
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric hydroxylative dearomatization of 2-naphthols: synthesis of lacinilene derivatives†
Yu
Zhang
 ,
Yuting
Liao
,
Xiaohua
Liu
*,
Xi
Xu
,
Lili
Lin
and
Xiaoming
Feng
*
,
Yuting
Liao
,
Xiaohua
Liu
*,
Xi
Xu
,
Lili
Lin
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
First published on 24th July 2017
Abstract
An enantioselective hydroxylative dearomatization of 2-naphthols with oxaziridines has been accomplished using a N,N′-dioxide–scandium(III) complex catalyst. Various substituted ortho-quinols could be obtained in high yields (up to 99%) and enantioselectivities (up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 er). This methodology could be applied in the synthesis of bioactive lacinilenes in a gram-scale reaction. Based on the experimental investigations and previous work, a possible catalytic model was proposed.
:5 er). This methodology could be applied in the synthesis of bioactive lacinilenes in a gram-scale reaction. Based on the experimental investigations and previous work, a possible catalytic model was proposed.
Introduction
Substituted ortho-quinols are essential structural motifs in a number of natural products and pharmaceuticals.1 For instance, chiral lacinilene derivatives (Fig. 1), a series of phytoalexines isolated from cotton plants, have been utilized for inhibiting the growth of cotton bacterial pathogens, such as Xanthomonas campestris or malvacearum.2 Studies have showed that the (S)-enantiomer of lacinilene C is more active than the (R)-enantiomer.2c While these biological activities provide a justification for the development of approaches to the synthesis of enantiomerically enriched lacinilene derivatives, novel catalytic enantioselective methods remain limited.2b,d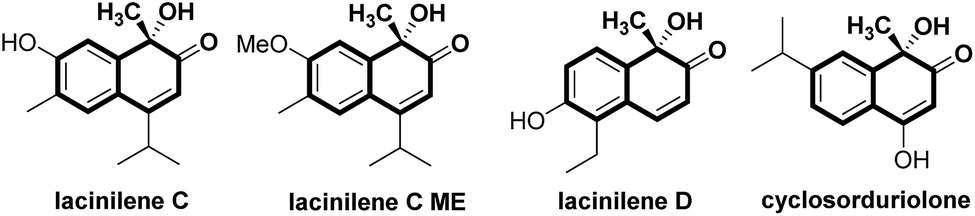 | ||
| Fig. 1 Representative active lacinilene derivatives bearing ortho-quinol structures. | ||
Optically active lacinilene derivatives in nature were proposed to be produced enzymically from the oxidation of dihydroxycadalenes, thus it is of practical interest to discover a catalytic asymmetric oxidative dearomatization route to the synthesis of these cadinanes.3 Compared with other successful dearomatization events of phenols or naphthols,4,5 controlling the chemo-, regio- and enantioselectivity of the asymmetric hydroxylative dearomatization is more difficult,6 as there might be serious side reactions in the presence of oxidants including overoxidation of alkene functions, competitive para-oxidation and homocoupling.6c,e Additionally, the ortho-quinol product could undergo an unexpected α-ketol rearrangement, which enhances the difficulty of controlling the reactivity and selectivity.6a,7 In this respect, only a few reports related to asymmetric hydroxylative dearomatization of phenols or naphthols have been reported. Asymmetric oxidative dearomatization of phenolate mediated by copper–sparteine–dioxygen complexes followed a [4 + 2] dimerization cascade, giving bicyclo[2.2.2]octenones as the final products.6a Several chiral hypervalent organoiodine compounds were developed for the asymmetric hydroxylative dearomatization of phenols and 1-naphthols.6b–e Taking these examples into account, we want to engage in discovering new enantioselective strategies for the synthesis of ortho-quinol moieties with improved efficiency and selectivity. Here, we present an efficient asymmetric hydroxylative dearomatization of 2-naphthols catalyzed by a chiral N,N′-dioxide–scandium(III) complex catalyst.8 The process could be applied to the synthesis of various 1-hydroxy-1-alkyl-naphthalen-2-one derivatives including lacinilene C methyl ether and lacinilene D, in high to excellent yields and good enantioselectivities under mild reaction conditions (Scheme 1).
