
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiosemicarbazone organocatalysis: tetrahydropyranylation and 2-deoxygalactosylation reactions and kinetics-based mechanistic investigation†
Dennis
Larsen‡
 ,
Line M.
Langhorn
,
Olivia M.
Akselsen
,
Bjarne E.
Nielsen
and
Michael
Pittelkow
*
,
Line M.
Langhorn
,
Olivia M.
Akselsen
,
Bjarne E.
Nielsen
and
Michael
Pittelkow
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark. E-mail: pittel@chem.ku.dk
First published on 6th October 2017
Abstract
The first use of thiosemicarbazone-based organocatalysis was demonstrated on both tetrahydropyranylation and 2-deoxygalactosylation reactions. The organocatalysts were optimised using kinetics-based selection. The best catalyst outperformed previously reported thiourea catalysts for tetrahydropyranylation by 50-fold. Hammett investigations of both the organocatalyst and the substrate indicate an oxyanion hole-like reaction mechanism.
Dual hydrogen-bonding organocatalysts have gained significant popularity recently, particularly the use of (thio)urea catalysts.1 Ever since Jacobsen's serendipitous 1998 landmark discovery of the thiourea-catalysed asymmetric Strecker reaction,2 building on pioneering urea work by Curran and others,3 chemists have been looking for new double hydrogen-bonding motifs to exploit for organocatalytic purposes.
Corey introduced guanidinium-based dual hydrogen bond donor catalysts as early as 1999,4 while Rawal demonstrated squaramide-based organocatalysis in 2008.5 Based on our recent development of the squaramide-related croconamide organocatalysts and our concurrent work with thiourea-like thiosemicarbazones,6 we became interested in developing organocatalysts structurally related to thioureas as well.
In this proof-of-principle study, we establish thiosemicarbazones (Fig. 1a) as a new class of organocatalysts that act as unique catalysts for the tetrahydropyranylation of alcohols under mild conditions, and we perform a double Hammett investigation of the reaction mechanism using a range of catalysts and a range of phenol substrates (Fig. 1b). The resulting double Hammett plot (Fig. 1c) gives experimentally-based insights into the electronic requirements of both the catalyst and phenol substrates in the transition state, and allows us to propose a mechanism of the reaction based solely on experimentally-gathered evidence.
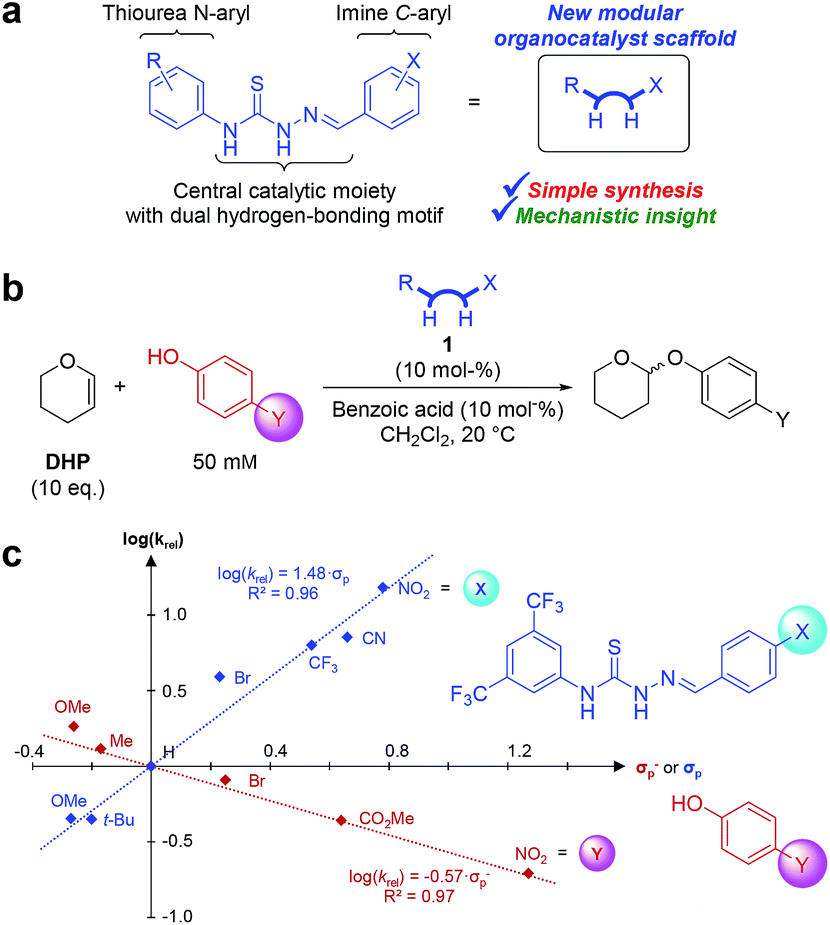 | ||
Fig. 1 (a) Thiosemicarbazone organocatalyst scaffold used in this study. (b) Optimised conditions for Hammett investigations of the thiosemicarbazone-catalysed tetrahydropyranylation of phenols. (c) Hammett plot for catalyst ( ) and substrate ( ) and substrate (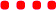 ). ). | ||
Inspired by the thiourea catalysts developed by Schreiner and co-workers,7 catalyst 1b (Fig. 2) was synthesised and used (10 mol%) to develop conditions for a kinetics-based catalyst screening. While 1b was able to afford turnover in several non-polar solvents (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DHP/solvent), CH2Cl2 proved to be most suitable (see Table S1 in the ESI†). Addition of benzoic acid co-catalyst further improved reactivity, allowing for full conversion within hours under relatively dilute conditions (10 eq. of DHP).
