Lithium sulfur and lithium oxygen batteries: new frontiers of sustainable energy storage
Received
30th December 2016
, Accepted 9th February 2017
First published on 10th February 2017
Abstract
Lithium sulfur and lithium oxygen batteries are predicted to be high-energy rechargeable systems of choice for emerging applications, such as modern electronics and electric vehicles. Despite the several issues hindering their diffusion, the two attractive systems are rapidly evolving, and achieving high performances and targets, which were only partially expected in the past few years. Lithium sulfur batteries have been recently introduced into the energy storage market, while practical prototypes of lithium oxygen cells are already emerging, thus indicating the high level achieved by these systems. Therefore, we report herein an overview of recent studies of the reaction mechanism which allowed the development of Li/S and Li/O2 batteries. We show and discuss the latest advances, in terms of electrochemical performances and characteristics, in order to shed light on the feasibility of the two important, cheap and environmentally compatible energy storage systems.
 Lorenzo Carbone | Lorenzo Carbone is a PhD student at Sapienza University of Rome in Mathematical Models for Engineering, Electromagnetics and Nanoscience, presently at the University of Ferrara Italy. He graduated from the same university in Chemical Engineering dealing with materials science, electrochemistry and energy storage. He obtained a fellowship at Volkswagen AG (Germany) and a visiting researcher at Hunter College of CUNY, New York City (USA). He is a co-author of more than 8 papers focusing on the new generation of lithium and lithium ion batteries. |
 Steve G. Greenbaum | Steve Greenbaum is a Professor of Physics at Hunter College (CUNY) and a Fellow of the American Physical Society. He earned his PhD in Physics from Brown University, was a Postdoc at the U.S. Naval Research Laboratory and spent sabbatical years as a Fulbright Scholar at the Weizmann Institute and a Senior Fellow at the Jet Propulsion Laboratory. He was a Visiting Professor at Stony Brook, Rutgers, Tel Aviv, Paris-Sud, Padova, and Rome. He studies disordered solids by magnetic resonance and synchrotron X-ray absorption, and has co-authored over 230 publications. He was a Jefferson Science Fellow at the U.S. State Department in 2014–15. |
 Jusef Hassoun | Jusef Hassoun is presently an Associate Professor of Physical Chemistry and Electrochemistry at the University of Ferrara, Italy. After graduating in Chemistry at the University of Rome Sapienza, he worked in an industrial company for 3 years before returning to academy to obtain the PhD degree in Materials Science in the field of advanced lithium ion batteries in 2009. He has been an Assistant Research Scientist at the Chemistry Department of the University of Rome from 2011 to 2015, and a visiting professor at the Hanyang University in Seoul, South Korea. He is a co-author of more than 145 papers in international journals, book and chapters. |
1 Introduction
The lithium-ion battery, nowadays the most popular and efficient energy storage system, has almost achieved the maximum performance expected from its theoretical potentiality, i.e., an energy density of about 250 W h kg−1.1 However, this value may be insufficient for emerging applications such as all-electric vehicles, which require high energy storage in order to extend the driving range of the car between charges.2 Among the various systems proposed to match this challenging target, lithium sulfur and lithium air batteries represent the most promising solutions. These emerging systems, suitable for achieving an energy density higher than 400 W h kg−1, have the additional bonuses of low expected cost and modest environmental impact.3 Recent progress has led to satisfactory performances with particular relevance for lithium sulfur batteries, while many efforts are still required to make these promising systems suitable for practical application. Room temperature lithium–sulfur batteries of practical interest have been developed only recently by replacing carbonate-based electrolytes, i.e., unsuitable media for the electrochemical reaction of sulfur with lithium, by new classes of solutions.4 The new electrolytes, mainly based on ether chains,5 glymes,6 polyethyleneoxide6 and solid, glass-type media,7 have triggered a remarkable improvement in Li/S batteries. However, additional efforts have been needed during the last few years to address various problems affecting the cell, such as the short cycle life, cell polarization8 and the shuttle reactions