
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDearomatization of electron poor six-membered N-heterocycles through [3 + 2] annulation with aminocyclopropanes†
Johannes
Preindl
,
Shyamal
Chakrabarty‡
and
Jérôme
Waser
 *
*
Laboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 25th August 2017
Abstract
Many abundant and highly bioactive natural alkaloids contain an indolizidine skeleton. A simple, high yielding method to synthesize this scaffold from N-heterocycles was developed. A wide range of pyridines, quinolines and isoquinolines reacted with donor–acceptor (DA)-aminocyclopropanes via an ytterbium(III) catalyzed [3 + 2] annulation reaction to give tetrahydroindolizine derivatives. The products were obtained with high diastereoselectivities (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as anti-isomers. Additionally, the formed aminals could be easily converted into secondary and tertiary amines through iminium formation followed by reduction or nucleophile addition. This transformation constitutes the first example of dearomatization of electron-poor six-membered heterocycles via [3 + 2] annulation with DA cyclopropanes.
:1) as anti-isomers. Additionally, the formed aminals could be easily converted into secondary and tertiary amines through iminium formation followed by reduction or nucleophile addition. This transformation constitutes the first example of dearomatization of electron-poor six-membered heterocycles via [3 + 2] annulation with DA cyclopropanes.
1. Introduction
The indolizidine skeleton is widely represented in bioactive alkaloids.1 For example, castanospermine (1, Scheme 1) is a potent inhibitor of α-glucosidase I, an enzyme with a critical role in viral maturation, and was the lead structure for celgosivir which is currently under investigation for treatment of hepatitis C virus infection and dengue fever.2 The indolizidine class of natural products also includes more complex polycyclic compounds incorporating further fused saturated or unsaturated rings.3 For instance, isoschizogaline (2) contains a reduced quinoline core structure,3a whereas jamtine (3)3b or haiderine (4)3c can be seen as isoquinoline derived alkaloids. The construction of these polycyclic scaffolds by dearomatization of quinolines, isoquinolines or pyridines is highly attractive, due to the broad availability of the unsaturated heterocycles. Classic dearomatization strategies most often rely on the formation of a single bond, starting from activated pyridinium or (iso)quinolinium intermediates.4 Dearomatization reactions through direct annulation via ring-extension of cyclopropanes would provide a more convergent synthesis (Scheme 1). Nevertheless, such processes are unknown.5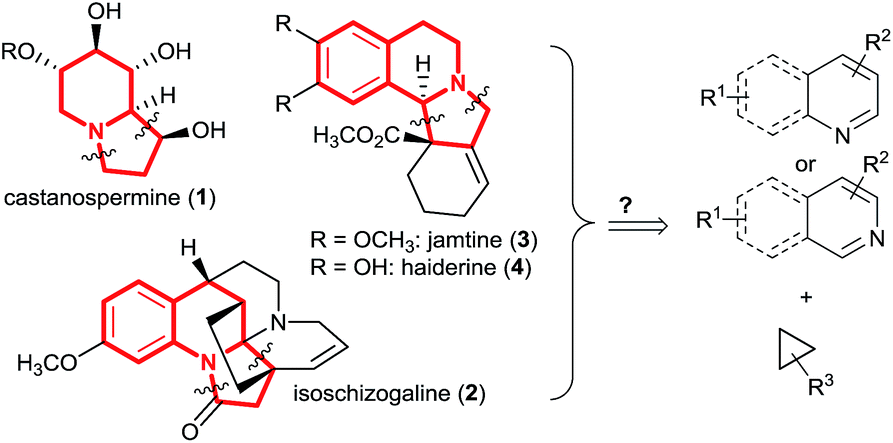 | ||
| Scheme 1 Examples for indolizidine containing natural products and general retrosynthetic scheme. | ||
In this context, Lewis acid (LA) catalyzed [3 + 2] annulation reactions of donor–acceptor (DA) cyclopropanes with dipolarophiles have been intensively studied.6 In particular, these reactions are highly successful with enol-ethers,7 nitrosoarenes,8 imines,9 heteroatom substituted alkynes,10 carbonyl compounds11 and nitrones.12 However, dearomative [3 + 2] annulation reactions were only intensively studied with indoles13 and a single example was reported for benzothiazoles (Scheme 2).14 Therefore, only [6,5,5] polycyclic ring systems can be currently accessed, although this approach would appear highly attractive for the synthesis of other polycyclic scaffolds as well. In fact, indole, with its high nucleophilicity and low aromatization energy (28 kcal mol−1 only for the pyrrole ring),15 constitutes an ideal case for dearomatization reactions: the nucleophilic character leads to a fast reaction with Lewis acid activated DA cyclopropanes, and the lower aromatization energy makes isolation of the saturated products easier.