:1 DHP/solvent), CH2Cl2 proved to be most suitable (see Table S1 in the ESI†). Addition of benzoic acid co-catalyst further improved reactivity, allowing for full conversion within hours under relatively dilute conditions (10 eq. of DHP).
 | ||
| Fig. 2 Catalysts 1a–h, 3a–b, and 4. | ||
Though benzoic acid gave a sizeable 1.5-fold rate increase in the presence of catalyst 1b (Fig. S1 and Table S2 in the ESI for details†), benzoic acid itself, i.e. without catalyst, did not afford any turnover, underscoring the positive catalytic effect of the thiosemicarbazone. Use of 2-(4-nitrophenyl)ethanol, 2, ensured simple reaction monitoring by HPLC-UV (290 nm). Kinetics studies confirmed first-order dependence with respect to both DHP and alcohol concentrations, and the overall second-order rate constant, k2, was determined by non-linear regression (see ESI for details†) for a range of thiosemicarbazones, 1a–h and 3a–b (Fig. 2). The best previously reported thiourea catalyst for this reaction, 4,7b was used for comparison.
Thiosemicarbazone catalysts are highly tuneable, and there is an apparent trend favouring catalysts that have electron-withdrawing groups (EWGs) (Table 1). This is in accordance with results described by the Schreiner lab, who found that EWGs improve the efficacy of thiourea catalysts, with 4 being the most prominent example.8 This indirectly suggests that the thiosemicarbazones used in this study function in a similar manner to thiourea catalysts, e.g. via dual hydrogen-bonding from the two NH-groups.7b
|
|
|||
|---|---|---|---|
| Catalyst | k 2 (10−6 M−1 s−1) | Catalyst | k 2 (10−6 M−1 s−1) |
| a Obtained by non-linear regression (see ESI). b Standard errors on best fit. | |||
| (None) | No conversion | 1f | Conv. too low |
| 1a | 6.8 ± 0.2 | 1g | Conv. too low |
| 1b | 37.7 ± 1.1 | 1h | Solubility too low |
| 1c | 129 ± 5 | 3a | 1.83 ± 0.05 |
| 1d | 53 ± 2 | 3b | 12.38 ± 0.07 |
| 1e | 101 ± 3 | 4 | 2.55 ± 0.03 |
The data presented in Table 1 indicate that EWGs on the imine C-phenyl are better able to afford turnover than EWGs on the thiourea N-phenyl. Thus, there is a modest rate increase going from 3a-catalysis to 1a-catalysis (3.7-fold), while a larger improvement is seen when going from 3a to 3b (6.8-fold). A possible explanation for this phenomenon might be inferred from the X-ray crystal structure of 1b (Fig. 3a and b). Due to the steric demand of the thiocarbonyl, the N-phenyl cannot be in the same plane as the central thiosemicarbazone moiety. The C-phenyl, on the other hand, forms a planar, fully conjugated system with the thiosemicarbazone moiety. Therefore, the EWGs on the C-phenyl withdraw electron-density via both inductive and mesomeric pathways, while the EWGs on the N-phenyl can only exert electron-withdrawing effects by way of induction.