due to the dissolution of polysulfide intermediates in the electrolyte during cell operation.9 Polysulfide shuttle represents one of the most severe issues of the lithium sulfur cell. This process consists of an electrochemical short-circuit reaction driven by the formation of soluble polysulfide species, i.e., Li2Sx, during charge of the Li/S cell.9 These dissolved species undergo oxidation at the cathode during subsequent charge, migrate to the lithium anode in which they are reduced and flow back to the cathode to be newly oxidized. This repeated process leads to an “apparent charge” process evolving at a constant voltage of about 2.5 V without energy storage,4 hence to a significant reduction of the coulombic efficiency, and final capacity fading by active material loss and resistance increase.10 The most relevant solutions proposed to solve this issue, taking into account the mechanism of the shuttle reaction above described, were represented by (i) the synthesis of a suitable cathode material in which the polysulfide dissolution is remarkably mitigated11–14 and (ii) the use of electrolyte additives, such as LiNO3, which sacrificially decompose and cover the lithium surface by a protecting solid electrolyte interphase (SEI) layer almost completely avoiding the reduction of the polysulfide at the anode side during Li/S cell charge.4,15 The issues affecting the sulfur cell have been recently mitigated and remarkably addressed by tuning the cathode morphology,11,12 chemical nature,13,14 the electrolyte composition15 and characteristics, as well as by setting-up a suitable cell configuration.16 These efforts have led to lithium sulfur cells of remarkable performances, comparable to lithium-ion batteries, with the additional bonus of high energy density, thus suggesting this system as well suitable for employment in the emerging applications.17 Furthermore, possible safety issues associated with the use of lithium metal in the cell have been addressed by successfully replacing this anode by alternative anodes, mainly based on carbon and lithium-alloying compounds, in a very efficient, high energy and safe lithium-ion sulfur configuration.18 Lithium–oxygen batteries, generally accepted as lithium–air batteries despite in large part using pure oxygen rather than air, attracted large interest since they are characterized by a very high energy density, even exceeding the one associated with lithium–sulfur.19 This outstanding system has been studied during the initial research stages in a common carbonate-based electrolyte eliciting an enthusiastic follow-up.20 So far, the high polarization of the cell, the very short cycle life and the anomalous charge/discharge of the battery have dampened the enthusiasm largely due to the reactivity of the nucleophilic peroxide and superoxide species formed during the Li/O2 reaction with carbonate solvents including the electrophilic carbonyl bond.21 As in the case of the sulfur cell, these issues have been partially mitigated by completely replacing the carbonate with more suitable ether-based electrolytes,22 polymers23 and ionic liquids.24 The change of the electrolyte, as well as a new and more suitable configuration of the cell, in terms of components and engineering,25 and new classes of catalysts26 shed new light on this appealing system. Indeed, these new findings have led to a decreased cell polarization and increased cycle life.27 Only recently, new configurations and compositions of the cathode used as the support for the Li/O2 process,28 including carbons of various natures and morphologies,29 nanostructures,30 doped porous composites,31 metal oxide catalysts and separators designed ad hoc for avoiding oxygen permeation to the anode,32 remarkably increased the lithium–oxygen cell performance in terms of delivered capacity, cycle life and low polarization.33 These breakthroughs, in addition to the development of new and very safe cell prototypes in which the lithium metal is replaced by a lithium-ion anode,34 suggested the lithium oxygen battery as a potential system for next generation energy storage.35