 | ||
| Scheme 2 Dearomatization via [3 + 2] annulations with DA-cyclopropanes. | ||
Dearomatizing electron-poor quinolines with higher aromatization energy (35 kcal mol−1 for the pyridine ring) is much more challenging. In 2006, Pagenkopf and coworkers reported a method for the formation of indolizines via [3 + 2] annulation of pyridines or quinolines with DA cyclopropanes.16a In this work, dihydroindolizines were observed as intermediates, but they could be only isolated in very low yield and partially oxidized to indolizines. Therefore, the authors decided to completely aromatize the crude product with manganese(IV) oxide to obtain single, clean products. Later, Wang and co-workers used a similar approach with iodine as oxidant for indolizine synthesis.16b
Compared to bicyclic aromatic compounds, the dearomatization of monocyclic aromatics is even more challenging due to increased resonance stabilization. It is therefore not surprising that no dearomatizing [3 + 2] annulation was yet reported for these compounds.
Herein we describe the dearomative [3 + 2] annulation of N-heterocycles with aminocyclopropanes to generate tetrahydroindolizines with high yield and stereoselectivity. Key for success were the exceptional properties of imido-substituted DA diester cyclopropanes, as other types of donor groups were not successful. A broad substrate scope including pyridines, quinolines, and isoquinolines is presented. The obtained aminals can be easily modified through iminium formation and subsequent reduction or nucleophile addition.
2. Results and discussion
2.1. Preliminary results and optimization
We started our investigations by examining the reactions of DA acceptor cyclopropanes with quinoline (8) using scandium triflate as a Lewis acid catalyst (Scheme 3). Under these conditions, no reactivity was observed using well-established aryl-substituted DA cyclopropane 13.6 We then wondered if DA cyclopropanes bearing a heteroatom donor group would be more reactive.7c Indeed, cyclopropane 14 bearing a phthalimide donor led to the formation of [3 + 2] annulation product 17 in 80% yield. Cyclopropane 14 is easily available in one step on multigram scale from N-vinylphthalimide and diazomalonates by Rh-catalyzed cyclopropanation.17 In contrast, no conversion was observed with cyclopropane 15 bearing an oxygen donating group. This results further highlight the unique reactivity of imido-substituted DA cyclopropanes. Gratifyingly, compound 17 was stable enough to be isolated and fully characterized. The cis-relationship of the phthalimide and the hydrogen at ring junction was confirmed by X-ray analysis (Fig. 1).18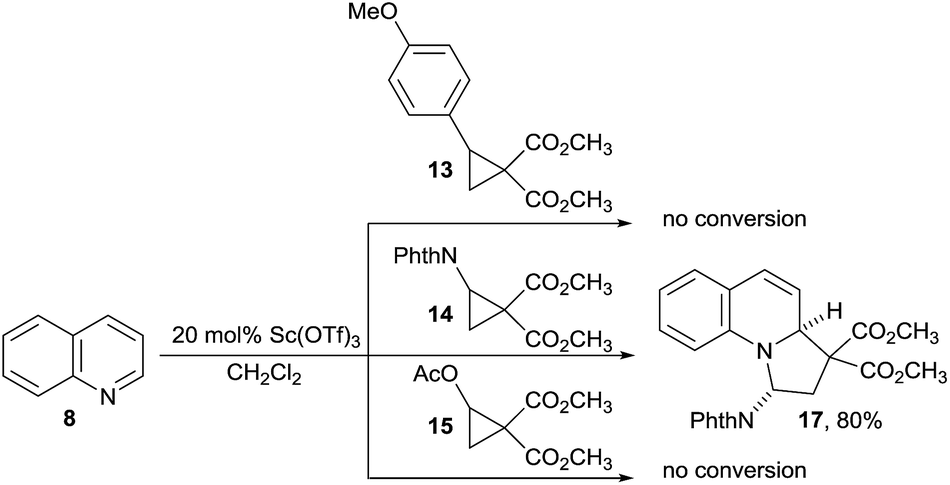 | ||
| Scheme 3 Preliminary results on the dearomatization of quinoline (8). | ||
 | ||
| Fig. 1 Structure of compound 17 as determined by X-ray analysis. Some hydrogen atoms are omitted for clarity. | ||
We then turned to the optimization of the [3 + 2] annulation. Product 17 was formed with >20:1 anti diastereoselectivity and 80% yield using Sc(OTf)3 as catalyst (Table 1, entry 1). Nevertheless, this result could only be obtained with 1.5 equiv. of cyclopropane 14 and relatively low molarity (0.05 M) to prevent decomposition. Furthermore, the amount of Sc(OTf)3 could not be reduced. Therefore, other Lewis acids were examined. No reaction was observed with In(OTf)3 or Cu(OTf)2 as catalysts (Table 1, entries 2 and 3) whereas the use of Hf(OTf)4 resulted in full decomposition of the DA-cyclopropane 14 (Table 1, entry 4). Better results were obtained using Yb(OTf)3 as catalyst. A first experiment gave 90% of the desired product 17 while the high diastereoselectivity was maintained (Table 1, entry 5). Furthermore, the mild conditions with Yb(OTf)3 allowed us to conduct the reaction more concentrated (0.5 M), with only 1.05 equivalents 14 and at lower catalyst loading (5 mol%) without observing any decrease in yield (Table 1, entry 6).19 The reaction proved to be easily scalable, as the yield did not change on 2 mmol scale. Eventual Brønsted acid catalysis of the reaction could be excluded by a control experiment with triflic acid (Table 1, entry 7). No reaction between 8 and para-methoxy phenyl or acetate substituted DA cyclopropanes (13 and 15) was again observed in presence of catalytic amounts of ytterbium(III) triflate (Table 1, entries 8 and 9).