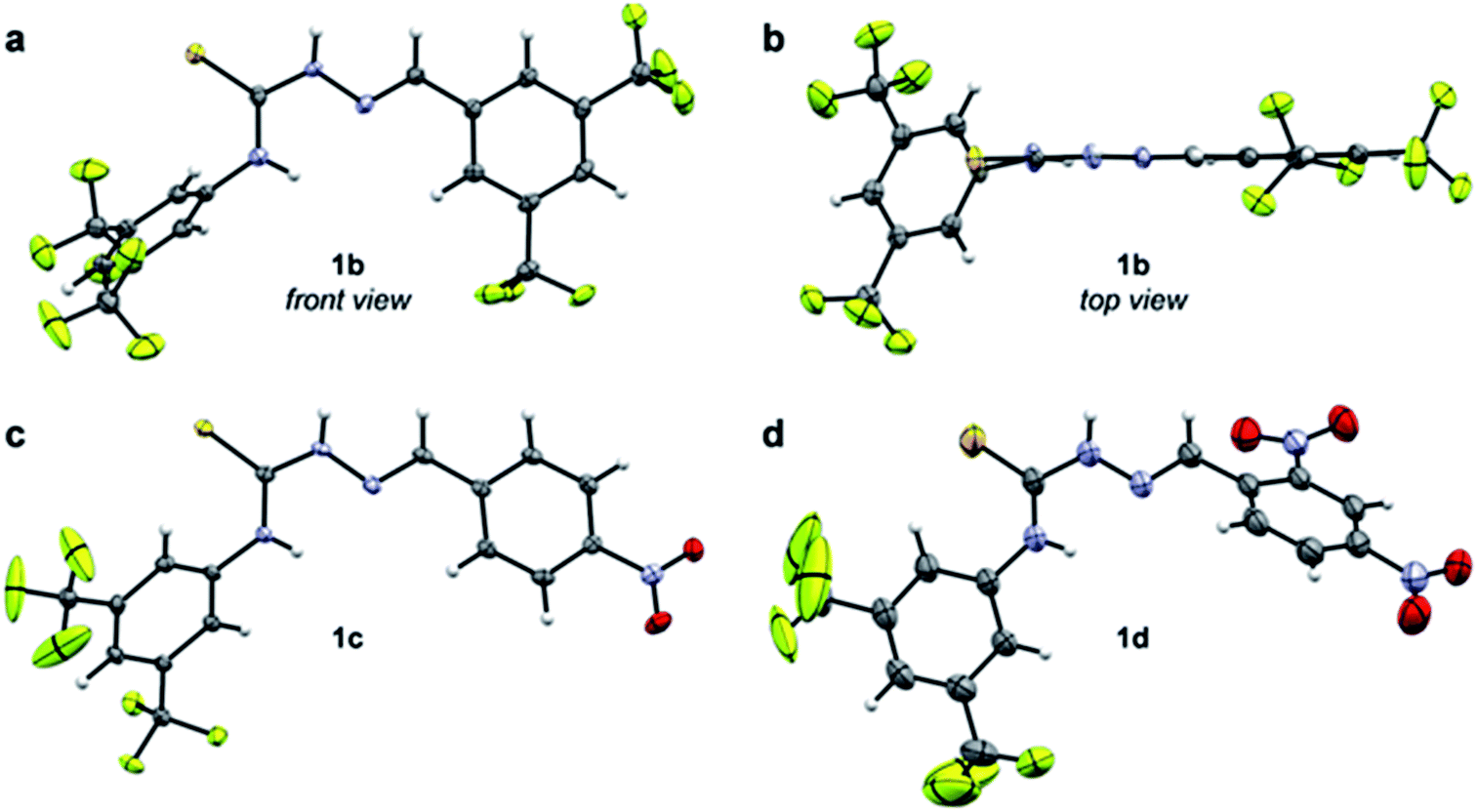 | ||
| Fig. 3 Crystal structures of selected catalysts, C: grey, H: white, F: green, N: blue, O: red, S: yellow. (a) Front and (b) top view of 1b (ethanol). (c) 1c (CH2Cl2/methanol). (d) 1d (ethanol, disorder of CF3 groups omitted for clarity). | ||
Despite having an additional EWG, the catalytic efficacy of 1d was found to be inferior to 1c. This is in contradiction to the hypothesis that more EWGs lead to higher catalytic effect, but as is evident from the X-ray crystal structures, the additional nitro group on 1d forces the imine C-phenyl out of the plane of the thiosemicarbazone moiety (Fig. 3c and d). It appears that the added electron-withdrawal from the additional nitro group is overcome by this perturbation of the conjugated system.
The C6F5-substituted catalyst 1e performs almost as well as 1c, while the 2-pyridinyl derivative 1f failed to give useable conversion. In accordance with the observed trend, catalyst 1g, with the strongly electron-donating dimethylamino group, failed to give appreciable turnover.
All but the slowest of the working thiosemicarbazone catalysts give higher k2's than previously reported thiourea 4. While 4 remains one of the most efficient organocatalysts reported under high reagent concentration conditions (as low as 0.001 mol% catalyst loading in neat DHP),7b the best thiosemicarbazone (1c) gives a 50-fold increase of k2 compared to thiourea 4 under the more dilute conditions in this study.
The transient nature of the intermediates in non-covalent hydrogen bond-catalysed reactions makes mechanistic investigations of reactions involving non-covalent organocatalysts difficult.9,10 The lack of tangible evidence of unique reaction intermediates to substantiate and validate mechanistic proposals has made indirect methods the main source of mechanistic insight, e.g. computational