
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGelation-driven selection in dynamic covalent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C/CN exchange†
C/CN exchange†
Chunshuang
Liang
ab,
Sirinan
Kulchat
ac,
Shimei
Jiang
 bd and
Jean-Marie
Lehn
*a
bd and
Jean-Marie
Lehn
*a
aInstitut de Science et d’Ingénierie Supramoléculaires (ISIS), Université de Strasbourg, 8 allée Gaspard Monge, Strasbourg 67000, France. E-mail: lehn@unistra.fr
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Avenue, Changchun 130012, P. R. China
cMaterials Chemistry Research Unit, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand
dInternational Center of Future Science, Jilin University, Changchun 130012, China
First published on 6th September 2017
Abstract
Knoevenagel barbiturate derivatives bearing long alkyl chains were proven to form organogels in suitable solvents based on supramolecular interactions. Their reaction with imines allows for component exchange through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN recombination. The effect of various parameters (solvents, chain length, and temperature) on the CC/CN exchange reaction has been studied. Mixing Knoevenagel compound K and imine I-16 in a 1
C/CN recombination. The effect of various parameters (solvents, chain length, and temperature) on the CC/CN exchange reaction has been studied. Mixing Knoevenagel compound K and imine I-16 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio generated a constitutional dynamic library containing the four constituents K, I-16, K′-16, and I′. The reversible exchange reaction was monitored by 1H-NMR, showing marked changes in the fractions of the four constituents on sol–gel interconversion as a function of temperature. The library composition changed from statistical distribution of the four constituents in the sol state to selective amplification of the gel forming K′-16 constituent together with that of its agonist I′. The process amounts to self-organization driven component selection in a constitutional dynamic organogel system undergoing gelation. This process displays up-regulation of the gel-forming constituent by component redistribution through reversible covalent connections.
:1 ratio generated a constitutional dynamic library containing the four constituents K, I-16, K′-16, and I′. The reversible exchange reaction was monitored by 1H-NMR, showing marked changes in the fractions of the four constituents on sol–gel interconversion as a function of temperature. The library composition changed from statistical distribution of the four constituents in the sol state to selective amplification of the gel forming K′-16 constituent together with that of its agonist I′. The process amounts to self-organization driven component selection in a constitutional dynamic organogel system undergoing gelation. This process displays up-regulation of the gel-forming constituent by component redistribution through reversible covalent connections.
Introduction
Dynamic covalent chemistry (DCC) rests on the designed introduction of covalent bonds, formed by reversible reactions, into molecules, thus allowing for spontaneous modification of their constitution through component exchange.1 On the other hand, the constitution of supramolecular entities is intrinsically dynamic in view of the lability of intermolecular interactions. Together, these two domains lead to the definition of a constitutional dynamic chemistry (CDC)1d,1i that covers the dynamic properties at both the molecular and supramolecular levels.DCC1 allows for the generation of dynamic covalent/combinatorial libraries (DCLs) of constituents from reversibly connecting components. The implementation of DCLs requires fast recombination of components in equilibrating systems under mild conditions so that the equilibrium may be perturbed by external physical or chemical agents causing the system to reorganize in response to and with adaptation to the perturbation. Among the reversible covalent reactions, the condensation between a carbonyl group and an amine to form an imine bond (CN) has been extensively used in DCC for generating DCLs1 and in materials science, in particular for covalent dynamic polymers (dynamers).2
Various chemical effectors and physical stimuli have been applied to drive dynamic supramolecular and molecular libraries. Among these agents, self-organization deserves special attention as it may be considered to play a crucial, arguably the most significant, role in the evolution towards states of matter of increasing complexity.1d,3 In self-organization-driven processes the formation of a structured phase drives the selection, among the components making up the dynamic library, of those that produce the constituent(s) leading to the most highly organized and most stable assembly.
The amplification of a given constituent of a constitutional dynamic system under the driving force of the formation of an organized phase has been demonstrated for preferential crystallization,4 surface adsorption5 and in particular for the present purposes, the generation of a gel.6–9
Gels are well known soft materials of both basic interest and with a wide range of applications for instance in the food industry, cosmetics, and medicines. They are based on (supra)molecular networks trapping the aqueous (hydrogels) or organic (organogels) solvent molecules. Their internal network structure may result from physical bonds (physical gel) or chemical bonds (chemical gel). A supramolecular gel may be composed of a low-molecular weight gelator (LMWG), in which gelator molecules self-assemble under suitable conditions through various non-covalent interactions, such as hydrogen bonding, electrostatic interactions, van der Waals forces and π–π stacking, to form micro- or nano-scale networks (fibers, ribbons, sheets, or spheres) which further result in the formation of higher order three-dimensional networks.10 Supramolecular gels formed from the self-assembly of LMWGs open a new area of functional materials, which demonstrates the power of supramolecular chemistry in materials science.11 Besides, due to the non-covalent interactions, supramolecular gels have their own dynamic and reversible character and can respond sensitively to external stimuli (temperature, pH, solvent, light and redox reactions).10d,11b,12 Thus, gels currently attract much interest for their potential as intriguing soft materials, and as media for biomedical applications for instance.