We report in the following pages an overview of the most recent lithium sulfur and lithium air batteries considered as efficient and suitable system for the next generation of sustainable energy storage. Sulfur and oxygen belong to the same group in the periodic table of elements (i.e., the 16th); however, the electrochemical mechanisms of Li/S and Li/O2 differ substantially. Both elements undergo a conversion reaction with lithium, owing to multiple steps including formation of radicals or intermediate species. Nevertheless, chemical nature, reactivity and stability of the intermediates are remarkably different for sulfur and oxygen reactions, which lead to a characteristic behavior in lithium cells in terms of charge–discharge polarization, cycle life and rate capability.19 This review clarifies through multiple examples the similarities and differences between the two appealing systems. Indeed, both for Li/S and for Li/O2 cells this review begins with a brief paragraph in which the reaction mechanism is illustrated by recent studies. Subsequently, various and very recent lithium–sulfur and lithium–oxygen batteries, with enhanced characteristics and performances, will be discussed in detail to elucidate the potentialities of these systems. Therefore, the aim of this work is to shed light on the possibility of the practical employment of these high energy-density batteries in emerging fields, such as electric vehicles and energy storage from renewable sources.
2 Lithium sulfur battery
2.1 The electrochemical process
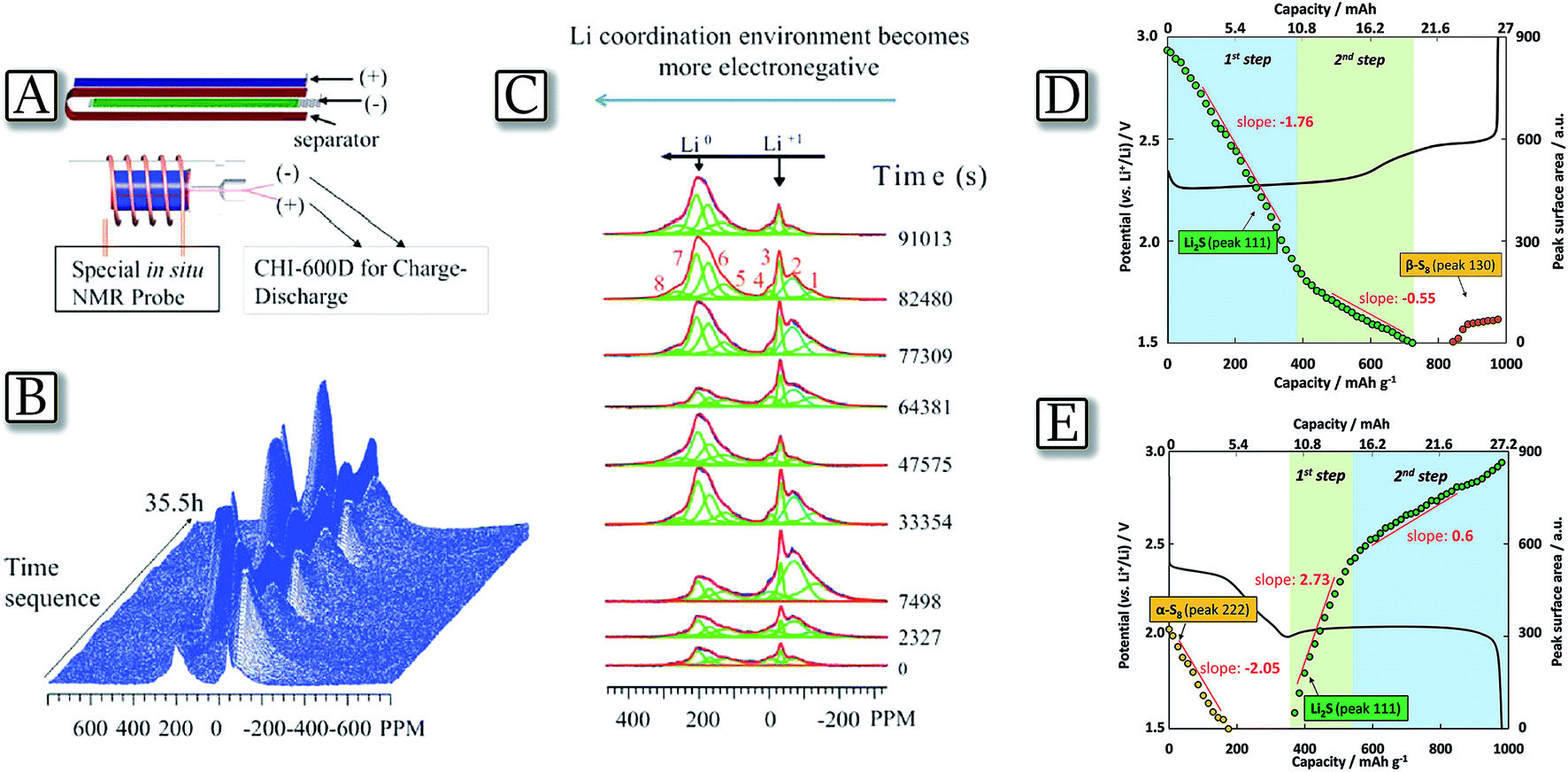
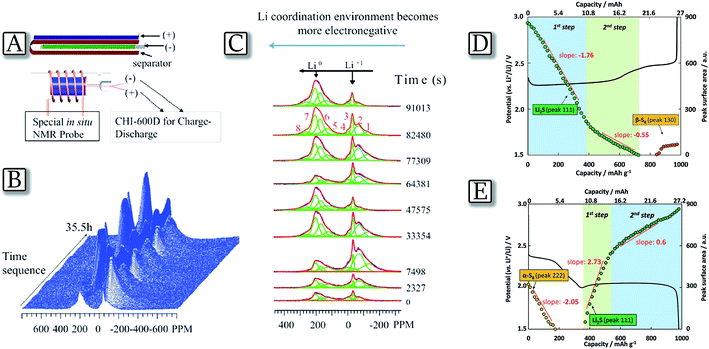
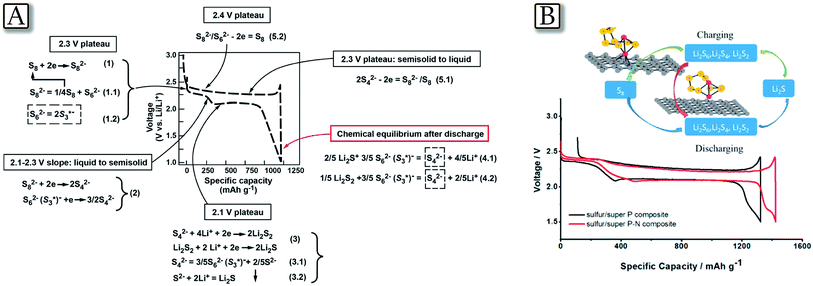
The lithium–sulfur electrochemical reaction proceeds at about 2.4 and 2.1 V vs. Li+/Li by the formation of various intermediates of a general formula of Li2Sx, where x ranges from 8 to 2.36,37 The overall process, i.e., 16Li + S8 ↔ 8Li2S, delivers a specific capacity of about 1700 mA h g−1 as referred to the pure sulfur mass which is reflected into a theoretical energy density as high as 3600 W h kg−1.38 However, the intermediate species, called polysulfides, are extremely soluble in organic liquids and, in the case of application in a lithium cell, they may cause loss of the active material from the sulfur cathode and directly react with the anode, thus leading to battery failure.10 A direct identification and quantification of the reaction species formed during discharge/charge cycling of the lithium–sulfur battery may be performed by an in situ7Li NMR technique which provides real time, semi-quantitative information related to the temporal evolution of lithium polysulfide allotropes.39Fig. 1(A–C)39 report the study of a cylindrical micro-battery used to probe the transient electrochemical and chemical reactions occurring during the cycling of a Li–S system. The in situ experiments provide information about the reaction sequences and reveal a reaction pathway composed of mixed species rather than discrete step-by-step reactions. The large line shifts and line broadening noted suggest a complex chemical environment, and potentially the important role of intermediate species. In particular, the study reveals that the polysulfide redox reactions involve charged free radicals as intermediate species that are difficult to detect in ex situ NMR studies.40 Additionally, the study uncovers information about the 7Li chemical environments during the electrochemical and parasitic reactions on the Li metal anode.41 For the anode side, the continuous evolution of the Li microstructure, SEI accumulation, and polysulfide contamination are simultaneously captured by 7Li in situ NMR, which are correlated with the Li–S cell degradation mechanisms. A quantitative evaluation of Li2S formation and consumption upon cycling may also be performed by means of XRD as reported in Fig. 1(D and E).42 The figure, obtained from a pouch Li/S cell using a nonwoven carbon sheet as a current collector and relatively high sulfur loading, along with a controlled electrolyte amount, shows the expected voltage profile, and charge/discharge capacities of 980 and 976 mA h g−1, respectively. The corresponding XRD pattern evolution indicates that as the discharge proceeds the peak intensities of the elemental orthorhombic α-S8 linearly decrease as a function of capacity. They vanish at the end of the first discharge plateau, proving a complete reduction of sulfur. This linear evolution can be associated with a one-step electrochemical process, based on the reduction reaction of S8 to S82−. Indeed, a two step process occurs, where crystalline Li2S is initially formed alone.43 Afterward, the specificity of the reduction reaction clearly decreases due to the complete coverage of the available conductive surface. The number of electrons exchanged is coherent with the formation of intermediate species such as Li2S2. During the lower discharge plateau, 24% of S42− is involved in the formation of Li2S, whereas 72% of S42− is assumed to be converted into Li2S2. During the following charge, Li2S is converted into a soluble polysulfide species in a two-step oxidation process, however, in a different reaction pathway as during discharge.