methods that account for solvent using continuum models.11 In spite of tremendous effort and great improvements, identifying the mechanistic pathway in organocatalytic reactions remains a difficult task,12 and reports of experimental methods to gain mechanistic insight into non-covalent organocatalysis are relatively few and relatively far between.9 Many rely on inferring catalytic properties from catalyst–substrate complexes identified by e.g. spectroscopy,13 while other methods rely on relatively cumbersome physico-chemical analyses of the catalysts.14
A Hammett analysis gives experimental insight into the nature of the transition state of a chemical reaction by determining the linear free energy relationship between the logarithm of relative reaction rate constants and Hammett's σ values.15 In spite of the widespread use of organocatalysts and the demand for insight into the way in which they operate, there are very few examples of Hammett analyses performed with focus on the organocatalyst, and none of these investigations were expanded to also include a Hammett analysis of the substrate.16
To gain mechanistic insight on this new class of organocatalysts, we exploited the ease of derivatisation of thiosemicarbazones. A range of thiosemicarbazone catalysts all bearing different substituents in the p-position on the imine C-phenyl were used to determine k2's for the tetrahydropyranylation of 4-methoxyphenol (i.e. different X's in Fig. 1b). In parallel, a range of p-substituted phenols were selected as substrates and tetrahydropyranylated using 1c (i.e. different Y's in Fig. 1b). This allowed us to perform a double Hammett analysis: one Hammett plot to investigate the effect of electron-withdrawing and electron-donating substituents on the catalyst and one Hammett plot to investigate the effect of the substituents on phenol substrates (Fig. 1c).