|
|
||||
|---|---|---|---|---|
| Entry | R | LA | Mol% | Yieldb |
| a Reactions were carried out on 0.10 mmol scale with 1.5 equiv. of 13–15 in CH2Cl2 (0.05 M). b Isolated yields. c Reaction was carried out on 0.20 mmol scale with 1.05 equiv. of 14 in CH2Cl2 (0.50 M). d Reaction was carried out on 2.00 mmol scale with 1.05 equiv. of 14 in CH2Cl2 (0.50 M). e 0.2 M in CH2Cl2. Phth = phthalimide, Tf = trifluoromethylsulfonyl, PMP = para methoxyphenyl. | ||||
| 1 | NPhth | Sc(OTf)3 | 20 | 80 |
| 2 | NPhth | In(OTf)3 | 20 | No conversion |
| 3 | NPhth | Cu(OTf)2 | 20 | No conversion |
| 4 | NPhth | Hf(OTf)4 | 20 | Decomposition |
| 5 | NPhth | Yb(OTf)3 | 20 | 90 |
| 6c | NPhth | Yb(OTf)3 | 5 | 96 (95)d |
| 7e | NPhth | TfOH | 20 | No conversion |
| 8 | PMP | Yb(OTf)3 | 20 | No conversion |
| 9 | OAc | Yb(OTf)3 | 20 | No conversion |

2.2. Scope of the [3 + 2] annulation
Next, the scope of the reaction was examined by submitting various quinolines to the optimized reaction conditions (Fig. 2). Substitution of the pyridine ring was examined first (Fig. 2A). Alkyl, alkynyl and halogen substituents were all well tolerated either in C3 or C4 position of the quinoline ring (products 20–25). To our delight, O-acetylated cinchonidine with its highly basic amine worked well and furnished compound 22 in 76% yield and 1:1 dr. A broad range of versatile substituents such as aryl, halogens, trifluoromethyl, ester, nitrile and nitro were also tolerated on the arene ring (Fig. 2B, products 26–34). Generally, no differences in term of reactivity were observed upon substitution of the benzene or the pyridine ring of the employed quinolines. Only 2- and 8-substituted quinolines did not react under the developed conditions (with the exception of the fluoro substituent, product 31), probably due to increased steric hindrance. As a limitation, dearomatization of highly electron-rich 6-methoxy quinoline was not successful and compound 33 could not be obtained. Overall, the tolerance of functional groups attached to the quinoline is extremely broad, including in particular:
 | ||
| Fig. 2 Scope of the [3 + 2] annulation with quinolines. Reaction conditions: quinoline (0.20 mmol), DA-cyclopropane (0.21 mmol), Yb(OTf)3 (5 mol%), CH2Cl2 (0.5 M). Unless noted otherwise products obtained with dr > 20:1. Phth = phthalimide, TBS = tert-butyldimethylsilyl. | ||
- Potentially sensitive π-systems, such as alkenes and alkynes (products 22, 24 and 25).
- Strongly electron-withdrawing groups, such as esters, cyano and nitro, which are useful precursors of amides or amines (products 29, 30 and 34).
- Halogens, which are easily further modified using cross-coupling chemistry (products 20 and 26) or useful to diminish the electron-density of the heterocycle for pharmaceutical or agrochemical purposes (especially fluorine, products 27 and 31)
- Highly basic tertiary amines (product 22).
The influence of different nitrogen substituents on the DA-cyclopropane was then investigated (Fig. 2C). Less electron donating phthalimide groups with chloro or nitro substituents gave the desired products 35 and 36 in good yields, but their stability was significantly lower compared to their unsubstituted relative 17. Furthermore, maleimide, succinimide or a 2,3-naphthimide substituted DA cyclopropanes could also be used under the developed conditions (products 37–39). Finally, different ester groups on the cyclopropane had low impact on the reaction outcome (Fig. 2D): replacing the methyl esters of 14 with benzyl or trifluoroethyl esters gave the desired products 40 and 41 in excellent yields. With a mixed diester, product 42 was isolated in 72% yield, and 3.5:1 dr at the additional stereogenic center.
