
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA β-Carbon elimination strategy for convenient in situ access to cyclopentadienyl metal complexes†
G.
Smits‡
a,
B.
Audic
 a,
M. D.
Wodrich
b,
C.
Corminboeuf
*b and
N.
Cramer
*a
a,
M. D.
Wodrich
b,
C.
Corminboeuf
*b and
N.
Cramer
*a
aLaboratory of Asymmetric Catalysis and Synthesis, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
bLaboratory for Computational Molecular Design, EPFL SB ISIC LCMD, BCH 5312, CH-1015 Lausanne, Switzerland. E-mail: clemence.corminboeuf@epfl.ch
First published on 24th August 2017
Abstract
The electronic and steric properties of tailored cyclopentadienyl (Cp) ligands are powerful handles to modulate the catalytic properties of their metal complexes. This requires the individual preparation, purification and storage of each ligand/metal combination. Alternative, ideally in situ, complexation protocols would be of high utility. We disclose a new approach to access Cp metal complexes. Common metal precursors rapidly react with cyclopentadienyl carbinols via β-carbon eliminations to directly give the Cp-metal complexes. An advantage of this is the direct and flexible use of storable pre-ligands. No auxiliary base is required and the Cp complexes can be prepared in situ in the reaction vessel for subsequent catalytic transformations.
Introduction
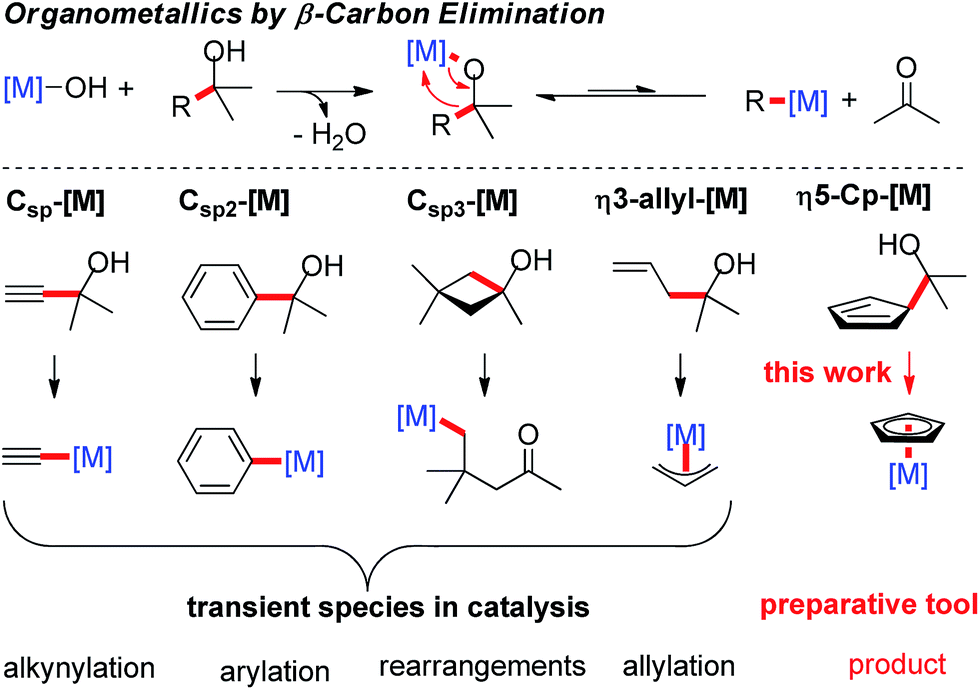
Cyclopentadienyl (Cp)-coordinated transition-metal complexes are ubiquitous and many of them are efficient catalysts for a broad range of versatile, atom-economic transformations.1 Many of these reactions have highly optimized conditions, but use commercially available complexes with a conserved Cp* or Cp ligand. Only recently, the modulation of the electronic and steric properties of tailored Cp ligands was recognized as a powerful tool to overcome sluggish reactivity,2 to address regio- and positional issues,3 and provide entry to enantioselective processes.4 In rapid reaction discovery and optimization, the ability to combine a library of ligands with a library of metal complexes, performing an in situ complexation to give the desired catalyst species is a relevant advantage. While this approach is very common for reactions involving, for instance, phosphine ligands, it is still elusive for Cp ligands. In this case, each ligand/metal combination has to be synthesized individually, purified and stocked prior to any use in catalysis. The typical complexation of common Cp* and Cp complexes involves the metal as a limiting reagent.5 However, highly elaborate Cp or chiral Cpx ligands require the use of the CpH derivative as the limiting reactant. In particular for chiral Cpx ligands, one has to rely on undesirable reaction conditions, for example involving thallium alkoxide in benzene.4d,6 These shortcomings make the development of complementary complexation strategies a priority. Ideally, such technology proceeds rapidly and in a quantitative manner, without the generation of inhibiting reaction byproducts. It should also be useable with a range of different transition-metals.β-Carbon elimination has been reported as a complementary method to access organometallic species in catalysis.7 Normally the forward reaction – the addition across a carbonyl group – is favored and reversing this pathway requires some additional driving force. This could be a combination of using the right transition-metal and the use of substrates leading to the formation of stronger Csp–[M]8 or Csp2–[M] bonds.9 The generation of Csp3–[M] bonds requires energy-rich starting materials, such as tert-cyclobutanols10, releasing strain upon β-C elimination.11 A particular class of substrate is that of homoallylic alcohols that can give π-bound allyl-metal species by retro-allylation.12 Along the same lines, a cyclopentadienyl carbinol would give a Cp–[M] species upon C–C bond cleavage. The very strong bond of the cyclopentadienyl anion to the transition-metal would be a very strong driving force. So far, the β-C elimination methodology has been used exclusively as an elementary step in catalytic transformations. Herein we exploit its potential for the preparation of Cp-metal complexes (Scheme 1).