 | ||
| Scheme 1 Catalytic asymmetric hydroxylative dearomatization of phenols and naphthols. | ||
Results and discussion
We selected the hydroxylative dearomatization of 1-methylnaphthalen-2-ol 1a as the model substrate using 3-phenyl-2-tosyl-1,2-oxaziridine 2a as the oxidant which was proven to be chemoselective as a phase-transfer-catalyst under basic conditions (Table 1).7a Initially, the catalytic asymmetric reaction was performed with 10 mol% of chiral N,N′-dioxide L-PiPr2–Sc(OTf)3 complex in DCM at 30 °C, and the desired product 3a could be obtained dominantly with 80:20 er while the α-ketol rearrangement byproduct 4a was isolated in around one-fourth of a 96% total yield (Table 1, entry 1). The evaluation of the structure of the N,N′-dioxides showed that L-PiPr2 was the optimal ligand in terms of the enantioselectivity albeit ligand L-PiMe2 and L-PiPr3 improved the yield of the desired product 3a (entries 2–5). Fortunately, changing the counterion of the scandium salt from −OTf to −NTf2 could suppress the α-ketol rearrangement, delivering the quinol 3a in a 99% yield with 92:8 er (Table 1, entry 6). Further optimization of the reaction conditions, such as decreasing the temperature and the catalyst loading to 5 mol%, resulted in slightly improved enantioselectivity with maintained efficiency (entry 7). Lowering the catalyst loading to 1 mol% or the amount of the oxidant 2a decreased either the yield or the selectivity a little (entries 8 and 9). We therefore chose the reaction conditions in Table 1, entry 7 for further studies.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | L* | Yieldb (%) | Ratio (3a/4a)c | er (3a)c |
|
a Unless otherwise noted, the reactions were performed with L*/Sc(III) (1:1, 10 mol%), 1a (0.10 mmol) and 2a (2.0 equiv.) in DCM (1.0 mL) under N2 at 30 °C for 3 h.
b Isolated yield by silica gel chromatography.
c Determined by chiral HPLC analysis.
d 5 mol% catalyst loading at 0 °C.
e 1 mol% catalyst loading at 0 °C for 4 h.
f
2a (1.5 equiv.) was used.
|
|||||
| 1 | Sc(OTf)3 | L-PiPr2 | 96 | 73:27 |
80:20 |
| 2 | Sc(OTf)3 | L-PrPr2 | 99 | 79:21 |
63:37 |
| 3 | Sc(OTf)3 | L-RaPr2 | 99 | 75:25 |
53.5:46.5 |
| 4 | Sc(OTf)3 | L-PiMe2 | 90 | >95:5 |
60:40 |
