
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting rhodium-catalysed ynamide hydroacylation as a platform for divergent heterocycle synthesis†
Robert N.
Straker
 ,
Manjeet K.
Majhail
and
Michael C.
Willis
*
,
Manjeet K.
Majhail
and
Michael C.
Willis
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 5th October 2017
Abstract
The first examples of ynamide hydroacylation are described. Using rhodium catalysis, linear β-enaminone products are generated in high yield and excellent regioselectivity from the combination of aldehydes and ynamides. The enaminone products are subsequently used as a platform to construct a diverse array of substituted pyrazoles, pyrimidines, and isoxazoles in a two-step, one-pot sequence. It was found that with judicious choice of catalyst system it was possible to overturn the regioselectivity of the hydroacylation reaction to generate α-enaminone products.
Introduction
The abundance of heterocycles in natural products and biologically active compounds has made them prime targets for the synthetic community.1 Despite many classical syntheses, the ability to construct these motifs in an efficient and atom-economical manner is of the utmost importance.2 Hydroacylation reactions enable the rapid assembly of diversely substituted carbonyl compounds, which can be further transformed into heterocycles.3 In this context, a number of strategies have been explored. Our laboratory has previously reported the synthesis of pyrroles,4 furans,5 and quinolines6 through intermolecular hydroacylation of alkynes, and subsequent intramolecular cyclisation of the enone product with a pendant nucleophile (Scheme 1a). The Dong group employed a similar tactic, in their report of vinylphenol-directed hydroacylation, to construct benzofurans via a cyclocondensation reaction (Scheme 1b).7 An alternative approach has been to incorporate the directing group, used to control the hydroacylation reaction and present in the aldehyde component, in the heterocyclic products (Scheme 1c). This method has been used to great effect to generate thiochroman-4-ones,8 4-quinolones, and chroman-4-ones.9 Although elegant, each of these previous syntheses required a specific substrate class in order to construct the desired heterocycle, as the “heteroatom” of each specific heterocycle is pre-installed in the hydroacylation product. We envisaged a conceptually new strategy, in which diverse heterocycles could be prepared from a single hydroacylation-derived scaffold; crucially, the heteroatom(s) of the heterocycle would be introduced using an initial intermolecular step (Scheme 1d). β-Enaminones serve as dipolar 1,3-dicarbonyl surrogates with defined reactivity,10 and as such they have been used in the synthesis of various valuable heterocycles, including uniquely substituted isoxazoles,11 pyrazoles,12 and pyrimidines.13 Typically, the synthesis of enaminones is achieved by the condensation of ketones with amides, which requires forcing conditions, and thus is limited to a small range of substituents and functional groups. This route also presents regioselectivity issues with ketones containing more than one enolisable position.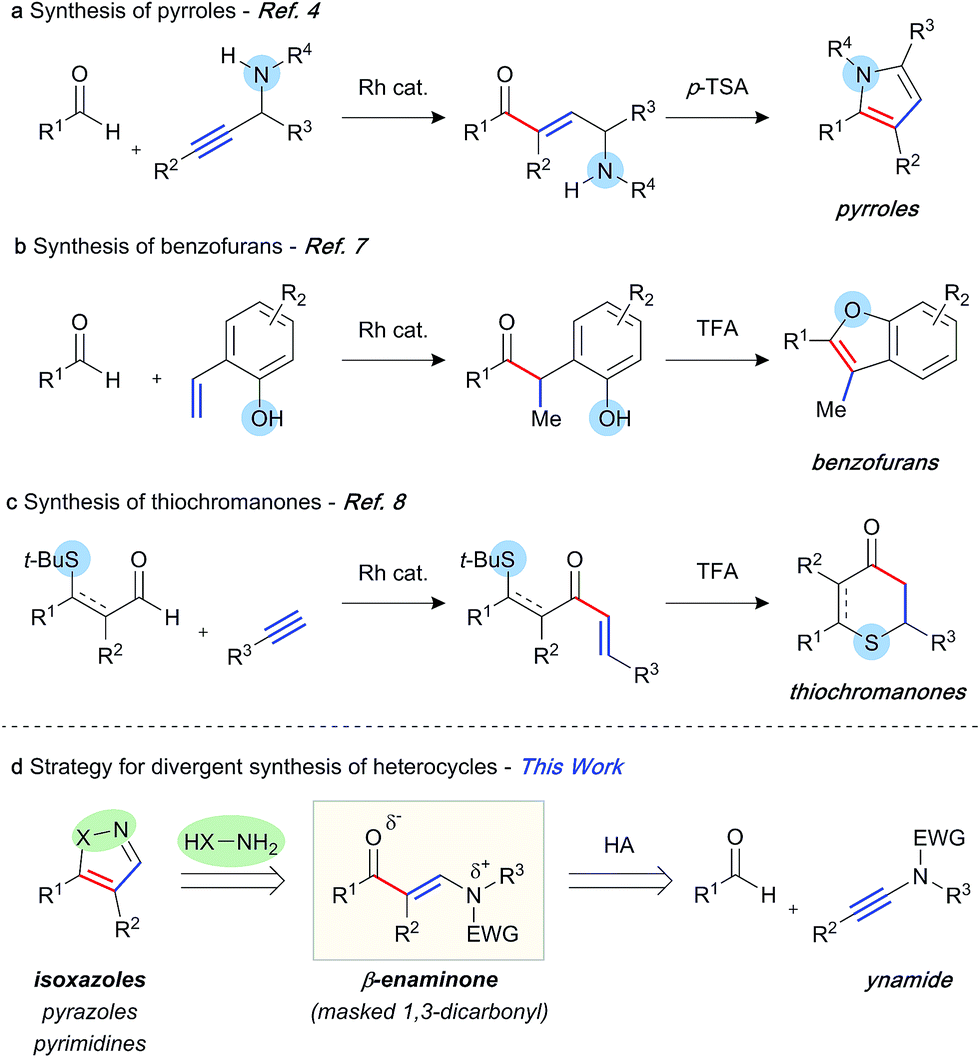 | ||
| Scheme 1 Hydroacylation strategies towards heterocycle synthesis. | ||
We proposed an unprecedented disconnection of the enaminone acyl-enamine bond, which could be achieved synthetically via the hydroacylation of an ynamide. The field of ynamide chemistry has burgeoned over the past decade owing to the unique properties and reactivity of this motif.14 Although ynamides have been employed in a number of transition metal-catalysed transformations, and the related enamides have previously been demonstrated as efficient hydroacylation substrates,15 ynamides remain novel substrates for hydroacylation reactions. In this capacity, ynamides have the potential to provide modular access to highly substituted enaminone products, and thus provide a platform for heterocycle synthesis. Herein, we report rhodium-catalysed intermolecular ynamide hydroacylation, and the synthesis of 4,5-disubstituted isoxazoles via a one-pot hydroacylation/cyclisation sequence. We also show the potential of this method in the formation of pyrazoles and pyrimidines.