 | ||
| Scheme 1 Generation of organometallics using the β-carbon elimination strategy. | ||
Results and discussion
The required cyclopentadienyl carbinol substrates for the pre-ligands are accessed in a straight forward manner by deprotonation and addition across the desired aldehyde or ketone (Scheme 2, see ESI† for details).13 In contrast to many lightly substituted cyclopentadienes which frequently undergo Diels–Alder dimerization, all prepared Cp-carbinols are stable and do not dimerize upon storage. Exemplarily for [Rh(cod)OH]2, ligand exchange of the hydroxy ligand by 2 would break up the dimer and release a molecule of water to give intermediate 3. In turn, 3 is predisposed for the final β-C elimination step to yield the Cp metal complex 4a and ketone 1.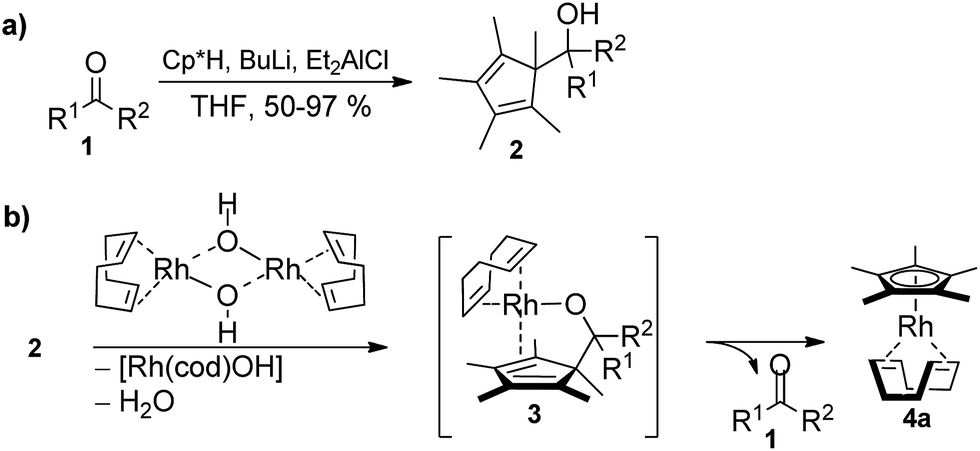 | ||
| Scheme 2 (a) Synthesis of Cp pre-ligand carbinols 2. (b) Envisioned β-C elimination to access Cp*Rh(I) complexes. | ||
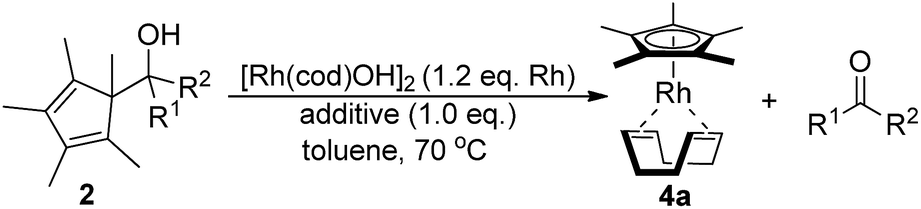
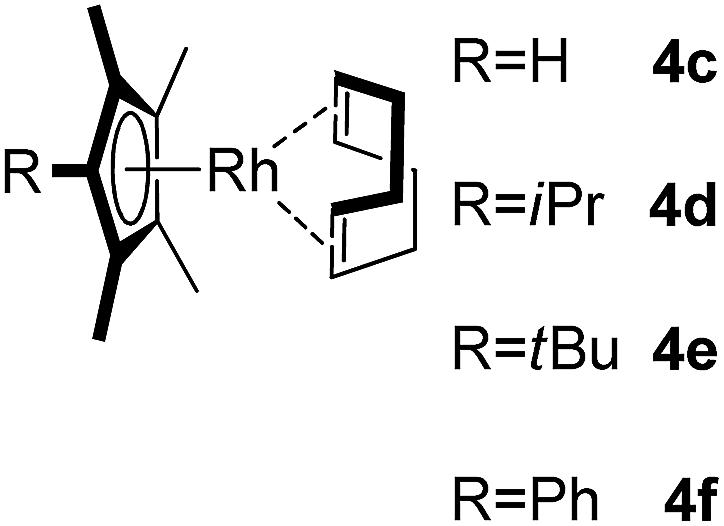
A selection of carbinols with sterically and electronically different substituents, R1 and R2, were initially evaluated (Table 1). A variety of factors influenced the reaction performance. An aromatic substituent R1 or R2 was found to be beneficial for yield and reactivity. A tertiary hydroxyl group is better than a secondary one. Strained ketone derived 2f and 2g (entries 6–7) partially underwent the alternative strain-release pathway,10 opening the four-membered ring instead of leading to the Cp-metal species. Most substrates required the addition of cesium carbonate for shorter reaction times. Without the rhodium complex, carbinols 2 are stable and no conversion to free Cp*H was observed. Notably, some substrates displayed significantly increased reactivity and did not require the addition of any base (entries 8, 10, 13, and 15). The two fastest reactions were 2h (Ph/CO2Me) and 2m (Ph/Me).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2 | R1 | R2 | Additive | Time | % yieldb |
| a Conditions: N2 atmosphere, 0.040 mmol 2, 0.024 mmol [Rh(cod)OH]2, 0.040 mmol additive, 0.2 M in toluene at 70 °C for the indicated time. b Yield determined by NMR with an internal standard. | ||||||
| 1 | 2a | H | nPentyl | Cs2CO3 | 3 h | 39 |
| 2 | 2b | H | Ph | Cs2CO3 | 1 h | 95 |
| 3 | 2c | H | CO2Et | Cs2CO3 | 3 h | 10 |
| 4 | 2d | Me | Me | Cs 2 CO 3 | 3 h | 99 |
| 5 | 2e | CH2(CH2)3CH2 | Cs2CO3 | 3 h | 93 | |
| 6 | 2f | CH2CH2CH2 | Cs2CO3 | 1 h | 30 | |
| 7 | 2g | CH2OCH2 | Cs2CO3 | 3 h | 20 | |
| 8 | 2h | CO 2 Me | P h | — | 15 min | 75 |
| 9 | 2i | CO2Me | Me | Cs2CO3 | 3 h | 75 |
| 10 | 2j | CO2Me | t Bu | — | 1 h | 45 |
| 11 | 2k | CO2Me | CF3 | Cs2CO3 | 1 h | <5 |
| 12 | 2l | CO2Et | CO2Et | Cs2CO3 | 3 h | <5 |