Recently, a novel exchange process leading to DCLs has been introduced. This process performs CC/CN exchange between a Knoevenagel derivative of a barbiturate and an imine.13 The reaction was found to occur especially fast in organic media of low polarity, probably through an organic metathesis mechanism.14
In the present work, such fast CC/CN exchange was implemented in the context of DCC-based selection driven by gelation, in order to examine the up-regulation of the constituent formed by the components generating the most stable gel, i.e. the materials with the highest state of self-organization. The exchange process was studied for compounds K, K′-n, I-n and I′ as illustrated in Scheme 1.
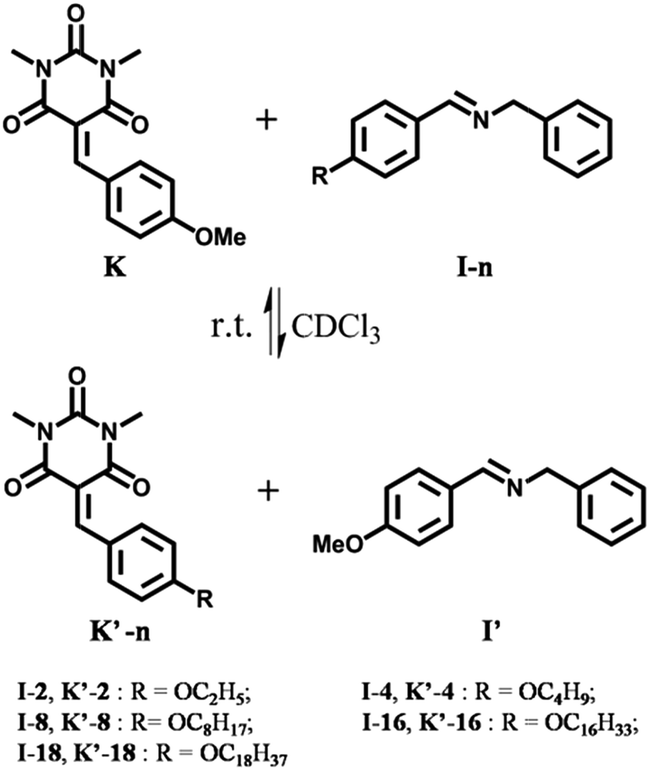 | ||
| Scheme 1 CC/CN exchange reactions between the barbiturate-derived Knoevenagel compounds K and K′-n and the imines I-n and I′. The benzaldehyde-derived components bear chains of different lengths. The reactions are performed in equimolar solutions of K, and I-n (10 mM each) in CDCl3 at room temperature, generating a constitutional dynamic library containing the four constituents K, I-n, K′-n, and I′. The OMe derivatives K and I′ are taken as reference compounds. | ||
Results and discussion
Gel formation and properties
The Knoevenagel benzylidene-barbiturates K and K′-n (Scheme 1) were synthesized by condensation between N,N′-dimethyl-barbituric acid and the corresponding aldehydes (see ESI†). We found that the Knoevenagel compounds with long chains (K′-16 and K′-18) do form a gel, while it was not the case with shorter chains. Besides, Knoevenagel compounds with two and three long chains (K′-(16)2 and K′-(10)3) were synthesized and they also formed a gel under proper conditions. The corresponding imines I-n and I′ were obtained from condensation between benzylamine and the corresponding aldehydes (see ESI†). None of them formed a gel in the studied solvents.The gel formation ability of these compounds was evaluated in different solvents such as acetonitrile, ethanol, methanol, DMSO, and chloroform (using 1% w/v of gelator in the solvent at 20.6, 19.5, 13.8, and 14.0 mM concentration for K′-16, K′-18, K′-(16)2, and K′-(10)3, respectively; see Table 1). The results showed that gel formation (Scheme 2) of K′-16, K′-18, and K′-(10)3 (Table 1, entries 1, 2, and 4) occured in both ethanol and acetonitrile, while a gel of K′-(16)2 (Table 1, entry 3) could only be obtained in DMSO. The gel to sol transition temperature (Tgel) was measured by the ‘inverse flow method’.15 The Tgel at 1% w/v was 56 °C, 56 °C, 51 °C, and 33 °C, respectively. When the same molar concentration of 14.0 mM was used, the Tgel was 53 °C, 53 °C, 51 °C, and 33 °C, respectively (see ESI†). For all the derivatives investigated, no gel formed in chloroform in which only solutions were obtained for all compounds. Furthermore, the Minimum Gel Concentration (MGC) was found to be 2.2, 2.3, 5.8, and 7.1 mg mL−1 (4.5, 4.4, 8.0 and 10.0 mM) for K′-16, K′-18, K′-(16)2 and K′-(10)3, respectively (see ESI†).