Results and discussion
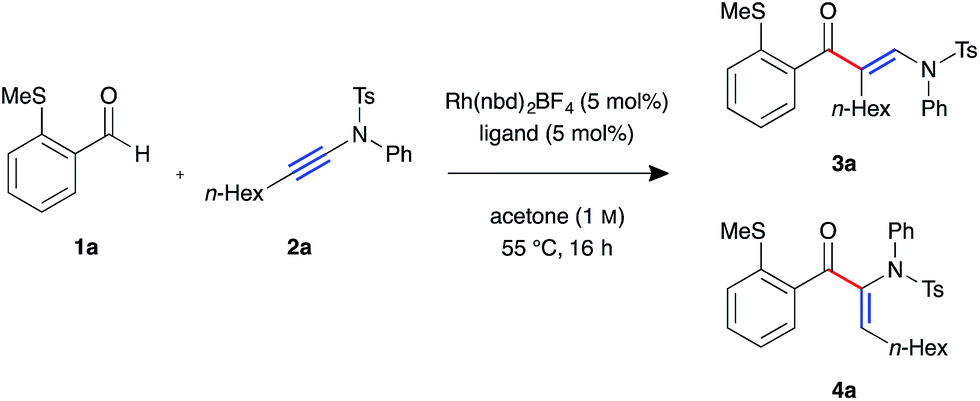
We began our investigation with S-substituted aldehyde 1a (Table 1), which we have previously shown to be an excellent substrate in a range of rhodium-catalysed alkene and alkyne hydroacylation reactions.16 Conscious of the requirements for subsequent heterocycle formation reactions, we chose to examine N-tosyl-aniline ynamides, as the resultant amine functionality would readily act as a leaving group. Ynamide 2a was submitted to rhodium-catalysed hydroacylation conditions with diphosphine ligands with varying bite-angle. Narrow bite-angle ligands dcpm and dppm, known to efficiently promote intermolecular alkene and alkyne hydroacylation,17 exhibited modest reactivity after 16 hours at 55 °C (entries 1 and 2). The electron-rich alkyl phosphine dcpm displayed a small preference for the linear β-enaminone product 3a over the branched α-enaminone 4a (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr). However, aryl phosphine dppm generated product 3a as a single regioisomer (>20:1). Maintaining a narrow bite angle but varying the nature of the tether, PNP(Cy) led to greatly increased reactivity and enhanced regioselectivity for α-enaminone 4a (1:2.5 rr) which was isolated in 60% yield (entry 3). Unfortunately, the aryl phosphine variant PNP(Ph) was not effective in promoting the reaction (entry 4). Increasing bite-angle with dcpe and dppe ligands, employed by Bosnich in intramolecular hydroacylation of cyclopentanones,18 resulted in lower levels of catalyst activity, but continued the trend of regioselectivity observed with alkyl and aryl phosphines (1:1 and >20:1 rr respectively, entries 5 and 6). Increasing bite-angle further with dppp and dppb resulted in loss of catalyst activity (entries 10 and 11). We next turned to ligands possessing hemi-labile O-tethers, which are known to minimise unwanted reductive decarbonylation in reactions of alkynes.19 DCEphos was found to be inactive (entry 13), however, DPEphos returned the catalyst activity, with the starting material entirely consumed after 16 hours, and the linear product 3a isolated in 90% yield (>20:1 rr, entry 14). Xantphos was not effective in promoting the reaction, which could perhaps be attributed to the reduced conformational freedom of the ligand backbone (entry 15).