At this point we wondered if the developed protocol for the dearomatization of quinoline could also be applied to other N-heterocycles. To our delight isoquinoline reacted equally well and furnished 43 with 83% yield and high diastereoselectivity (>20:1, Fig. 3A). Cyano and ester substituted isoquinolines reacted also well under the developed conditions (products 44 and 45). The scope of the reaction could be extended to benzothiazole and benzoxazole (Fig. 3B, products 46 and 47).
 | ||
| Fig. 3 Reaction conditions: N-heterocycle (0.20 mmol), 14 (0.21 mmol), Yb(OTf)3 (5 mol%), CH2Cl2 (0.5 M). Unless noted otherwise products obtained with dr > 20:1. aChanges from normal reaction conditions: Yb(OTf)3 (10 mol%), CH2Cl2 (1 M). b1 mmol scale. Phth = phthalimide, TBS = tert-butyldimethylsilyl. | ||
Further expansion of the scope to pyridines proved to be more difficult. Unsubstituted pyridines or pyridines with electron donating substituents did not react to form the desired products under the developed conditions. It is known, that nucleophilic ring opening of acceptor substituted cyclopropanes with pyridine furnishes betaine products.20 Ring closure was expected to be more favored with electron deficient pyridines, as the positive charge of the betaine intermediate is then less stabilized. Indeed, the desired dearomative [3 + 2] annulation products of electron-deficient pyridines and 6a were isolated with good yield and high diastereoselectivity (>20:1) when the catalyst loading was raised to 10 mol% and the concentration to 1 M (Fig. 3C). Nicotinonitrile as well as isonicotinonitrile gave the desired products 48 and 49 with high yield. 4-Methyl, bromo-, or alkyl-substituted nicotinonitrile could also be used (products 50–52). Remarkably, the annulation reaction was completely regioselective for the less sterically hindered position. Such a high selectivity has been only rarely reported for addition to pyridinium salts.4h Ethyl nicotinate derivative 53 could not be obtained, but 3,4- and 3,5-diester substituted pyridines gave the desired products 54 and 55 in good yields. Alternatively, installation of a more electron-withdrawing HFIP ester was also successful (product 56). These active esters can be readily converted into different amides and esters.21 Other electron-withdrawing groups on the nicotinate also led to good yields (products 57 and 58). Finally, nitro-substituted pyridines also furnished the desired products 59–61 in 74–80% yield.
2.3. Product functionalization