 |
| | Fig. 1
In situ NMR probe and the acquired spectra as a function of discharge/charge time. (A) Cylindrical micro-battery, fitted into the in situ NMR probe. (B) Stacked plot of the in situ7Li NMR spectra in a functioning Li–S cell vs. time during discharge/charge. (C) Major peaks of interest within the range of −260 to 300 ppm extracted at different times by fitting the spectra. Reproduced with permission.39 Copyright American Chemical Society 2015. (D) Investigation of the Li2S consumption during the charge process and (E) proposed mechanism for Li2S/Li2S2 formation upon discharge. Integrated surface area of (111) reflection as a function of capacity, superimposed with the experimentally obtained galvanostatic cycling profile. Reproduced with permission.42 Copyright Wiley 2015. | |
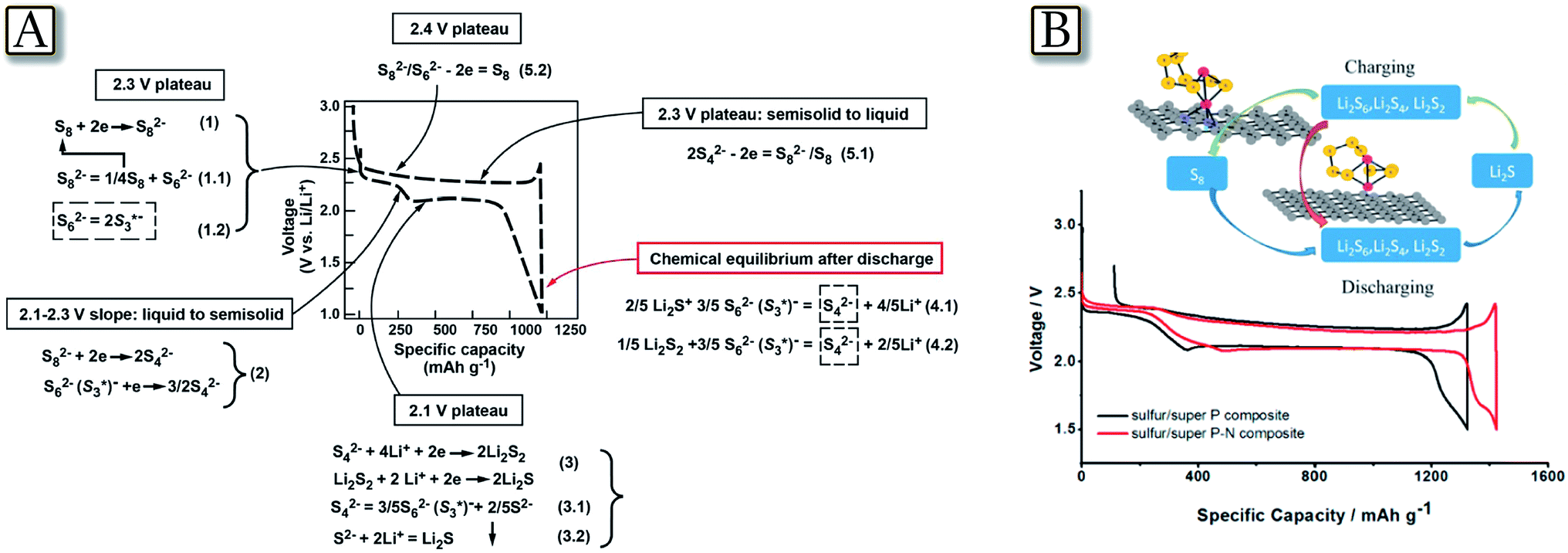
The discharge/charge pathways of the Li/S cell may also be directly observed by monitoring the sulfur radicals using an in situ electron paramagnetic resonance (EPR) technique.44Fig. 2(A)44 reveals that the chemical and electrochemical reactions in the Li–S cell are driving each other to proceed through sulfur radicals, leading to two completely different reaction pathways during discharge and charge. The different reaction pathways for the discharge and charge processes of the Li–S battery match with their voltage profiles and help to describe the origin of the high overpotential of pure Li2S. In situ EPR identifies S3*− throughout the whole cycling, thus indicating the temporal equilibrium among different polysulfides instead of producing a single polysulfide component at each different potential. The EPR study also suggests that the electrolyte selection for the Li–S system should consider the reactivity of sulfur radicals to enable the long-term cycling stability of the cells.45 Combined operando Raman spectroscopy and density functional theory (DFT) methods have been used to understand the complicated sulfur redox chemistry at the electrolyte/carbon interface.46Fig. 2(B)46 illustrates this study and shows that the higher polysulfide, Li2S8, is absent during the whole redox route, whereas the charging process of the Li–S battery ends up with Li2S6. DFT calculations suggest that Li2S8 accepts electrons more readily than S8 and Li2S6 so that it is thermodynamically and kinetically unstable. Meanwhile, the study indicates the poor adsorption behavior of Li2Sn on the carbon surface further as a factor preventing the oxidization of Li2Sn back to S8 upon charging. Periodic DFT calculations show that the N-doped carbon surface can serve as a conductive Lewis base “catalyst” matrix to enhance the adsorption energy of Li2Sn (n = 4–8). Therefore, the theoretical calculation suggests N-doped carbons for significantly improving S utilization and cycle stability even at a high sulfur loading.47
 |
| | Fig. 2 (A) Proposed reaction mechanism for Li–S batteries during cycling. The equilibrium between S62− and S*3− always exists and shifts at different potentials. S62− prefers to stay at relatively high potential (>2.1 V) while S*3− is the preferred form at low potential (≤2.1 V). Different reaction pathways are suggested for discharge and charge processes. The Li–S cell was tested at C/5 rate between 1 V and 3 V in typical button cells. Reproduced with permission.44 Copyright ECS 2015. (B) Comparative study of the first discharging–charging curves at 0.1 A g−1 for super P carbon with or without N doping. The inset picture demonstrates the influence of the Lewis base conductive matrix of N-doped super P carbon on the polysulfide redox route. Reproduced with permission.46 Copyright American Chemical Society 2015. | |