| Entry | Compoundsa | Solventsb | ||||
|---|---|---|---|---|---|---|
| CH3CN | EtOH | MeOH | DMSO | CHCl3 | ||
| a [Gelator] = 1% w/v; [K′-16] = 20.6 mM, [K′-18] = 19.5 mM, [K′-(16)2] = 13.8 mM, and [K′-(10)3] = 14.0 mM. b G, gel; S, solution; P, precipitate. c T gel = 56 °C. d T gel = 56 °C. e T gel = 51 °C. f T gel = 33 °C. | ||||||
| 1 | K′-16 | G | Gc | P | P | S |
| 2 | K′-18 | G | Gd | P | P | S |
| 3 | K′-(16)2 | P | P | P | Ge | S |
| 4 | K′-(10)3 | G | Gf | P | S | S |
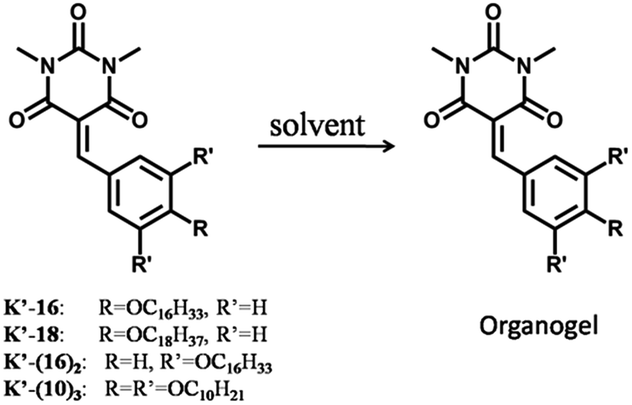 | ||
| Scheme 2 Representation of the benzylidene barbiturate compounds (K′-n) which were used in the present gelation investigations. | ||
The gels obtained were observed using Scanning Electron Microscopy (SEM). The SEM images of the gels formed by organogelators K′-n showed micrometer-scale fibers (Fig. 1). The width of these fibers ranged from 0.5 to 1.5 μm, and their length can be extended to tens of micrometers, which accounted for the opacity of the organogels (Fig. 1a, b, e and f, insets). The optical aspect in the visible wavelength range (400–800 nm) may depend on the crystallinity of the gel16 and on a matching between the index of refraction of the fibers and of the solvent. The fibers formed from gelators K′-(16)2 and K′-(10)3 (Fig. 1e and f) containing two or three alkyl chains were cross-linked with each other, and not as straight as those formed from gelators K′-16 and K′-18 (Fig. 1a and b) containing only one alkyl chain. In Fig. 1c and d, it can be seen that the fibers were very thin as seen from the overlap of strap fibers (marked by red dash line). These long fibers were entangled or crosslinked to form a 3D network, which is typical of gel structures. This self-assembling property leads to the formation of a self-supporting soft solid-like supramolecular material.
 | ||
| Fig. 1 SEM images of the gels formed by the organogelators in EtOH (K′-16, K′-18, and K′-(10)3) and DMSO (K′-(16)2): (a) and (c) K′-16; (b) and (d) K′-18; (e) K′-(16)2; (f) K′-(10)3; insets: photographic images of the gels. | ||
In order to investigate the packing mode of the molecules in the supramolecular gel state, X-ray scattering experiments were carried out on these organogels. Fig. 2 shows the XRD patterns of xerogels obtained from K-16, K-18, K-(16)2 and K-(10)3. In Fig. 2a and b, five diffraction peaks with d-spacings of 28.2, 14.0, 9.3, 7.0 and 5.6 Å for K-16 and 29.9, 15.0, 10.0, 7.4 and 6.0 Å for K-18 were found, corresponding to a ratio of 1:1/2:1/3:1/4:1/5. These data illustrate the formation of a layered structure.17 Similarly, diffraction peaks with d-spacings of 25.4, 12.5, and 8.3 Å (1:1/2:1/3) for K-(16)2 and 37.6, 18.7, 12.6, and 7.9 Å (1:1/2:1/3:1/4) for K-(10)3 were also observed, again indicative of a lamellar structure (Fig. 2c and d). Moreover, diffraction peaks with a d-spacing of 3.4–3.9 Å were found in the wide-angle region, corresponding to the distance of π–π interactions.17a,18
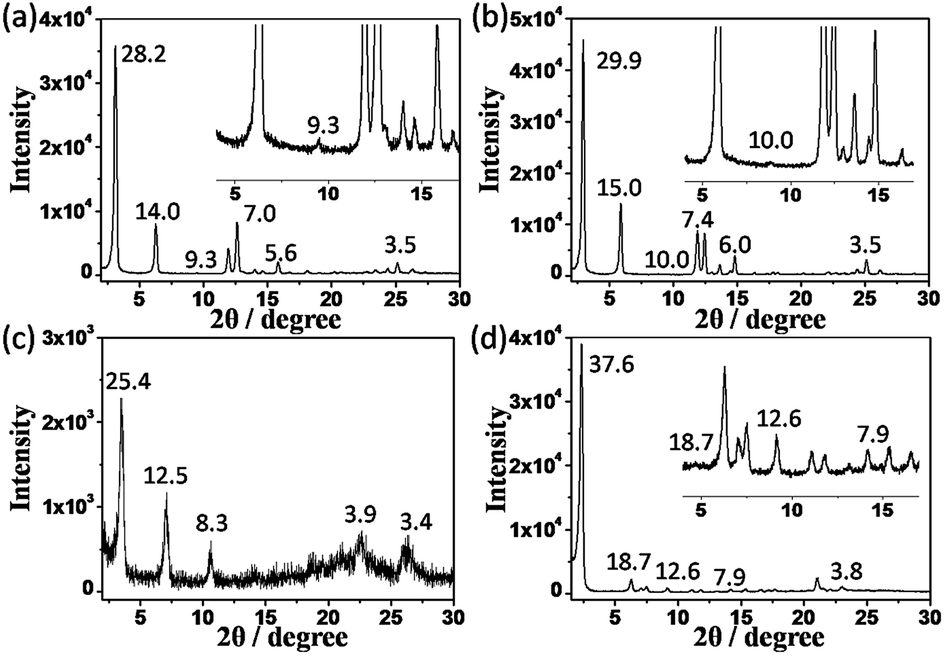 | ||
| Fig. 2 XRD patterns of the xerogels obtained from (a) K-16, (b) K-18, (c) K-(16)2, and (d) K-(10)3. | ||
Based on the results of the SEM and XRD, a schematic representation of this hierarchical self-assembly is proposed in Scheme 3. The gelators self-assemble into a fibril structure probably through π–π interaction between the aromatic rings and van der Waals interaction between the outside alkyl chains. The fibers then weave into a 3D network and fix the solvents to form a gel through surface tension.10a
