Effect of spacers and anchoring groups of extended π-conjugated tetrathiafulvalene based sensitizers on the performance of dye sensitized solar cells†
Received
20th October 2016
, Accepted 30th November 2016
First published on 9th January 2017
Abstract
Four new extended π-conjugated tetrathiafulvalene (ex-TTF) based dyes featuring a donor–π–acceptor (D–π–A) configuration with varying π-spacers and anchoring groups were synthesized and characterized. The sensitizer having the 4-ethynyl phenyl π-spacer (G4) shows red shifted absorption maxima in comparison with the sensitizer having only the phenyl π-spacer (G2). All four sensitizers undergo reversible oxidations to form stable radical cations. TDDFT calculations highlighted that the LUMO of the G4 sensitizer is more stabilized by the incorporation of the ethynyl group between the π-spacer and the cyanoacrylic acid anchoring group that aid to inject electrons efficiently into TiO2 thereby resulting in an enhanced power conversion efficiency of 6.36% when compared to the other derivatives, which is also confirmed by the intensity-modulated photovoltage spectroscopy (IMVS) method. Kinetic studies demonstrated that fast regeneration of the oxidized dye by the redox couple is an important factor behind enhanced efficiencies in solar cells. Finally, the performance of the present sensitizers is compared to that of previously reported sensitizers.
Introduction
Dye-Sensitized Solar Cells (DSSCs) have attracted interest for over two and half decades because of their ease of fabrication, flexibility and low environmental impact compared to conventional solid-state p–n junction photovoltaic devices.1–8 The sensitizer is one of the vital components in achieving high efficiency and durability of the device. Among various classes of sensitizers, Ru(II) polypyridyl complexes are widely used in view of their broad absorption through the metal-to-ligand charge transfer (MLCT) transition and their long excited state lifetimes.9–11 However, the rarity of Ru metal in the Earth’s crust and cost due to intricate synthesis and purification steps might impede commercialization of the technology. Recently, a new class of compounds i.e., metal-free organic dyes have been found to be alternatives based on the tunability of their optical and electrochemical properties in a desired manner through suitable molecular design strategies.12–15 Organic dyes showing good efficiencies are composed of a donor (D) and an acceptor (A) linked through a π-bridge and one can efficiently tune their optical properties.16–18 By adopting the D–π–A approach, great varieties of metal-free organic dyes have been reported with a record efficiency of 14%.19
Tetrathiafulvalene (TTF) scaffolds have a wide range of applications in materials chemistry towards molecular optoelectronics.20,21 A few TTF scaffold based sensitizers have been reported in the literature.22–24 Grätzel and co-workers reported extended π-conjugated tetrathiafulvalene (ex-TTF) based sensitizers for DSSCs with an efficiency of 3.8%.24 The low efficiency of ex-TTF sensitizers is due to their energetically unfavourable HOMO level, in which dye-regeneration after electron-injection is thermodynamically not feasible. Geng et al. studied the effect of π-linkers between the donor tetrathiafulvalene scaffold and anchoring groups on optical and electronic properties, achieving an overall device conversion efficiency of 0.87%.22 Recently, our group have reported ex-TTF based sensitizers by introducing long alkyl chains and changing the π-linker, achieving an overall conversion efficiency of 7.15%.25 Motivated by these results, we have designed four new D–π–A sensitizers constituted with ex-TTF as the donor and either cyanoacrylic acid or rhodanine acetic acid as the acceptor. The change of π-linkers between the donor and acceptor units of dyes might tailor their frontier orbital energy levels to be beneficial to red shift the charge-transfer transition. In the present manuscript, we report the synthesis, characterization, optical and electrochemical properties, and recombination/dye regeneration kinetic study of four dyes as well as their application in DSSCs. Finally, the performance of the present ex-TTF sensitizers is compared to that of previously reported sensitizers.
Experimental section
Materials
Unless otherwise noted, all commercial reagents and chemicals were procured from Sigma-Aldrich. Analytical reagent (AR) grade solvents were used for the reactions while laboratory reagent (LR) grade solvents were used for purifications and column chromatography. Dichloromethane (DCM), chloroform (CHCl3), acetonitrile (ACN), N,N-dimethyl formamide (DMF), and ethylacetate (EtOAC) were dried in the presence of calcium hydride under a nitrogen atmosphere. Hexane was purified by Na/benzophenone refluxing overnight, then distilled under vacuum and stored over 4 Å molecular sieves. Triethylamine (TEA) was distilled over NaOH pellets. ACME silica gel (100–200 mesh) was used for column chromatography. Thin-layer chromatography was performed on Merck-precoated silica gel 60-F254 plates. Either gravity or flash chromatography was performed for purification of all compounds. All the reactions were carried out under a nitrogen or argon atmosphere using dry and degassed solvents.
Synthesis
The compounds 2,2′-(2-iodoanthracene-9,10-diylidene)bis(4,5-bis(hexylthio)-1,3-dithiole (1) and 5-(9,10-bis(hexylthio)-1,3-dithiol-2-ylidene)-9,10-dihydroanthracene-2-yl)thiophene-2-carbaldehyde (2a) were synthesized as per literature reports.24,25 The detailed synthetic procedures are as follows.
Synthesis of 5-((9,10-bis(hexylthio)-1,3-dithiol-2-ylidene)-9,10-dihydroanthracene-2-yl)benzaldehyde (2b).
4-Formylphenylboronic acid (0.075 g, 0.48 mmol), compound 1 (0.048 g, 0.5 mmol), Pd(PPh3)4 (0.0114 mmol), and a 2 M aqueous solution of Na2CO3 (2 mL) in dry THF (20 mL) were refluxed overnight under an inert atmosphere. The reaction mixture was extracted with DCM, and washed with water and brine solution and the organic layer was dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation and the obtained residue was purified by column chromatography on silica gel using hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate (10:1, v/v) as the eluent to get the desired compound in 81% yield. Anal. calcd for C51H64OS8 % (949.57): C, 64.51; H, 6.79. Found C, 64.54; H, 6.81. 1H NMR (400 MHz, CDCl3): δ ppm, 0.88 (t, 12H), 1.69–1.61 (m, 24H), 2.90–2.78 (m, 8H), 7.32 (dd, 2H), 7.45 (d, 2H), 7.72 (d, 2H), 7.84 (dd, 3H), 8.0 (d, 2H), 10.1 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 190.78, 146.28, 141.60, 135.08, 134.81, 134.31, 130.57, 129.78, 127.18, 126.95, 126.37, 123.20, 122.57, 112.59, 36.29, 31.05, 29.70, 28.25, 22.57, 14.22; MALDI-TOF MS calcd for 949.6, found 949.28.
:ethylacetate (10:1, v/v) as the eluent to get the desired compound in 81% yield. Anal. calcd for C51H64OS8 % (949.57): C, 64.51; H, 6.79. Found C, 64.54; H, 6.81. 1H NMR (400 MHz, CDCl3): δ ppm, 0.88 (t, 12H), 1.69–1.61 (m, 24H), 2.90–2.78 (m, 8H), 7.32 (dd, 2H), 7.45 (d, 2H), 7.72 (d, 2H), 7.84 (dd, 3H), 8.0 (d, 2H), 10.1 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 190.78, 146.28, 141.60, 135.08, 134.81, 134.31, 130.57, 129.78, 127.18, 126.95, 126.37, 123.20, 122.57, 112.59, 36.29, 31.05, 29.70, 28.25, 22.57, 14.22; MALDI-TOF MS calcd for 949.6, found 949.28.
Synthesis of 5-((9,10-bis(hexylthio)-1,3-dithiol-2-ylidene)-9,10-dihydroanthracene-2-yl)ethynyl)benzaldehyde (2c).
4-Ethynylbenzaldehyde (0.5 mmol) was dissolved in THF (10 mL) followed by addition of copper(I)-iodide (CuI) (0.04 mmol), dichlorobis(triphenylphosphine) palladium(II) (0.02 mmol) and triethylamine (1 mL). To this, 1 (0.5 mmol) was added and the reaction mixture was warmed at 30 °C with stirring for 45 min at room temperature for 20 h. Then the solvent was removed under vacuum and the residue was purified by silica gel column chromatography (hexane:ethyl acetate: 8:2, v/v as eluent) to give the desired compound 2c in 62.4% yield. Anal. calcd for C53H64OS8 % (973.57): C, 65.39; H, 6.63. Found C, 65.34; H, 6.61. 1H NMR (400 MHz, CDCl3): δ ppm, 0.92 (t, 12H), 1.27–1.41 (m, 24H), 1.60–1.66 (m, 8H), 2.77–2.86 (m, 8H), 7.32 (dd, 2H), 7.47 (dd, 1H), 7.60 (m, 3H), 7.70 (d, 2H), 7.73 (d, 1H), 7.87–7.90 (d, 1H), 10.03 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 188.27, 140.61, 139.25, 138.98, 136.78, 135.36, 134.53, 134.30, 133.30, 132.03, 131.56, 129.12, 126.40, 125.31, 124.03, 123.32, 121.27, 93.77, 90.72, 36.37, 31.33, 26.25, 22.58, 17.85, 14.08; MALDI-TOF MS calcd for 973.6, found 974.24.
