biologically relevant ones. These chiral 3,3′-substituted SPINOL phosphoric acids were shown to be highly efficient at the resolution of this type of substrates reaching unprecedented levels of selectivity. The operational selectivity of the extractions is highly dependent on the solvent, the pH and the temperature. We have also validated these systems towards multistage processes by demonstrating their catalytic efficiency in a U-tube system and have taken advantage of its unique properties to develop a novel triphasic extraction system. We conclude that these hosts, in view of the results obtained, could be used in an industrial-scale racemate separation process. We are currently continuing our studies on the mode of action of these systems based on the initial observation made.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from STW through project no. 11404 (Chiral Separations by Kinetic Extractive Resolution in Microfluidic Devices), from the Swiss National Science Foundation (SNSF) and from the Ministry of Education, Culture and Science (Gravitation Program 024.001.035 to BLF) are all gratefully acknowledged.Notes and references
-
D. J. Ager, Handbook of Chiral Chemicals, Marcel Dekker, New York, 2005 Search PubMed
.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed
- P. K. Ajikumar, K. Tyo, S. Carlsen, O. Mucha, T. H. Phon and G. Stephanopoulos, Mol. Pharm., 2008, 5, 167–190 CrossRef CAS PubMed
-
M. J. Waites, Industrial Microbiology, Blackwell Science, Oxford, 2001 Search PubMed
- D. Cascaval, C. Oniscu and A. I. Galaction, Biochem. Eng. J., 2001, 7, 171–176 CrossRef CAS
- M. Reschke and K. Schügerl, Chem. Ing. Tech., 1984, 56, 141 CrossRef CAS
-
J. G. de Vries, G. A. Molander and P. A. Evans, Science of Synthesis, Stereoselective Synthesis, Georg Thieme Verlag KG, Stuttgart, 2011, vol. 1–3 Search PubMed
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1996, 67, 1–14 CrossRef CAS
-
A. N. Collins, G. N. Sheldrake and J. Crosby, Chirality in Industry II: The Commercial Manufacture and Applications of Optically Active Compounds, Wiley and Sons, New York, 1997 Search PubMed
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr. A, 2001, 906, 3–33 CrossRef CAS PubMed
- J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 5, 799–811 CrossRef
-
K. Reuter, WO 97/32644, 1997
-
A. Bruggink, Rational Design in Resolutions, in Chirality in Industry II, ed. A. N. Collins, G. N. Sheldrake and J. Crosby, John Wiley & sons Ltd., Chichester, 1997 Search PubMed
-
D. Kozma, CRC Handbook of Optical Resolutions Via Diastereomeric Salt Formation, CRC Press LLC Boca Raton, 2002 Search PubMed
- E. Fogassy, M. Nogradi, D. Kozma, G. Egri, E. Palovics and V. Kiss, Org. Biomol. Chem., 2006, 4, 3011–3030 CAS
- F. Faigl, E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 19, 519–536 CrossRef CAS
- M. Leeman, G. Brasile, E. Gelens, T. Vries, B. Kaptein and R. Kellogg, Angew. Chem., Int. Ed., 2008, 47, 1287–1290 CrossRef CAS PubMed
-
G. Subramanian, Chiral Separation Techniques: A Practical Approach, Wiley-VCH, Weinheim, 2001 Search PubMed
-
K. W. Busch and M. A. Busch, Chiral Analysis, Elsevier, Amsterdam, 2004 Search PubMed
-
F. Toda, Enantiomer Separation: Fundamentals and Practical Methods, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed
-
G. Guebitz and M. G. Schmid, Chiral Separations, Humana Press, Totowa (NJ), 2004 Search PubMed
-
S. Ahuja, Chiral Separations: Application and Technology, ACS, Washington DC, 1997 Search PubMed
- T. Jira, A. Bunke, M. G. Schmid and G. Gübitz, J. Chromatogr. A, 1997, 761, 269–275 CrossRef CAS
- V. A. Davankov, J. Chromatogr. A, 1994, 666, 55–76 CrossRef CAS
- M. Steensma, N. J. M. Kuipers, A. B. De Haan and G. Kwant, Chirality, 2006, 18, 314–328 CrossRef CAS PubMed
-
G. B. Cox, Preparative Enantioselective Chromatography, Blackwell Publishing Ltd, 2007 Search PubMed
- E. Gavioli, N. M. Maier, C. Minguillón and W. Lindner, Anal. Chem., 2004, 76, 5837–5848 CrossRef CAS PubMed
- R. Xie, L.-Y. Chu and J.-G. Deng, Chem. Soc. Rev., 2008, 37, 1243–1263 RSC