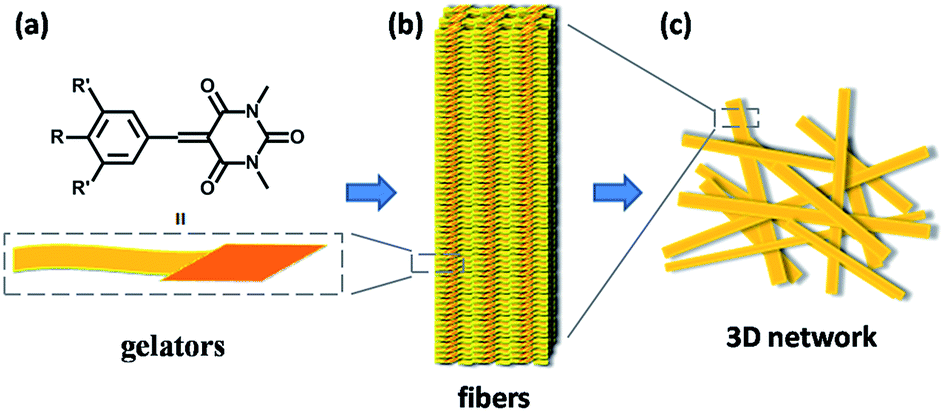 | ||
| Scheme 3 Hierarchical structure model of the K′-n gels: (a) structure model of the gelators; (b) self-assembly of gelators to form fibers as the secondary structure; and (c) 3D network weaved from fibers as the tertiary structure. | ||
Furthermore, to confirm the formation of the gels, the mechanical properties of these organogels were characterized by mechanical rheometry at 20 °C, as shown in Fig. S1.† The storage modulus (G′) describes the ability of the deformed materials to store energy, and the loss modulus (G′′) corresponds to the ability of the material to dissipate energy.19 Strain amplitude sweeps, as shown in Fig. S1†, revealed the elastic response of the gels. The elastic moduli (G′) for all of the gels gradually decreased with increasing strain and above the critical strain region there was a crossover of G′ and G′′, indicating a disruption and collapse of the gel state.20 These results indicate that the studied gels had clear elastic characteristics with a very stable self-assembled structure.
Factors influencing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C/CN exchange
C/CN exchange
Several factors may have an impact on the CC/CN exchange, such as molecular structure, solvent, temperature, etc.13 They have been studied for the reactions of the present compounds (Scheme 1).
Influence of different solvents
It has been found earlier that CC/CN exchange between imines and Knoevenagel derivatives of 1,3-dimethylbarbituric acid was fast and reversible in CDCl3 solution at room temperature in the absence of a catalyst, leading to rapid thermodynamical equilibration.13 Here, we also compared the exchange reaction in different solvents, including chloroform, acetonitrile, DMSO and acetonitrile–ethanol mixture solution.
As imine I-2 bearing a –C2H5 chain was suitably soluble in chloroform, acetonitrile and DMSO, exchange experiments between K and I-2 were monitored by 1H-NMR (see chemical shifts in Table S1, ESI†) in different solvents (Table 2, Fig. S2†). The time to reach equilibrium is different in CDCl3 and CD3CN–EtOD (9:1), being fastest in the medium of lowest polarity, in agreement with earlier results,13 but the same compound distribution was finally reached. Thus, for the component exchange between the present K and I-n constituents, the solvent affects the reaction kinetics, but not the thermodynamics within experimental accuracy. This result serves as a basis for the identification of an eventual gelation-driven perturbation of the compound distribution (see below).
| Solvents | t f | t 1/2 | Compound distribution [%] | ||||
|---|---|---|---|---|---|---|---|
| K | I-2 | K′-2 | I′ | Hydrolysis | |||
|
a CD3CN:EtOD = 9:1 (v/v). Error in 1H-NMR determination by signal integration: ±5%.
|
|||||||
| CDCl3 | 15–20 min | 7 min | 23.8 | 25.2 | 26.2 | 24.8 | — |
| CD3CN | 200–220 min | 70 min | 24.3 | 25.0 | 25.6 | 25.1 | — |
| CD3CN–EtODa | 6–6.5 h | 1.8 h | 25.1 | 24.8 | 25.1 | 25.0 | <1 |
| DMSO | 30 h | 12.5 h | 23.9 | 25.3 | 22.0 | 22.1 | ∼6 |
Influence of chain length on CC/CN exchange
From the data already published13 and further studies,14 it results that both the reactivity and the final compound distribution at equilibrium are strongly influenced by substitution. Here, the para-alkoxy-substituted imines I-n were reacted with the benzyl Knoevenagel compound K (Scheme 1) to check whether chain length would have any influence on the exchange reaction in CDCl3.