2.2 Optimization of the electrolyte and electrodes for practical lithium–sulfur cell application
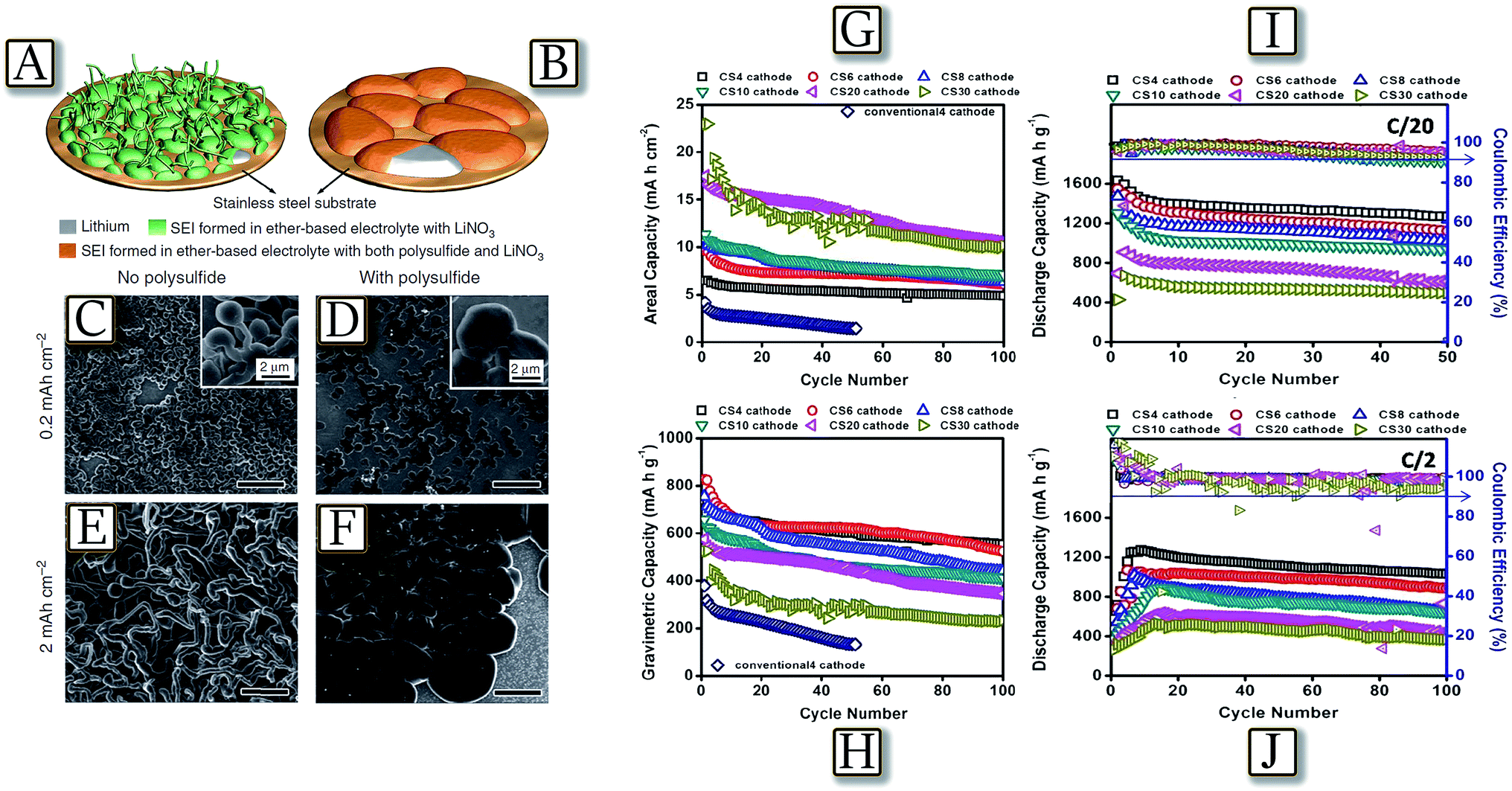
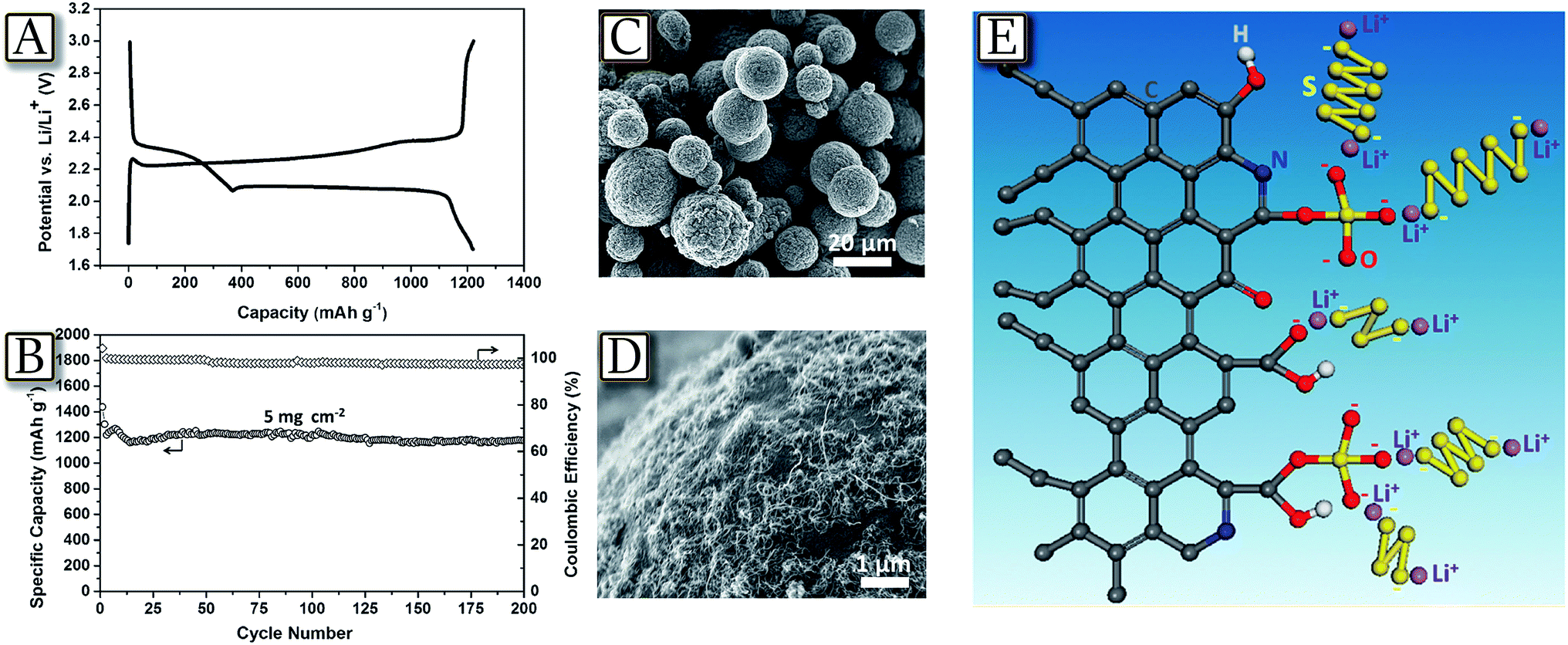
Uncontrolled dendritic lithium growth,41 as well as electrolyte decomposition in lithium sulfur batteries,48 can be efficiently suppressed by the formation of a stable and uniform solid electrolyte interphase layer,49 due to a synergetic effect of both lithium polysulfide and lithium nitrate as additives in an ether-based electrolyte.50 Indeed, Fig. 3(A and B)50 schematize how lithium dendrite growth can be effectively suppressed via manipulating the chemical reactions of lithium, lithium polysulfide and LiNO3. By controlling the concentration of polysulfide and LiNO3 in the electrolyte, a stable and uniform SEI can be formed on the lithium surface, which greatly minimizes electrolyte decomposition and prevents dendrite growth.51,52 The observations, made in the edge regions of the deposition area where dendrite growth is most relevant due to the absence of physical constraints, Fig. 3(C–F), actually revealed no lithium dendrite formation at practical currents and coulombic efficiency and cycle life of the lithium metal cells significantly improved. A further concern of the lithium sulfur battery may be found in active material loading, which may allow a high practical energy density. High loadings, leading to high practical energy of the cell, may be achieved by combining electrolyte optimization illustrated above and suitable electrode morphology and design. Core–shell cathodes, with a sulfur core shielded within a conductive shell-shaped electrode, may actually allow Li–S cells to load with a high amount of sulfur (up to 30 mg cm−2 of electrode powder with sulfur content approaching 70 wt%).53 Indeed, the core–shell high-loading cathodes studied within the polysulfide-trap cell configuration reported in Fig. 3(G–J)53 exhibit a high sulfur utilization of up to 97%, at C/20–C/2 rates, and low self-discharge during long-term cell storage for a three-month rest period under various cell-storage conditions. Therefore, an optimal cell design and analytical approach can both overcome the current technical limitations of Li–S batteries and provide a better detection method for understanding the key challenges of Li–S cells.54
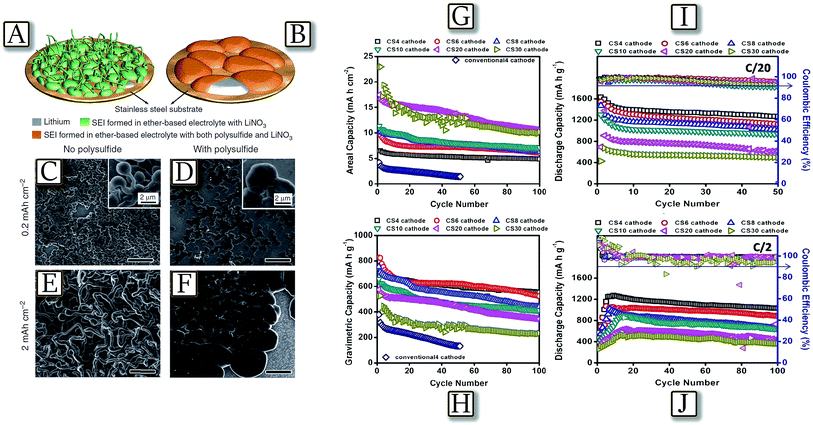 |
| | Fig. 3 Schematic illustration showing the morphology difference of lithium deposited on the stainless-steel substrate in the two electrolytes (both contain lithium nitrate), but (A) without lithium polysulfide (B) containing lithium polysulfide. SEM images of lithium metal deposited onto a bare stainless steel substrate (the edge region of the whole deposition area) in the electrolyte (C and E) with the addition of only LiNO3 (1 wt%) and (D and F) with the addition of both Li2S8 (0.18 M) and LiNO3 (1 wt%) at a current density of 2 mA cm−2 and deposition capacities of 0.2 and 2 mA h cm−2, respectively. Scale bars, 20 μm. Reproduced with permission.50 Copyright Nature Publishing Group 2015. Battery performance of the core–shell cathodes with various sulfur loadings from 4 to 30 mg cm−2 at C/10 rate: (G) areal capacity (mA h cm−2) and (H) gravimetric capacity (mA h g−1). Battery performance of the core–shell cathodes with various sulfur loadings from 4 to 30 mg cm−2 at various cycling rates: (I) C/20 and (J) C/2 rates. Reproduced with permission.53 Copyright Royal Society of Chemistry 2016. | |