The obtained building blocks contain highly interesting functionalities for further modification, including in particular reactive alkenes and aminals. To further establish the synthetic potential of the method, a few transformations of the dearomatization products were therefore examined (Scheme 4). Hydrogenation of the benzylic olefin and aminal of tetrahydro-benzoindolizine 17 and removal of one methyl ester group was achieved using Pearlman's catalyst, followed by Krapcho decarboxylation to give amine 62 in 60% overall yield. The phthalimido and the diester groups can therefore be considered as traceless activating and directing groups for the annulation reaction.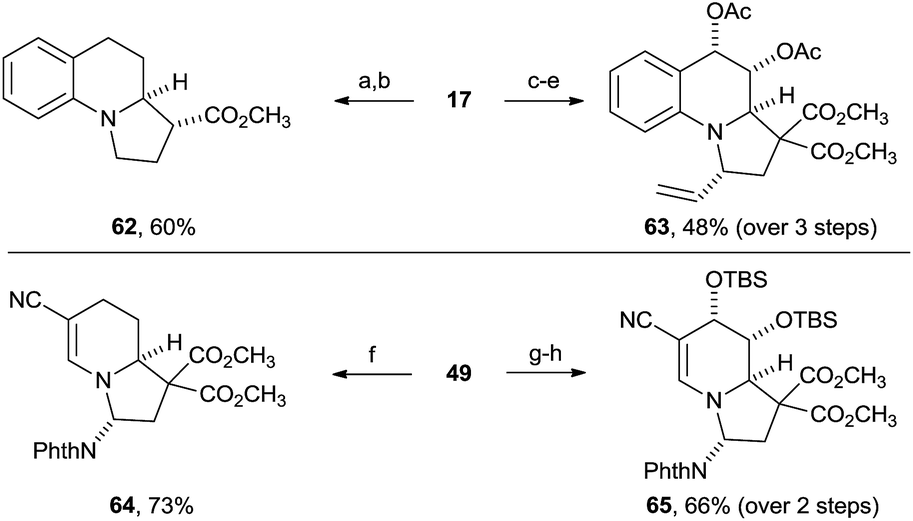 | ||
| Scheme 4 Functionalization of products 17 and 49. Reaction conditions: (a) H2, Pd(OH)2 (10% w/w), CH3OH, 70%; (b) LiCl (5 equiv.), DMSO:H2O 10:1, 140 °C, 85%; (c) OsO4 (5 mol%), NMO·H2O (1.2 equiv.), THF:acetone:H2O (2:2:1); (d) Ac2O (3 equiv.), DMAP (10 mol%), NEt3 (4 equiv.), CH2Cl2, 71% dr > 20:1 (over 2 steps); (e) vinylMgBr (4 equiv.), ZnCl2 (10 equiv.), THF, 50 °C, 68%, dr > 20:1; (f) H2, Pd(OH)2 (10% w/w), CH3OH, 73%; (g) OsO4 (5 mol%), NMO·H2O (1.2 equiv.), acetone:H2O (20:1), 0 °C; (h) TBSOTf, pyridine, CH2Cl2, 66%, dr > 20:1 (over 2 steps). Phth = phthalimide, TBS = tert-butyldimethylsilyl, NMO = N-methylmorpholine-N-oxide, THF = tetrahydrofuran, DMAP = N,N-dimethylpyridin-4-amine. | ||
Selective dihydroxylation of the benzylic olefin from the convex side of the molecule was possible (dr > 20:1). After acetylation of the alcohols, the aminal was converted with high diastereoselectivity (dr > 20:1) into tertiary amine 63 through alkylation of the intermediary iminium with a vinyl zinc reagent, resulting in the stereoselective installation of four stereocenters around the tricyclic system.
Selective reduction of the more electron rich olefin of tetrahydroindolizine 49 furnished compound 64 in 73% yield. Moreover, selective dihydroxylation via osmium(VIII) catalysis and subsequent silylation of the diol gave compound 65 in high yield and high diastereoselectivity (dr > 20:1). Our methodology is therefore highly suited for accessing polysubstituted indolizidine rings frequently encountered in natural products (Scheme 1).
2.4. Speculative reaction mechanism
Three experiments gave first insights into the reaction mechanism (Scheme 5A):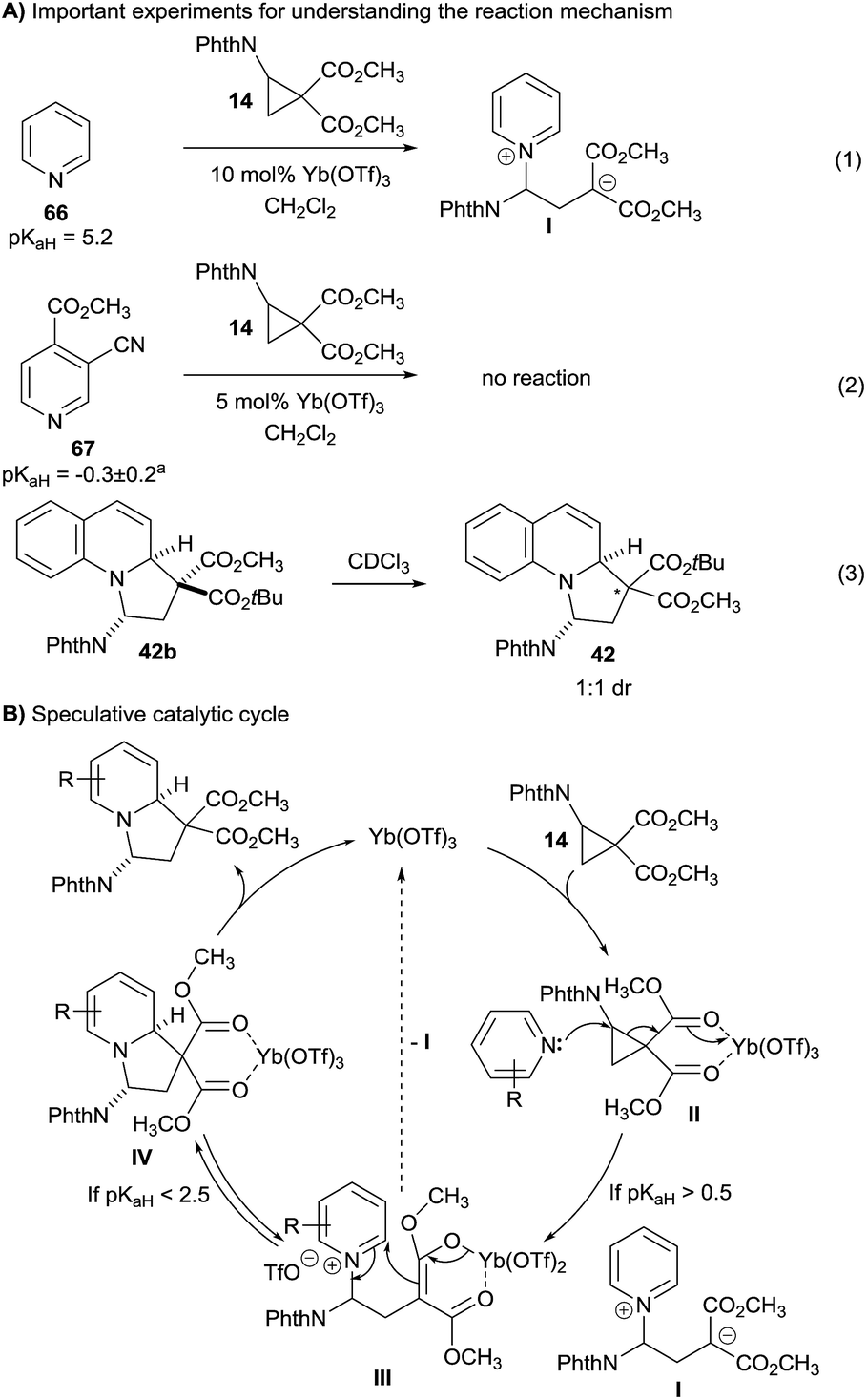 | ||
| Scheme 5 Key experiments and speculative mechanism. aPredicted with ACD Labs. | ||
(1) When pyridine (66) was reacted with cyclopropane 14 using ytterbium triflate as catalyst, the desired product was not obtained. Full conversion of cyclopropane 14 was observed, but no pure product could be isolated from the reaction mixture. Nevertheless, a molecular ion corresponding to zwitterion I could be detected by mass spectroscopy.
(2) When highly electron-poor pyridine 67 was used, no reaction was observed (eqn (2)).