For the catalysts, Hammett's σp values were used to obtain a plot with good linear correlation ( , Fig. 1c).17 The positive slope (ρ = 1.48) is in agreement with the previous notion that EWGs on the catalyst leads to faster reactions.
, Fig. 1c).17 The positive slope (ρ = 1.48) is in agreement with the previous notion that EWGs on the catalyst leads to faster reactions.
For the phenols, a similar plot was made against the Hammett σp− values to obtain a plot with good linear correlation (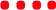 , Fig. 1c).17 The negative slope (ρ = −0.57) reveals a decrease in electron-density (build-up of positive charge or loss of negative charge) on the phenol in the transition state.
, Fig. 1c).17 The negative slope (ρ = −0.57) reveals a decrease in electron-density (build-up of positive charge or loss of negative charge) on the phenol in the transition state.
Based on these double Hammett investigations, several possible reaction pathways were considered. Traditional Brønsted acid-catalysed tetrahydropyranylation via an oxocarbenium ion formed by protonation of DHP by the thiosemicarbazone catalyst was ruled out. The best catalyst (1c) was found to have a pKa value of 11.5 ± 0.1 (DMSO) (see ESI for details†), and since this is less acidic than benzoic acid (pKa 11.1, DMSO),18 which itself does not promote the reaction, this type of mechanism was rejected.
1H NMR titrations in CDCl3 revealed that 1c forms a 1:1 adduct with 4-methoxyphenol with a low binding constant, Ka, of 1.71 ± 0.03 m−1, while DHP was found to have a significantly weaker interaction with 1c (Ka below the detection limit afforded by 1H NMR titrations) (details in ESI†). This suggests a mechanism in which DHP does not interact with the catalyst itself, but rather with a catalyst–substrate adduct.
Based on a thorough evaluation of the available data, plausible mechanistic pathways (see further details in the ESI†), and comparison to similar reactions,7b we suggest a mechanism in which the catalyst–phenol complex reacts with DHP to form the product via a not fully synchronous cyclic proton transfer, which in its entirety results in the same product that would be formed via a formally forbidden [2 + 2] cycloaddition mechanism. After initial build-up of negative charge on the phenol oxygen, stabilised by double H-bonding from the organocatalyst (pre-TS, Fig. 4) in the so-called “oxyanion hole”,1b,7b,11a the partial negative charge on the phenol oxygen is lost in the ensuing transition state (TS, Fig. 4). This is in agreement with the Hammett analysis of the phenol substrates, which supports a mechanism going via a loss of (partial) negative charge on the phenol in the transition state.
 | ||
| Fig. 4 Proposed catalytic cycle for the thiosemicarbazone-catalysed tetrahydropyranylation of phenols. | ||
The best catalysts are the strongest hydrogen bond donors, since they most efficiently afford preorganisation of the reactants to form the pre-TS and stabilisation of the transition state, TS, in accordance with the oxyanion stabilisation concept.1b,7b,11a Further 1H NMR investigations revealed a 1:1 interaction (Ka = 46 ± 4 m−1) as well as chemical shift changes consistent with an electrophilic interaction upon titration of 1c with benzoic acid. Such an interaction results in a more electron-deficient catalyst and thus can seemingly explain the slight increase in reaction rate (1.5-fold with 1b and 2-(4-nitrophenyl)ethanol, Table S2, ESI†) seen with addition of benzoic acid.