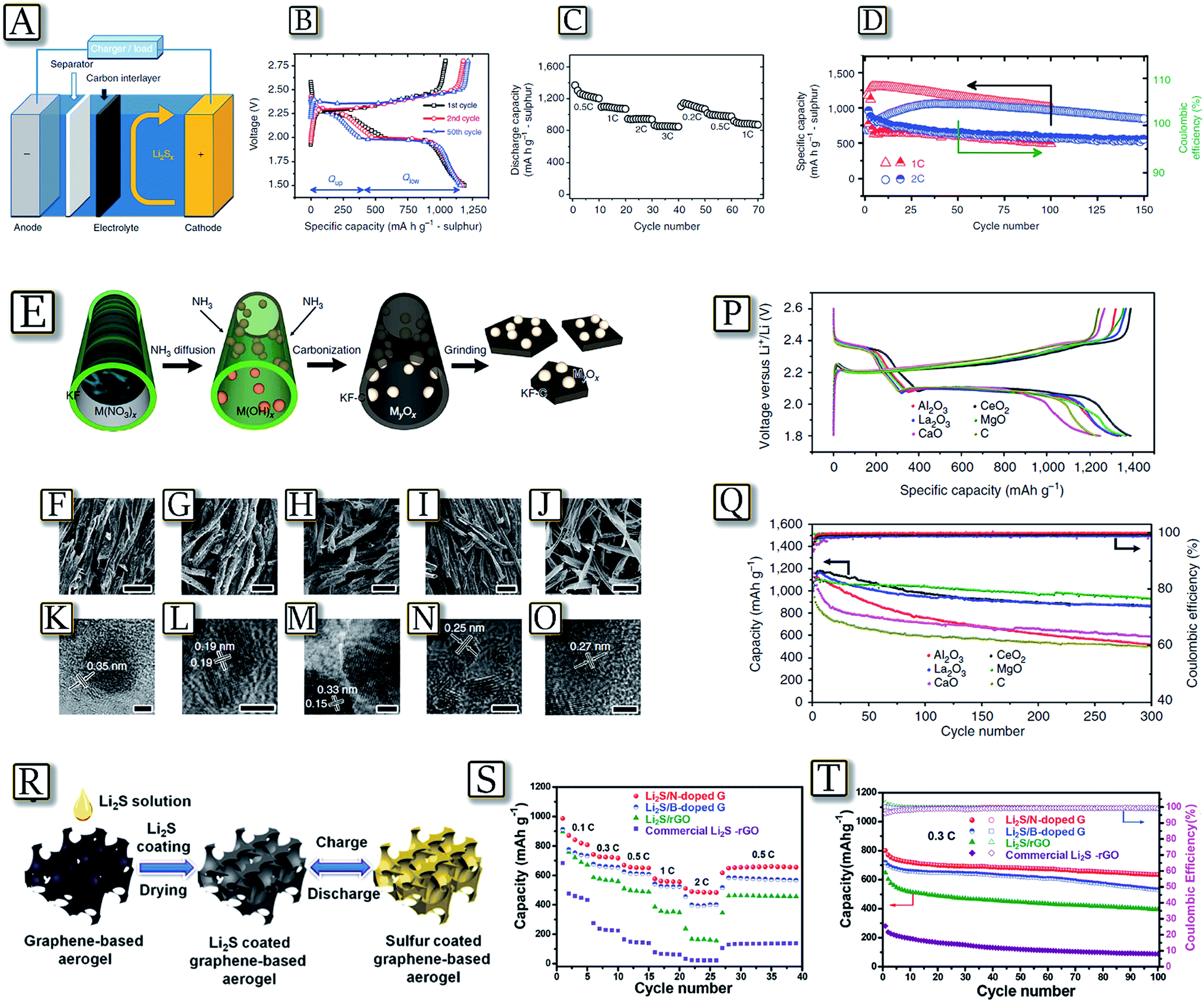
A bi-functional microporous carbon paper between the cathode and separator may be a simple adjustment16 to the traditional lithium–sulfur battery configuration to achieve high capacity with a long cycle life and rapid charge rate.55 The example in Fig. 4(A–D)55 shows a significant improvement not only in the active material utilization but also in capacity retention, without involving complex synthesis or surface modification. The insertion of a microporous carbon interlayer decreases the internal charge transfer resistance and localizes the soluble polysulfide species, facilitating a commercially feasible means of fabricating lithium–sulfur batteries. The flexible microporous carbon interlayer substantially improves the cycle performance of the Li–S batteries by providing a fine contact with the cathode surface, offering electron pathways through the insulating S/Li2S and accommodating the migrating polysulfide intermediates. This bi-functional carbon interlayer with micro-pores/small-mesopores operates not only as an “upper current collector” to enhance the active material utilization but also as a “polysulfide stockroom” to retain the cycle life. Furthermore, nonconductive metal-oxide nanoparticle decorated carbon flakes synthesized via a bio-template method have been proposed for high performance Li–S batteries.56Fig. 4(E–Q)56 shows that the series composite sulfur electrodes based on nonconductive metal oxide (MgO, Al2O3, CeO2, La2O3 and CaO) nanoparticles anchored on porous carbon nano-flakes allow enhanced cycling performance. Electrochemical tests, adsorption experiments and DFT calculations revealed that polysulfide capture by the oxides proceeds via mono-layered chemisorption. In addition, comprehensive oxide selection criteria referring to the strong binding, high surface area and good surface diffusion properties were proposed. A fully lithiated lithium sulfide (Li2S) cathode represents a valid candidate characterized by a mitigated volume expansion during the electrochemical process, and can be easily paired with anode materials in a full cell configuration.57 However, the bulk Li2S electrode suffers from a large activation barrier during the initial charge, low active-material utilization, poor electrical conductivity, and fast capacity fade, thus preventing practical application. The development of a self-supported, high capacity and long-life cathode is presented for the Li/S battery by coating Li2S onto N- or B-doped graphene aerogels using a simple liquid infiltration/evaporation method as reported in Fig. 4(R).58Fig. 4(S and T)58 shows that the cathode material has a high specific capacity, good rate capability, and excellent cycling stability in the lithium cell. The improved performance was attributed to: (i) cross-linked, porous graphene network enabling fast electron/ion transfer; (ii) complete Li2S coating on flexible graphene which accommodates stress or volume change during cycling, allows high active material utilization and lowers the energy barrier;59 and (iii) doped heteroatoms with a strong binding affinity toward Li2S/Li2Sx intermediate species which limit the polysulfide dissolution.60 Therefore, the 3D-functionalized graphene aerogel strongly binds Li2S and leads to a highly performing cathode material for application in rechargeable Li/S batteries.
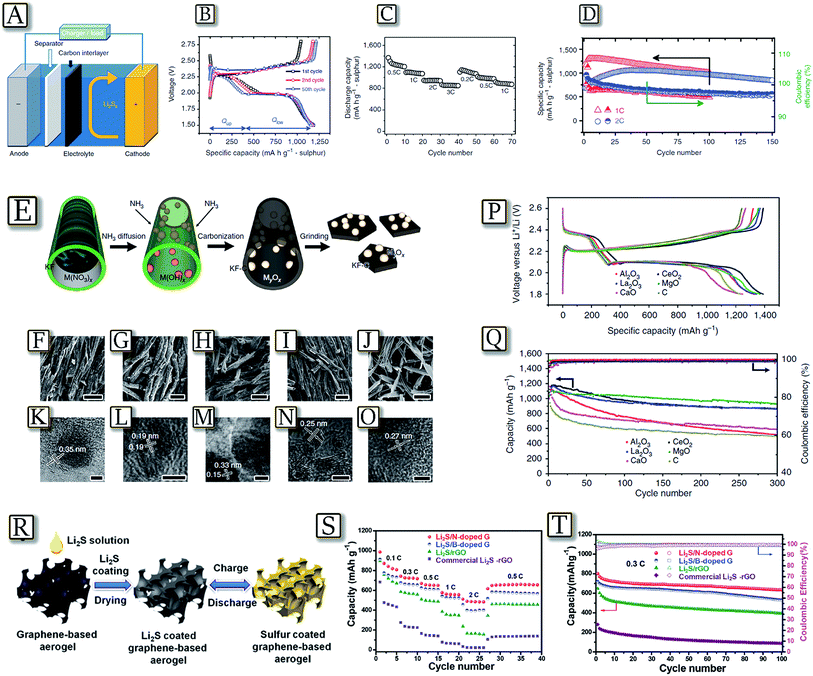 |