The synthesized p-ethoxyl, p-butoxyl, p-octyloxyl, p-hexadecyloxy and p-octadecyloxyl imines (I-n) were reacted with the p-methoxybenzyl Knoevenagel compound (K) in CDCl3 to generate the equilibrium shown in Scheme 1. After recording the 1H-NMR signals (at 3.91 ppm for –OCH3 of K; 4.08 ppm for –OCH2 of I-2; 3.99 ppm for –OCH2 of I-4, I-8, I-16 and I-18; 4.16 ppm for –OCH2 of K′-2; 4.06 ppm for –OCH2 of K′-4, K′-8, K′-16and K′-18; 3.85 ppm for –OCH3 of I′; or see chemical shifts in Table S1†) as a function of time for each exchange reaction, the time to reach equilibrium and the equilibrium distributions were compared (Table S2 and Fig. S3†). The reaction kinetics with different chain length are shown in Fig. S3.† No matter how long the chains are, the time to reach thermodynamic equilibrium was about 20 minutes in all cases and the distributions for both the starting compounds (K, and I-n) and final compounds (K′-n, and I′) were almost the same, around the 25% statistical value. That is to say, chains in the para-position will not cause steric hindrance. Therefore, the length of the chains does not affect (within measurement accuracy) the kinetics and thermodynamic equilibrium of the CC/CN exchange reactions studied here.
Influence of temperature on CC/CN exchange
The effect of temperature on the CC/CN exchange reaction was investigated in acetonitrile where the reaction is neither too fast nor too slow. Specifically, exchange experiments for the K, I-2 pair were recorded in acetonitrile at different temperatures by monitoring the 1H-NMR signals (see chemical shifts in Table S1, ESI†). The changes of distribution with time at 5 °C, 25 °C and 60 °C were followed by monitoring the decrease of starting compound I-2 and the increase of product I′ (Fig. 3). The times for full and half exchange as well as the final distribution are given in Table 3. The time of half exchange at 5 °C, 25 °C and 60 °C was about 112 min, 72 min and 18 min, respectively. The time to reach equilibrium at 5 °C was more than 7 hours, while it was about 1 h at 60 °C. The final distribution for all compounds was the same regardless of the temperature. So, as expected, temperature markedly affected the reaction rates but no change in the thermodynamic equilibrium was observed.
 | ||
| Fig. 3 CC/CN exchange reactions between K and I-2 (10 mM each) in acetonitrile at different temperatures (60 °C, 25 °C, and 5 °C). Error in 1H-NMR determination by signal integration: ±5%. | ||
C/CN exchange reactions between K and I-2, 10 mM each in acetonitrile at different temperatures. The time for full exchange/reaching equilibrium tf is estimated from the moment where no further change is observed. The time for half-exchange t1/2 is obtained from integration of the corresponding signals (see text). Error in 1H-NMR determination by signal integration: ±5%
| Temperature/°C | t f/min | t 1/2/min | Compound distribution [%] | ||||
|---|---|---|---|---|---|---|---|
| K | I-2 | K′-2 | I′ | Hydrolysis | |||
| 5 | ∼450 | 112 | 23.9 | 25.3 | 26.2 | 24.6 | — |
| 25 | ∼240 | 72 | 24.5 | 24.7 | 26.2 | 24.6 | — |
| 60 | 60 | 18 | 24.2 | 25.5 | 25.8 | 24.5 | — |
Gelation-driven component selection in a CC/CN dynamic library
The studies above show that fast exchange occurs under mild conditions at room temperature between compound K and a series of imines I-n in CDCl3 solution. On the other hand, it was found that K′-16, which contains a single –C16H33 chain, forms a gel in acetonitrile and ethanol. It thus appeared to be of much interest to investigate component selection driven by self-organization in the case of the formation of organogels via the present CC/CN covalent dynamic exchange process.
It was first necessary to select a specific solution environment for both the exchange reaction and the gel formation. As I-16 did not dissolve well in pure acetonitrile but dissolved better in ethanol, a 9:1 mixture of acetonitrile and ethanol was used for a 1:1 mixture of compounds K and I-16 at a concentration of 10 mM each, which was compatible with gel formation. The distribution of the constituents was determined as a function of time by monitoring the 1H-NMR signals (see chemical shifts in Table S1, ESI†). In the gel phase, the signals of the gelator K′-16 were very broad as expected (Fig. 4). So, the fractions of the different constituents were first obtained by integration of the signals (for comparison of the whole NMR spectra, see Fig. S4†) of the other compounds (K, I-16, and I′). The amount of K′-16 generated by the exchange reaction is the same as that measured for I′.