This mechanistic proposal runs parallel to the previously proposed mechanism for thiourea-catalysed tetrahydropyranylations by Kotke and Schreiner, who, based on computational results, identified a mechanism going via a cyclic transition state, though they emphasised that the overall addition must be “highly asynchronous” since a thermal [2 + 2] cycloaddition is formally forbidden.7b The study presented in this report represents the first kinetics-based experimental evidence for the importance of oxyanion stabilisation in the mechanism for this reaction.
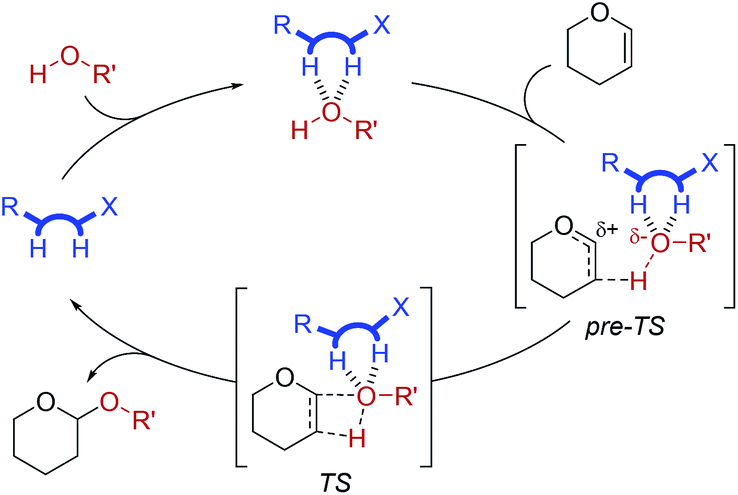
While tetrahydropyranylations are an important chemical tool,19 an interesting analogous reaction is formation of 2-deoxyglycosides by use of glucal enol ether substrates. Inspired by McGarrigle and co-workers,20 we used 1c at 1 mol% to afford 2-deoxygalactosylation of alcohol 2 using galactal 5 (Fig. 5).
 | ||
Fig. 5 Formation of 2-deoxygalactoside 6 as a function of time when using catalyst 1c in combination with benzoic acid (both 1 mol%) ( ), 1c (1 mol%) only ( ), 1c (1 mol%) only ( ), or benzoic acid (1 mol%) only ( ), or benzoic acid (1 mol%) only ( ). ). | ||
It was found that catalyst 1c affords formation of the desired product, 6, by HPLC-UV-MS. Full turnover was achieved in ca. 40 hours when using only catalyst 1c, while use of benzoic acid and catalyst 1c in combination resulted in full turnover in ca. 28 hours. No appreciable turnover was detected when using benzoic acid in the absence of 1c. This demonstrates that thiosemicarbazones are also attractive catalysts for other reactions.
In summary, we have illustrated the first use of thiosemicarbazones as organocatalysts. Guided by kinetics, an optimised catalyst structure, 1c, was identified. A double Hammett analysis, along with NMR and pKa data, allowed us to suggest an asynchronous cyclic transition state. The fact that thiosemicarbazones function in a similar manner to the well-proven thiourea catalysts gives rise to optimism regarding the future use of thiosemicarbazone catalysts in other reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Lundbeck Foundation (Young Group Leader Fellowship), the Danish Council for Independent Research (Sapere Aude – DFF 4181-00206), and the Department of Chemistry, University of Copenhagen.Notes and references
- (a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (b) M. Kotke and P. R. Schreiner in Hydrogen bonding in organic synthesis, ed. P. M. Pihko, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 141–351 Search PubMed; (c) S. J. Connon, Synlett, 2009, 354 CAS; (d) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593 CrossRef CAS PubMed.
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef CAS.
- (a) D. P. Curran and L. H. Kuo, J. Org. Chem., 1994, 59, 3259 CrossRef CAS; (b) M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 CrossRef CAS; (c) R. M. Tel and J. B. F. N. Engberts, J. Chem. Soc., Perkin Trans. 2, 1976, 483 RSC.