| | Fig. 4 (A) Schematic configuration of a Li–S cell with a bifunctional microporous carbon interlayer inserted between the sulphur cathode and the separator, (B) discharge/charge profiles at various cycles, (C) rate capability up to 3C and (D) cycle life and coulombic efficiency of the cell with MCP at 1C and 2C for long cycles. Reproduced with permission.55 Copyright Nature Publishing Group 2012. (E) Schematic illustration of synthesis of oxides/carbon using the kapok tree fibres (KFs) as both the template and carbon sources. (F–J) SEM images of Al2O3/C, CeO2/C, La2O3/C, MgO/C and CaO/C composites, respectively (scale bar = 50 μm). (K–O) The corresponding HRTEM images of Al2O3/C, CeO2/C, La2O3/C, MgO/C and CaO/C, respectively (scale bar = 3 nm). (P) Representative charge–discharge profiles of the composite electrodes based on different oxide/carbon nanostructures at 0.1C. (Q) Specific capacity and the corresponding coulombic efficiency of the composite electrodes upon prolonged 300 charge–discharge cycles at 0.5C. Reproduced with permission.56 Copyright Nature Publishing Group 2016. (R) Illustrations of the Li2S coating process and the in situ charge/discharge process of the graphene-based electrode. (S) Rate capability of the commercial Li2S–rGO hybrid, Li2S/rGO, Li2S/B-doped graphene, and Li2S/N-doped graphene electrodes. (T) Cycling performance and coulombic efficiency of the commercial Li2S–rGO hybrid, Li2S/rGO, Li2S/B-doped graphene, and Li2S/N-doped graphene electrodes at 0.3C rate for 100 cycles. Reproduced with permission.58 Copyright Wiley 2016. | |
2.3 Nitrogen doping and metal oxide addition for avoiding polysulfide dissolution
Functional carbon composites, such as carbon-nanotubes,61 nano-spheres,62,63 mesoporous carbons,64,65 graphene composites,66 carbon nanosheets,67 nitrogen-doped carbon spheres and MNCS/CNT, can strongly adsorb lithium polysulfides acting as a sulfur host.68Fig. 5(A–D)68 demonstrates that the nitrogen functional groups of this composite enable the effective trapping of lithium polysulfides on electroactive sites within the cathode, leading to a much improved electrochemical performance. The enhancement in adsorption can be attributed to the chemical bonding of lithium ions by nitrogen functional groups in the MNCS/CNT framework due to the electron-donating ability of the doped N atoms. Indeed, the MNCS/CNT composite can significantly inhibit the dissolution of polysulfides and retard the loss of the active material,69 and as a result, it can deliver a high initial specific capacity (1480 mA h g−1) and excellent cycling stability (90% retention over 200 cycles after activation) at a high sulfur content of 70 wt% and a high sulfur loading of approximately 5 mg cm−2. An alternative explanation of the pronounced polysulfide-attracting characteristics of nitrogen doped carbons has been given by X-ray absorption near-edge structure (XANES) spectroscopy focusing on the coordination structures of C, O, and N in a carbon cathode before and after the sulfur loading.70 The study, with the schematic result displayed in Fig. 5(E),70 revealed that the presence of nitrogen promotes the enhanced reactivity of surface oxygen functional groups, and the formation of thiosulfate and sulfate species fixed on the carbon cathode. These sulfur-containing functional groups provide excellent adsorption sites for sulfur species that are generated during the electrochemical processes characteristic of battery operation. These observations reveal that strong interaction between the nitrogen-doped carbon and sulfur is through oxygen functional groups, and nitrogen doping probably makes oxygen functional groups more reactive toward sulfur. Accordingly, the transformation of oxygen functional groups was observed in O K-edge XANES spectra when mesoporous nitrogen-doped carbon (MPNC) materials adsorbed sulfur species, whereas mesoporous carbon (MPC) materials without nitrogen doping did not show such a trend. In addition, S K-edge XANES showed that the interaction between elemental sulfur (S8) and nitrogen-doped carbon produces more thiosulfate and sulfate species than the carbon without nitrogen doping.