| 13 | 2m | Me | P h | — | 15 min | 89 |
| 14 | 2n | Me | Cy | Cs2CO3 | 3 h | 80 |
| 15 | 2o | CF3 | Ph | — | 1 h | 95 |
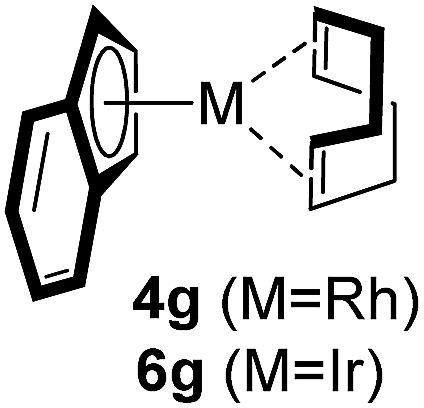
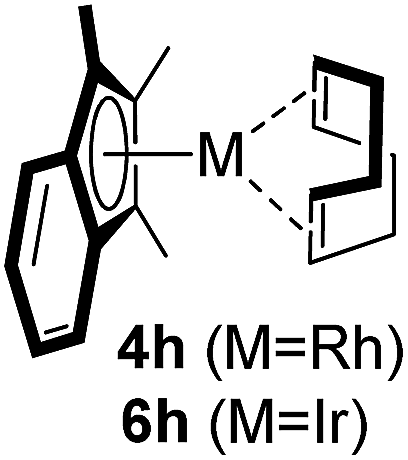
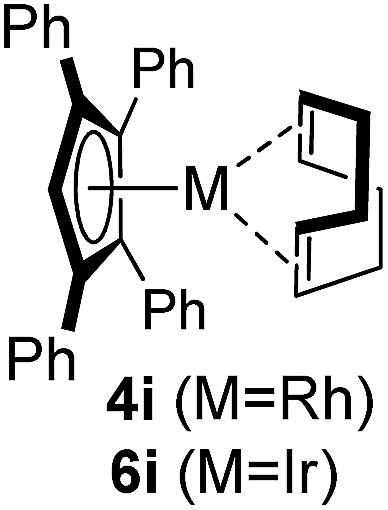
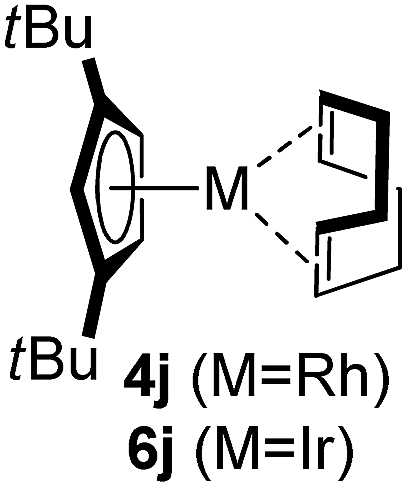
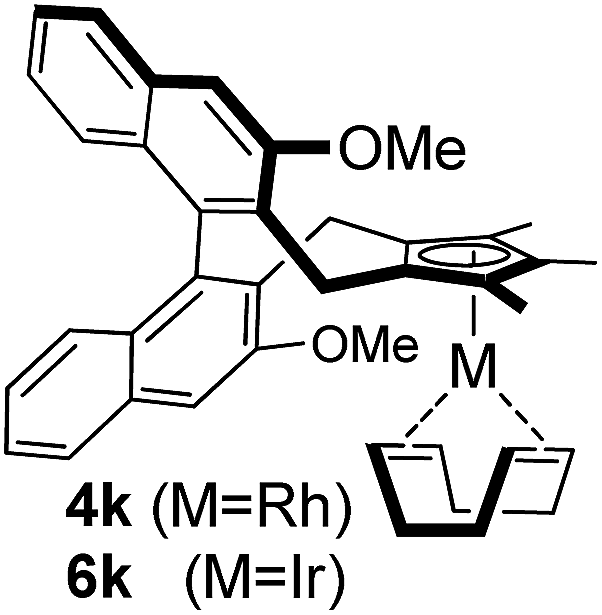
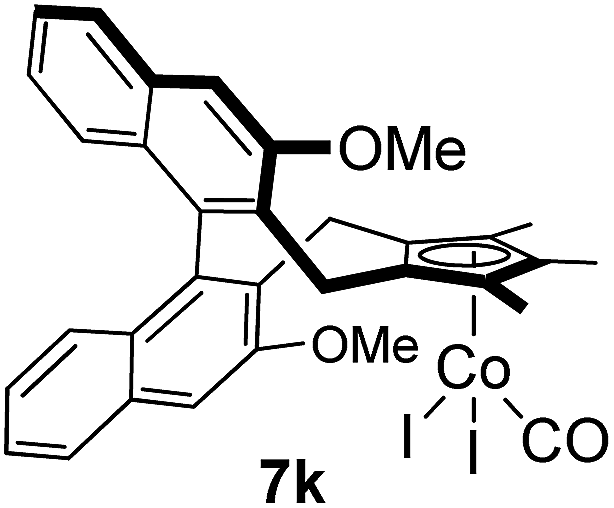
Among the tested substrates, the dimethyl-type carbinol moiety was further investigated, despite its lower reactivity, as it releases only volatile acetone as a byproduct (Table 2). It provides a clean complexation giving a range of complexes 4 with different bulks, ranging from Me3Cp (entry 1) to tBuMe4Cp (entry 5). While substrates with bulky groups react very smoothly, less substituted ones like the trimethyl Cp precursor 5b react less efficiently due to some fulvene byproduct formation. Indenyl complexes can be prepared with similar efficiency (entries 6–7). Methyl phenylglyoxylate and acetophenone derived pre-ligands 5i and 5j are easier to access and better suited in the complexation giving, as before, fast and clean transformations without the requirement of a basic additive (entries 8–9). Importantly, this complexation strategy is equally successful for chiral Cp* ligands equipped with our atropchiral backbone.4f–i Importantly, the β-C eliminative complexation is not limited to rhodium. For instance, using [Ir(cod)OH]2 as the metal salt provided access to indenyl Ir(cod) and cyclopentadienyl complexes 6g–6j in comparable yields (entries 6–9). Exposure of the chiral Cpx* pre-ligand 5k to the reaction conditions induced a smooth complexation, providing rhodium complex 4k in 93% yield (entry 10). This is particularly noteworthy as classical complexation methods4d,f for a di-substituted chiral Cpx ligand failed completely for this chiral penta-substituted analog. Analogously, the chiral iridium complex 6k could be obtained in 87% yield. Moreover, we could extend the method for the preparation of a chiral Cpx*CoIII complex 7k (entry 11). For the first time, chiral fully penta-substituted cyclopentadienyl rhodium, iridium and cobalt complexes are now accessible. This sets the stage for future applications in asymmetric catalysis, especially for transformations where previously reported complexes with the disubstituted chiral variants4a,b failed to provide adequate reactivity.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 5 | R1 | R2 | 4 | % yieldb |
| a Conditions: 0.04 mmol 5, 0.024 mmol of [Rh(cod)OH]2, 0.040 mmol Cs2CO3, 0.2 mL toluene at 70 °C, 15 min–10 h. b Determined by NMR with an internal standard. c 4 Å molecular sieves instead of Cs2CO3 at 23 °C. d With [Ir(cod)OH]2 for 6. e Isolated yield. f Conditions: 0.06 mmol 5k and 36 μmol Co2(CO)8, 0.4 mL CH2Cl2 at 40 °C, then I2 (0.06 mmol). | |||||