:1 rr). However, aryl phosphine dppm generated product 3a as a single regioisomer (>20:1). Maintaining a narrow bite angle but varying the nature of the tether, PNP(Cy) led to greatly increased reactivity and enhanced regioselectivity for α-enaminone 4a (1:2.5 rr) which was isolated in 60% yield (entry 3). Unfortunately, the aryl phosphine variant PNP(Ph) was not effective in promoting the reaction (entry 4). Increasing bite-angle with dcpe and dppe ligands, employed by Bosnich in intramolecular hydroacylation of cyclopentanones,18 resulted in lower levels of catalyst activity, but continued the trend of regioselectivity observed with alkyl and aryl phosphines (1:1 and >20:1 rr respectively, entries 5 and 6). Increasing bite-angle further with dppp and dppb resulted in loss of catalyst activity (entries 10 and 11). We next turned to ligands possessing hemi-labile O-tethers, which are known to minimise unwanted reductive decarbonylation in reactions of alkynes.19 DCEphos was found to be inactive (entry 13), however, DPEphos returned the catalyst activity, with the starting material entirely consumed after 16 hours, and the linear product 3a isolated in 90% yield (>20:1 rr, entry 14). Xantphos was not effective in promoting the reaction, which could perhaps be attributed to the reduced conformational freedom of the ligand backbone (entry 15).
|
|
|||
|---|---|---|---|
| Entry | Ligand | Yieldb/% |
3a:4ab |
| a Reaction conditions: Rh(nbd)2BF4 (5 mol%), ligand (5 mol%), aldehyde (0.3 mmol, 1.0 equiv.), ynamide (1.1 equiv.), acetone (1.0 M), 55 °C for 16 h. b Determined by 1H NMR spectroscopic analysis of the crude reaction mixture, using 1,3,5-trimethoxybenzene as the internal standard. c Isolated yield of 4a. d Isolated yield of 3a. | |||
| 1 | dcpm | 28 | 2:1 |
| 2 | dppm | 36 | >20:1 |
| 3 | PNP(Cy) | 92 (60)c | 1:2.5 |
| 4 | PNP(Ph) | 0 | — |
| 5 | dcpe | 30 | 1:1 |
| 6 | dppe | 46 | >20:1 |
| 7 | dape | 36 | 17:1 |
| 8 | dtfpe | 23 | 17:1 |
| 9 | dppe(o-iPr) | 0 | — |
| 10 | dppp | 0 | — |
| 11 | dppb | 0 | — |
| 12 | dppf | 26 | 3:1 |
| 13 | DCEphos | 0 | — |
| 14 | DPEphos | 93 (90)d | >20:1 |
| 15 | Xantphos | 0 | — |
|
|||
In order to elucidate the origin of the observed change in regioselectivity between alkyl and aryl phosphines, electron-rich and electron-poor aryl phosphine ligands dape and dtfpe were tested (entries 7 and 8). However, both ligands led to the generation of the β-enaminone product 3a with identical selectivity (17:1 rr). The more sterically encumbered analogue dppe(o-iPr) was ineffectual in the reaction (entry 9), with no product formation observed.
In addition, control experiments were performed with an electronically neutral but sterically biased internal alkyne 2aa, using DPEphos and PNP(Cy) ligands (Scheme 2). Interestingly, the linear enone product 3aa was formed as a single regioisomer in the presence of DPEphos (>20:1 rr). However, in contrast to the ynamide substrate which gave branched selectivity with PNP(Cy) (1:2.5 rr), the alkyne substrate led to the linear product being formed but with lower regioselectivity (5:1 rr).
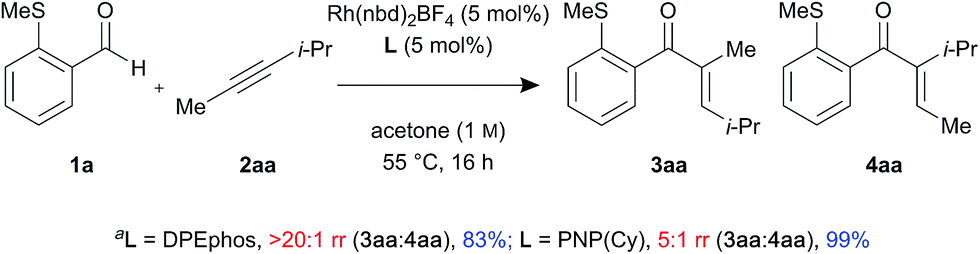 | ||
| Scheme 2 Control experiments with sterically biased internal alkyne (adetermined by 1H NMR spectroscopic analysis of the crude reaction mixture, using 1,3,5-trimethoxybenzene as the internal standard). | ||
A general mechanism for ynamide hydroacylation, based on these results and our previous studies of alkene and alkyne systems,17b is illustrated in Scheme 3. Upon ynamide coordination, hydrometallation may proceed via one of two regioisomeric intermediate complexes; II-L leading to the linear product 3, and II-B to the branched product 4. Owing to their π-acidity, aryl phosphines result in an electron-poor rhodium metal centre, which is compensated for by stronger coordination of the ynamide. This effect is expected to exacerbate steric interactions between the substrate and ligand substituents, favouring intermediate II-L, and leading to the linear product 3. In contrast, strongly σ-donating alkyl phosphines increase electron density on the metal, resulting in a more weakly bound substrate. This, paired with an electronically biased ynamide would allow for the formation of increasing amounts of the branched isomer 4.
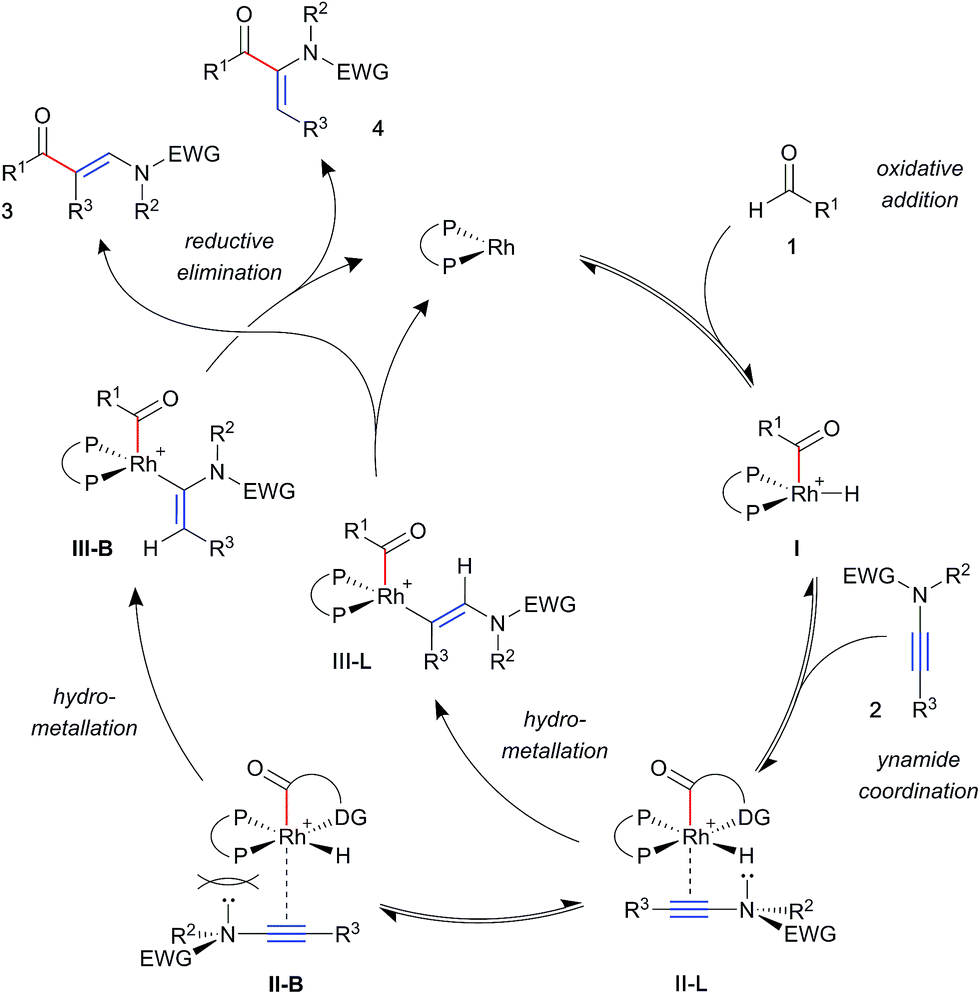 | ||
| Scheme 3 Proposed mechanism for regioselectivity in ynamide hydroacylation. | ||
With a suitable catalyst system in hand, we examined the tolerance of the linear-selective ynamide hydroacylation reaction towards various ynamide substituents (Fig. 1a). Ynamides 2a–l were synthesised via copper-catalysed oxidative coupling of N-protected amines with the corresponding alkynes,20 and submitted to the optimised reaction conditions with aldehyde 1a. Pleasingly, in addition to sulfonamides, the reaction also tolerated carbamate substrates, with carboxybenzyl-protected β-enaminone product 3b generated in excellent yield, albeit with slightly reduced regioselectivity (7:1 rr). Boc-protected ynamide 1c was less reactive, requiring increased concentration (2 M) to give 3c in moderate yield. Mesyl-protected methylamine ynamide 1d performed well, generating the linear product 3d in high yield. However, here, again, lower levels of regioselectivity were observed (5:1 rr), perhaps due to reduced steric bias of the ynamide. Both sp3 and sp2 hybridised ynamide substituents were well tolerated, with the former providing higher linear:branched selectivities. It was found that under the mild reaction conditions, primary alkyl halides 3e and silyl ethers 3f were tolerated, both exhibiting perfect regioselectivity and isolated in >85% yield. Ynamides 2i and 2j, bearing electron-poor aromatic groups, gave higher yields compared to that of the neutral and electron-rich aryl substituted ynamides 2g and 2h. However, there was little observed change in regioselectivity between the para-substituted aryl ynamides (6:1 rr). Thiophenyl and cyclohexenyl substituted ynamides 2k and 2l also gave the corresponding β-enaminone products 3k and 3l in high yields. In order to assess the practicability of the methodology, reaction of ynamide 2b was performed on a 4 mmol scale, using only 1 mol% catalyst, which successfully generated enaminone 3b as a single regioisomer (>20:1 rr) in 88% yield (1.79 g) after 40 hours at 55 °C.
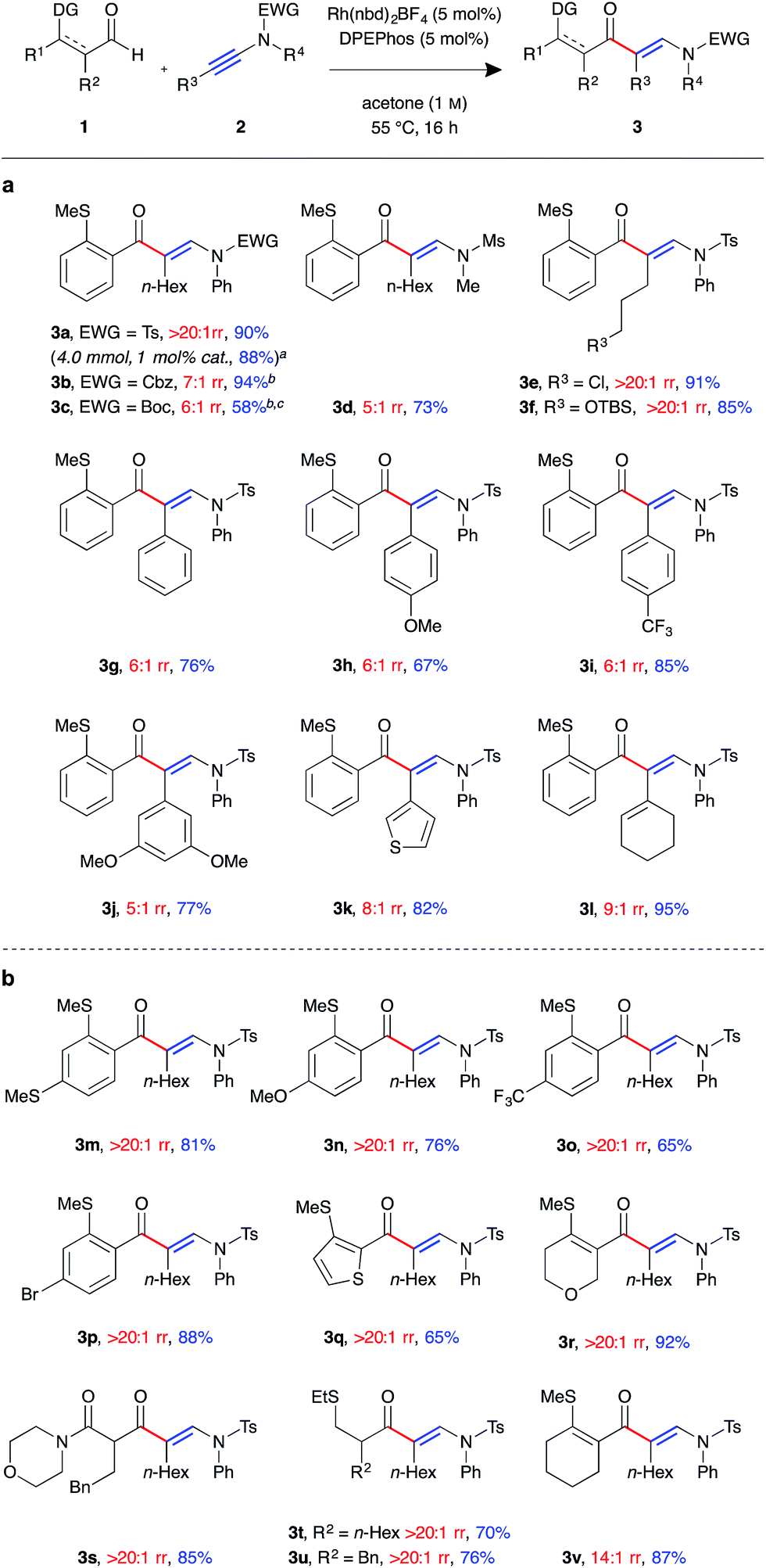 | ||
| Fig. 1 Linear-selective ynamide hydroacylation (a) scope of ynamide component. (b) Scope of aldehyde component. (Rh(nbd)2BF4 (5 mol%), DPEphos (5 mol%), aldehyde (0.30 mmol, 1.0 equiv.), ynamide (1.1 equiv.), acetone (1.0 M), 55 °C for 16 h. Regioisomeric ratio determined by 1H NMR spectroscopic analysis of the crude reaction mixture. aPerformed with 4 mmol of aldehyde, using 1 mol% catalyst, with the reaction mixture stirred for 40 h. bIsolated as an inseparable mixture of isomers. cReaction performed at 2 M concentration). | ||
We next examined the scope of the reaction with respect to the aldehyde component using various substituted aldehydes 1 (Fig. 1b). Electron-rich aryl aldehydes performed well, with products 3m and 3n both obtained in high yields. Electron-poor aryl aldehydes exhibited the desired reactivity, however, β-enaminone 3o was only isolated in moderate yield. In contrast, bromo-substituted product 3p was obtained in excellent yield. Thiophenyl aldehyde 1q was found to be less reactive, with the reaction not reaching completion after 16 h at 55 °C. As a result the product 3q was isolated in 65% yield. Dihydropyran 3r was formed in excellent yield. Our laboratory recently reported the use of β-carbonyl-substituted aldehydes in alkene and alkyne hydroacylation reactions,21 which here too demonstrated as efficient substrates; β-enaminone 3s was obtained in an 85% yield as a single regioisomer (>20:1 rr). Pleasingly, α-substituted alkyl aldehydes also underwent the desired C–H oxidative addition, to yield hydroacylation products 3t and 3u in good yield. β-Substituted alkyl aldehydes were found to be unreactive using the current methodology. Cyclohexenyl aldehyde 1v was the only example to exhibit lower than perfect levels of regioselectivity when combined with an alkyl substituted ynamide (14:1 rr); despite this, the product 3v was isolated in excellent yield.
To demonstrate the utility of the requisite sulfide directing group present in the β-enaminone products, three-component ynamide hydroacylation/Suzuki-type coupling reactions were performed (Fig. 2).22 Upon consumption of the aldehyde starting material, the reaction mixture was transferred to a second reaction vessel containing a solution of Rh-dcpm catalyst, boronic acid, and silver carbonate in acetone, and the reaction mixture stirred for a further 16 hours at 55 °C. The coupled β-enaminone products 5a–c were formed in high yield over two steps.
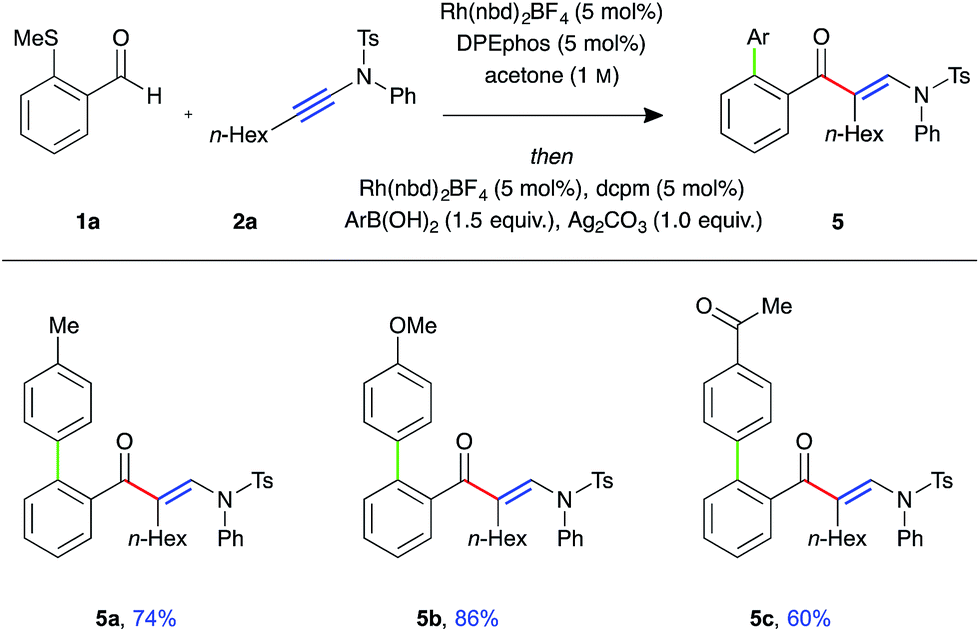 | ||
| Fig. 2 Three-component ynamide hydroacylation/Suzuki-type coupling (Rh(nbd)2BF4 (5 mol%), DPEphos (5 mol%), aldehyde (0.3 mmol, 1 equiv.), ynamide (1.1 equiv.), acetone (1.0 M), 55 °C for 16 h; then Rh(nbd)2BF4 (5 mol%), dcpm (5 mol%), silver carbonate (1.0 equiv.), boronic acid (1.5 equiv.), acetone (0.3 M), 55 °C for 16 h). | ||
In the process of optimising the linear-selective hydroacylation reaction we observed a reversal in regioselectivity with the use of the PNP(Cy) ligand, which led to the formation of the α-enaminone product 4a. These have been shown as valuable precursors for the synthesis of chiral α-amino acid derivatives via asymmetric reduction.23 As such we decided to examine the scope of the branched-selective reaction with a range of aldehydes and ynamides (Fig. 3). Overall, a lower level of regioselectivity was observed than in the linear selective reaction. Nevertheless, it was possible to separate, using simple silica column chromatography, and isolate the branched products 4 in moderate to good yields. For example, formation of branched product 4a was achieved on a 4 mmol scale, with 2.5 mol% catalyst loading, and was isolated as a single regoisomer in 68% yield (1.38 g) after 16 h at 55 °C. The linear isomer 3b was also isolated from this reaction in 25% yield (0.51 g). As in the linear-selective reaction, aryl substituted ynamide 2e exhibited the lowest level of regioselectivity (1:1 rr), with the branched isomer isolated in 42%.
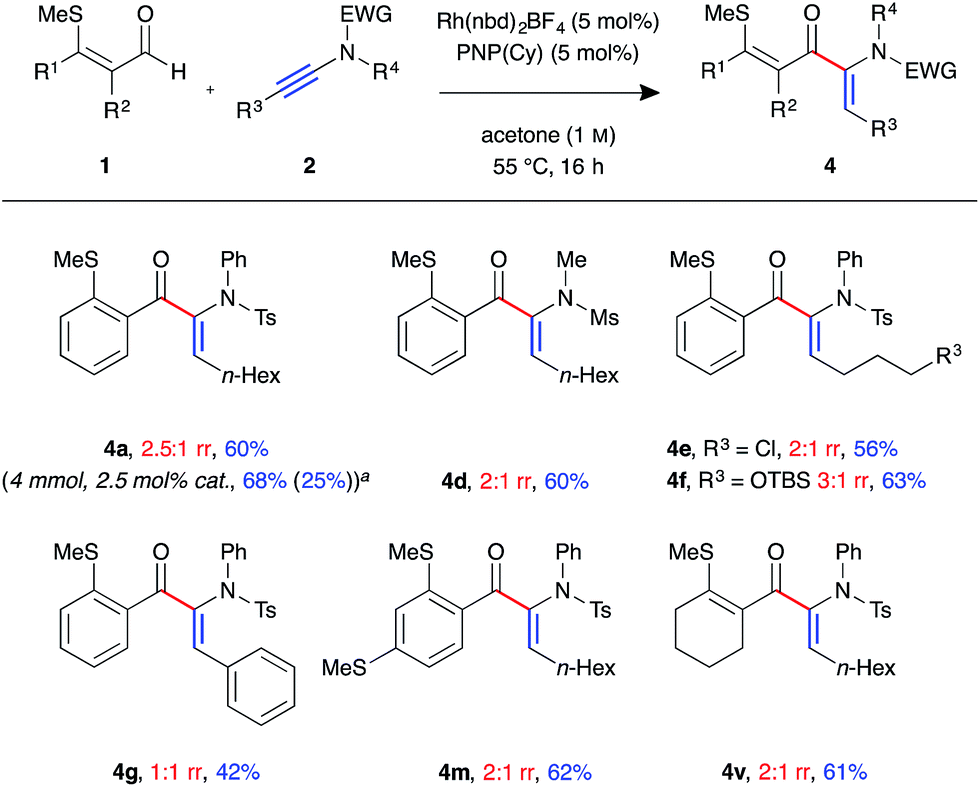 | ||
| Fig. 3 Scope of branched-selective ynamide hydroacylation (Rh(nbd)2BF4 (5 mol%), PNP(Cy) (5 mol%), aldehyde (0.3 mmol, 1 equiv.), ynamide (1.1 equiv.), acetone (1.0 M), 55 °C for 16 h; regioisomeric ratio determined by 1H NMR spectroscopic analysis of the crude reaction mixture; yields of isolated single regioisomers. aPerformed with 4 mmol of aldehyde, using 2.5 mol% catalyst, with the reaction mixture stirred for 16 h, value in parentheses is the isolated yield of linear isomer 3a). | ||
Having established a robust protocol for ynamide hydroacylation, we turned our attention to utilising the β-enaminone products in the generation of heterocyclic compounds (Fig. 4). It was found that under acidic conditions, in the presence of an external nucleophile, these species indeed behave as 1,3-dicarbonyl surrogates. Furthermore, isoxazole products 6 could be obtained directly in a one-pot hydroacylation/nucleophilic addition/cyclisation process. Upon consumption of the aldehyde starting material, hydroxylamine hydrochloride and ethanol were added, and the reaction mixture stirred for a further 16 hours at 80 °C. The protic solvent was crucial for reactivity as the reaction was found to proceed through an enol ether adduct of the β-enaminone and alcohol, which could be isolated from the reaction mixture when the reaction was conducted at room temperature. In general, the isoxazole products 6a–u were isolated in high yields and near quantitative conversion from β-enaminone intermediates 3 in a one-pot procedure. The reactions were highly selective, with a single regioisomer observed in almost all cases. The products were determined to be 4,5-disubstituted isoxazoles by observation of nOe interactions of the N-methylated isoxazole derivative of 6g (see ESI†). Formation of heterocycle 6a could be achieved using either N-tosyl-aniline ynamide 2a or N-mesyl-methylamine ynamide 2d, the former providing the product in higher yield (85%) due to both higher selectivity in the hydroacylation reaction and better reactivity of the β-enaminone intermediate 3a compared to 3d. Primary alkyl halide 6e was isolated in relatively low yield, likely due to unwanted side reaction arising from nucleophilic substitution by hydroxylamine. Similarly, under the acidic reaction conditions, the silyl-protected primary alcohol was deprotected, and the free alcohol product 6f isolated in 79% yield.
 | ||
| Fig. 4 Tandem ynamide hydroacylation/isoxazole formation (a) scope of ynamide component. (b) Scope of aldehyde component (Rh(nbd)2BF4 (5 mol%), DPEphos (5 mol%), aldehyde (0.3 mmol, 1 equiv.), ynamide (1.1 equiv.), acetone (1.0 M), 55 °C for 16 h, then hydroxylamine hydrochloride (5 equiv.), ethanol (0.5 M), 80 °C for 16 h). aIsolated along with the regioisomeric isoxazole (<10%). | ||
Finally, enaminone 3b could be further derivatised with a variety nucleophiles to generate an array of heterocyclic products (Fig. 5). Use of N-substituted hydrazines gave 4,5-disubstituted pyrazoles 7a–c in high yields under the above cyclisation reaction conditions (Fig. 5a). A single regioisomer was observed in all cases, with the product regiochemistry of 7b determined by nOe experiment (see ESI†). Under the same reaction conditions, the synthesis of 3,4-disubstituted pyrimidine 8a was achieved with benzamidine, with the product isolated in excellent yield (Fig. 5b). Reaction with guanidines was only possible under basic conditions, requiring addition of K2CO3 as base and DMF as solvent, yielding amino-pyrimidines 8b and 8c in good yield.
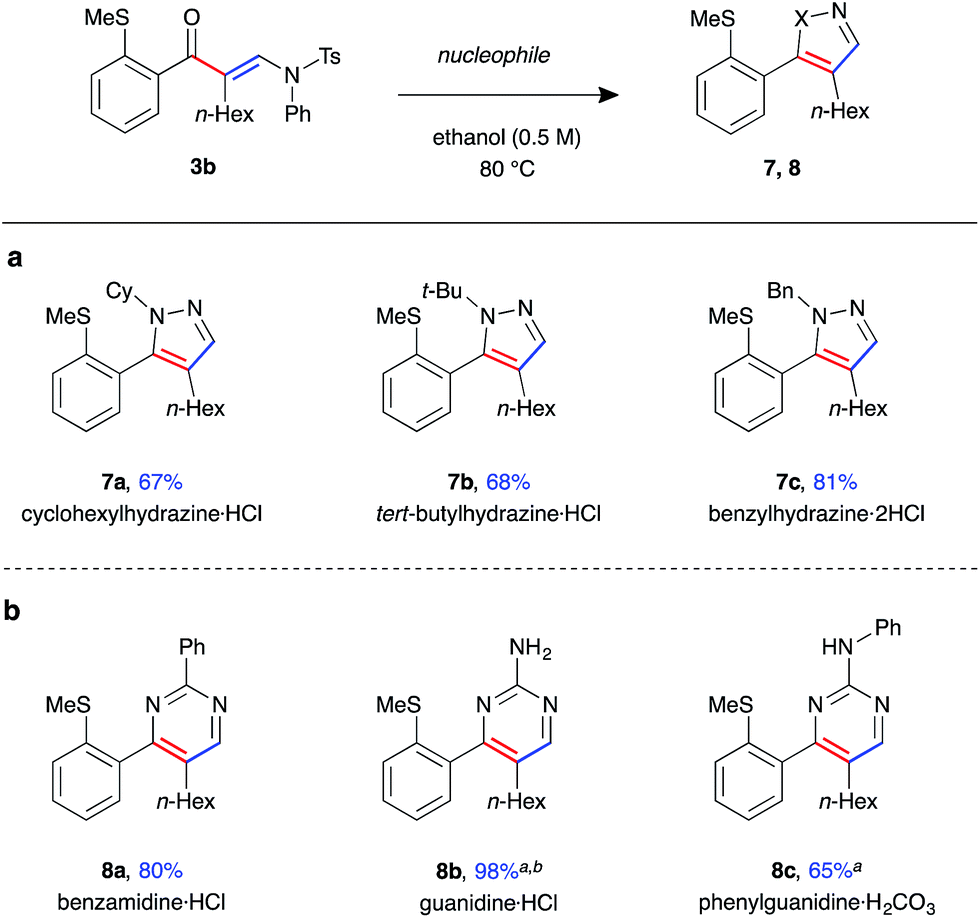 | ||
| Fig. 5 Scope of heterocycle formation from β-enaminone 3b (a) pyrazoles. (b) Pyrimidines (enaminone (0.2 mmol, 1.0 equiv.), nucleophile (5 equiv.), ethanol (0.5 M), 80 °C for 16 h). aAdditional K2CO3 (6 equiv.), reaction performed in DMF (0.5 M) at 100 °C for 16 h. bIsolated after work-up without purification. | ||
Conclusions
In summary, we have developed the first examples of ynamide hydroacylation, which yield substituted α- and β-enaminones with tunable regioselectivity determined by the choice of catalyst system. Using this methodology, it was possible to synthesise a number of heterocyclic products, from a single set of hydroacylation starting materials and a selected nucleophile, in tandem hydroacylation/nuclophilic addition/cyclisation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for financial support for this research.Notes and references
- (a) A. R. Katritzky, Chem. Rev., 2004, 104, 2125–2126 CrossRef CAS; (b) L. D. T. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, Wiley, 2010 Search PubMed.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed.
- M. K. Majhail, P. M. Ylioja and M. C. Willis, Chem.–Eur. J., 2016, 22, 7879–7884 CrossRef CAS PubMed.
- P. Lenden, D. A. Entwistle and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 10657–10660 CrossRef CAS PubMed.
- J. D. Neuhaus, S. M. Morrow, M. Brunavs and M. C. Willis, Org. Lett., 2016, 18, 1562–1565 CrossRef CAS PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Angew. Chem., Int. Ed., 2014, 53, 2455–2459 CrossRef CAS PubMed.
- A. Bouisseau, J. Glancy and M. C. Willis, Org. Lett., 2016, 18, 5676–5679 CrossRef CAS PubMed.
- (a) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed; (b) X.-W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed.
- (a) P. Lue and J. V. Greenhill, in Advances in Heterocyclic Chemistry, ed. R. K. Alan, Academic Press, 1996, vol. 67, pp. 207–343 Search PubMed; (b) A. Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS; (c) G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461–491 CrossRef CAS; (d) C. B. d. K. J. P. Michael, D. Gravestock, G. D. Hosken, A. S. Howard, C. M. Jungmann, R. W. M. Krause, A. S. Parsons, S. C. Pelly and T. V. Stanbury, Pure Appl. Chem., 1999, 71, 979–988 CrossRef.
- (a) Y.-I. Lin and S. A. Lang, J. Org. Chem., 1980, 45, 4857–4860 CrossRef CAS; (b) E. Domínguez, E. Ibeas, E. Martínez de Marigorta, J. K. Palacios and R. SanMartín, J. Org. Chem., 1996, 61, 5435–5439 CrossRef; (c) R. Olivera, R. SanMartin, E. Domínguez, X. Solans, M. K. Urtiaga and M. I. Arriortua, J. Org. Chem., 2000, 65, 6398–6411 CrossRef CAS PubMed.
- L. J. Missio, H. S. Braibante and M. E. F. Braibante, J. Heterocycl. Chem., 1996, 33, 1243–1245 CrossRef CAS.
- T. J. Müller and A. S. Karpov, Synthesis, 2003, 2815–2826 CrossRef.
- (a) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (c) R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Nat. Commun., 2016, 7, 10109 CrossRef CAS PubMed.
- H. J. Zhang and C. Bolm, Org. Lett., 2011, 13, 3900–3903 CrossRef CAS PubMed.
- M. C. Willis, S. J. McNally and P. J. Beswick, Angew. Chem., Int. Ed., 2004, 43, 340–343 CrossRef CAS PubMed.
- (a) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed; (b) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- D. P. Fairlie and B. Bosnich, Organometallics, 1988, 7, 936–945 CrossRef CAS.
- (a) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2006, 45, 7618–7622 CrossRef CAS PubMed; (b) G. L. Moxham, H. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem.–Eur. J., 2008, 14, 8383–8397 CrossRef CAS PubMed.
- (a) N. Riddell, K. Villeneuve and W. Tam, Org. Lett., 2005, 7, 3681–3684 CrossRef CAS PubMed; (b) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS PubMed; (c) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833–835 CrossRef CAS PubMed.
- T. J. Coxon, M. Fernandez, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2017, 139, 10142–10149 CrossRef CAS PubMed.
- J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC.
- W. Gao, Q. Wang, Y. Xie, H. Lv and X. Zhang, Chem.–Asian. J., 2016, 11, 231–233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterisation for all compounds. See DOI: 10.1039/c7sc03795c |
| This journal is © The Royal Society of Chemistry 2017 |