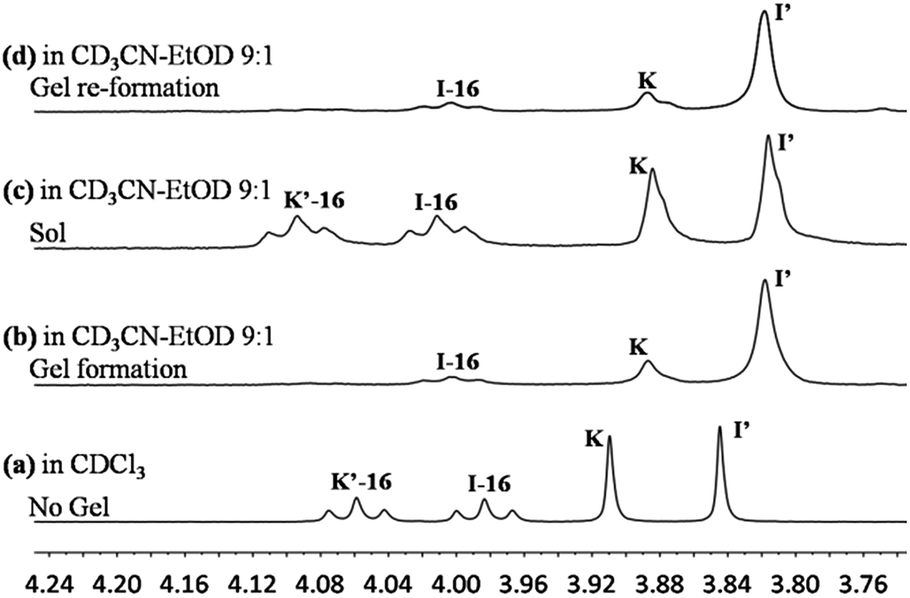 | ||
| Fig. 4
1H-NMR spectra of the exchange reaction of K and I-16 (10 mM each) measured in different states at equilibrium: (a) solution in CDCl3; (b) gel at 22 °C; (c) sol at 60 °C; (d) gel at 22 °C; (b), (c) and (d) in acetonitrile–ethanol 9:1. | ||
The exchange was slow, reaching equilibrium in about 11 hours (Fig. 5). The gel formed from about 3 hours after mixing. After waiting for 14 h to ensure that equilibrium was reached, the mixture of the exchange reaction was analysed, giving the compound distribution as K (10.3%), I-16 (9.4%), K′-16 (40.2%), and I′ (40.1%) (Fig. 4 and 5; gel state). Gel–sol interconversion was performed along temperature cycles (Fig. 4 and 5). Thereafter, the melting temperature of the gel to give a sol was determined to be 52 °C by visual observation. The gel was then heated above that temperature, kept at 60 °C and the change in distribution of the constituents was monitored by 1H-NMR (see chemical shifts in Table S1, ESI†). The compound distribution changed quickly due to faster exchange at this higher temperature, and after about 90 minutes it had the following values: K (24.2%), I-16 (24.8%), K′-16 (25.3%), and I′ (25.7%) (Fig. 4 and 5; sol state). Finally, the temperature was lowered down to 22 °C so that the gel could form again. After reaching equilibrium, the constituent distribution was: K (10.2%), I-16 (10.1%), K′-16 (39.8%), and I′ (39.8%) in the gel state (Fig. 4 and 5; re-gel state), i.e. the same as that when the gel formed for the first time. Considering these results, it is clear that the exchange reaction is reversible, but the more important and interesting point is to determine the reason for the different distributions at the two temperatures.
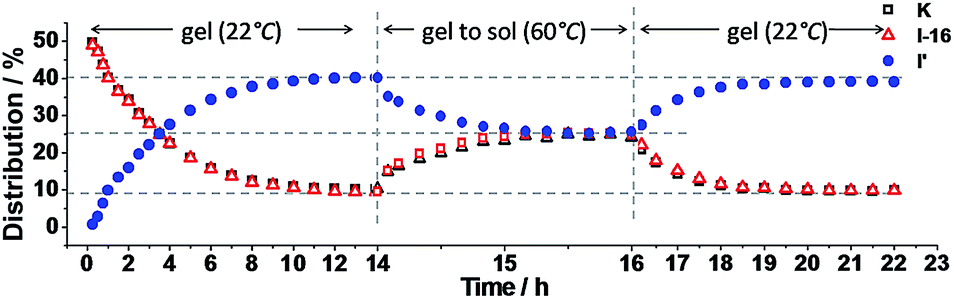 | ||
| Fig. 5 Distribution of K, I-16 and I′ as a function of time. Left: gel formation from K and I-16 (10 mM each in acetonitrile–ethanol 9:1) at 22 °C; middle: from gel state to sol state at 60 °C; right: from sol state to gel state on cooling to 22 °C. Error in 1H-NMR determination by signal integration: ±5% of spectra in Fig. 4. | ||
Comparing the exchange reactions in chloroform and in the acetonitrile–ethanol mixture, the distributions were very different in the gel state (Fig. 6a and b). However, the previous results indicated that different solvents influenced the time to reach equilibrium, but had no influence to the final distribution for the same exchange reaction. That is to say, there was no observable change in the thermodynamic equilibrium when the solvent was changed.