(3) Quinolizine 42 could be isolated with 3.5:1 dr at the diester center. However, after separation the minor isomer 42a equilibrated to a 1:1 mixture just upon standing in deuterated chloroform (eqn (3)).
Based on these experiments and the well-established activation of DA diester cyclopropanes with Lewis acids,11a a first speculative mechanism can be proposed (Scheme 5B). Coordination of cyclopropane 14 by the Lewis acid led to activated intermediate II. Only sufficiently electron-rich pyridine (pKaH > 0.5) are nucleophilic enough to react with this intermediate and give pyridinium III. At this point, reversible ring closure can occur to give coordinated product IV. The equilibrium lays on the product side for quinolines. For pyridines, this is the case only if the heterocycle is sufficiently electron poor (pKaH < 2.5). If this is not the case, decoordination of the Lewis acid would free zwitterion I,20 which could be the detected by mass spectroscopy. The high diastereoselectivity observed in the reaction is probably due to the higher stability of the products having the phthalimide group in the convex face of the polycyclic systems (thermodynamic control). From IV, the catalytic cycle is then closed by a simple ligand exchange on ytterbium.
3. Conclusion
In summary, a highly efficient method for the preparation of tetrahydroindolizine derivatives by dearomative [3 + 2] annulation reactions of pyridines, isoquinolines or quinolines and 2-aminocyclopropanes was developed. The fine modulation of the reactivity by the phthalimido group was essential for the success of this process. Excellent yields, high diastereoselectivities and a very broad substrate scope was achieved by employing ytterbium(III) triflate as catalyst. The reaction proved to be scalable and further functionalization of the obtained products was easily possible, setting the base for the synthesis of more complex bioactive compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPFL for financial support. Dr Rosario Scopelliti, Dr Euro Solari and Dr Farzaneh Fadaei Tirani are acknowledged for the X-ray study.Notes and references
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC.
- (a) F. Wong-Staal, G. Liu and M. Jeffrey, in Antiviral Drugs: From Basic Discovery through Clinical Trials, ed. W. M. Kazmierski, John Wiley & Sons, Inc., Hoboken, NJ, 2011, pp. 329–337 Search PubMed; (b) S. J. F. Kaptein and J. Neyts, Curr. Opin. Pharmacol., 2016, 30, 1 CrossRef CAS PubMed.
- (a) R. M. Kariba, P. J. Houghton and A. Yenesew, J. Nat. Prod., 2002, 65, 566 CrossRef CAS PubMed; (b) V. U. Ahmad, A.-u. Rahman, T. Rasheed and H.-u. Rehman, Heterocycles, 1987, 26, 1251 CrossRef CAS; (c) V. U. Ahmad and S. Iqbal, Nat. Prod. Lett., 1993, 2, 105 CrossRef CAS.