- E. J. Corey and M. J. Grogan, Org. Lett., 1999, 1, 157 CrossRef CAS PubMed.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed.
- (a) A. Jeppesen, B. E. Nielsen, D. Larsen, O. M. Akselsen, T. I. Sølling, T. Brock-Nannestad and M. Pittelkow, Org. Biomol. Chem., 2017, 15, 2784 RSC; (b) D. Larsen, A. Jeppesen, C. Kleinlein and M. Pittelkow, J. Org. Chem., 2017, 82, 8580 CrossRef CAS PubMed.
- (a) P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 217 CrossRef CAS; (b) P. R. Schreiner and M. Kotke, Synthesis, 2007, 779 CrossRef.
- A. Wittkopp and P. R. Schreiner, Chem.–Eur. J., 2003, 9, 407 CrossRef CAS PubMed.
- M. Žabka and R. Šebesta, Molecules, 2015, 20, 15500 CrossRef PubMed.
- (a) T. Marcelli, in Ideas in chemistry and molecular sciences, ed. B. Pignataro, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 115–141 Search PubMed; (b) S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101 CrossRef CAS PubMed.
- (a) K. Etzenbach-Effers and A. Berkessel, in Asymmetric organocatalysis, ed. B. List, Springer, 2009, pp. 38–69 Search PubMed; (b) A. Jalan, R. W. Ashcraft, R. H. West and W. H. Green, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2010, 106, 211 RSC; (c) D. Ardura, R. López and T. L. Sordo, J. Phys. Chem. B, 2005, 109, 23618 CrossRef CAS PubMed; (d) J. N. Harvey, Faraday Discuss., 2010, 145, 487 RSC.
- (a) G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed; (b) R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811 CrossRef CAS PubMed.
- (a) T. Azuma, Y. Kobayashi, K. Sakata, T. Sasamori, N. Tokitoh and Y. Takemoto, J. Org. Chem., 2014, 79, 1805 CrossRef CAS PubMed; (b) S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817 CrossRef CAS PubMed; (c) K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 5919 CrossRef CAS.
- (a) X. Li, H. Deng, B. Zhang, J. Li, L. Zhang, S. Luo and J.-P. Cheng, Chem.–Eur. J., 2010, 16, 450 CrossRef CAS PubMed; (b) R. R. Walvoord, P. N. H. Huynh and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 16055 CrossRef CAS PubMed; (c) A. R. Nödling, G. Jakab, P. R. Schreiner and G. Hilt, Eur. J. Org. Chem., 2014, 6394 CrossRef.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96 CrossRef CAS.
- (a) S. Chen, M. S. Hossain and F. W. Foss, Org. Lett., 2012, 14, 2806 CrossRef CAS PubMed; (b) C. J. Rogers, T. J. Dickerson, A. P. Brogan and K. D. Janda, J. Org. Chem., 2005, 70, 3705 CrossRef CAS PubMed; (c) F. Eisenreich, P. Viehmann, F. Müller and S. Hecht, Macromolecules, 2015, 48, 8729 CrossRef CAS; (d) P. Ménová, H. Dvořáková, V. Eigner, J. Ludvík and R. Cibulka, Adv. Synth. Catal., 2013, 355, 3451 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006 CrossRef CAS.
- B. Kumar, M. A. Aga, A. Rouf, B. A. Shah and S. C. Taneja, RSC Adv., 2014, 4, 21121 RSC.
- E. I. Balmond, D. M. Coe, M. C. Galan and E. M. McGarrigle, Angew. Chem., Int. Ed., 2012, 51, 9152 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary data, tables, figures, mechanistic discussions, and crystal structures. CCDC 1473889–1473891 and 1473916. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03366d |
| ‡ Current address: Department of Chemistry, Technical University of Denmark, Kemitorvet, DK-2800 Kongens Lyngby, Denmark. |
| This journal is © The Royal Society of Chemistry 2017 |