General procedure for the synthesis of G2 and G4
Under a N2 atmosphere, the mixture of the corresponding aldehydes (2b or 2c) (1 mmol), cyanoacetic acid (5 mmol) and ammonium acetate (3 mmol) was dissolved in acetic acid (10 mL) and refluxed for 12 h. The reaction was monitored by TLC and the reaction mixture was poured onto ice-cold water and extracted with chloroform. The organic layers were evaporated and dried. The obtained solid residue was purified by column chromatography using silica gel with MeOH:CHCl3 (1:10, v/v) as the eluent to give the corresponding G2 and G4.
G2
.
Yield: 70.5%. Anal. calcd for C54H65NO2S8 % (1016.60): C, 63.80; H, 6.45; N, 1.38. Found C, 63.84; H, 6.46; N, 1.40. 1H NMR (400 MHz, CDCl3): δ ppm, 0.90 (t, 12H), 1.20–1.29 (m, 24H), 1.60–1.64 (m, 8H), 2.75–2.87 (m, 8H), 7.32 (dd, 2H), 7.44 (d, 2H), 7.72 (d, 2H), 7.45 (s, 1H), 7.81 (d, 2H), 7.85 (d, 1H), 7.99 (d, 2H); 13C NMR (100 MHz, CDCl3): δ ppm, 160.77, 158.11, 143.13, 141.58, 135.40, 133.28, 132.75, 130.62, 128.47, 127.66, 126.04, 125.51, 123.64, 122.57, 116.68, 91.29, 36.81, 31.47, 30.17, 27.76, 22.46, 14.49. MALDI-TOF MS calcd for 1016.6, found 1016.40.
G4
.
Yield: 62%. Anal. calcd for C56H65NO2S8 % (1040.62): C, 64.64; H, 6.30; N, 1.35. Found C, 64.64; H, 6.34; N, 1.40. 1H NMR (400 MHz, CDCl3): δ ppm, 0.91 (t, 12H), 1.22–1.38 (m, 24H), 1.59–1.69 (m, 8H), 2.80 (t, 8H), 7.31 (t, 2H), 7.44 (t, 2H), 7.51 (d, 1H), 7.59 (dd, 2H), 7.72 (d, 2H), 7.80 (d, 2H), 7.99 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 161.22, 160.78, 144.96, 138.12, 137.20, 136.52, 135.77, 134.30, 132.89, 131.31, 130.90, 127.85, 126.74, 124.85, 123.32, 121.40, 118.11, 94.92, 91.12, 89.78, 36.62, 31.33, 29.70, 28.22, 21.78, 14.98; MALDI-TOF MS calcd for 1040.6, found 1041.14.
General procedure for the synthesis of G5 and G6
Compound 2a or 2b (1 mmol), rhodanine acetic acid (5 mmol) and ammonium acetate (3 mmol) were dissolved in 10 mL of acetic acid. The resulting reaction mixture was refluxed for 6 h and then cooled to room temperature and extracted with chloroform. The organic layer was evaporated and dried. The obtained solid compound was purified by column chromatography with silica gel using MeOH:CHCl3 (1:10, v/v) as the eluent to give the corresponding G5 and G6.
G5
.
Yield: 69%. Anal. calcd for C54H65NO3S11 % (1128.78): C, 57.46; H, 5.80; N, 1.24. Found C, 57.42; H, 5.83; N, 1.30. 1H NMR (400 MHz, CDCl3): δ ppm, 0.91 (t, 12H), 1.26–1.39 (m, 24H), 1.59–1.69 (m, 8H), 2.92–20.81 (m, 8H), 4.12 (s, 2H), 7.30–7.35 (m, 4H), 7.43 (d, 1H), 7.55 (d, 1H), 7.77 (d, 1H), 7.91 (d, 2H), 8.10 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 195.61, 172.85, 163.95, 144.96, 137.72, 137.20, 135.04, 134.53, 133.83, 132.68, 131.87, 128.54, 126.85, 126.40, 126.18, 125.31, 121.97, 118.11, 113.74, 91.12, 49.83, 37.28, 34.06, 29.00, 26.04, 22.31, 14.08; MALDI-TOF MS calcd for 1128.8, found 1129.51.
G6
.
Yield: 76%. Anal. calcd for C56H67NO3S10 % (1122.76): C, 59.91; H, 6.02; N, 1.25. Found C, 59.92; H, 6.00; N, 1.27. 1H NMR (400 MHz, CDCl3): δ ppm, 0.89 (t, 12H), 1.26–1.40 (m, 24H), 1.59–1.69 (m, 8H), 2.82–2.90 (m, 8H), 4.18 (s, 2H), 7.32–7.37 (m, 2H), 7.44 (dd, 2H), 7.51 (d, 1H), 7.59 (d, 1H), 7.71 (dd, 2H), 7.81 (dd, 2H), 7.85 (s, 1H), 8.08 (s, 1H); 13C NMR (100 MHz, CDCl3): δ ppm, 193.20, 170.94, 166.64, 148.06, 141.42, 140.79, 138.28, 137.20, 136.43, 134.43, 134.86, 134.42, 133.83, 133.37, 132.03, 131.72, 130.87, 126.85, 126.30, 125.31, 121.97, 119.94, 46.87, 36.71, 28.22, 26.41, 22.58, 14.46; MALDI-TOF MS calcd for 1124.8, found 1125.4.
Methods and instrumentation
1H-NMR and 13C-NMR spectra were recorded on a 400 MHz AVANCE spectrometer. Differential pulse voltammetric measurements were performed on a PC-controlled electrochemical analyser (CH instruments model CHI620C). All these experiments were conducted with 1 mM concentration of compounds in DCM at a scan rate of 10 mV s−1 in which tetrabutylammonium hexafluorophosphate (TBAP) was used as a supporting electrolyte. The working electrode was glassy carbon; a standard calomel electrode (SCE) was the reference electrode and Pt-wire was the auxiliary electrode. The optical absorption spectra were recorded on a Shimadzu (Model UV-3600) spectrophotometer. Steady-state fluorescence spectra were recorded on a Fluorolog-3 spectrofluorometer (Spex model, JobinYvon) for solutions with optical density at excitation wavelength (λex) ≈ 0.05. Thermogravimetric measurements were carried out on a Mettler Toledo TGA/SDTA 851e instrument at a heating rate of 10 °C min−1 with 10 mg of sample under a nitrogen atmosphere.
Computational methodology
All the calculations have been carried out using a Gaussian 09 package26 on a personal computer. The ground state geometry of four sensitizers G2, G4, G5 and G6 was optimized using density functional theory (DFT), while time-dependent DFT (TDDFT) was employed for the estimation of ground to excited-state transitions. The B3LYP method and 6-31G (d, p) basis set27,28 were used to optimize the geometries of the dyads to be genuine global minimum energy structures and were used as the input for further calculations. The geometries were then used to obtain frontier molecular orbitals (FMOs) and were also subjected to single-point TDDFT studies (first 15 vertical singlet–singlet transitions) to obtain the UV-Vis spectra of the dyes. The integral equation formalism polarisable continuum model (PCM)29,30 within the self-consistent reaction field (SCRF) theory was used in the TDDFT calculations to describe the solvation of the dyes in dichloromethane. The software Gauss Sum 2.2.5 was employed to simulate the major portions of the absorption spectra and to interpret the nature of transitions.31,32 The contribution percentages of individual units present in the dyes to the respective molecular orbitals were calculated.
Transient absorption spectroscopy
A laser flash photolysis spectrometer (model LP920, Edinburg) has been used. It was equipped with a Continuum Nd-YAG laser (Surelite; 10 Hz repetition rate; FWHM 5 ns). A Surelite optical parametric oscillator permits conversion of the second harmonic to a visible spectrum of 400–820 nm. The respective dyes were excited at 445 nm. The excited state decay of the dye and the recovery of its fundamental state were monitored by the change in the absorbed continuous wave light. A satisfactory signal-to-noise ratio was obtained by averaging absorption spectra for 100–200 laser shots.
CEM and IMVS measurements
The photovoltaic response induced by the modulated light was studied by using intensity-modulated photovoltage spectroscopy (IMVS). A potentiostat (Solartron1287) equipped with a frequency response analyzer (Solartron1255B) under an open-circuit condition, based on monochromatic illumination (420 nm) controlled by a Lab view system, was used to measure IMVS. The modulated light was driven with a 10% AC perturbation current superimposed on a DC current in the frequency range from 0.1 to 106 Hz. The charge extraction method (CEM) was performed with the same monochromatic light source. The solar cell was illuminated under an open-circuit condition for 5 s to attain a steady state and then the light source was switched off when the device simultaneously switched to a short-circuit condition to extract the charges generated at that light intensity.
Device fabrication and photovoltaic performance measurements
The TiO2 photoanode was prepared as reported previously.25,33 A fluorine-doped tin oxide (FTO) conducting glass substrate with a resistance of ∼10 ohm m−2 was used. Screen printed double layer TiO2 films of (8 + 5) μm thickness (0.25 cm2 cell area) with an 8 μm transparent layer of TiO2 particles (approximately 20 nm in diameter) and a 5 μm scattering layer of TiO2 particles (approximately 400 nm in diameter) were prepared. The films were sintered at 500 °C for 1 hour. The thickness of the films was measured with a Surfcom 1400A surface profiler (Tokyo Seimitsu Co. Ltd.). A 0.2 mM solution of dye in 1:1 (v/v) acetonitrile/tert-butyl alcohol was used to coat the TiO2 films. The TiO2 films were immersed in the dye solutions and then kept at 25 °C for 15 hours. To assemble each cell, each dye-coated TiO2 film and a platinum-coated conducting glass were separated by using a Surlyn spacer (40 μm thick) and sealed by heating the polymer frame at 100 °C. An electrolyte consisting of a mixture of 0.6 M dimethylpropyl-imidazolium iodide, 0.05 M I2, 0.1 M LiI, and 0.5 M tert-butylpyridine in acetonitrile was used in each cell. G2, G4, G5 and G6 were desorbed from the TiO2 film by immersing the TiO2 film in 0.1 M TBAOH solution (1:1 mixture of H2O and ethanol). The amount of adsorbed dye was estimated from the absorption peak of each resulting solution.
The current–voltage characteristics were measured using a black metal mask with an area of 0.25 cm2 under AM 1.5 sunlight (100 mW cm−2, WXS-155S-10: Wacom Denso Co. Japan). The IPCE spectra were measured with a monochromatic incident light of 1 × 1016 photons cm−2 in direct current mode (CEP-2000BX, Bunko-Keiki).
Results and discussion
Synthesis
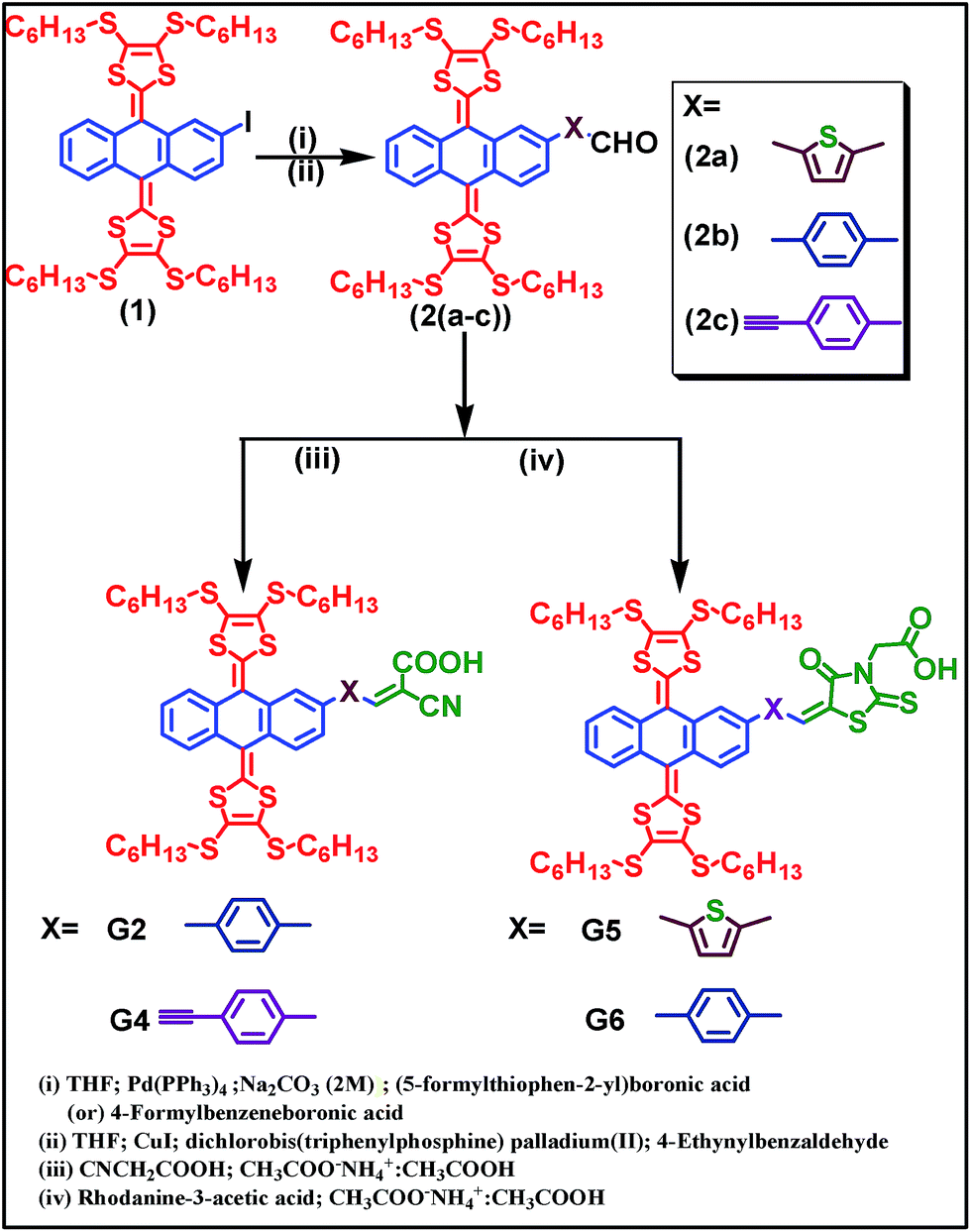
Scheme 1 illustrates the synthetic approach to the ex-TTF based sensitizers G2, G4, G5 and G6. Treatment of compound 1 with either (5-formylthiophen-2-yl)boronic acid or 4-formylphenylboronicacid or 4-ethynylbenzaldehyde in the presence of the Pd(PPh3)4/aq. Na2CO3 or CuI/Pd(PPh3)2Cl2 catalyst to give 2(a–c) followed by Knoevenagel condensation with cyanoacrylic acid or rhodanine 3-acetic acid yields G2, G4, G5, and G6. The resultant precursors and final compounds are purified using standard chromatographic techniques and characterized by elemental analyses, MALDI-TOF-MS, 1H and 13C NMR (Fig. S1–S18†). The elemental analysis data in the Experimental section are found to be satisfactory.
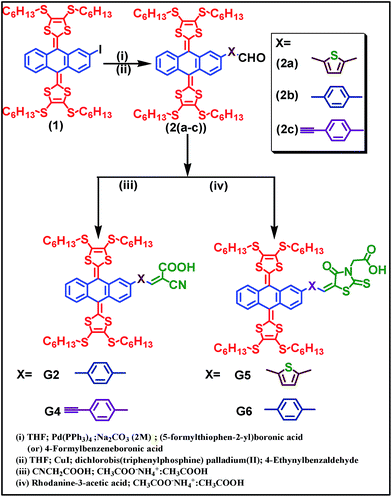 |
| | Scheme 1 Synthetic scheme of G2, G4, G5 and G6 sensitizers. | |
Optical & electrochemical properties
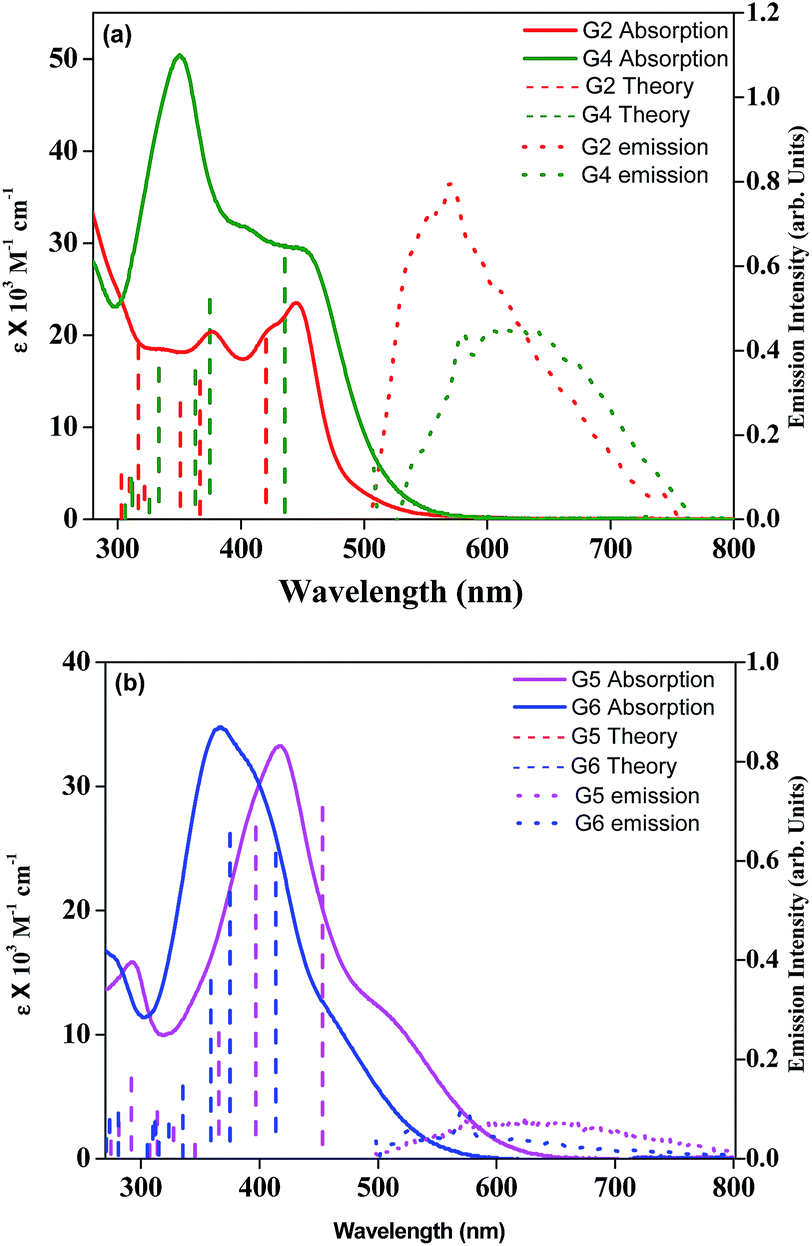
Fig. 1a and b illustrate the optical absorption spectra of newly synthesized G2, G4, G5, and G6 and were measured in dichloromethane solvent. The corresponding wavelength of absorption maxima and molar extinction coefficient values are presented in Table 1. As can be seen from Fig. 1, all four sensitizers except G6 exhibit prominent absorption peaks near the visible region, at about 440 nm, which are assigned to the π–π* transition. Table 1 and Fig. 1 suggest that the absorption maximum of G4 is red-shifted, when compared to the G2 sensitizer due to the presence of an additional triple bond in the π-spacer. In contrast, the absorption maximum of G6 is blue-shifted, when compared to G5 probably due to the participation of the lone pair of electrons on the sulfur atom of the thiophene moiety in conjugation. Moreover, these four compounds displayed a significant bathochromic shift, particularly G4, compared to previously reported G1 and G3 sensitizers,25 which is due to the extended π-conjugation on incorporation of an ethynyl bridge between the π-spacer and the acceptor. Fig. 1 also exemplifies the emission spectra of all new sensitizers in DCM solvent at room temperature and the corresponding emission maxima are presented in Table 1. Based on the optical properties, the E0–0 energy (singlet state) of G2, G4, G5, and G6 is found to be 2.65 ± 0.05, 2.52 ± 0.05, 2.05 ± 0.05, and 2.30 ± 0.05 eV, respectively. A quenched emission spectrum was observed when all these sensitizers were adsorbed onto a 2 μm thick TiO2 layer as a result of electron injection from the excited state of the sensitizer into the TiO2 conduction band. Similarly, the simulated absorption spectra of all four sensitizers obtained from TDDFT calculations (Fig. 1a and b) are also plotted in Fig. 1.
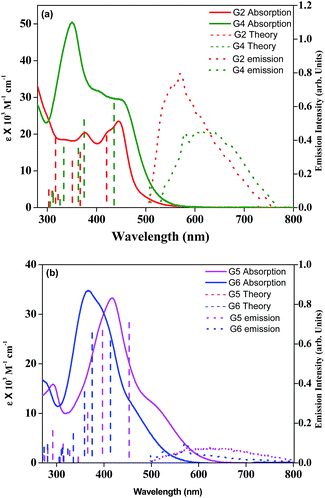 |
| | Fig. 1 (a) Absorption and emission spectra of G2 and G4 in DCM solvent (λex = 450 nm). (b) Absorption and emission spectra of G5 and G6 in DCM solvent (λex = 440 nm). Simulated absorption bands are shown as vertical bars. | |
Table 1 Photophysical properties
| Compound |
λ
max
(nm)/(ε M−1 cm−1) |
λ (abs. edge on TiO2)a (nm) |
λ
em
nm |
OX |
Red (E1/2 V vs. SCE)c |
E
0–0
(eV) |
E
*Ox
(E1/2 V vs. SCE) |
Adsorbed amount of dye (mol cm−2) |
|
Absorption spectra were recorded in DMF solution, error limits: λmax, ±1 nm, ε ±10%.
Solvent: ethanol, λmax, ±1 nm.
Solvent: CH2Cl2, error limits: E1/2 ±0.03 V, 0.1 M TBAP.
E
0–0 was determined from the intersection of absorption and emission spectra, as shown in Fig. 1. Error limits: ±0.05 eV.
Ref. 25.
Excited state oxidation potential, E*Ox = E0–0 − OX; error limits: ±0.05 eV.
|
|
G1
|
421 (18700) |
720 |
606 |
0.50 |
−1.56 |
2.50 |
−2.00 |
1.27 × 10−7 |
| 374 (18350) |
|
G2
|
445 (23496) |
690 |
570 |
0.53 |
−1.62 |
2.65 |
−2.12 |
1.23 × 10−7 |
| 375 (20417) |
|
G3
|
417 (17850) |
720 |
610 |
0.48 |
−1.69 |
2.48 |
−2.00 |
1.10 × 10−7 |
| 375 (18224) |
|
G4
|
455 (28973) |
690 |
610 |
0.49 |
−1.55 |
2.52 |
−2.03 |
1.30 × 10−7 |
| 350 (50194) |
|
G5
|
440 (33251) |
770 |
600 |
0.51 |
−1.36 |
2.05 |
−1.54 |
1.19 × 10−7 |
|
G6
|
385 (34112) |
700 |
580 |
0.52 |
−1.37 |
2.30 |
−1.78 |
8.63 × 10−8 |
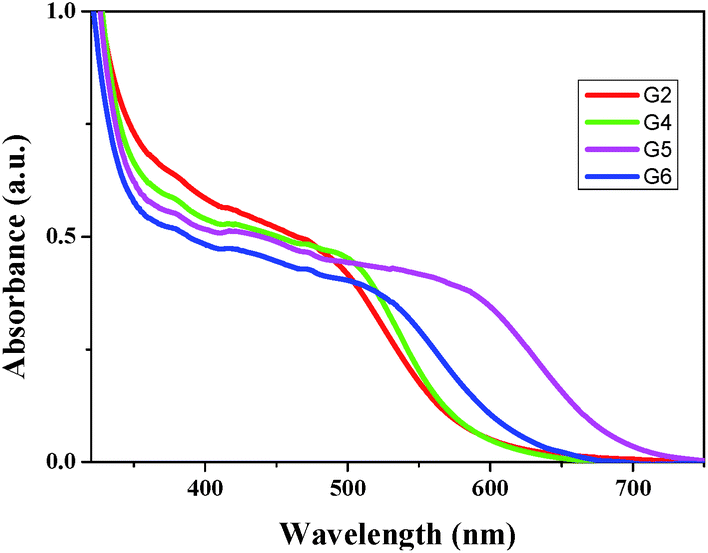
In order to understand the dye loading concentration on TiO2, we have carried out adsorption studies. For this reason, a 6 μm thick transparent TiO2 layer was soaked into 0.2 mM dye solution (1:1 (v/v) acetonitrile/tert-butyl alcohol) at room temperature for 15 h and the photoanode was rinsed with fresh solvent to remove the unanchored sensitizer. Fig. 2 illustrates the absorption spectra of all four sensitizers adsorbed on TiO2. The absorption features of these sensitizers in solution as well as when anchored onto TiO2 are identical except for a slight red shift in the absorption maxima due to the interaction of anchoring groups with the semiconductor surface. From Fig. 2, it is clear that the absorption intensities (in the visible region) of three sensitizers are identical except for G6 where the absorption intensity is low despite the molar extinction coefficient of G6 being higher than those of the remaining three sensitizers. This suggests that the loading of the G6 dye is lower compared to other sensitizers. The results of the desorption experiment showed that the amounts of loaded dye are almost the same for all G series dyes and are in the range of (1.10–1.30) × 10−7 mol cm−2 except for G6 which shows a lower value of dye loading than that (8.63 × 10−8 mol cm−2) of the other G dyes, as listed in Table 1. These data are consistent with the earlier absorption results when anchored onto TiO2.
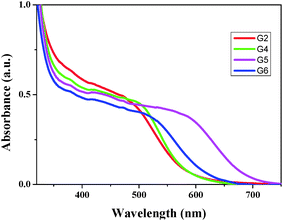 |
| | Fig. 2 Absorption spectra of G2, G4, G5 and G6 on TiO2 films. | |
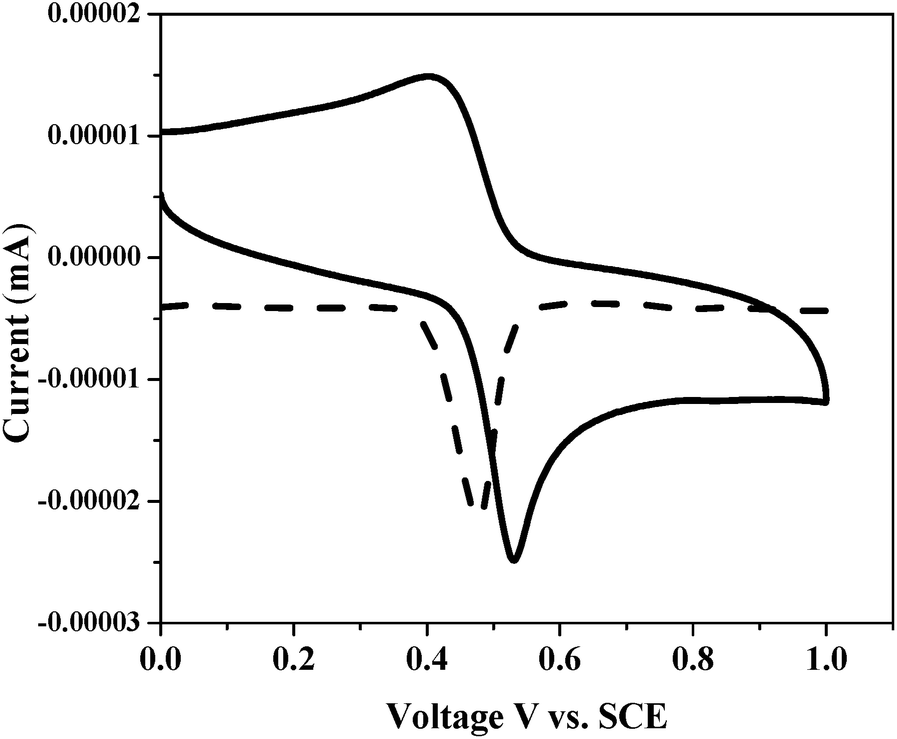
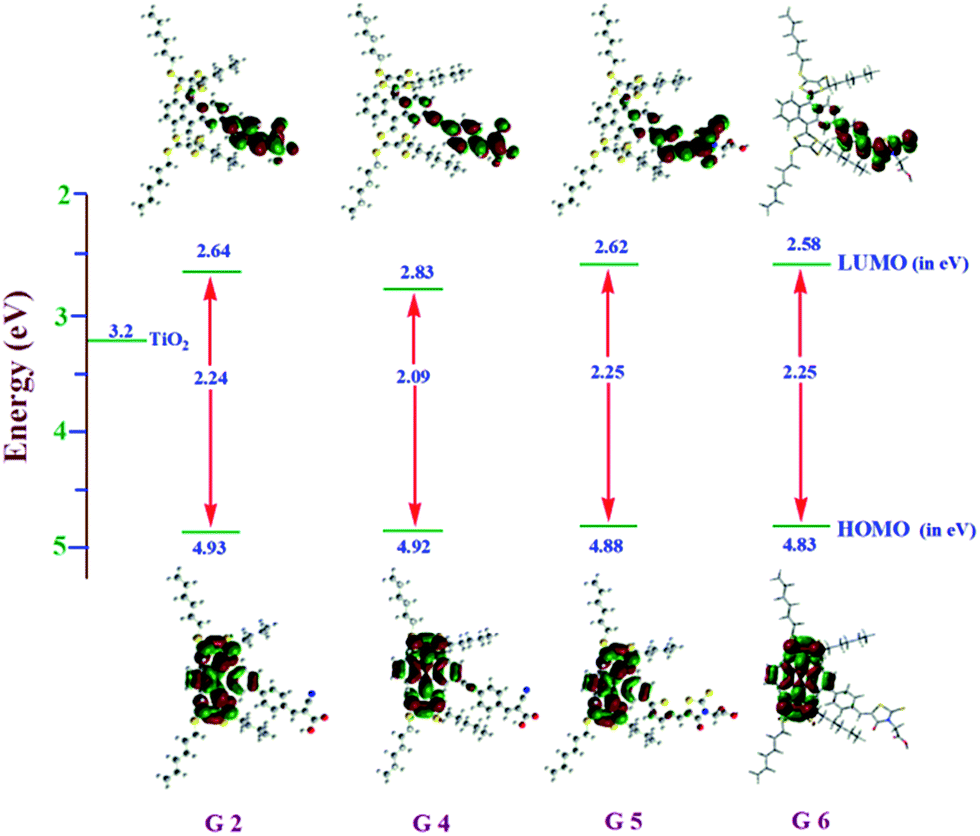
With a view to evaluate the HOMO–LUMO levels of newly designed ex-TTF sensitizers, we performed electrochemistry by using a differential pulse voltammetric technique in DCM solvent by using tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte. Fig. 3 presents the cyclic voltammogram (CV) of G5 and the corresponding redox data are presented in Table 1. Each sensitizer has shown reversible one-electron oxidations G2 (0.53 V), G4 (0.49), G5 (0.51 V) and G6 (0.52 V). The experimental HOMO energy values for G2 (−5.48 eV), G4 (−5.26 eV), G5 at (−5.32 eV) and G6 (−5.45 eV) were also measured using a photoemission yield spectrometer, Riken Keiki AC-3E (Fig. S19–S22†).
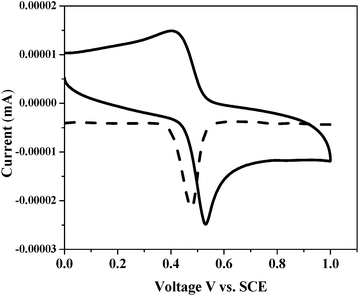 |
| | Fig. 3 Cyclic voltammogram of G5 in dichloromethane solvent. | |
As a result, all these dyes could be regenerated by the I−/I3− redox mediator (−0.21 V vs. Fc+/Fc in ionic liquid).34 On the other hand G5 and G6 are easy to reduce when compared to other sensitizers probably due to the presence of more electron withdrawing rhodanine acetic acid moieties in its molecular structure. The reduction potential (Ered) of the dyes, indicating their lowest unoccupied molecular orbital (LUMO), is considerably more negative than that of the TiO2 conduction band, providing ample driving force for electron transfer.
Computational study
In the study of electron transfer processes from sensitizers to the TiO2 conduction band, the electron distribution of the frontier molecular orbitals plays a major role. Thus, density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were performed using a Gaussian 09 package with a functional basis set of the B3LYP/6-31G (d, p) level. Fig. 4 illustrates the orbital energy levels in a vacuum; the minimum energy conformation suggests that optimized dyes resemble flying bird-like structures similar to G1 and G3 (Table S1†). These structures contain hexyl-substituted dithiafulvalene at the 9th and 10th positions of anthracene and anchoring groups at the 2nd position and π-conjugated spacers (thiophene or phenyl or ethynylbenzene) are present in between them. The 9th and 10th positions of the anthracene moiety of these sensitizers are tilted with an angle of ∼67° because of steric hindrance of hexyl-substituted dithiafulvalene. This steric hindrance retards the recombination of electrons in the TiO2 conduction band (3.2 eV) with the oxidized dye. The energy level diagram revealed that the HOMOs of all four dyes are the same and delocalized over the anthracene and dithiole units. However, the LUMO of G4 is lower than that of the remaining dyes (G2, G5 and G6) which again proved that extended π-conjugation in the spacer influencing the electronic structures of G4 offers efficient electron transfer to TiO2. Therefore, these results are in rational agreement with the experimental values (Fig. 1), and the resultant singlet state properties of all dyes are presented in Table S2.†
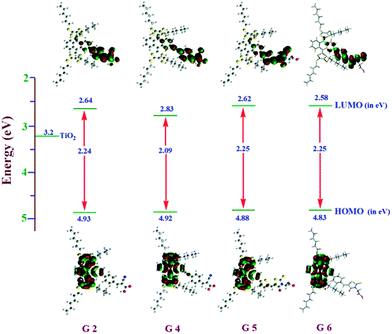 |
| | Fig. 4 Electronic distribution computed in acetonitrile of the first occupied/unoccupied molecular orbitals of the studied species G2, G4, G5, and G6 and the CB energy level of TiO2 in a vacuum. | |
Photovoltaic studies
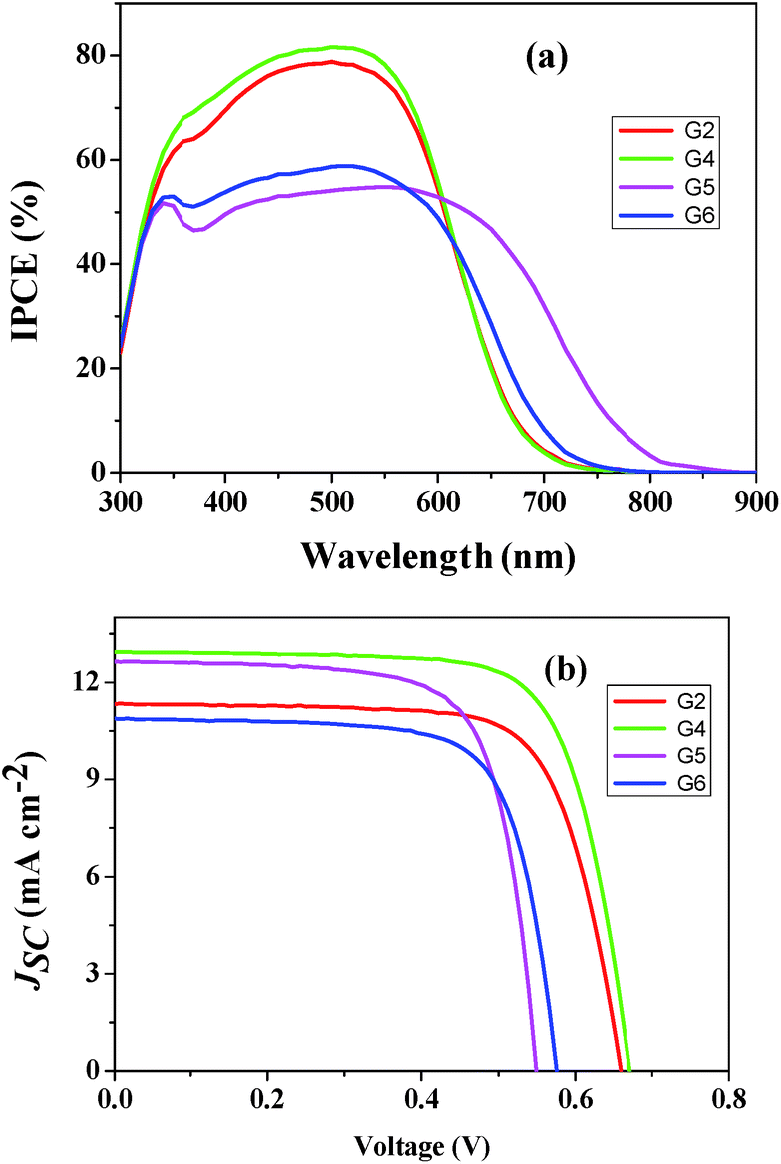
Based on the optical and electrochemical properties and energy levels of G2, G4, G5, and G6 sensitizers, photovoltaic performances i.e., incident photon-to-current conversion efficiency (IPCE) and photocurrent–voltage (J–V) using a volatile acetonitrile-based electrolyte composition of 0.6 M 1,2-dimethyl-3-propylimidazoliumiodide (DMPII), 0.05 M I2, 0.1 M LiI and 0.5 M 4-tert-butylpyridine (TBP) were tested. The incident photon-to-current conversion efficiency (IPCE) of G2 and G4 sensitizers showed a maximum value of 80% at 550 nm similar to G1 and G3 sensitizers. On the other hand, G5 and G6 sensitizers displayed only ∼55% at 550 nm (Fig. 5a). In all these sensitizers, thin films showed a broad spectral response covering the entire visible spectrum up to 800 nm. Fig. 5b demonstrates current–voltage characteristics of the devices and the resultant efficiencies of G2, G4, G5 and G6 are 5.45%, 6.36%, 5.04% and 4.55%, respectively under standard global Air Mass (AM) 1.5 solar conditions (Table 2). Under similar test cell conditions G1 and G3 have shown a conversion efficiency of 6.60 and 7.15%, respectively.25 Both the open-circuit voltage (Voc) and short circuit current density (Jsc) of G4 are higher than those of G2, which is due to incorporation of the π-conjugated phenylethynyl group between anthracene and the anchoring group. Thus, the resultant power conversion efficiency of G4 was enhanced by 14% when compared to G2. But, in both G2 and G4 the efficiency is low when compared to the previously reported G1 and G3 due to the presence of the electron rich thiophene group in later sensitizers.25 On the other hand, G5 displayed a significantly better efficiency over 10% than G6, attributed to the presence of the electron rich thiophene group which not only effectively promotes the electron injection into TiO2 compared to substituted phenyl in G6 but also extends IPCE spectra up to 850 nm that improves Jsc. The IPCE maxima of G2, G4, G5 and G6 are 79%, 83%, 55% and 59%, respectively. The dye desorption experiment showed that the amounts of adsorbed G2, G4, and G5 dyes are almost the same. This strongly suggests that the low IPCE of the G5 dye compared to that of G2 and G4 is due to the aggregation of the G5 dye molecules with each other during the dye loading process. Among all the sensitizers, G4 has shown the highest value of Jsc probably due to the stabilization of the LUMO level by incorporation of the ethynyl group between the π-spacer and the cyanoacrylic acid anchoring group that aid to inject electrons efficiently into TiO2 and also the relatively larger molar absorptivity.
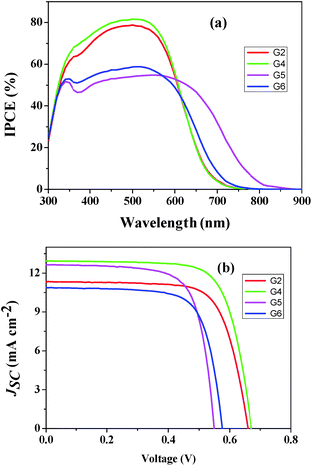 |
| | Fig. 5 (a) Photocurrent action spectra and (b) current–voltage characteristics of G2 ( ), G4 ( ), G4 ( ), G5 ( ), G5 ( ) and G6 ( ) and G6 ( ). ). | |
Table 2 Photovoltaic performance of ex-TTF sensitizersa
| Dye |
J
sc
[mA] |
V
oc
[V] |
FFb |
E
ff
[%] |
|
Photoelectrode: TiO2 (8 + 5 mm and 0.25 cm2).
Error limits: Jsc = ±0.20 mA cm−2, Voc = ±30 mV, FF = ±0.03.
Ref. 25.
|
|
G1
|
15.48 |
0.601 |
0.709 |
6.60 |
|
G2
|
11.42 |
0.659 |
0.724 |
5.45 |
|
G3
|
15.09 |
0.650 |
0.729 |
7.15 |
|
G4
|
13.03 |
0.671 |
0.728 |
6.36 |
|
G5
|
12.71 |
0.549 |
0.721 |
5.04 |
|
G6
|
10.92 |
0.576 |
0.724 |
4.55 |
CEM and IMVS measurements
Overall, this result suggests that the spacer and anchoring group greatly influenced the opto-electronics and device performance of ex-TTF sensitizers.
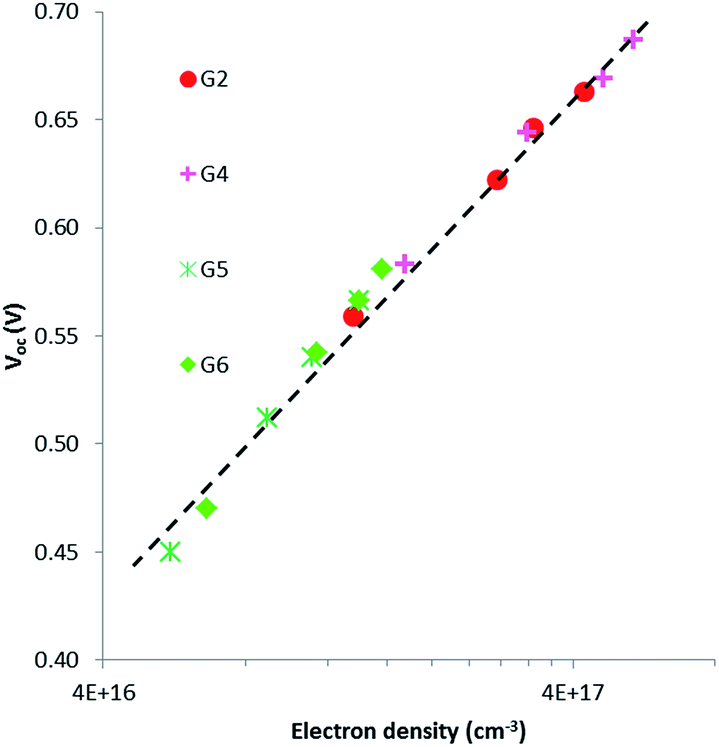
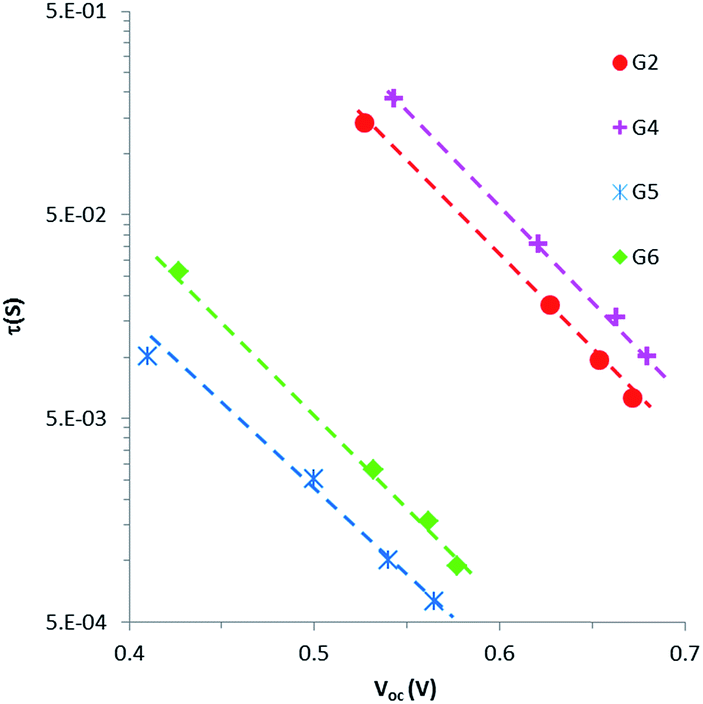
Intensity-modulated photovoltage spectroscopy (IMVS) is a powerful tool to investigate the charge recombination kinetics and the band edge level of TiO2 in DSSCs. A charge trapping–detrapping mechanism during the charge diffusion in the semiconductor and charge transfer from the TiO2 conduction band (CB) to redox species (I−/I3−) at the semiconductor/electrolyte interface was considered in the IMVS model. This model is invaluable in elucidating the change in the open-circuit photovoltage (Voc) because of the surface modification of the semiconductor oxide. IMVS measurements of G2, G4, G5 and G6 show a linear increase in Voc as a function of electron density for all sensitizers (Fig. 6). This result indicated that dyes' contribution to the band edge shift is almost similar as the plots for all dyes overlapped with each other, regardless of different molecular structures. This implies that the open-circuit photovoltage (Voc) is more likely dependent on charge recombination reactions rather than molecular structures of the dyes. Hence, the Voc for all DSSCs containing G2, G4, G5 and G6 should be attributed to the extent of charge recombination which is ultimately related to the electron lifetime (τ) in TiO2. Fig. 7 shows the electron lifetime (τ) as a function of Voc. At a certain electron density, τ follows the following order: G4 > G2 > G6 > G5. G4 and G2 exhibited much longer τ than G5 and G6 at the same electron density, implying that the recombination between electrons on the TiO2 surface and I3− ions in the electrolyte was considerably suppressed in G2 and G4 dyes with cyanoacrylic acid as an anchoring group. The amounts of the adsorbed dye for G2 and G4 are almost equal to those for G5. This result suggests that in cyanoacrylic acid based G2 and G4 cells the dyes most likely arrayed regularly and formed blocking layers on the TiO2 surface to suppress the electron recombination between the injected electron in the TiO2 conduction band and I3− in the electrolyte; while in the rhodanine acetic acid based G5 cell, the dye molecules were likely aggregated with each other on the TiO2 surface leaving some bare area for the electrolyte and thus accelerated the electron transfer back to the dyes or the electrolyte. The different molecular structures/anchoring groups of G series dyes greatly affected the τ and thus influenced the Voc of the corresponding DSSCs.
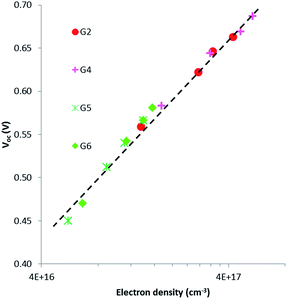 |
| | Fig. 6 Electron density as a function of open-circuit photovoltage for DSSCs sensitized with G2, G4, G5 and G6. | |
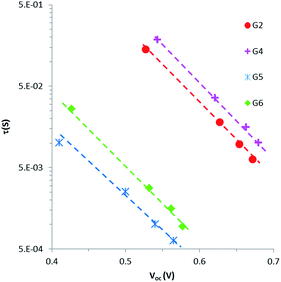 |
| | Fig. 7 Electron lifetime (t) as a function of Voc for DSSCs sensitized with G2, G4, G5 and G6. | |
Transient absorption study
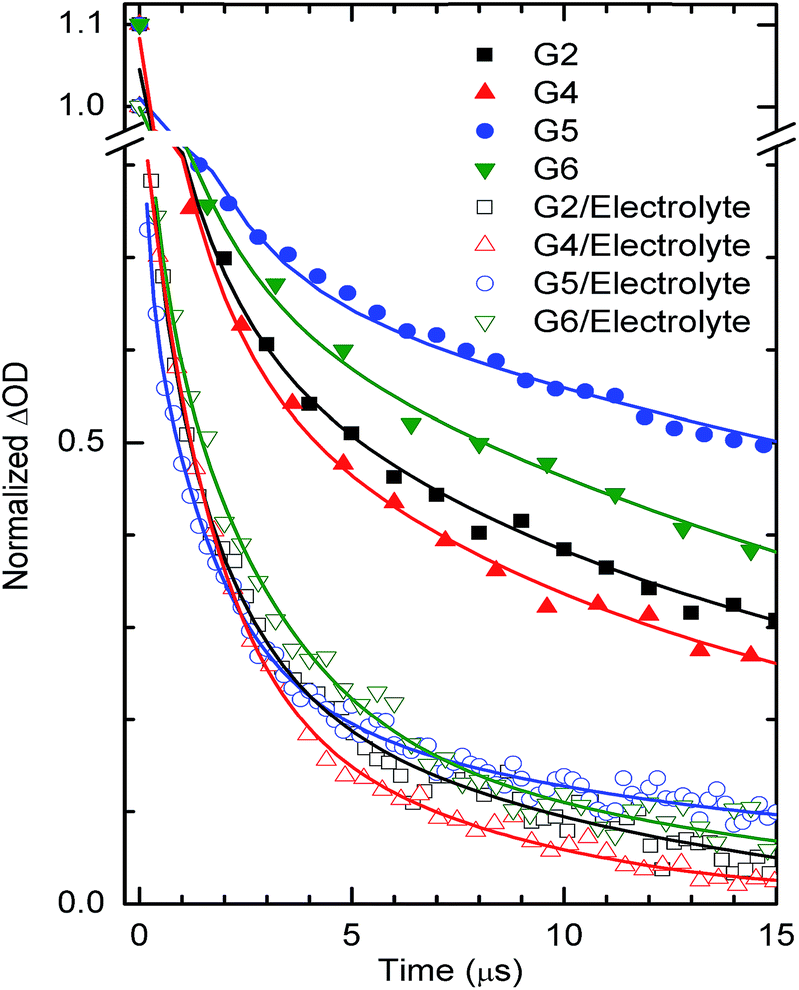
In order to support and understand the above characteristics and performances of the above dyes, we have adopted nanosecond laser flash photolysis spectroscopy to investigate and compare the kinetics competition between waste recombination and dye regeneration by the electrolyte among G2, G4, G5 & G6 dyes (Fig. 8). These kinetics are considered to be one of the crucial steps for the DSSC performance.35,36
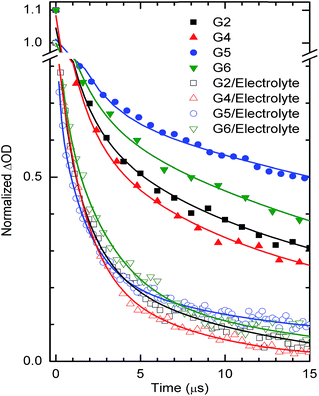 |
| | Fig. 8 Transient absorbance decay profiles obtained upon nanosecond pulsed laser excitations (bandwidth 6 ns) on mesoporous TiO2 films sensitized with G2, G4, G5, G6 dyes at a laser excitation of 445 nm and monitored at 700 nm in the presence and in the absence of the LiI/I2 electrolyte (No TBP). | |
Recombination decay times recorded are one-fold lower than the typical time constants (10−4 s) of DSSCs under working conditions (1 Sun).37 The relatively lower decay times obtained in our case are mainly due to the use of a high energy laser pulse. In the absence of the LiI/I2 electrolyte, the transient decays of the maximum absorption signals (ΔOD) of the dye (also called the bleaching of the ground states of the dye) reflect mostly the dynamics of, namely, excited state relaxation and recombination of photo injected electrons in TiO2 with the oxidized dye. Due to very small ΔOD amplitudes of transient absorbance signals of the ground state bleaching, we measured, instead, the lifetime of the oxidative dye formation decays at red shifted absorption spectra (here 700 nm). This is also shown in our previous work,38 and also with N719.39 Importantly, the time needed for dye regeneration is quite similar to the time needed to decay the transient oxidative dye product.39 The transient absorption spectra of G2, G4, G5 and G6 dyes on TiO2 (no electrolyte added) were fitted using an exponential decay function, y = A1exp(−t/τ1) + A2exp(−t/τ2) + y0, where A corresponds to the amplitude, τ1 denotes the dye excited state relaxation time (a few to hundreds of ns); τ2 the decay time for dye regeneration by TiO2 injected electrons (tens to hundreds of μs). In the presence of an electrolyte, the spectra were fitted using the function, y = A1exp(−t/τ1) + A2exp(−t/τ2) + A3exp(−t/τ3) + y0. Here, we mostly employ τ2 (a few μs) for dye regeneration by I− which has a large control on DSSC performance by reducing to the maximum the electron recombination with oxidative dyes. In the presence of an electrolyte, the fitting of the decays led to dye regeneration times 1.41, 1.90, 1.92 and 2.20 μs which are much faster compared to the waste recombination times in the presence of air (no electrolyte added), 11.22, 12.35, 33.30 and 25.35, respectively for the dyes G4, G2, G5 and G6 dyes; thus, it allows a better collection of photo injected electrons into an external circuit.
A good corroboration is found here, where faster regeneration decay has allowed higher power efficiencies of 6.36, 5.45, 5.04 and 4.55% for the dyes G4, G2, G5 and G6, respectively. Most important is the corroboration with current density, as dye regeneration should have a direct effect on the current density collected. Here, a good corroboration was found with G4, G2, and G6 dyes (1.41, 1.90, and 2.20 μs), except for the G5 dye (1.92 μs) where dye regeneration shows a slower or similar decay to the G2 dye (1.90 μs), despite its higher current density collected (12.71 mA−2) compared to the G2 dye (11.42 mA−2). This might be explained by the fact that the G5 dye has a broader and red shifted absorption spectrum; adding to this its little higher LUMO which allows higher electron injection (see Fig. 5a for IPCE of G5). Its lower power efficiency compared to the G2 dye originates from the lower measured Voc. In addition, this good corroboration between recombination/dye-regeneration kinetics and produced photocurrent indicates a good anchoring between G2, G4, G5 and G6 dyes and TiO2 substrate films.
Conclusions
In summary, we focused on a rational approach for systematically improving the voltage (Voc) and current (Jsc), thus pursuing high efficiencies for DSSC applications by changing the π-spacers and anchoring groups. The push–pull structure in G2, G4, G5 and G6 dyes facilitated broad light harvesting ability, more stability and good photovoltaic properties and showed efficiencies ranging from 4.55% to 6.36%. Among the four sensitizers, G4 achieved the highest efficiency of 6.36% with a Jsc of 13.0 mA cm−2, a Voc of 0.671 V and a FF of 0.728. The improved efficiency of cyanoacrylic acid based G4 might be due to the formation of a compact monolayer of G4 dye on the TiO2 surface that suppresses the electron recombination between the injected electron in the TiO2 conduction band and I3− in the electrolyte, thus increasing the Voc. This class of sensitizers is thermally stable up to 200 °C that may improve the durability of the device.
Acknowledgements
We thank CSIR-NISE and Department of Science & Technology (DST), Government of India under major project DST-UK (‘APEX’) for financial support to carry out this work. Author ND thanks CSIR for a junior research fellowship. A. Islam and I. Bedja extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP #0019.
Notes and references
- Z.-S. Huang, H. Meier and D. Cao, J. Mater. Chem. C, 2016, 4, 2404–2426 RSC.
- M. Ye, X. Wen, M. Wang, J. Locozzia, N. Zhang, C. Lin and Z. Lin, Mater. Today, 2015, 18, 155–162 CrossRef CAS.
- V. Sugathan, E. John and K. Sudhakar, Renewable Sustainable Energy Rev., 2015, 52, 54–64 CrossRef CAS.
- F. Bella, C. Gerbaldi, C. Barloo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- V. K. Singh, R. K. Kanaparthi and L. Giribabu, RSC Adv., 2014, 4, 6970–6984 RSC.
- S. Zhang, X. Yang, Y. Numata and L. Han, Energy Environ. Sci., 2013, 6, 1443–1464 CAS.
- R. K. Kanaparthi, J. Kandhadi and L. Giribabu, Tetrahedron, 2012, 68, 8383–8393 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comete, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed.
- L. Giribabu, R. K. Kanaparthi and V. Velkannan, Chem. Rec., 2012, 12, 306–328 CrossRef CAS PubMed.
- N. Duvva, D. Raptis, C. V. Kumar, E. N. Koukara, L. Giribabu and P. Lianos, Dyes Pigm., 2016, 134, 472–479 CrossRef CAS.
- G. H. Rao, A. V. Rao, L. Giribabu and S. P. Singh, Photochem. Photobiol. Sci., 2016, 15, 287–296 CAS.
- F. M. Jradi, D. O'Neil, X. Kang, J. Wong, P. Szymanski, T. C. Parker, H. L. Anderson, M. A. El-Sayad and S. R. Marder, Chem. Mater., 2015, 27, 6305–6313 CrossRef CAS.
- A. Mishra, M. K. R. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- N. P. Liyanage, A. Yella, M. Nazeeruddin, M. Grätzel and J. H. Delcamp, ACS Appl. Mater. Interfaces, 2016, 8, 5376–5384 CAS.
- K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanava, Chem. Commun., 2014, 50, 6379–6381 RSC.
- P. Dai, L. Yang, M. Liang, H. Dong, P. Wang, C. Zhang, Z. Sun and S. Xue, ACS Appl. Mater. Interfaces, 2015, 7, 22436–22447 CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J.-I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef CAS PubMed.
- J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS PubMed.
- Y. Geng, F. Pop, C. Yi, N. Avarvari, M. Grätzel, S. Decurtins and S.-X. Liu, New J. Chem., 2014, 38, 3269–3274 RSC.
- K. Guo, K. Yan, X. Lu, Y. Qiu, Z. Liu, J. Sun, F. Yan, W. Guo and S. Yang, Org. Lett., 2012, 14, 2214–2217 CrossRef CAS PubMed.
- S. Wenger, P. A. Bout, Q. Chen, J. Teuscher, D. D. Censo, R. Hmphry-Baker, J.-E. Moser, J. L. Delgado, N. Martin, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 5164–5169 CrossRef CAS PubMed.
- L. Giribabu, N. Duvva, S. P. Singh, L. Han, I. M. Bedja, R. K. Gupta and A. Islam, Mater. Chem. Front., 2017 10.1039/c6qm00070c.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117–129 CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
-
R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
- T. Jella, M. Srikanth, R. Bolligarla, Y. Soujanya, S. P. Singh and L. Giribabu, Dalton Trans., 2015, 44, 14697–14706 RSC.
- R. Kawano and M. Watanabe, Chem. Commun., 2003, 330–331 RSC.
- G. Boschloo and A. Hagfeldt, Chem. Phys. Lett., 2003, 370, 381 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- G. H. Rao, A. Venkateswararao, L. Giribabu, L. Han, I. Bedja, R. K. Gupta, A. Islam and S. P. Singh, Phys. Chem. Chem. Phys., 2016, 18, 14279–14285 RSC.
- G. Boschloo and A. Hagfeldt, Chem. Phys. Lett., 2003, 370, 381 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic details. See DOI: 10.1039/c6se00014b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ), G4 (
), G4 ( ), G5 (
), G5 ( ) and G6 (
) and G6 ( ).
).