| 1 | 5b | Me | Me |
|
42 |
| 2 | 5c | Me | Me |
|
80 |
| 3 | 5d | Me | Me | 95 | |
| 4 | 5e | Me | Me | 90 | |
| 5 | 5f | Me | Me | 95 | |
| 6d | 5g | Me | Me |
|
90 (4g) |
| 88 (6g) | |||||
| 7d | 5h | Me | Me |
|
95 (4h) |
| 86 (6h) | |||||
| 8c,d | 5i | Ph | CO2Me |
|
60 (4i) |
| 44(6j) | |||||
| 9c,d | 5j | Ph | Me |
|
95 (4j) |
| 63 (6j) | |||||
| 10d | 5k | Ph | CO2Me |
|
93 (4k)e |
| 87 (6k)e | |||||
| 11f | 5k | Ph | CO2Me |
|
32e |
Besides the hydroxyl bridged dimer, other [Rh(cod)X]2 complexes (X = OMe, OAc, and Cl) can be used (Table 3, entries 1–3). The chloride containing complexes require the addition of KOH for an in situ exchange. Moreover, [Rh(nbd)X]2 (nbd = norbornadiene) and [Rh(CO)2Cl]2 work similarly (entries 4–6). Without a change in protocol, the corresponding iridium(I) complex 6a and cobalt(0) complex 8 were prepared (entries 7–8). Solely the rhodium ethylene congener is not well suited for these conditions (entry 9). However, an improved protocol (vide infra) solved this issue and provided 4n and 4o (entries 10 and 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | CpM | [M] | Additive | Time | % yieldb |
| a Conditions: 0.04 mmol 2d, 24 μmol [M], 0.04 mmol Cs2CO3 or 0.12 mmol KOH, 0.2 mL toluene. b Determined by NMR with an internal standard. c Conditions: 0.06 mmol 2d and 36 μmol Co2(CO)8, 0.4 mL CH2Cl2 at 40 °C. d With 2m instead of 2d at 23 °C. e With 2m instead of 2d. f Isolated yield. | |||||
| 1 | 4a | [Rh(cod)OMe]2 | Cs2CO3 | 3 h | 70 |
| 2 | 4a | [Rh(cod)OAc]2 | Cs2CO3 | 3 h | 70 |
| 3 | 4a | [Rh(cod)Cl]2 | KOH | 3 h | 90 |
| 4 | 4l | [Rh(nbd)Cl]2 | KOH | 2 h | 80 |
| 5 | 4l | [Rh(nbd)OH]2 | Cs2CO3 | 3 h | 99 |
| 6 | 4m | [Rh(CO)2Cl]2 | KOH | 15 min | 49 |
| 7 | 6a | [Ir(cod)OH]2 | CS 2 CO 3 | 5 h | 92 |
| 8c | 8 | [Co2(CO)8] | — | 6 h | 60 |
| 9 | 4n | [Rh(C2H4)2OAc]2 | Cs2CO3 | 1 h | 10 |
| 10d | 4n | [Rh(C2H4)2Cl]2 | KOH, 4 Å MS | 5 h | 70 |
| 11e | 4o | [Rh(C2H3TMS)Cl]2 | KOH, 4 Å MS | 1 h | 66f |
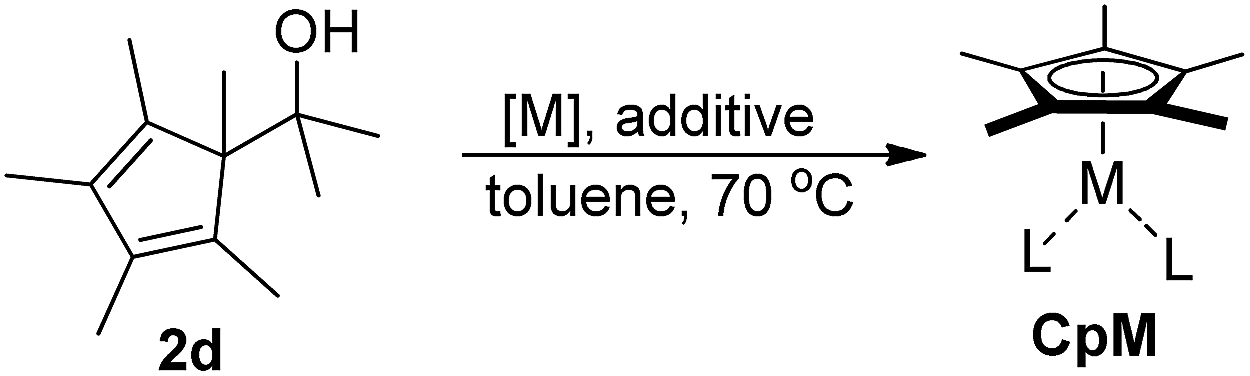
For the Ph/Me substrate 2m, DFT computations (at the PBE014-dDsC15/TZ2P//M0616/def2-SVP level in implicit toluene solvent using COSMO-RS,17 see ESI† for additional details) provided some more details on the reaction profile of the complexation reaction (Fig. 1). First, the [Rh(cod)OH] fragment forms complex 3m with the substrate carbinol. Cleavage of the C–C bond proceeds viaTS1 with a barrier of only 16.3 kcal mol−1, which aligns well with the experimentally observed fast reaction. After C–C cleavage, a metastable species (Int 1) is formed which quickly dissociates to the products 4a and 1m after dissociation of the ketone (TS2). The overall process is thermodynamically favourable, being exergonic by 34.5 kcal mol−1.
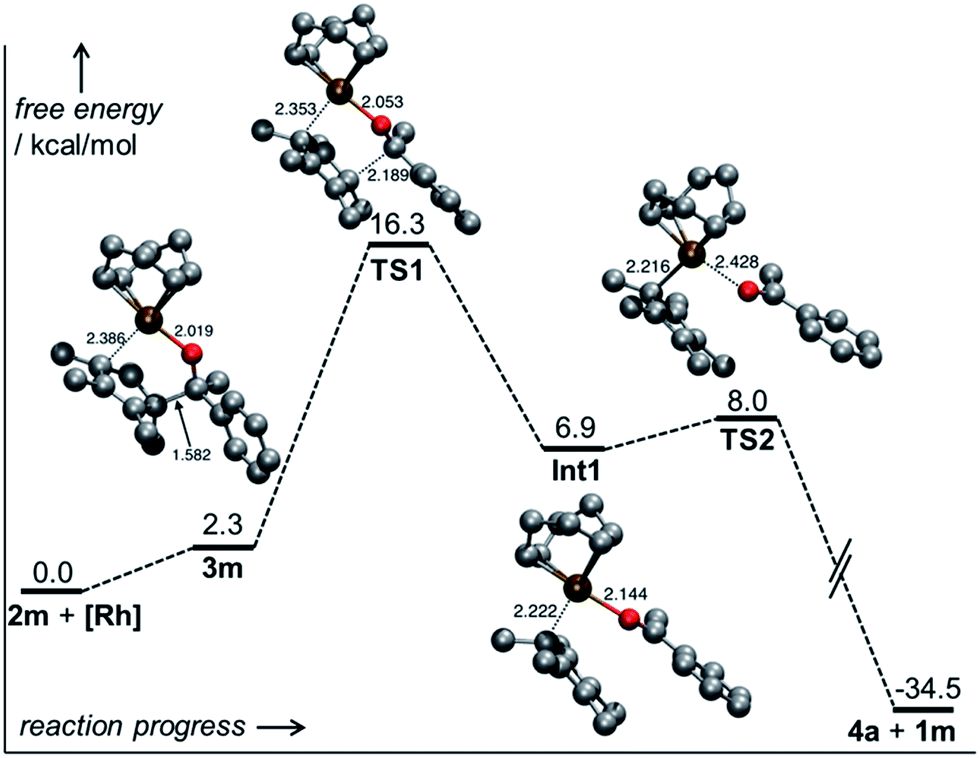 | ||
| Fig. 1 Reaction profile of the β-C elimination of carbinol 2m. | ||
While the outlined protocol works very well for robust diene containing metal precursors, some more sensitive ones, e.g. [Rh(C2H4)2OAc]2 resulted in unsatisfactory yields despite full conversion. DFT computations indicated that the height of the β-C elimination barrier was not the issue. We hypothesized that the reaction rate could be negatively impacted by the increasing amount of formed water. This could compete with the sterically hindered cyclopentadienyl carbinol and be the limiting factor in the ligand exchange of the metal hydroxy complex. Indeed, the addition of 4 Å molecular sieves as traps for the generated water byproduct accelerates the reaction significantly (Table 3, entry 10). Instead of 70 °C, fast and complete complexation occurs at ambient temperature. Under these conditions, the β-carbon elimination was monitored by 1H-NMR spectroscopy (Fig. 2). From all of the tested substrates, the initially selected Cp* dimethyl carbinol 2d was the slowest one. Methyl phenylglyoxylate derived substrate 2h initially reacts very fast, but stalls at around 80% yield. Acetophenone derived substrate 2m reacts smoothly and cleanly, producing the desired complex in 90% yield after 30 minutes and quantitatively after 1 hour at ambient temperature. Moreover, some dependence on the substitution pattern of the aryl group was found. For instance, the p-nitro derivative 2p reacts fastest and the p-methoxy congener 2q is slower than the parent substrate 2m. DFT results of this substrate set confirmed the observed reactivity trends. Fig. 2b clearly shows that the substitution pattern strongly influences the TS1 barrier height, with the slower substrate 2d (21.1 kcal mol−1) having the highest barrier and the fastest substrate, p-NO2 bearing 2p, having the lowest (15.4 kcal mol−1).