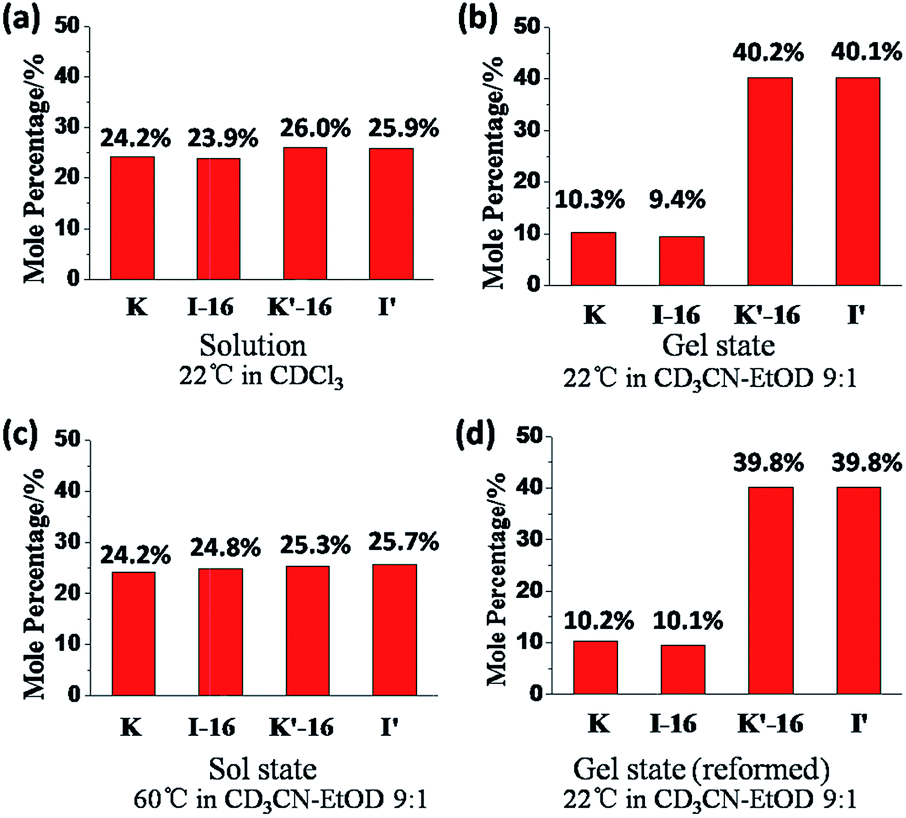 | ||
| Fig. 6 Constituent distribution (K, I-16, K′-16, and I′) in different states after reaching equilibrium: (a) sol at 22 °C in chloroform, solution; (b) gel at 22 °C; (c) sol at 60 °C; (d) gel state reformed on cooling from 60 °C; (b), (c) and (d) in acetonitrile–ethanol 9:1. Error in 1H-NMR determination by signal integration: ±5% of spectra in Fig. 4. | ||
One may conclude that the formation of the gel by the reversible exchange reaction breaks the original thermodynamic equilibrium and led to another thermodynamic equilibrium state corresponding to a different distribution containing more I′ as well as K′-16 with an increase of ∼15%, from ∼25% to ∼40%. Thus, gelation was indeed the driving force for component selection.
At higher temperature, the gel gave a solution and the driving-force of gel formation disappeared, so that the distribution returned to the statistical one of ∼25% for each constituent (Fig. 6c). After cooling down, the sol gave a gel again and the gelation driven force gave back the initial distribution (Fig. 6d).
The previous gelation tests showed that K′-18, K′-(16)2 and K′-(10)3 could also form a gel in a suitable solvent. The exchange experiments using I-18, which contains a –C18H37 chain, in acetonitrile–ethanol gave similar results, as shown in Fig. S5.† However, though K′-(16)2 formed a gel in DMSO, the corresponding imine I-(16)2 was only slightly soluble in this solvent. Similarly, K′-(10)3 formed a gel in acetonitrile and ethanol, but again the corresponding imine I-(10)3 did not dissolve well in these two solvents. So, I-(16)2 and I-(10)3 could unfortunately not be used for exchange reactions.
SEM measurements were performed on the system produced after reaching equilibrium from a mixture of K and I-16 or I-18 (Fig. 7). They again showed strips, which were however not as long and regular as those formed by the corresponding pure gelators (Fig. 1). Probably, the other molecules formed in the exchanging system affected the ordered self-assembly of the gelators.
 | ||
| Fig. 7 SEM pictures of the gels formed after reaching equilibrium from mixing K and (a) I-16 or (b) I-18; insets: photographic images of the gels formed in NMR tubes. | ||
 | ||
| Scheme 4 Constitutional dynamic network representing the agonistic and antagonistic links between the constituents in the process of selection driven by gelation with reversibility upon sol/gel interconversion. The self-organization of the gel phase enforces the formation of the constituent that forms the gel. | ||
Experimental
Experimental procedures and chemical characterization can be found in the ESI.†Conclusions
We have designed and demonstrated a new approach to generating organogels through the dynamic covalent exchange of CC/CN bond recombination. The thermodynamic equilibrium of the CC/CN exchange reactions was not influenced by solvents, chain length, and temperature. In the dynamic network, the optimal gel-forming constituent (the long alkyl chain-containing K′-16 compound) was amplified under the effect of the gelation process. The selective up-regulation of the K′-16 gelator constituent indicated that self-organization acted as a driving force towards higher organization. The present results demonstrate the ability of a highly organized phase (here, an organogel), to direct a constitutional dynamic system towards the selection of the “fittest” components in a function-driven process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ERC (Advanced Research Grant SUPRADAPT 290585) for financial support. Chunshuang Liang would like to acknowledge the funding of the CSC (China Scholarship Council) for a doctoral fellowship. Chunshuang Liang and Shimei Jiang gratefully acknowledge the National Natural Science Foundation of China (51673082). Sirinan Kulchat thanks Khon Kaen University (Thailand) and the Franco-Thai scholarship program for a doctoral fellowship.Notes and references
- (a) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (b) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (c) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (d) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (e) S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 RSC; (f) Dynamic Combinatorial Chemistry, ed. B. L. Miller, Wiley, Chichester, 2010 Search PubMed; (g) Dynamic Combinatorial Chemistry, ed. J. N. H. Reek and S. Otto, Wiley-VCH, Weinheim, 2010 Search PubMed; (h) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC; (i) J.-M. Lehn, in Constitutional Dynamic Chemistry, Topics Curr. Chem, ed. M. Barboiu, Springer, Berlin, 2012, vol. 322, pp. 1–32 Search PubMed.
- (a) J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS; (b) Z. Rodriguez-Docampo and S. Otto, Chem. Commun., 2008, 42, 5301–5303 RSC; (c) J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC; (d) E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC; (e) Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (f) J.-M. Lehn, Adv. Polym. Sci., 2013, 261, 155–172 CrossRef CAS; (g) N. Roy, B. Bruchmannand and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- P. N. W. Baxter, J.-M. Lehn and K. Rissanen, Chem. Commun., 1997, 1323–1324 RSC.
- A. Ciesielski, M. El Garah, S. Haar, P. Kovaříček, J.-M. Lehn and P. Samorì, Nat. Chem., 2014, 6, 1017–1023 CrossRef CAS PubMed.
- N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS PubMed.
- (a) G. T. Wang, J. B. Lin, X. K. Jiang and Z. T. Li, Langmuir, 2009, 25, 8414–8418 CrossRef CAS PubMed; (b) L. Marin, B. Simionescu and M. Barboiu, Chem. Commun., 2012, 48, 8778–8780 RSC.
- L. Hu, Y. Zhang and O. Ramström, Sci. Rep., 2015, 5, 11065 CrossRef CAS PubMed.
- S. K. M. Nalluri and R. V. Ulijn, Chem. Sci., 2013, 4, 3699–3705 RSC.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed; (b) J. H. Jung, J. H. Lee, J. R. Silverman and G. John, Chem. Soc. Rev., 2013, 42, 924–936 RSC; (c) M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC; (d) G. C. Yu, X. Z. Yan, C. Y. Han and F. H. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC; (e) D. K. Smith, Molecular Gels – Nanostructured Soft Materials, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, Weinheim, 2008 Search PubMed; (f) Molecular Gels. Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006 Search PubMed.
- (a) D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS; (b) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- (a) Z. F. Sun, Q. Y. Huang, T. He, Z. Y. Li, Y. Zhang and L. Z. Yi, ChemPhysChem, 2014, 15, 2421–2430 CrossRef CAS PubMed; (b) Y. P. Zhang and S. M. Jiang, Org. Biomol. Chem., 2012, 10, 6973–6979 RSC; (c) L. B. Zang, H. X. Shang, D. Y. Wei and S. M. Jiang, Sens. Actuators, B, 2013, 185, 389–397 CrossRef CAS; (d) Y. Ma, M. Cametti, Z. Džolić and S. M. Jiang, J. Mater. Chem. C, 2016, 4, 10786–10790 RSC.
- S. Kulchat, K. Meguellati and J.-M. Lehn, Helv. Chim. Acta, 2014, 97, 1219–1236 CrossRef CAS.
- K. Flidrova, R. R. Gu and J.-M. Lehn, in preparation.
- J. E. Eldridge and J. D. Ferry, J. Phys. Chem., 1954, 58, 992–995 CrossRef CAS.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS.
- (a) X. H. Lan, L. M. Chen, Y. Fan and T. Yi, Langmuir, 2014, 30, 11753–11760 CrossRef PubMed; (b) J. Peng, J. B. Sun, P. Gong, P. C. Xue, Z. Q. Zhang, G. H. Zhang and R. Lu, Chem.–Asian J., 2015, 10, 1717–1724 CrossRef CAS PubMed; (c) Q. T. Liu, Y. L. Wang, W. Li and L. X. Wu, Langmuir, 2007, 23, 8217–8223 CrossRef CAS PubMed.
- (a) G. H. Hong, J. B. Sun, C. Qian, P. C. Xue, P. Gong, Z. Q. Zhang and R. Lu, J. Mater. Chem. C, 2015, 3, 2371–2379 RSC; (b) H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., Int. Ed., 2010, 49, 7688–7691 CrossRef CAS PubMed.
- H. A. Barnes, J. F. Hutton and F. R. S. Walters, An Introduction To Rheology, P. O. Box 211, 1000, AE Amsterdam, The Netherlands, 3rd edn, 1993 Search PubMed.
- (a) S. K. Samanta, A. Pal, S. Bhattacharya and C. N. R. Rao, J. Mater. Chem., 2010, 20, 6881–6890 RSC; (b) Y. Zhang, H. Ding, Y. F. Wu, C. X. Zhang, B. L. Bai, H. T. Wang and M. Li, Soft Matter, 2014, 10, 8838–8845 RSC.
- G. Vantomme, S. M. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS PubMed.
- J. Holub, G. Vantomme and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02827j |
| This journal is © The Royal Society of Chemistry 2017 |