| 5 | Sc(OTf)3 | L-PiPr3 | 96 | 89:11 |
73:27 |
| 6 | Sc(NTf2)3 | L-PiPr2 | 99 | >95:5 |
92:8 |
| 7d | Sc(NTf2)3 | L-PiPr2 | 99 | >95:5 |
95:5 |
| 8e | Sc(NTf2)3 | L-PiPr2 | 86 | >95:5 |
93.5:6.5 |
| 9d,f | Sc(NTf2)3 | L-PiPr2 | 99 | >95:5 |
94.5:5.5 |
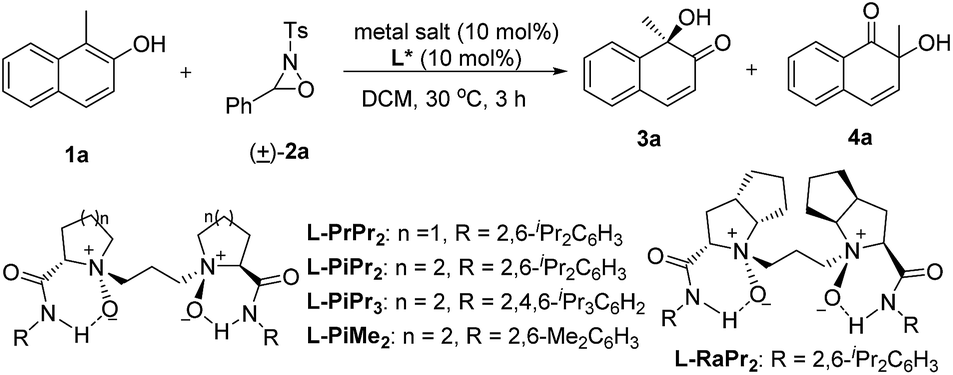
We next explored the substrate scope of 2-naphthols (Table 2). The introduction of bromo or methoxyl groups at the C6-position of 2-naphthols had no obvious effect on the result. The 6-aryl substituted 2-naphthol derivatives 1d–1l tethering various electron-donating and electron-withdrawing substituents could undergo the transformations smoothly, providing the products 3d–3l in 95–99% yield and 93.5:6.5–95:5 er. It was noteworthy that 6-alkenyl and alkynyl substituted substrates 1m–1q were compatible with the reaction conditions, and no aminohydroxylation of the unsaturated carbon–carbon bond occurred, giving the hydroxylative dearomatization products 3m–3q in good to excellent yields and enantioselectivities.9 Additionally, 6-alkyl substituted 2-naphthols 3r–3w bearing methyl, ethyl, and butyl groups were well tolerated, accomplishing the asymmetric hydroxylative reaction with the outcomes of 95–99% yield and 94:6–95:5 er. The installation of substituents to the 5- and 7-positions did not influence the reaction efficiency (3x–3z). The MOM-protected substrate 1z could deliver the desired product 3z with good results without any deprotection process occurring under the reaction conditions. However, the increase of steric hindrance at the ortho-position of 2-naphthol was harmful as a consequence (3aa and 3ab).

To show the synthetic utility of the current catalyst system, asymmetric synthesis of bioactive lacinilenes was carried out (Scheme 2). Initially, the direct deprotection of the product 3z under acidic conditions formed the optically active lacinilene D, but an aromatization side product 1-ethyl-5-methylnaphthalene-2,6-diol was obtained.2d,10 It was anticipated that the TBS protecting group could be easily removed under neutral conditions, which might avoid the occurrence of the aromatization process. As expected, the TBS-substituted 2-naphthol 1ae could be easily synthesized from 9 in 66% yield after 3 steps, which was further