- Reviews: (a) S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (b) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (c) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807 RSC; selected examples: (d) K. Frisch, A. Landa, S. Saaby and K. A. Jorgensen, Angew. Chem., Int. Ed., 2005, 44, 6058 CrossRef CAS PubMed; (e) M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700 CrossRef CAS PubMed; (f) Z. Sun, S. Yu, Z. Ding and D. Ma, J. Am. Chem. Soc., 2007, 129, 9300 CrossRef CAS PubMed; (g) M. A. Fernandez-Ibanez, B. Macia, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2009, 48, 9339 CrossRef CAS PubMed; (h) N. Christian, S. Aly and K. Belyk, J. Am. Chem. Soc., 2011, 133, 2878 CrossRef CAS PubMed; (i) O. García Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823 CrossRef PubMed; (j) G. Bertuzzi, A. Sinisi, L. Caruana, A. Mazzanti, M. Fochi and L. Bernardi, ACS Catal., 2016, 6, 6473 CrossRef CAS; (k) K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338 CrossRef CAS PubMed; for intramolecular approaches, see: (l) Z.-P. Yang, Q.-F. Wu and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 6986 CrossRef CAS PubMed; (m) Z.-P. Yang, Q.-F. Wu, W. Shao and S.-L. You, J. Am. Chem. Soc., 2015, 137, 15899 CrossRef CAS PubMed.
- For selected rare examples of other type of annulation processes, see: (a) R. Huisgen, M. Morikawa, K. Herbig and E. Brunn, Chem. Ber., 1967, 100, 1094 CrossRef CAS; (b) O. Tsuge, S. Kanemasa and S. Takenaka, Bull. Chem. Soc. Jpn., 1985, 58, 3320 CrossRef CAS; (c) V. Nair, A. R. Sreekanth, N. Abhilash, M. M. Bhadbhade and R. C. Gonnade, Org. Lett., 2002, 4, 3575 CrossRef PubMed; (d) V. Nair, S. Devipriya and E. Suresh, Tetrahedron, 2008, 64, 3567 CrossRef CAS; (e) V. Nair, S. Devipriya and E. Suresh, Synthesis, 2008, 2008, 1065 CrossRef; (f) A. Shaabani, A. H. Rezayan, A. Sarvary and H. R. Khavasi, Tetrahedron Lett., 2008, 49, 1469 CrossRef CAS; (g) D. B. Huple, S. Ghorpade and R.-S. Liu, Chem.–Eur. J., 2013, 19, 12965 CrossRef CAS PubMed; (h) T. Onnagawa, Y. Shima, T. Yoshimura and J.-i. Matsuo, Tetrahedron Lett., 2016, 57, 3050 CrossRef CAS.
- Selected reviews: (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (b) F. De Simone and J. Waser, Synthesis, 2009, 3353 CAS; (c) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (d) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (e) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655 RSC.
- Selected examples: (a) J.-P. Qu, C. Deng, J. Zhou, X.-L. Sun and Y. Tang, J. Org. Chem., 2009, 74, 7684 CrossRef CAS PubMed; (b) J.-P. Qu, Y. Liang, H. Xu, X.-L. Sun, Z.-X. Yu and Y. Tang, Chem.–Eur. J., 2012, 18, 2196 CrossRef CAS PubMed; (c) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 12075 CrossRef CAS PubMed; (d) S. Racine, F. de Nanteuil, E. Serrano and J. Waser, Angew. Chem., Int. Ed., 2014, 53, 8484 CrossRef CAS PubMed.
- S. Chakrabarty, I. Chatterjee, B. Wibbeling, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2014, 53, 5964 CrossRef CAS PubMed.
- Selected examples: (a) C. A. Carson and M. A. Kerr, J. Org. Chem., 2005, 70, 8242 CrossRef CAS PubMed; (b) V. S. Korotkov, O. V. Larionov, A. Hofmeister, J. Magull and A. de Meijere, J. Org. Chem., 2007, 72, 7504 CrossRef CAS PubMed; (c) R. Tombe, T. Kurahashi and S. Matsubara, Org. Lett., 2013, 15, 1791 CrossRef CAS PubMed.
- Selected examples: (a) X. Qi and J. M. Ready, Angew. Chem., Int. Ed., 2008, 47, 7068 CrossRef CAS PubMed; (b) W. D. Mackay, M. Fistikci, R. M. Carris and J. S. Johnson, Org. Lett., 2014, 16, 1626 CrossRef CAS PubMed; (c) F. G. Perrin, G. Kiefer, L. Jeanbourquin, S. Racine, D. Perrotta, J. Waser, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 13393 CrossRef CAS PubMed; (d) S. Racine, B. Hegedüs, R. Scopelliti and J. Waser, Chem.–Eur. J., 2016, 22, 11997 CrossRef CAS PubMed.