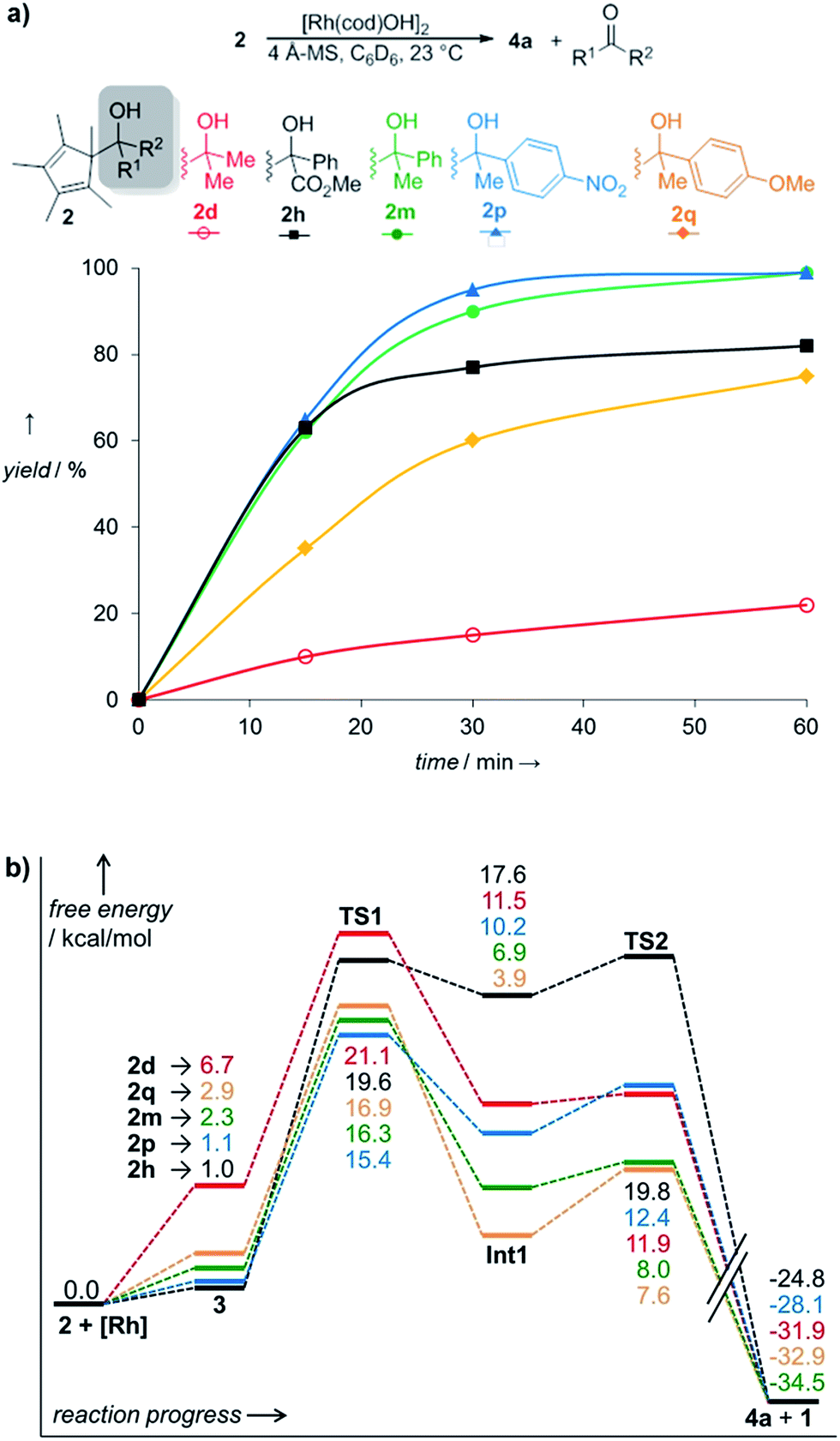 | ||
| Fig. 2 (a) Complexation rates of different carbinols 2; yields of 4a after 60 min: 2d: 22%; 2h: 82%; 2m: 99%; 2p: 99%; 2q: 75%. (b) Reaction profiles of carbinols 2 during complexation. | ||
Besides base-free complexation avoiding toxic solvents and bases, the instant preparation and direct subsequent use in catalysis of the formed Cp-metal complex is a salient feature of this protocol (Scheme 3). As an example for Rh(III)-catalysis, the regioselective dihydroisoquinolone formation with terminal 1-decene is shown using the in situ formed CptRh(I) complex 4p. This protocol provides 10 in similar yield and regioselectivity as previously reported by Rovis with an isolated CptRh complex.3a,b The simple precatalyst 2d can be used to access 4a, which in turn was used for the synthesis of isoquinolone 12 with reversed regioselectivity, again with comparable yield and regioselectivity as previously observed by us.18 The vinyl-TMS bearing complex 4o is hardly accessible from the reduction of [Cp*RhCl2] and simplifies applications in Cp*Rh(I) catalysis. For instance, it is a competent catalyst for intramolecular transfer olefinations19 of piperidinyl amide 13 providing enamide 14. In addition, chiral CpxIr(I) complex 6k could be smoothly oxidized with iodine to the Ir(III) species 15. This catalyst provides promising preliminary results for oxime-directed asymmetric C(sp3)–H functionalizations of substrate 16 yielding the tosylamide product 17.20
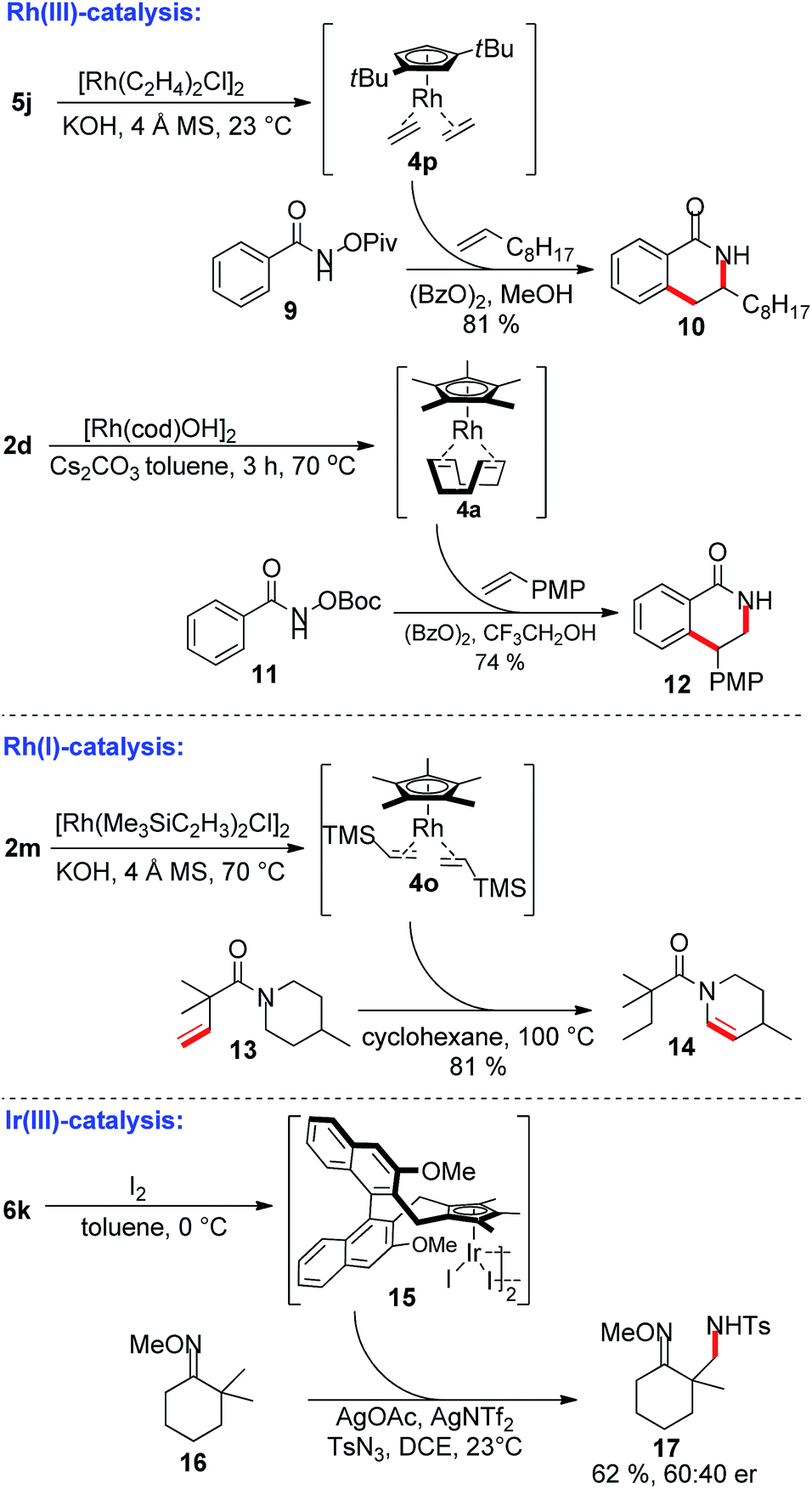 | ||
| Scheme 3 In situ Cp metal complex preparation and direct application in Rh(III), Rh(I) and Ir(III) catalysis. PMP = 4-methoxyphenyl. | ||
Conclusions
In summary, we have reported a new complexation strategy to access late transition-metal Cp complexes. Rapid β-carbon elimination of cyclopentadienyl carbinols in the presence of a common metal precursor provides access to a wide range of Cp metal complexes. The advantages of this process are the direct and flexible use of storable pre-ligands. No auxiliary base is required and the Cp complexes can be prepared in situ in the reaction vessel. This protocol should enhance the convenience in applying these complexes in catalysis and allow for more rapid exploitation of the untapped potential of tailored Cp ligands. In particular, we expect this is a door-opener for further application of chiral Cp* metal complexes in catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the EPFL and the Swiss National Science Foundation Consolidator Grant no. 157741. G. S. thanks the EC 7th Framework Program project REGPOT-CT-2013-316149-InnovaBalt.Notes and references