enantioselectively oxidized into the product 3ae in quantitative yield and 95:5 er, even when it was performed at the gram scale. The absolute configuration of 3ae from L-PiPr2–Sc(NTf2)3 complex catalysis was determined to be (R) by X-ray crystal diffraction analysis.11 For the benefit of the further differential biological activity study on each enantiomer of the chiral lacinilenes,2c (S)-lacinilene D was synthesized using an ent-L-PiPr2–Sc(NTf2)3 complex with a comparable result of 99% yield and 95:5 er. Next, the synthesis of optically active lacinilene C methyl ether was explored. The synthetic route began from 1,2-dihydronaphthalene 12, which could be easily accessed from 2-methoxytoluene through a four-step protocol.2d Subsequent two-step oxidation could afford the 2-naphthol derivative 1y in 49% yield, which underwent hydroxylative dearomatization catalyzed by 0.1 mol% of the Sc(NTf2)3/rac-L-PiPr2 complex to produce racemic lacinilene 3y in 90% yield.2d After trimethylsilylation and copper catalyzed 1,4-addition/aromatization, 2-naphthol 1af could be attained in 45% yield after two steps. By treatment with oxaziridine 2a in the presence of Sc(OTf)3/L-RaPr2, chiral lacinilene C methyl ether could be obtained in quantitative yield and 83:17 er, which could further transform to lacinilene C according to the literature.2b
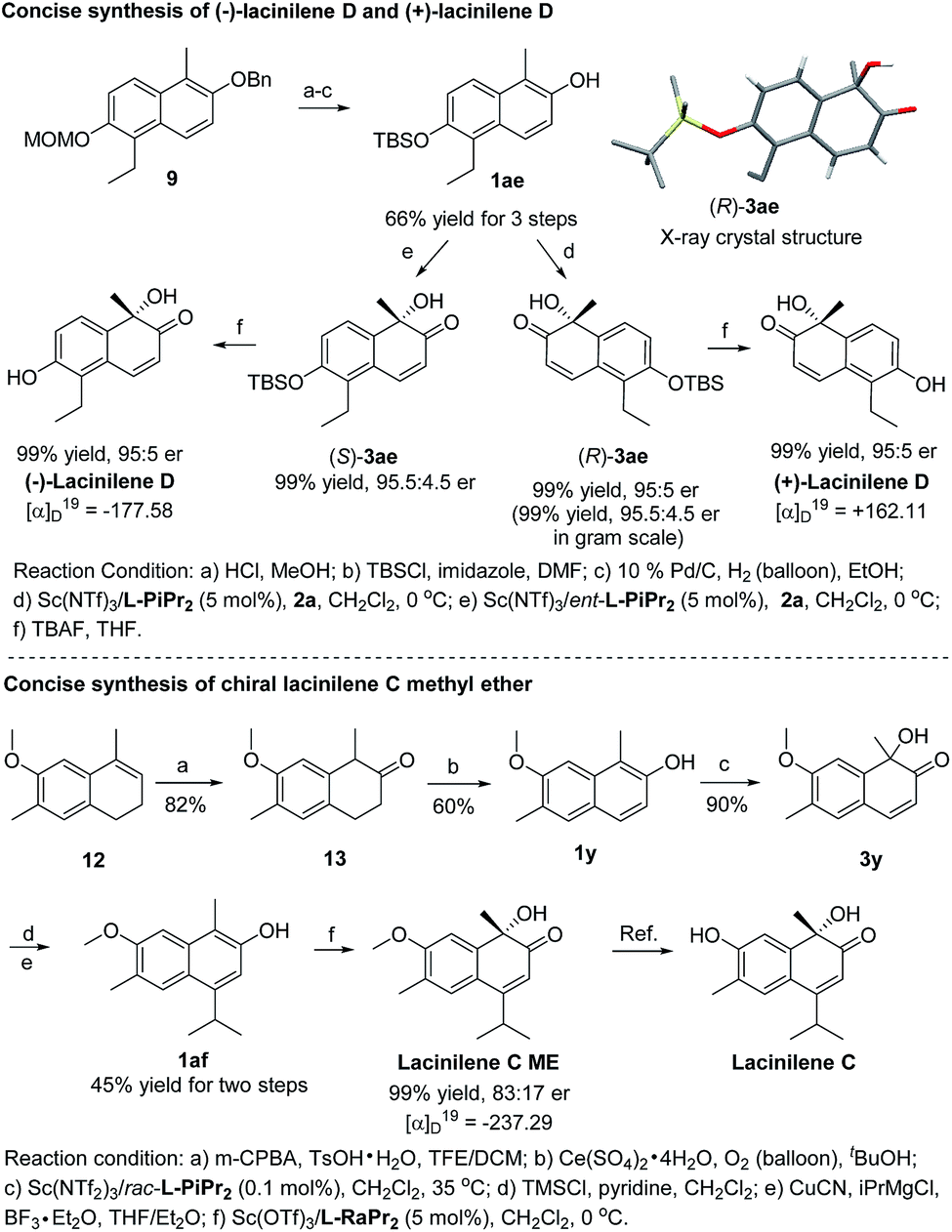 | ||
| Scheme 2 Concise synthesis of chiral lacinilene C methyl ether, (−)-lacinilene D and (+)-lacinilene D. | ||
To elucidate the stereochemical course of the oxidation process, some control experiments were conducted (Scheme 3). The optically pure oxaziridine (S)-2a reacted with 2-naphthol 1a in the presence of the Sc(NTf2)3/L-PiPr2 complex, affording the (R)-quinol 3a in 49% yield and 95.5:4.5 er with the recovered oxaziridine (S)-2a in 45% yield.12d Using ent-L-PiPr2 as the ligand, (S)-quinol 3a was obtained in 46% yield and 90:10 er with the recovered oxaziridine (S)-2a in 52% yield. This indicates that the chiral matched and mis-matched effect between chiral ligand and chiral oxaziridine was not obvious in this case compared to previous reports,12 and there might be negligible interaction between the chiral catalyst and oxaziridine.
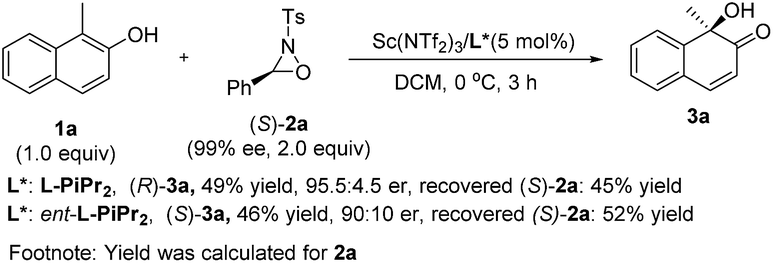 | ||
| Scheme 3 Control experiments. | ||
To probe into the interaction between the catalysts and 2-naphthol, 1H NMR analysis of the mixture of components was carried out (see ESI† for details). The chemical shift of 1-methyl 2-naphthol 1a remained nearly unchanged after Sc(NTf2)3 was added. There was an obvious high-field shift for most signals of 1a after mixing with the Sc(NTf2)3/L-PiPr2 catalyst. This indicates that the chiral catalyst makes the 2-naphthol reactive for hydroxylative reactions. Based on these results and our previous study on the chiral N,N′-dioxide–metal complex catalysts,8,13 we suggested an enantioselective catalytic model as shown in Fig. 2. The ligand L-PiPr2 binds to the scandium(III) center via four oxygens to form a polycyclic octahedral metal complex catalyst. The 2-naphthol coordinates to the metal center at one of the vacant sites, with its Re-face shielded by one amide unit of the ligand. Therefore, 2a preferably attacked the α-position of 2-naphthol from the Si-face to generate the corresponding R-configured product 3ae and imine byproduct. If a substituent was introduced into the C3 or C4 positions of 2-naphthol, the steric hindrance discrimination between the two sides of the hydroxyl group decreases, thus it is difficult to control the face-selection. As a result, the enantioselectivity for the generation of product 3aa and lacinilene C methyl ether is lower than that for the others.
 | ||
| Fig. 2 Proposed enantioselective catalytic model. | ||
Conclusions
In summary, we have described a highly chemo- and enantioselective hydroxylative dearomatization of 2-naphthol derivatives with oxaziridine catalyzed by a chiral N,N′-dioxide–Sc(NTf2)3 complex catalyst. The desired substituted ortho-quinols with one quaternary carbon stereogenic center were afforded with high enantioselectivities and reactivity (up to 99% yield and 95:5 er). The α-ketol rearrangement byproducts were efficiently suppressed. This new procedure has been successfully applied to the catalytic asymmetric synthesis of the phytoalexines lacinilenes. The application of the N,N′-dioxide/metal catalyst system in the synthesis of other bioactive molecules will be explored.
Acknowledgements
The study was funded by the National Natural Science Foundation of China (No. 21290182, 21432006 and 21625205).Notes and references
- (a) D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS PubMed; (b) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed.
- (a) P. W. Jeffs and D. G. Lynn, J. Org. Chem., 1975, 40, 2958 CrossRef CAS; (b) J. P. McCormick, T. Shinmyozu, J. P. Pachlatko, T. R. Schafer and J. W. Gardner, J. Org. Chem., 1984, 49, 34 CrossRef CAS; (c) R. D. Stipanovic, J. P. McCormick, E. O. Schlemper, B. C. Hamper, T. Shinmyozu and W. H. Pirkle, J. Org. Chem., 1986, 51, 2500 CrossRef CAS; (d) K. Krohn and G. Zimmermann, J. Org. Chem., 1998, 63, 4140 CrossRef CAS.
- For selected examples of asymmetric oxidative dearomatization in the biosynthesis of natural products: (a) A. Bérubé, I. Drutu and J. L. Wood, Org. Lett., 2006, 8, 5421 CrossRef PubMed; (b) L. H. Mejorado and T. R. R. Pettus, J. Am. Chem. Soc., 2006, 128, 15625 CrossRef CAS PubMed; (c) S. P. Cook, A. Polara and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 16440 CrossRef CAS PubMed; (d) J. Gagnepain, F. Castet and S. Quideau, Angew. Chem., Int. Ed., 2007, 46, 1533 CrossRef CAS PubMed; (e) A. Rudolph, P. H. Bos, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 5834 CrossRef CAS PubMed.
- For selected reviews on dearomatization reactions of phenols and naphthols: (a) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed; (b) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC; (c) W. Sun, G. Li, L. Hong and R. Wang, Org. Biomol. Chem., 2016, 14, 2164 RSC. For selected recent examples, see: (d) Q. Yin, S.-G. Wang, X.-W. Liang, D.-W. Gao, J. Zheng and S.-L. You, Chem. Sci., 2015, 6, 4179 RSC; (e) S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647 CAS; (f) D. Yang, L. Wang, F. Han, D. Li, D. Zhao and R. Wang, Angew. Chem., Int. Ed., 2015, 54, 2185 CrossRef CAS PubMed; (g) J. Nan, J. Liu, H. Zheng, Z. Zuo, L. Hou, H. Hu, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 2356 CrossRef CAS PubMed; (h) D. Yang, L. Wang, M. Kai, D. Li, X. Yao and R. Wang, Angew. Chem., Int. Ed., 2015, 54, 9523 CrossRef CAS PubMed; (i) S.-G. Wang, X.-J. Liu, Q.-C. Zhao, C. Zheng, S.-B. Wang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14929 CrossRef CAS PubMed; (j) L. Yang, H. Zheng, L. Luo, J. Nan, J. Liu, Y. Wang and X. Luan, J. Am. Chem. Soc., 2015, 137, 4876 CrossRef CAS PubMed; (k) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880 CrossRef CAS PubMed; (l) Q. Cheng, Y. Wang and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 3496 CrossRef CAS PubMed; (m) H.-F. Tu, C. Zheng, R.-Q. Xu, X.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 3237 CrossRef CAS PubMed; (n) D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Angew. Chem., Int. Ed., 2017, 56, 3242 CrossRef CAS PubMed.
- For selected reviews on recent asymmetric oxidative dearomatization of phenols and naphthols: (a) M. Uyanik and K. Ishihara, Asymmetric Oxidative Dearomatization Reaction, in Asymmetric Dearomatization Reactions, ed. S.-L. You, Wiley-VCH, Weinheim, 2016, ch. 6, pp. 129–152 Search PubMed. For examples: (b) T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787 CrossRef CAS PubMed; (c) M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175 CrossRef CAS PubMed; (d) M. Uyanik, T. Yasui and K. Ishihara, Tetrahedron, 2010, 66, 5841 CrossRef CAS; (e) T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2012, 134, 20017 CrossRef CAS PubMed; (f) T. Dohi, N. Takenaga, T. Nakae, Y. Toyoda, M. Yamasaki, M. Shiro, H. Fujioka, A. Maruyama and Y. Kita, J. Am. Chem. Soc., 2013, 135, 4558 CrossRef CAS PubMed; (g) M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215 CrossRef CAS PubMed; (h) T. Oguma and T. Katsuki, Chem. Commun., 2014, 50, 5053 RSC; (i) S. J. Murray and H. Ibrahim, Chem. Commun., 2015, 51, 2376 RSC; (j) D.-Y. Zhang, L. Xu, H. Wu and L.-Z. Gong, Chem.–Eur. J., 2015, 21, 10314 CrossRef CAS PubMed; (k) N. Jain, S. Xu and M. A. Ciufolini, Chem.–Eur. J., 2017, 23, 4542 CrossRef CAS PubMed.
- For selected examples of asymmetric hydroxylative dearomatization: (a) S. Dong, J. Zhu and J. A. Porco Jr, J. Am. Chem. Soc., 2008, 130, 2738 CrossRef CAS PubMed; (b) J. K. Boppisetti and V. B. Birman, Org. Lett., 2009, 11, 1221 CrossRef CAS PubMed; (c) S. Quideau, G. Lyvinec, M. Marguerit, K. Bathany, A. Ozanne-Beaudenon, T. Buffeteau, D. Cavagnat and A. Chénedé, Angew. Chem., Int. Ed., 2009, 48, 4605 CrossRef CAS PubMed; (d) K. A. Volp and A. M. Harned, Chem. Commun., 2013, 49, 3001 RSC; (e) C. Bosset, R. Coffinier, P. A. Peixoto, M. El Assal, K. Miqueu, J.-M. Sotiropoulos, L. Pouységu and S. Quideau, Angew. Chem., Int. Ed., 2014, 53, 9860 CrossRef CAS PubMed.