- A. Maximini, H. Chmiel, H. Holdik and N. W. Maier, J. Membr. Sci., 2006, 276, 221–231 CrossRef CAS
- C. A. M. Afonso and J. G. Crespo, Angew. Chem., Int. Ed., 2004, 43, 5293–5295 CrossRef CAS PubMed
- G. Zenoni, F. Quattrini, M. Mazzotti, C. Fuganti and M. Morbidelli, Flavour Fragrance J., 2002, 17, 195–202 CrossRef CAS
- E. Francotte, T. Leutert, L. La Vecchia, F. Ossola, P. Richert and A. Schmidt, Chirality, 2002, 14, 313–317 CrossRef CAS PubMed
- J. T. F. Keurentjes, L. J. W. M. Nabuurs and E. A. Vegter, J. Membr. Sci., 1996, 113, 351–360 CrossRef CAS
- B. Baragaña, A. G. Blackburn, P. Breccia, A. P. Davis, J. de Mendoza, J. M. Padrón-Carrillo, P. Prados, J. Riedner and J. G. de Vries, Chem.–Eur. J., 2002, 8, 2931–2936 CrossRef
- R. M. C. Viegas, C. A. M. Afonso, J. G. Crespo and I. M. Coelhoso, Sep. Purif. Technol., 2007, 53, 224–234 CrossRef CAS
- M. Steensma, N. J. M. Kuipers, A. B. de Haan and G. Kwant, Chem. Eng. Sci., 2007, 62, 1395–1407 CrossRef CAS
- J. Koska and C. A. Haynes, Chem. Eng. Sci., 2001, 56, 5853–5864 CrossRef CAS
-
A. B. de Haan and B. Simandi, Extraction Technology for the Separation of Optical Isomers, in Ion Exchange and Solvent Extraction, ed. Y. Marcus, M. M. Sharma and J. A. Marinsky, Marcel Dekker, Inc, New York, 2001, pp. 255–294 Search PubMed
- P. J. Pickering and J. B. Chaudhuri, Chem. Eng. Sci., 1997, 52, 377–386 CrossRef CAS
-
J. C. Godfrey and M. J. Slater, Liquid–Liquid Extraction Equipment, John Wiley & Sons, New York, 1994 Search PubMed
-
E. Eliel, S. Wilen and L. Mander, Stereochemistry of Organic Compounds, John Wiley & Sons, New York, 1994 Search PubMed
- M. Steensma, N. J. M. Kuipers, A. B. de Haan and G. Kwant, Chemical Engineering and Processing: Process Intensification, 2007, 46, 996–1005 CrossRef CAS
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. de Vries, H. J. Heeres and B. L. Feringa, Org. Biomol. Chem., 2011, 9, 36–51 CAS
- A. J. Hallett, G. J. Kwant and J. G. de Vries, Chem.–Eur. J., 2009, 15, 2111–2120 CrossRef CAS PubMed
- B. Schuur, A. J. Hallett, J. G. M. Winkelman, J. G. de Vries and H. J. Heeres, Org. Process Res. Dev., 2009, 13, 911–914 CrossRef CAS
- See Methods section.
- B. J. V. Verkuijl, J. G. de Vries and B. L. Feringa, Chirality, 2011, 23, 34–43 CrossRef CAS PubMed
- B. Schuur, M. Blahušiak, C. R. Vitasari, M. Gramblička, A. B. De Haan and T. J. Visser, Chirality, 2015, 27, 123–130 CrossRef CAS PubMed
- P. Zhang, C. Liu, K. Tang, J. Liu, C. Zhou and C. Yang, Chirality, 2014, 26, 79–87 CrossRef CAS PubMed
-
J.-M. Lehn, Supramolecular Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed
-
D. J. Cram and J. M. Cram, Container Molecules and Their Guests, The Royal Society of Chemistry, 1997 Search PubMed
- X.-H. Huo, J.-H. Xie, Q.-S. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2007, 349, 2477–2484 CrossRef CAS
- F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677–8680 CrossRef CAS PubMed
- S. C. Peacock, L. A. Domeier, F. C. A. Gaeta, R. C. Helgeson, J. M. Timko and D. J. Cram, J. Am. Chem. Soc., 1978, 100, 8190–8202 CrossRef CAS
- C. Yang, X.-S. Xue, J.-L. Jin, X. Li and J.-P. Cheng, J. Org. Chem., 2013, 78, 7076–7085 CrossRef CAS PubMed
- F. Hein and F. Meier, Z. Anorg. Allg. Chem., 1970, 376, 296–302 CrossRef CAS
- This is supported by host-less blank extractions run at higher pH's where the guest can be obsereved to partition between phases.
- E. Grouzmann, J.-B. Gualtierotti, S. Gerber-Lemaire, K. Abid, N. Brakch, A. Pedretti, B. Testa and G. Vistoli, Chirality, 2013, 25, 28–34 CrossRef CAS PubMed
- M. Newcomb, J. L. Toner, R. C. Helgeson and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 4941–4947 CrossRef CAS
- Value obtained by back-extraction of the organic phase via aquous HCl followed by HPLC of the obtained aquous phase. Control extractions showed this method resulted in >98% extraction of guest into aquous phase.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data regarding the synthesis the hosts as well as procedures and raw data and for ELLE experiments. See DOI: 10.1039/c7sc02783d |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |