
Asymmetric tin–vanadium redox electrolyte for hybrid energy storage with nanoporous carbon electrodes†
Juhan
Lee
ab,
Aura
Tolosa
ab,
Benjamin
Krüner
ab,
Nicolas
Jäckel
ab,
Simon
Fleischmann
b,
Marco
Zeiger
ab,
Daekyu
Kim
ac and
Volker
Presser
 *ab
*ab
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Technology, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cSchool of Energy, Materials and Chemical Engineering, Korea University of Technology and Education, Chungjeol-ro 1600, 31253 Cheonan, Republic of Korea
First published on 5th January 2017
Abstract
In recent decades, redox-active electrolytes have been applied in stationary energy storage systems, benefitting from Faradaic reactions of the electrolyte instead of the electrode material. One of the challenging tasks is to balance the redox activities between the negative and positive electrode. As a possible solution, a mixed electrolyte with vanadyl and tin sulfate was previously suggested; however, a low power performance is a great challenge to be overcome. Here, we found that the origin of the poor power performance in the mixture electrolyte system (vanadium complex and tin solution) is the reduction of the pore volume at the positive electrode via irreversible tin dioxide formation. To prevent the latter, we introduce a hybrid energy storage system exhibiting both battery-like and supercapacitor-like features via asymmetric redox electrolytes at the microporous activated carbon electrodes; SnF2 solution as anolyte and VOSO4 as catholyte. By employing an anion exchange membrane, the irreversible SnO2 formation at the positive electrode is effectively suppressed; thus, an asymmetric 1 M SnF2|3 M VOSO4 system provides a high maximum specific power (3.8 kW kg−1 or 1.5 kW L−1), while still exhibiting a high maximum specific energy up to 58.4 W h kg−1 (23.4 W h L−1) and a high cycling stability over 6500 cycles.
Introduction
Ranging from intercalation batteries and redox flow systems to supercapacitors and hybrid devices: electrochemical energy storage is getting more and more integrated in our daily lives.1,2 Achieving high storage capacities combined with rapid charge/discharge rates and performance longevity remains a particular challenge. For example, batteries demonstrate very high energy storage capacities with a rather limited power and cycling longevity. In contrast, supercapacitors, especially double-layer capacitors, are characterized by particularly high power and performance stability owing to the physical mechanism of ion electrosorption at the interface between high surface area of carbons and the electrolyte. Enhancing the energy storage capacity by adding redox active electrolytes to porous carbon has emerged as a promising strategy for hybrid energy storage systems combining the principles of supercapacitors and batteries.3–5 Unlike other hybrid approaches, redox electrolytes can be directly used in combination with conventional nanoporous carbons as they benefit from Faradaic reactions of the electrolyte instead of the electrode material.After the pioneering work, for example, by Li et al. (2007),6 Lota et al. (2009),7 and Roldán (2011),8 redox-active electrolyte aided hybrid energy storage (REHES) systems have quickly evolved to high performance devices.3–5 The specific energy of REHES systems is close to that of lead acid batteries, reaching values of ca. 10–20 W h kg−1, while keeping the power performance as high as that of supercapacitors (>5 kW kg−1) with a high cycling stability over 5000 cycles.4 The most frequently studied REHES systems rely on soluble redox couples like quinone/hydroquinone (H/HQ)9–11 ferricyanide/ferrocyanide,12–16p-phenylenediamine,17–19 vanadium complexes,20,21 and halides.22–24 Advanced systems approach energy storage capacities nearing lithium ion batteries (∼100 W h kg−1), such as the mixture electrolytes of vanadyl and tin sulfate (75 W h kg−1)21 or solutions of cupric chloride (73 W h kg−1).25 As most REHES systems are based on aqueous solutions, they also have strong advantages regarding cost, safety, and environmental issues.4
The reason of the high performance of REHES systems is the synergetic charge storage enhancement by the redox activities of the soluble ions and the rapid charge transfer across short distances within the confinement of carbon nanopores.12,26 In addition to the redox-activity (i.e., Faradaic charge transfer), fast electrostatic physisorption of the ions on the electrode occurs (i.e., electric double-layer (EDL) formation).26 The aspect of rapid charge transfer in nanopores distinguishes REHES systems from flow battery technologies, where confinement of redox electrolytes in the micropores causes a pressure drop for the circulation of the electrolyte in flow mode.27
A key challenge for REHES is to balance the redox activities occurring at the negative and positive electrode. For instance, iodide is a promising redox ion at the positive electrode;7 however, so far, only the vanadyl sulfate system has been introduced at the negative electrode to balance the iodide's enormous redox capacity at the positive electrode.20 This system could also work without introducing a redox electrolyte at the negative electrode because reversible hydrogen storage at the negative electrode can be utilized as a balance to the iodine redox activities.22 However, since the electrochemical hydrogen storage shows a huge separation potential between anodic and cathodic reactions, the energy efficiency of the system is observed to be rather low (<80%).28 Furthermore, the hydrogen redox activity cannot fully compensate for the huge redox capacity of the iodine at the positive electrode.29 Pioneering works can be found regarding the balance of the redox activities from viologen/bromide23,24 and vanadyl sulfate/potassium iodide20 systems reaching specific energies up to 50 W h kg−1. Even higher energy storage capacities exceeding 70 W h kg−1 can be obtained for a well-balanced REHES employing SnSO4·VOSO4, as recently demonstrated by us.21 Though the latter redox electrolyte system exhibited a highly promising energy storage capacity (∼75 W h kg−1), the limited power performance (1.5 kW kg−1 or 0.6 kW L−1) must be overcome to achieve high energy storage capacity and high power handling.
In this work, as a starting point for designing a power-enhanced system, we will first explore the origin of the low power performance for the mixture electrolyte system (SnSO4·VOSO4) by post mortem analysis of positive and negative electrodes via scanning electron microscopy (SEM), nitrogen gas sorption analysis (GSA), and X-ray diffraction (XRD). As a solution to the pore volume reduction at the positive electrode via tin oxide formation, we then introduce an asymmetric electrolyte design by applying SnF2 solution as electrolyte at the negative electrode (anolyte) and VOSO4 solution as electrolyte at the positive electrode (catholyte). The electrochemical performance of the asymmetric electrolyte system (SnF2|VOSO4) will be discussed as well as the post mortem analysis with electron micrographs, GSA, and XRD to understand the morphological and electrochemical state change of the positive and negative electrode after electrochemical operation.
Experimental section
Materials
For the free-standing film electrodes, we applied a mechanical rolling process to mixture paste of YP-80F type activated carbon (Kuraray Chemicals) as active electrode material, ethanol as solvent, and polytetrafluoroethylene (PTFE) as binder material. Finally, 200 ± 6 μm thick electrodes were obtained with 5 mass% PTFE after being dried in a vacuum oven (120 °C, 2 × 103 Pa) for 48 h.The commercially available activated carbon electrode exhibits a specific surface area of 2105 m2 g−1 by applying the Brunauer–Emmett–Teller (BET)30 equation or density-functional theory (DFT) surface area of 1672 m2 g−1 using quenched-solid density functional theory.31Via quenched-solid density functional theory with a slit model and pore size in the range of 0.56–37.5 nm, a total pore volume of 1.01 cm3 g−1 and an average pore size of 1.6 nm were calculated.
Type FAS15 (FuMA-TECH) anion exchange membranes (dry thickness of 14 ± 1 μm) were activated by being soaked in deionized water for 8 h, then, in 0.5 M H2SO4 for at least 72 h while changing the electrolyte more than three times. The selectivity of the FAS15 membrane was determined to be higher than 95% by the manufacturer. In case of half-cell investigation, glassy fiber filter (GF/A, Whatman) was applied.
For the electrolyte, vanadyl sulfate (VOSO4, 97% purity), tin sulfate (SnSO4, 95% purity), tin fluoride (SnF2, 99% purity), sodium sulfate (Na2SO4) with 99% purity, and sulfuric acid (H2SO4, 99.99% purity based on trace metal analysis) were purchased from Sigma Aldrich and mixed with deionized water.
Cell design and assembly
For the half-cell experiments, a spring-loaded two-piston cell was applied with the Ag/AgCl reference electrode (saturated, 3 M NaCl); the scheme of our cell design can be found elsewhere.32,33 While two graphite pistons served as current collectors, the working (4 mm diameter, 200 μm thickness) and counter electrode (12 mm diameter, 500 μm thickness) were separated by GF/A glassy fiber filter (Whatman). We used two cell designs for full-cell operation: (i) spring-loaded two-piston cells (see ref. 33), and (ii) cells which allow the complete sealing of the ion exchange membrane and to limit the excess volume of the redox electrolyte; here, excessive volume means the volume of the electrolyte in the cell except the geometrical volume of the film electrode. The drawing of the latter cell can be found in our previous reports.12,21,22 For the latter cell, the electrolyte was dropped to the gasket channel having a size of 1.33 cm2 where the circular film electrode (12 mm diameter, 200 μm thickness) was placed above the glassy carbon current collectors, and the cell was tightly sealed afterward. To prevent the electrolyte shortage via the electrolyte adsorption during the electrochemical operation, the electrodes were previously soaked in the electrolyte and degassed by applying vacuum for 5 min. For the cells with reference electrode, thin string of FKS15 membrane was additionally mounted at the negative electrode as an ion bridge to the reference electrode (Ag/AgCl). Through the tight gap between the gaskets this porous membrane string was extended to the glass container with H2SO4 (1 M) aqueous solution where Ag/AgCl reference electrode was mounted. In case of spring-loaded cells, electrolyte is injected by syringe after assembly.Electrochemical characterization
For the electrochemical characterizations, a VMP300 potentiostat/galvanostat (Bio-Logic) was used. The cells were stored at 25 °C in a climate chamber.For the normalization of the specific energy (E), specific power (P), and specific current, the total mass of activated carbon material is considered to provide a comparative data basis with battery and supercapacitor materials in the literature. Values for the specific energy were calculated by eqn (1):
 | (1) |
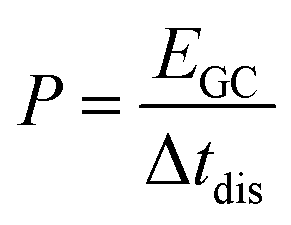 | (2) |
To condition the cell, cyclic voltage sweeping was carried out at 1 mV s−1 for 10 cycles at the voltage from 0–1.4 V cell voltage prior to further characterization. Subsequently, cyclic voltammetry was carried out at the scan rates from 1 mV s−1 to 2.5 V s−1 for 3 cycles at each scan rate at the cell voltage from 0 V to 1.4 V. Afterwards, galvanostatic charge/discharge with potential limitation (GCPL) was performed at 100 mA g−1 to 30 A g−1 with the voltage range from 0–1.4 V. Next, open circuit voltage measurements over 10 h were carried out after the cell had been discharged to 0 V for 30 min, then, a constant specific current of 50 mA g−1 was applied to reach a cell voltage of 1.4 V. For the cyclic stability test, galvanostatic cycling was applied to the full-cell at 1 A g−1 at the voltage window of 1.4 V.
In case of the full-cell with the reference electrode, the cell was charged by applying a potential from 0 V to 1.4 V between the negative and the positive electrode at 0.5 A g−1 while measuring the potential difference between the negative and the reference electrode. Initially, the cell was conditioned over 20 cycles at 0.5 A g−1.
Material characterization
The nitrogen gas sorption analyses were carried out with a Quantachrome Autosorb iQ system. The electrodes were degassed at 150 °C for up to 20 h at a relative pressure of 0.1 Pa. The analysis was performed in liquid nitrogen at −196 °C by applying relative pressure of 1 × 10−6 to 1.0 in 56 steps. The pore size distribution (PSD) was derived using the quenched-solid density functional theory (QSDFT), assuming slit-like pores.31,34 Additionally, the specific surface area (SSA) was also calculated using the Brunauer–Emmett–Teller equation (BET) in the linear regime of the measured isotherms, typically 3 × 10−2 to 2 × 10−1 in relative pressure,30 with a Quantachrome Autosorb 6B. All calculations were performed with the ASiQwin-software 3.0.The X-ray diffraction measurements were carried out with a Bruker D8 Discover using Cu-Kα radiation with a Goebel mirror, a point focus (diameter: 0.5 mm), and a 2D-detector (Vantec-500). The device was calibrated with a corundum standard and the sample moved during the measurement to obtain a better statistic (amplitude in x and y: 5 mm; speed in x: 0.5 mm s−1; speed in y: 0.2 mm s−1). The diffraction pattern was fitted with TOPAS (version 5 from Bruker AXS).
Changes in the morphology of the electrodes were examined by a JEOL JSM 7500F field emission scanning microscope (FE-SEM) at 3 kV. The post mortem electrodes were fixed on steel sample holders with sticky carbon tape. The composition of the electrodes was determined by energy dispersive X-ray spectroscopy (EDX) using an X-Max-150 detector from Oxford Instruments attached to the SEM chamber. The spectra of 10 particles were measured using an accelerating voltage of 10 kV and averaged. For standardization, silicon crystal was used. For high resolution analysis, a JEOL JEM-2100F high resolution transmission electron microscope (TEM) was employed at 200 kV. TEM samples were prepared by mechanically dispersing the electrodes in isopropanol, and drop casting them on a carbon film on copper grid. Using TEM, we also carried out selected area electron diffraction (SAED).
Thermogravimetric analysis (TGA) was carried out with a TG 209 F1 Libra system (Netzsch) in oxygen containing synthetic air (80% N2, 20% O2). The temperature was increased at a rate of 19 °C min−1 to 250 °C and help for 2 h to remove surface groups. Afterwards, the temperature was further increased up to 900 °C at a rate of 5 °C min−1 to burn off the carbon and to determine the mass of residual tin oxide.
For the post mortem analysis, full-cells were initially conditioned with 20 cycles in GCPL mode (0.5 A g−1 0–1.4 V) and subsequently charged to 1.4 V or discharged to 0 V in chronoamperometry mode for 30 min. Afterwards, the negative and positive electrode samples were soaked in 1 M H2SO4 aqueous solution for a day after the cell disassembly, subsequently soaked into deionized water for a day, and dried in a desiccator to remove the dissolved ions.
Results and discussion
Issues and limitations of the SnSO4·VOSO4 redox electrolyte system
As we have recently demonstrated,21 the specific energy of a well-balanced redox-active electrolyte aided hybrid energy storage (REHES) can exceed 70 W h kg−1 by employing a semi-soluble redox system like Sn2+/Sn(s) redox couple as a balance to the activities of vanadium complexes at the positive electrode (see ESI,† Fig. S1A). For the latter study, the mixture of 0.75 M SnSO4 and 2 M VOSO4 in 0.1 H2SO4 aqueous solution was applied as redox electrolyte. When a constant current is applied to a full-cell in the potential range from 0 V to 1.4 V, two distinctive voltage plateaus arise: one due to the redox activities of vanadyl sulfate, namely V3+/VO2+ and VO2+/VO2+ redox couples at the positive electrode, and one related to tin redox activities and the V2+/V3+ redox couple (see ESI,† Fig. S1B) at the negative electrode. Chemical analyses with X-ray photoelectron spectroscopy (XPS) after electrochemical operation (post mortem) confirm the reversible Sn(s)/Sn2+ redox reaction for the negative electrode in charged state (−0.57 V vs. Ag/AgCl).21 The strong redox activities of the Sn2+/Sn(s) redox couple prevents the irreversible hydrogen evolution reaction which would onset at a potential below −0.5 V vs. Ag/AgCl.The starting point to design a redox electrolyte system with improved power handling ability was our previous work on the SnSO4·VOSO4 redox electrolyte system,21 where tin electroplating occurs at the negative electrode when a full-cell system is charged up to 1.4 V. As metallic tin is formed in the pore network of the activated carbon electrode, the pore volume of the electrode decreases when the negative electrode is in charged state since the metal tin is formed in the pores (see ESI,† Fig. S2A). When the negative electrode is in a discharged state, most of the pore volume is recovered as the metallic tin is dissolved in the solution again, indicating that the Sn2+/Sn(s) redox reaction occurs reversibly in the pore network at the negative electrode. The pore volume of the charged and discharged positive electrodes are significantly lower compared to that of its initial value which implies that some of the pores are filled and possibly blocked at the positive electrode.
From the post mortem XRD results, there is clear evidence of stannic oxide (SnO2) at both electrodes (see ESI,† Fig. S2B). Therefore, SnO2 formation can be the origin of the irreversible pore volume reduction at the positive electrode, as also supported by the morphological changes seen on post mortem samples in scanning electron micrographs (ESI,† Fig. S2C–F). While the discharged negative electrode maintains the initial morphology with smooth activated carbon particles (Fig. S2E†), the positive electrode presents a non-uniform coating, which implies the possibility of blocked micropores (Fig. S2D and F†). Given that the SnSO4·VOSO4 redox electrolyte system relies mostly on the reversible redox reaction of the vanadium complex at the positive electrode and the tin redox activities at the negative electrode, the stannous oxide formation at the positive electrode blocks the active surface area and energy storage mechanism is limited. Also, the transition between dissolved and solid state of tin is a key limiting factor which prevents this specific redox electrolyte system to achieve rapid charge and discharge rates.
Electrochemistry of the new SnF2|VOSO4 redox electrolyte system
To prevent the reduction of the pore volume at the positive electrode via tin oxide formation, we designed a new system based on separate electrolytes at the positive and negative electrode by using an anion ion exchange membrane (AEM): a tin containing solution as anolyte for the negative electrode and vanadium containing solution as catholyte for the positive electrode. Stannous fluoride (SnF2) was selected as solute for the anolyte due to its even higher solubility compared to SnSO4 which can be beneficial for the stability of the solution.35 For the catholyte, VOSO4 was selected as a solute.First, the individual redox activities of VOSO4 and SnF2 can be identified in the cyclic voltammograms at 1 M concentration in 1 M H2SO4 solution (Fig. 1A). In the potential range from −0.17 V to +0.9 V vs. Ag/AgCl, two pairs of redox reactions can be seen for 1 M VOSO4 in 1 M H2SO4 solution which can be identified as the following redox reactions (eqn (3) and (4)):
| [VO](aq)2+ + 2H+ + e− ⇌ V(aq)3+ + H2O | (3) |
| [VO2](aq)+ + 2H+ + e− ⇌ [VO](aq)2+ + H2O | (4) |
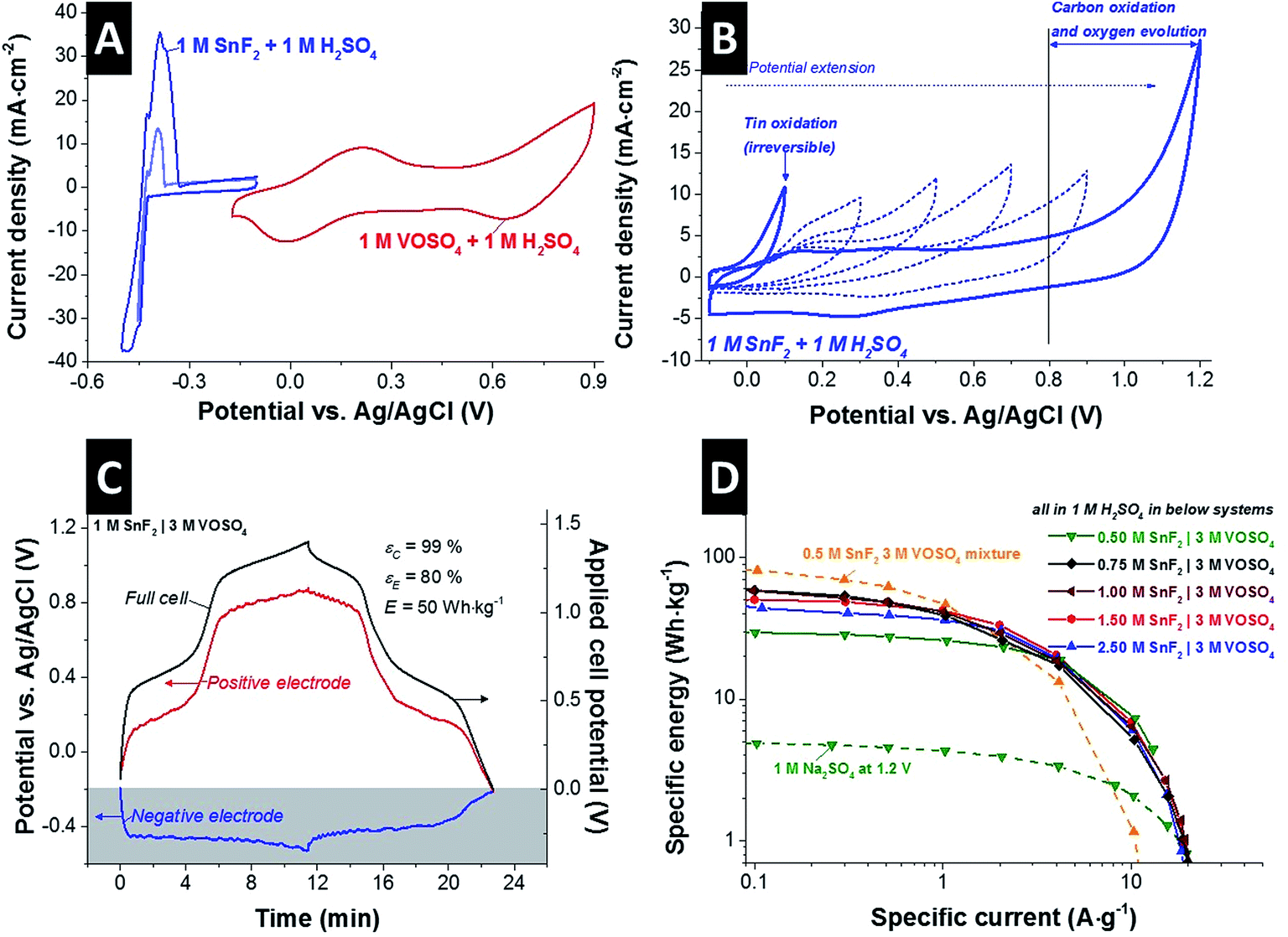 | ||
| Fig. 1 (A) Cyclic voltammograms obtained for individual 1 M SnF2 + 1 M H2SO4 and 1 M VOSO4 + 1 M H2SO4. (B) Electrochemical behavior of 1 M SnF2 + 1 M H2SO4 was investigated by cyclic voltammetry in the positive potential range. (C) The potential developments of the positive and the negative electrode were measure for the 1 SnF2|3 M VOSO4 system in 1 M H2SO4. (D) Rate handling comparison with the separate electrolyte system (SnF2|VOSO4) by various SnF2 concentration and the mixture electrolyte system (SnF2·VOSO4). | ||
For aqueous 1 M SnF2, there is a significant, redox-related current emerging at −423 mV vs. Ag/AgCl, indicative of tin electroplating viaeqn (5). Without having a clear peak, the reduction current shows a slight plateau and a subsequent second current increase at around −453 mV vs. Ag/AgCl (see also ESI,† Fig. S3A) which indicates the possibility of tin fluoride redox reaction viaeqn (6)36,37 and/or hydrogen gas evolution viaeqn (7).
Sn(aq)2+ + 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e− ⇌ Sn(s) e− ⇌ Sn(s) | (5) |
| [SnF6](aq)2− + 4e− ⇌ Sn(s) + 6F(aq)− | (6) |
| 2H+ + 2e− ⇌ H2(g) | (7) |
In the anodic scan, two distinctive oxidations peaks (at −426 mV and −386 mV) and a small shoulder (at −371 mV) appear which are also indications for the possible redox reactions viaeqn (5)–(7). For reversible energy storage, hydrogen evolution in such a low potential range (around −500 mV vs. Ag/AgCl) is not preferable. However, the cathodic stability limit for the solution can be slightly extended to −500 mV vs. Ag/AgCl via reversible hydrogen storage (Fig. S3B†) in the carbon micropores and the tin induced overpotential; which was demonstrated by our earlier work by rigorous stability test with voltage cycling and floating tests.21
The cyclic voltammograms (Fig. 1B) show the irreversible redox behavior of the tin fluoride solution in the potential range from −0.1 V to +1.2 V which is the possible potential range for the positive electrode. The onset potential of ca. −40 mV vs. Ag/AgCl for the increasing oxidation current implies a change of the tin oxidation state from 2+ to 4+ which does not seem to be reversible because no clear reduction current can be seen during backward scan. As the cycle number increases and the potential window is extended, we see that the oxidation potential is shifting to a higher potential until carbon oxidation and oxygen generation occur. However, as illustrated by our post mortem findings (Fig. S2†), this irreversible transition to Sn4+ seems to be the origin of the active surface area blocking in the carbon electrode via the formation of SnO2 which can be the major reason for the low power performance of the mixture electrolyte system (SnSO4·VOSO4). Our results also indicate that SnO2 is a much more favorable form than tin hydroxides in acidic solution which also line with previous reports.38–41
To prevent the unfavorable SnO2 formation at the positive electrode, an anion exchange membrane (AEM) was applied to effectively separate SnF2 anolyte and VOSO4 catholyte. The AEM prevents also the redox shutting via V3+, VO2+, and VO2+ cations which causes a low energy efficiency of the system.11,12Fig. 1C shows the full-cell performance of the separate electrolytes system in acidic media (1 M H2SO4) as exemplified with 1 M SnF2|3 M VOSO4 during galvanostatic cycling, while also monitoring the potential of the negative electrode by a spectator Ag/AgCl reference electrode. As the cell is charged to 1.4 V, the potential of the positive electrode changes from −0.2 V to +0.88 V vs. Ag/AgCl having two clear potential plateaus corresponding to the redox reactions of the vanadium complex viaeqn (3) and (4). At the negative electrode, the potential develops from −0.2 V to −0.53 V vs. Ag/AgCl showing a single potential plateau via the redox activities of SnF2 (eqn (5) and (6)) and the reduction of protons. Capitalizing on tin and vanadium redox activities, a specific energy of 50 W h kg−1 (20 W h L−1) was achieved at 0.5 A g−1 for 1 M SnF2|3 M VOSO4 system with high efficiencies (Coulombic efficiency: 99%; energy efficiency: 80%).
The improved rate behavior of the system with separated electrolytes (SnF2|VOSO4) can be seen in Fig. 1D as compared to the mixture electrolyte system (SnF2·VOSO4). The concentration of separated SnF2 anolyte was varied from 0.5 M to 2.5 M, and 1 M SnF2 was found to be the optimized system in terms of maximum specific energy as well as high Coulombic and energy efficiency (ESI,† Fig. S4A). At a high current density of 10 A g−1, the optimized SnF2|VOSO4 system retains a high specific energy of 5–7 W h kg−1. All investigated SnF2|VOSO4 systems, surveying different concentrations, exhibit a significantly higher power performance than the mixture electrolyte (exemplified for 0.5 M SnF2 and 3 M VOSO4 in Fig. 1D). Since the V3+/V2+ redox reaction does not occur in the separate electrolyte system, 0.5 M SnF2|3 M VOSO4 shows a lower maximum specific energy (29.5 W h kg−1) compared to 0.5 M SnF2·3 M VOSO4 system (81.5 W h kg−1) but still far more than the constituent double-layer capacitor (1 M Na2SO4: 2.1 W h kg−1).
The highest specific energy (at 0.1 A g−1) of 81.5 W h kg−1 was achieved for mixed 0.5 M SnF2·3 M VOSO4, which is about 1.4 times higher than that the separated 1 M SnF2|3 M VOSO4 (58.4 W h kg−1) and over one magnitude higher than the electric double-layer system (1 M Na2SO4: 5 W h kg−1). Considering the maximum specific power of 1.6 kW kg−1 (640 W L−1) for mixed 0.5 M SnF2·3 M VOSO4 and 3.8 kW kg−1 (1.5 kW L−1) for separated 1 M SnF2|VOSO4, the power enhancement of ca. 138% by separating the catholyte and anolyte is promising as seen more clearly when using a Ragone plot applying various normalization methods (ESI,† Fig. S5).
Fig. 2A shows the long-term stability of the optimized SnF2|VOSO4 system in a full-cell configuration by GCPL mode; the cell was charged to 1.4 V and discharged to 0 V at 1 A g−1. After a rapid performance degradation after 100 cycles, the system exhibited a stable performance over 6500 cycles (ca. 40 days) showing a 20% capacitance loss as compared to the stabilized value after 100 cycles (75.7 mA h g−1 or 46 W h kg−1) with almost 100% Coulombic efficiency throughout the test. The initial performance degradation might have been caused by the pore volume reduction via formation of irreversible solid phase since post mortem GSA (Fig. 3A and B) shows slightly lower pore volume from the positive and the negative electrodes as compared to the pristine electrode. The inset shows the GCPL curves obtained after the stabilization period. The small iR drop (ca. 42 mV; ESI† Fig. S4B) assures that the actual stability window applied for the stability test was close to 1.4 V.42 As compared to the reported long-term stability of the other redox electrolyte hybrid energy storage systems,12 capacity retention over 80% after 6500 cycles is promising.
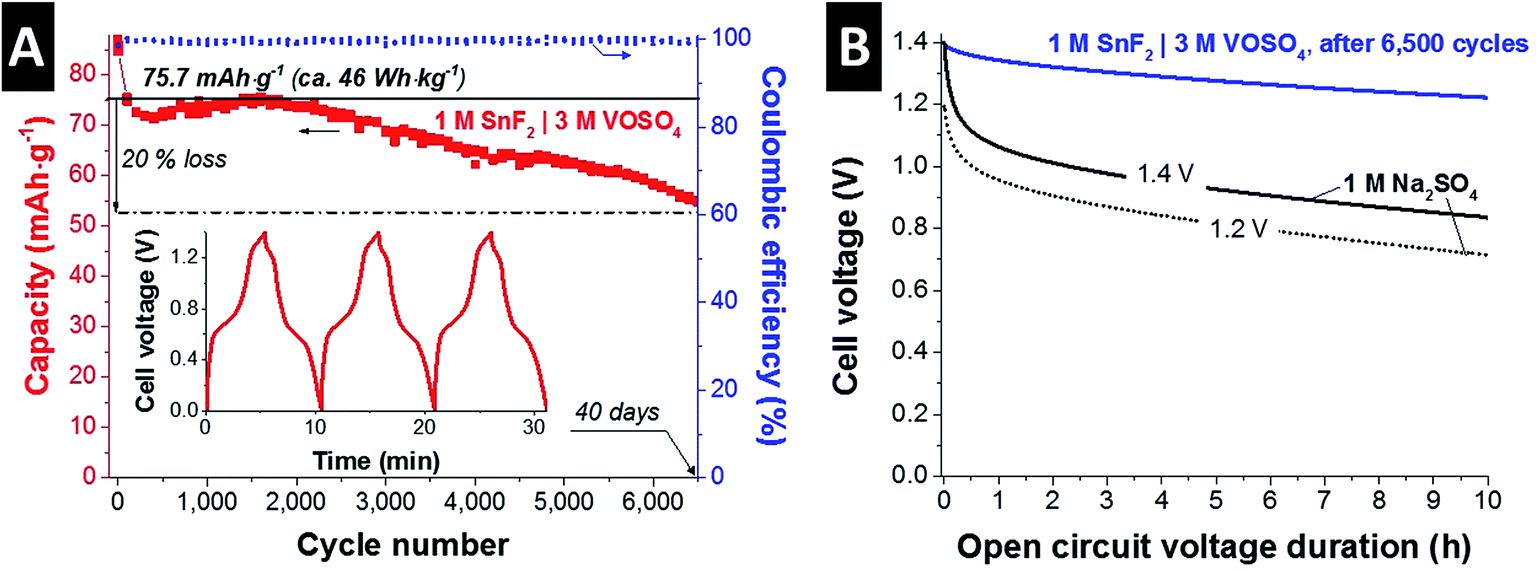 | ||
| Fig. 2 (A) Cycling stability test for 1 M SnF2|3 M VOSO4 system in 1 M H2SO4. A full-cell was charged (1.4 V) and discharged (0 V) at 1 A g−1. Inset shows the GCPL curves for three initial cycles. (B) Self-discharging performance obtained from 1 M SnF2|3 M VOSO4 system in 1 M H2SO4 and a double-layer capacitor (1 M Na2SO4) which was charged up to 1.2 V and 1.4 V. | ||
 | ||
| Fig. 3 Nitrogen gas sorption isotherms (A) and cumulative pore volume (B) as well as X-ray diffractograms obtained for the negative and positive electrodes for 1 M SnF2|3 M VOSO4 system in 1 M H2SO4 after electrochemical operations (C). (D) Scanning electron micrograph and transmission electron micrograph (inset) of the carbon electrode before electrochemical testing. | ||
Fig. 2B shows the self-discharging (voltage decay) performance of the SnF|VOSO4 system compared to a conventional double-layer capacitor system (1 M Na2SO4). Aqueous electric double-layer capacitors suffer from high self-discharging rate; yet, not the entire voltage decay is caused by actual loss of charge, but is associated with ion redistribution and consequently there is a dependency of the charging duration prior to open circuit operation.43–46 In case of aqueous 1 M Na2SO4, we see a rapid, initial voltage decay after charging to 1.2 V or 1.4 V, most likely linked to ion redistribution. Over time, the slope decreases and a continued linear voltage decrease is caused by actual self-discharging (loss of charge rather than just loss of voltage). In case of 1 M SnF2|3 M VOSO4, the potential loss is significantly lower than that of 1 M Na2SO4; 87% potential retention for 1 M SnF2|3 M VOSO4 after 10 h and 60% (both 1.2 V and 1.4 V) for 1 M Na2SO4 as compared to the initial potential. The lower level of the potential loss can be since the hybrid system has more battery-like feature than that of double-layer capacitors as also demonstrated in our earlier works.12,21
Post mortem analysis of electrodes after using the SnF2|VOSO4 redox electrolyte system
So far, we have shown the improved power handling performance by using a system with separated electrolytes (SnF2|VOSO4) as compared to the mixed electrolyte system (SnF2·VOSO4). Yet, the mechanism triggering the power enhancement needs further clarification. For the first step, post mortem nitrogen gas sorption analysis was carried out for the positive and negative electrodes which were electrochemically operated in a full-cell to have different charge states (charged state: cell voltage of 1.4 V; discharged state: cell voltage of 0 V). The activated carbon electrode before the electrochemical operation exhibits a type I(b) isotherm; the high amount of adsorbed nitrogen at very low pressures indicates a predominantly microporous system with pore sizes below 2 nm (Fig. 3A). The pore size distribution pattern calculated by DFT confirm that most pores of the pristine carbon electrode are below 3 nm (Fig. 3B).All nitrogen gas sorption isotherms of the operated positive and negative electrode are of a similar shape as the pristine activated carbon electrode (Fig. 3A). The only main difference lies within changes in the total pore volume (i.e., total amount of adsorbed nitrogen). Therefore, the pore structure of the operated electrodes does not seem to be significantly changed with the only exception of the charged negative electrode. The latter exhibits the largest loss in pores of the size above 1 nm which implies the deposition of SnO2 in the micro/mesopore space. This feature can be better seen when the pore volume is normalized and by assessing the different pore size distribution pattern (dV/dd) as shown in ESI,† Fig. S6A–C. In case of the charged negative electrode, the number of pores > 1 nm decreases compared to the pristine electrode (Table 1). This is in line with the growth of SnO2, as previous studies have shown preferential oxide growth in pores above 1 nm.47 The formation of a pore blocking, non-carbon phase (like SnO2) also further supported by thermogravimetric analysis and the occurrence of a significant residual mass after heat treatment in synthetic air up to 900 °C (ESI,† Fig. S6D).
| Sample | SSA DFT (m2 g−1) | SSA BET (m2 g−1) | Average pore size (nm) | Total pore volume (cm3 g−1) | Volume of micropores (cm3 g−1) |
|---|---|---|---|---|---|
| Initial | 1648 | 2094 | 1.4 | 1.05 | 0.81 |
| Negative, charged | 717 | 837 | 1.2 | 0.41 | 0.33 |
| Negative, discharged | 1449 | 1909 | 1.4 | 0.97 | 0.75 |
| Positive, charged | 1426 | 1864 | 1.4 | 0.97 | 0.73 |
| Positive, discharged | 1261 | 1703 | 1.5 | 0.96 | 0.64 |
Post mortem XRD analysis reveals that loss in pore volume in the charged negative electrode was caused by rutile-type SnO2 formation (Fig. 3C). All other samples showed no indication of SnO2 and the corresponding XRD pattern only exhibited signals from amorphous carbon and PTFE binder. The very broad peak width SnO2 is related to a very small domain size of ca. 2 nm which further supports that the tin dioxides are formed in the pores with the size over 1 nm.
For the detailed investigation on the SnO2 formation, post mortem scanning and transmission electron micrographs were obtained. Apparently different morphologies of the electrode surface can be seen from SEM images for the negative electrode between charged and discharged state (Fig. 4A–B). As compared to the smooth morphology of the pristine electrode (Fig. 3D) and the discharged negative electrode (Fig. 4B), the charged negative electrode (Fig. 4A) exhibits relatively rough surface most probably due to SnO2 formation. As obtained from the post mortem EDX analysis, the atomic percent of oxygen to tin ratio (ca. 2.4, Table 2) further confirms the stoichiometry of SnO2 (excess oxygen correlates with oxygen-containing carbon functional groups). No tin was found by EDX analysis for the discharged negative electrode nor charged and discharged positive electrode. This indicates that metallic tin is reversibly dissolved and the electrolyte separation via the ion exchange membrane is effectively functioning. The electron micrograph from TEM (Fig. 3A, inset) shows the presence of distributed SnO2 nanocrystals in the size range of 2–4 nm and rutile-type SnO2 is further confirmed by the SAED pattern (see ESI,† Fig. S7). From the positive electrode in both charged and discharged state as well as the discharged negative electrode, no SnO2 was found in TEM images or SAED patterns.48 Thus, the results from the post mortem analyses show the presence of SnO2 (Sn[IV]) nanocrystals only at the charged negative electrode in intraparticle micro/mesopores and interparticle macropores.
 | ||
| Fig. 4 Post mortem analysis by SEM and TEM (inset) for the positive and negative electrode in charged and discharged state in 1 M SnF2|3 M VOSO4 in 1 M H2SO4 system. | ||
| Sample | C (atom%) | C (mass%) | O (atom%) | O (mass%) | F (atom%) | F (mass%) | S (atom%) | S (mass%) | Sn (atom%) | Sn (mass%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Initial | 96.4 ± 0.5 | 95.1 ± 0.6 | 3.2 ± 0.3 | 4.2 ± 0.5 | 0.4 ± 0.2 | 0.7 ± 0.4 | — | — | — | — |
| Negative, charged | 78.4 ± 4.5 | 51.5 ± 8.4 | 13.0 ± 3.2 | 11.2 ± 2.2 | 2.6 ± 0.6 | 2.7 ± 0.8 | 0.5 ± 0.1 | 0.9 ± 0.2 | 5.4 ± 1.9 | 33.7 ± 7.9 |
| Negative, discharged | 95.6 ± 1.4 | 93.8 ± 1.9 | 3.5 ± 0.9 | 4.5 ± 1.2 | 0.8 ± 0.5 | 1.2 ± 0.8 | 0.2 ± 0.0 | 0.4 ± 0.1 | — | — |
| Positive, charged | 93.5 ± 0.8 | 91.2 ± 1.0 | 5.6 ± 0.7 | 7.6 ± 0.8 | 0.6 ± 0.3 | ±1.0 ± 0.5 | 0.1 ± 0.0 | 0.3 ± 0.1 | — | — |
| Positive, discharged | 94.3 ± 0.7 | 94.2 ± 0.9 | 5.0 ± 0.4 | 6.5 ± 0.6 | 0.6 ± 0.4 | 0.9 ± 0.6 | 0.1 ± 0.0 | 0.4 ± 0.1 | — | — |
Conclusion
We have demonstrated that the tin and vanadium complex mixture electrolyte shows limited power performance due to pore blocking of the positive electrode via tin oxide formation, as proven by post mortem gas sorption, XRD, and electron microscopy analyses. To prevent tin oxide formation at the positive electrode, a system with separated electrolytes was demonstrated, namely SnF2 + H2SO4 solution as anolyte and VOSO4 + H2SO4 solution as catholyte. As the anion exchange membrane effectively prevents the mixing of the electrolytes, no significant level of pore blocking occurred at the positive electrode during cycling. Therefore, a high specific power (3.8 kW kg−1 or 1.5 kW L−1) was achieved with the 1 M SnF2|3 M VOSO4 system while providing still a high specific energy up to 58.4 W h kg−1 (23.4 W h L−1). This system also showed a highly promising cycling stability over 6500 cycles with a capacity retention over 80%.Acknowledgements
We acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. The authors thank Prof. Eduard Arzt (INM) for his continuing support, FuMA-Tech (esp. Dr Bauer and Dr Klicpera) for kindly providing membranes and helpful discussions. We also thank Dr Choonsoo Kim, Sathyamoorthi Sethuraman, Pattarachai Srimuk, and Dr Soumyadip Choudhury (all at INM) for useful discussions.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054–A5059 CrossRef CAS.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386–12394 CAS.
- E. Frackowiak, M. Meller, J. Menzel, D. Gastol and K. Fic, Faraday Discuss., 2014, 172, 179–198 CAS.
- Q. Li, K. Li, C. Sun and Y. Li, J. Electroanal. Chem., 2007, 611, 43–50 CrossRef CAS.
- G. Lota and E. Frackowiak, Electrochem. Commun., 2009, 11, 87–90 CrossRef CAS.
- S. Roldán, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Angew. Chem., Int. Ed., 2011, 50, 1699–1701 CrossRef PubMed.
- D. Gastol, J. Walkowiak, K. Fic and E. Frackowiak, J. Power Sources, 2016, 326, 587–594 CrossRef CAS.
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS.
- L. Chen, H. Bai, Z. Huang and L. Li, Energy Environ. Sci., 2014, 7, 1750–1759 CAS.
- J. Lee, S. Choudhury, D. Weingarth, D. Kim and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 23676–23687 CAS.
- Y. Tian, J. Yan, R. Xue and B. Yi, J. Electrochem. Soc., 2011, 158, A818–A821 CrossRef CAS.
- L. H. Su, L. Y. Gong and Y. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 681–684 RSC.
- L. H. Su, X. G. Zhang, C. H. Mi, B. Gao and Y. Liu, Phys. Chem. Chem. Phys., 2009, 11, 2195–2202 RSC.
- A. Shanmugavani, S. Kaviselvi, K. V. Sankar and R. K. Selvan, Mater. Res. Bull., 2015, 62, 161–167 CrossRef CAS.
- Z. J. Zhang, Y. Q. Zhu, X. Y. Chen and Y. Cao, Electrochim. Acta, 2015, 176, 941–948 CrossRef CAS.
- H. Yu, J. Wu, L. Fan, S. Hao, J. Lin and M. Huang, J. Power Sources, 2014, 248, 1123–1126 CrossRef CAS.
- J. Wu, H. Yu, L. Fan, G. Luo, J. Lin and M. Huang, J. Mater. Chem., 2012, 22, 19025–19030 RSC.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, Chemsuschem, 2012, 5, 1181–1185 CrossRef CAS PubMed.
- J. Lee, B. Kruner, A. Tolosa, S. Sathyamoorthi, D. Kim, S. Choudhury, K.-H. Seo and V. Presser, Energy Environ. Sci., 2016, 9, 3392–3398 CAS.
- B. Krüner, J. Lee, N. Jäckel, A. Tolosa and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 9104–9115 Search PubMed.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- B. Evanko, S. J. Yoo, S.-E. Chun, X. Wang, X. Ji, S. W. Boettcher and G. D. Stucky, J. Am. Chem. Soc., 2016, 138, 9373–9376 CrossRef PubMed.
- L.-Q. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y.-L. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923 Search PubMed.
- R. Narayanan and P. R. Bandaru, J. Electrochem. Soc., 2014, 162, A86–A91 CrossRef.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, J. Appl. Electrochem., 2011, 41, 1137–1164 CrossRef CAS.
- A. Laheäär, P. Przygocki, Q. Abbas and F. Béguin, Electrochem. Commun., 2015, 60, 21–25 CrossRef.
- K. Fic, M. Meller and E. Frackowiak, J. Electrochem. Soc., 2015, 162, A5140–A5147 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 11 CrossRef.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- J. Lee, D. Weingarth, I. Grobelsek and V. Presser, Energy Technol., 2015, 4, 75–84 CrossRef.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- V. Presser, J. McDonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 CAS.
- J. D. Donaldson and W. Moser, J. Chem. Soc., 1960, 789, 4000–4003 RSC.
- W. B. Schaap, J. A. Davis and W. H. Nebergall, J. Am. Chem. Soc., 1954, 76, 5226–5229 CrossRef CAS.
- F. Séby, M. Potin-Gautier, E. Giffaut and O. F. X. Donard, Geochim. Cosmochim. Acta, 2001, 65, 3041–3053 CrossRef.
- A. J. Berry, Qualitative Inorganic Analysis, Cambridge University Press, 2016 Search PubMed.
- B. Giannetti, P. Sumodjo and T. Rabockai, J. Appl. Electrochem., 1990, 20, 672–676 CrossRef CAS.
- S. S. Abd El Rehim, H. H. Hassan and N. F. Mohamed, Corros. Sci., 2004, 46, 1071–1082 CrossRef CAS.
- B. Stirrup and N. Hampson, Surf. Technol., 1977, 5, 429–462 CrossRef CAS.
- D. Weingarth, A. Foelske-Schmitz and R. Kötz, J. Power Sources, 2013, 225, 84–88 CrossRef CAS.
- M. Kaus, J. Kowal and D. U. Sauer, Electrochim. Acta, 2010, 55, 7516–7523 CrossRef CAS.
- J. Kowal, E. Avaroglu, F. Chamekh, A. Šenfelds, T. Thien, D. Wijaya and D. U. Sauer, J. Power Sources, 2011, 196, 573–579 CrossRef CAS.
- L. García-Cruz, P. Ratajczak, J. Iniesta, V. Montiel and F. Béguin, Electrochim. Acta, 2016, 202, 66–72 CrossRef.
- H. A. Andreas, J. Electrochem. Soc., 2015, 162, A5047–A5053 CrossRef CAS.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- N. Jäckel, D. Weingarth, A. Schreiber, B. Krüner, M. Zeiger, A. Tolosa, M. Aslan and V. Presser, Electrochim. Acta, 2016, 191, 284–298 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical measurements, selected area electron diffraction pattern. See DOI: 10.1039/c6se00062b |
| This journal is © The Royal Society of Chemistry 2017 |