- Selected examples: (a) P. D. Pohlhaus and J. S. Johnson, J. Am. Chem. Soc., 2005, 127, 16014 CrossRef CAS PubMed; (b) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642 CrossRef CAS PubMed; (c) A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 3122 CrossRef CAS PubMed; (d) S. Xing, W. Pan, C. Liu, J. Ren and Z. Wang, Angew. Chem., Int. Ed., 2010, 49, 3215 CrossRef CAS PubMed; (e) F. Benfatti, F. de Nanteuil and J. Waser, Chem.–Eur. J., 2012, 18, 4844 CrossRef CAS PubMed; (f) F. Benfatti, F. de Nanteuil and J. Waser, Org. Lett., 2012, 14, 386 CrossRef CAS PubMed; (g) Y. Miyake, S. Endo, T. Moriyama, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2013, 52, 1758 CrossRef CAS PubMed; (h) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed; (i) S. Racine, J. Vuilleumier and J. Waser, Isr. J. Chem., 2016, 56, 566 CrossRef CAS.
- Selected examples: (a) I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 42, 3023 CrossRef CAS PubMed; (b) I. S. Young and M. A. Kerr, Org. Lett., 2004, 6, 139 CrossRef CAS PubMed; (c) I. S. Young, J. L. Williams and M. A. Kerr, Org. Lett., 2005, 7, 953 CrossRef CAS PubMed; (d) C. A. Carson and M. A. Kerr, Angew. Chem., Int. Ed., 2006, 45, 6560 CrossRef CAS PubMed; (e) K. Sapeta and M. A. Kerr, J. Org. Chem., 2007, 72, 8597 CrossRef CAS PubMed; (f) M. P. Sibi, Z. H. Ma and C. P. Jasperse, J. Am. Chem. Soc., 2005, 127, 5764 CrossRef CAS PubMed; (g) Y. B. Kang, X. L. Sun and Y. Tang, Angew. Chem., Int. Ed., 2007, 46, 3918 CrossRef CAS PubMed.
- Selected examples: (a) M. A. Kerr and R. G. Keddy, Tetrahedron Lett., 1999, 40, 5671 CrossRef CAS; (b) D. B. England, T. D. O. Kuss, R. G. Keddy and M. A. Kerr, J. Org. Chem., 2001, 66, 4704 CrossRef CAS PubMed; (c) C. Venkatesh, P. P. Singh, H. Ila and H. Junjappa, Eur. J. Org. Chem., 2006, 2006, 5378 CrossRef; (d) B. Bajtos, M. Yu, H. Zhao and B. L. Pagenkopf, J. Am. Chem. Soc., 2007, 129, 9631 CrossRef CAS PubMed; (e) M. M. A. R. Moustafa and B. L. Pagenkopf, Org. Lett., 2010, 12, 3168 CrossRef PubMed; (f) S. M. Wales, M. M. Walker and J. S. Johnson, Org. Lett., 2013, 15, 2558 CrossRef CAS PubMed; (g) H. Xiong, H. Xu, S. Liao, Z. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed; (h) C. C. Dulin, K. L. Murphy and K. A. Nolin, Tetrahedron Lett., 2014, 55, 5280 CrossRef CAS.
- D.-C. Wang, M.-S. Xie, H.-M. Guo, G.-R. Qu, M.-C. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 14111 CrossRef CAS PubMed.
- C. W. Bird, Tetrahedron, 1996, 52, 9945 CrossRef CAS.
- (a) N. A. Morra, C. L. Morales, B. Bajtos, X. Wang, H. Jang, J. Wang, M. Yu and B. L. Pagenkopf, Adv. Synth. Catal., 2006, 348, 2385 CrossRef CAS; (b) J. Liu, L. Zhou, W. Ye and C. Wang, Chem. Commun., 2014, 50, 9068 RSC.
- F. González-Bobes, M. D. B. Fenster, S. Kiau, L. Kolla, S. Kolotuchin and M. Soumeillant, Adv. Synth. Catal., 2008, 350, 813 CrossRef.
- X-ray data is available at the Cambridge Crystallographic Data Center (CCDC number: 1556244).† The relative configuration for other compounds was assumed to be the same on the basis of the similarity in their NMR-spectra.
- Preliminary investigations showed that an enantioselective process should be possible, but highly challenging to develop, as so far no enantioselectivity higher than 18% was obtained. See Table S8 in the ESI.†.
- (a) S. Danishefsky and R. K. Singh, J. Am. Chem. Soc., 1975, 97, 3239 CrossRef CAS; (b) E. M. Budynina, O. A. Ivanova, E. B. Averina, T. S. Kuznetsova and N. S. Zefirov, Tetrahedron Lett., 2006, 47, 647 CrossRef CAS.
- (a) J.-M. Vatèle, Synlett, 2015, 26, 2280 CrossRef; (b) Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 3191 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1556244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03197a |
| ‡ Dr Chakrabarty has decided to stop his scientific career and cannot be contacted any more. He therefore did not see the final version of this manuscript. Based on his important contribution to the project, both J. P. and J. W. agree to include him as co-author and are convinced that he would agree to be included if he knew about this submission. |
| This journal is © The Royal Society of Chemistry 2017 |