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- (a) T. A. Davis, C. Wang and T. Rovis, Synlett, 2015, 26, 1520 CrossRef CAS PubMed; (b) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292 CrossRef CAS PubMed; (c) T. Piou and T. Rovis, Nature, 2015, 527, 86 CrossRef CAS PubMed; (d) F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892 CrossRef CAS PubMed; (e) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS; (f) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (g) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10917 CrossRef CAS PubMed.
- (a) T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2015, 6, 254 RSC; (b) T. K. Hyster and T. Rovis, Chem. Sci., 2011, 2, 1606 RSC; (c) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 CrossRef CAS PubMed; (d) M. D. Wodrich, B. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem. –Eur. J., 2014, 20, 15409 CrossRef CAS PubMed.
- For a recent review see: (a) C. G. Newton, D. Kossler and N. Cramer, J. Am. Chem. Soc., 2016, 138, 3935 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMedfor selected examples, see: (c) G. Erker and A. A. H. van der Zeijden, Angew. Chem., Int. Ed., 1990, 29, 512 CrossRef; (d) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (e) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed; (f) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636 CrossRef CAS PubMed; (g) D. Kossler and N. Cramer, J. Am. Chem. Soc., 2015, 137, 12478 CrossRef CAS PubMed; (h) G. Song, W. N. O. Wylie and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed; (i) M. Dieckmann, Y.-S. Jang and N. Cramer, Angew. Chem., Int. Ed., 2015, 127, 12317 CrossRef.
- (a) R. Dubois, US4962253 A, The Dow Chemical, 1990; (b) R. B. King, Inorg. Chem., 1963, 2, 528 CrossRef; (c) M. D. Rausch and R. A. Genetti, J. Org. Chem., 1970, 35, 3888 CrossRef CAS; (d) N. Weding, R. Jackstell, H. Jiao, A. Spannenberg and M. Hapke, Adv. Synth. Catal., 2011, 353, 3423 CrossRef CAS; (e) C. White, A. Yates, P. M. Maitlis and D. M. Heinekey, in Inorg. Synth., John Wiley & Sons, Inc., 2007, p. 228 Search PubMed; (f) H. Chen, J. Hartwig and T. C. Semple, WO 01/64689 A1, Shell Internationale Research Maatschappij B.V., Yale University, 2001.
- (a) T. E. Bitterwolf, T. L. Hubler and A. L. Rheingold, J. Organomet. Chem., 1992, 431, 199 CrossRef CAS; (b) B. G. Conway and M. D. Rausch, Organometallics, 1985, 4, 688 CrossRef CAS; (c) P. G. Gassman and C. H. Winter, J. Am. Chem. Soc., 1986, 108, 4228 CrossRef CAS; (d) S. S. Jones, M. D. Rausch and T. E. Bitterwolf, J. Organomet. Chem., 1993, 450, 27 CrossRef CAS.
- For a review, see: (a) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272 CrossRef CAS; (b) T. Seiser and N. Cramer, Synlett, 2011, 4, 449 Search PubMed; (c) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (d) K. Ruhland, Eur. J. Org. Chem., 2012, 14, 2683 CrossRef; (e) M. Miura and T. Satoh, in Palladium in Organic Synthesis, ed. J. Tsuji, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 1–20 Search PubMed.
- For selected examples, see: (a) R. Shintani, K. Takatsu, T. Katoh, T. Nishimura and T. Hayashi, Angew. Chem., Int. Ed., 2008, 47, 1447 CrossRef CAS PubMed; (b) A. Horita, H. Tsurugi, A. Funayama, T. Satoh and M. Miura, Org. Lett., 2007, 9, 2231 CrossRef CAS PubMed; (c) T. Nishimura, T. Katoh, K. Takatsu, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 14158 CrossRef CAS PubMed; (d) R. Shintani, K. Takatsu, T. Katoh, T. Nishimura and T. Hayashi, Angew. Chem., Int. Ed., 2008, 47, 1447 CrossRef CAS PubMed; (e) A. Funayama, T. Satoh and M. Miura, J. Am. Chem. Soc., 2005, 127, 15354 CrossRef CAS PubMed; (f) T. Nishimura, H. Araki, Y. Maeda and S. Uemura, Org. Lett., 2003, 5, 2997 CrossRef CAS PubMed.
- For selected examples, see: (a) X. Yu, J. Wang, W. Guo, Y. Tian and J. Wang, Organometallics, 2016, 35, 1876 CrossRef CAS; (b) E. Ozkal, B. Cacherat and B. Morandi, ACS Catal., 2015, 5, 6458 CrossRef CAS; (c) J. R. Bour, J. C. Green, V. J. Winton and J. B. Johnson, J. Org. Chem., 2013, 78, 1665 CrossRef CAS PubMed; (d) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244 CrossRef CAS PubMed; (e) T. Uto, M. Shimizu, K. Ueura, H. Tsurugi, T. Satoh and M. Miura, J. Org. Chem., 2008, 73, 298 CrossRef CAS PubMed; (f) M. Nakano, T. Satoh and M. Miura, J. Org. Chem., 2006, 71, 8309 CrossRef CAS PubMed; (g) P. Zhao, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 3124 CrossRef CAS PubMed; (h) Y. Terao, M. Nomoto, T. Satoh, M. Miura and M. Nomura, J. Org. Chem., 2004, 69, 6942 CrossRef CAS PubMed; (i) Y. Terao, H. Wakui, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2001, 123, 10407 CrossRef CAS PubMed.
- (a) N. Ishida, N. Ishikawa, S. Sawano, Y. Masuda and M. Murakami, Chem. Commun., 2015, 51, 1882 RSC; (b) L. Souillart and N. Cramer, Chem. Sci., 2014, 5, 837 RSC; (c) N. Ishida, S. Sawano and M. Murakami, Chem. Commun., 2012, 48, 1973 RSC; (d) N. Ishida, S. Sawano, Y. Masuda and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17502 CrossRef CAS PubMed; (e) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed; (f) T. Seiser and N. Cramer, Chem.–Eur. J., 2010, 16, 3383 CrossRef CAS PubMed; (g) T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320 CrossRef CAS PubMed; (h) M. Shigeno, T. Yamamoto and M. Murakami, Chem.–Eur. J., 2009, 15, 12929 CrossRef CAS PubMed; (i) S. Matsumura, Y. Maeda, T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2003, 125, 8862 CrossRef CAS PubMed; (j) R. C. Laroc, Ch and K. Reddy, Org. Lett., 2000, 1, 3325 CrossRef; (k) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010 CrossRef CAS.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103 CrossRef CAS.
- For a review see: (a) H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 778 CrossRef CASfor selected examples, see: (b) M. Waibel and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 4455 CrossRef CAS PubMed; (c) R. Wakabayashi, D. Fuijino, S. Hayashi, H. Yorimitsu and K. Oshima, J. Org. Chem., 2010, 75, 4337 CrossRef CAS PubMed; (d) M. Iwasaki, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 4463 CrossRef CAS PubMed; (e) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2006, 128, 2210 CrossRef CAS PubMed; (f) T. Kondo, K. Kodoi, E. Nishinaga, T. Okada, Y. Morisaki, Y. Watanbe and T. Mitsudo, J. Am. Chem. Soc., 1999, 120, 5587 CrossRef; (g) J. Nokami, K. Yoshizane, H. Matsuura and S. Sumida, J. Am. Chem. Soc., 1998, 120, 6609 CrossRef CAS.
- M. Uemura, K. Yagi, M. Iwasaki, K. Nomura, H. Yorimitsu and K. Oshima, Tetrahedron, 2006, 62, 3523 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- (a) S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2010, 6, 1990 CrossRef CAS PubMed; (b) S. N. Steinmann and C. Corminboeuf, Chimia, 2011, 65, 240 CrossRef CAS PubMed; (c) S. N. Steinmann and C. Corminboeuf, J. Chem. Phys., 2011, 134, 044117 CrossRef PubMed; (d) S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2011, 7, 3567 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- A. Klamt, WIREs Comput. Mol. Sci., 2011, 1, 699 CrossRef CAS.
- M. D. Wodrich, B. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem. Eur. J., 2014, 20, 15409 CrossRef CAS PubMed.
- (a) B. DeBoef, S. J. Pastine and D. Sames, J. Am. Chem. Soc., 2004, 126, 6556 CrossRef CAS PubMed; (b) C. P. Langes, P. S. White and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 4385 CrossRef.
- T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation of all new compounds. See DOI: 10.1039/c7sc02986a |
| ‡ Present address: Laboratory of CNS Active Compounds, Latvian Institute of Organic Synthesis, Aizkraukles 21, LV-1006, Riga, Latvia. |
| This journal is © The Royal Society of Chemistry 2017 |