- (a) C. Grandclaudon and P. Y. Toullec, Eur. J. Org. Chem., 2016, 260 CrossRef CAS; (b) M. Uyanik, T. Mutsuga and K. Ishihara, Angew. Chem., Int. Ed., 2017, 56, 3956 CrossRef CAS PubMed.
- For reviews on chiral N,N′-dioxides: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; for recent examples: (c) Q. Yao, Y. T. Liao, L. L. Lin, X. B. Lin, J. Ji, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 1859 CrossRef CAS PubMed; (d) Y. Xia, L. L. Lin, F. Z. Chang, Y. T. Liao, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 12228 CrossRef CAS PubMed; (e) Y. Zhang, Y. T. Liao, X. H. Liu, Q. Yao, Y. H. Zhou, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2016, 22, 15119 CrossRef CAS PubMed; (f) Y. T. Liao, X. H. Liu, Y. Zhang, Y. L. Xu, Y. Xia, L. L. Lin and X. M. Feng, Chem. Sci., 2016, 7, 3775 RSC.
- (a) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866 CrossRef CAS PubMed; (b) D. J. Michaelis, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 6610 CrossRef CAS PubMed.
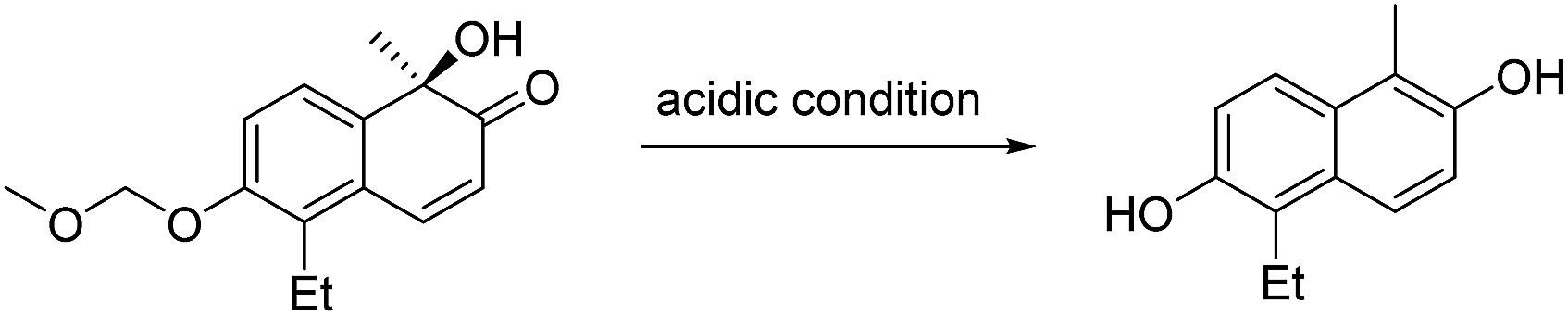
- We tested HCl, AlCl3/NaI, BBr3, and TMSBr to remove the MOM-protecting group, but the corresponding lacinilene D was not observed. The reactions proceeded as follows:
 see ref. 2d for details.
see ref. 2d for details. - CCDC 1536822 (3ae) contains the ESI crystallographic data for this paper.†.
- (a) P.-L. Shao, X.-Y. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412 CrossRef CAS PubMed; (b) S. X. Dong, X. H. Liu, Y. Zhu, P. He, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2013, 135, 10026 CrossRef CAS PubMed; (c) K. S. Williamson, J. W. Sawicki and T. P. Yoon, Chem. Sci., 2014, 5, 3524 RSC; (d) X. B. Lin, S. Ruan, Q. Yao, C. K. Yin, L. L. Lin, X. M. Feng and X. H. Liu, Org. Lett., 2016, 18, 3602 CrossRef CAS PubMed.
- X-ray single-crystal structure of N,N′-dioxide–Sc(III): Y. L. Liu, D. J. Shang, X. Zhou, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2009, 15, 2055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1536822. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02809a |
| This journal is © The Royal Society of Chemistry 2017 |