 |
| | Fig. 5 (A) A typical discharge–charge voltage–capacity profile of an MNCS/CNT–S cathode. (B) Cycling performance of MNCS/CNT–S at a current density of 0.84 mA cm−2 for the first cycles and 1.68 mA cm−2 for the subsequent cycles. SEM images of the MNCS/CNT composite with low (C) and high (D) magnification. Reproduced with permission.68 Copyright Wiley 2015. (E) Schematic illustration of the possible interaction between the nitrogen-doped carbon and sulfur through oxygen functional groups. The nitrogen doping probably makes oxygen functional groups more reactive toward sulfur. Reproduced with permission.70 Copyright American Chemical Society 2014. | |
Further progress in avoiding polysulfide dissolution has been achieved by incorporating cobalt disulfide (CoS2) into carbon/sulfur cathodes, leading to a strong interaction between lithium polysulfides and CoS2 under working conditions.71Fig. 6(A–E)71 shows that the interfaces between CoS2 and electrolyte serve as strong adsorption and activation sites for polar polysulfides and therefore accelerated redox reactions of polysulfides. The high polysulfide reactivity not only guarantees effective polarization mitigation and augmented energy efficiency by 10% but also high discharge capacity and stable cycling performance during 2000 cycles. This improvement was attributed to strongly coupled CoS2–polysulfide interaction, and to dynamically enhanced polysulfide redox reactions, confirmed by both theoretical and experimental results.72,73 A similar approach for suppression of the polysulfide shuttle while maintaining a high sulfur content is represented by using 2D early transition-metal carbide conductive M-Xene phases, reported to be also impressive supercapacitor materials, and also performing as excellent sulfur battery hosts owing to their inherently high underlying metallic conductivity and self-functionalized surfaces.74Fig. 6(F–H)74 reveals that composites of sulfur and highly conductive carbide Ti2C/2D nanosheets with a hydrophilic surface (70 wt% S/Ti2C) exhibit stable long-term cycling performance because of strong interaction of the polysulfide species with the surface Ti atoms. The cathodes show excellent cycling performance with specific capacity close to 1200 mA h g−1 at a five-hour charge/discharge (C/5) current rate. A capacity retention of 80% is achieved over 400 cycles at a two-hour charge/discharge (C/2) current rate. The existence of S–Ti–C bonding at the interface, determined by XPS, suggested strong interaction and chemisorption of polysulfides onto the “acid” Ti sites and hydroxy surface groups. In a cell, the initially adsorbed polysulfides undergo conversion to Li2S either by electron transfer via the Ti2C or by disproportionation to form multiple Li2S nucleation sites on the surface. Progressive consumption of sulfur generates more polysulfides that are reduced to sulfide preferentially deposited epitaxially on the existing Li2S nuclei, thus effectively mitigating the dissolution of polysulfides into the electrolyte and providing very good cycling performance with a capacity fade rate of 0.05% per cycle.
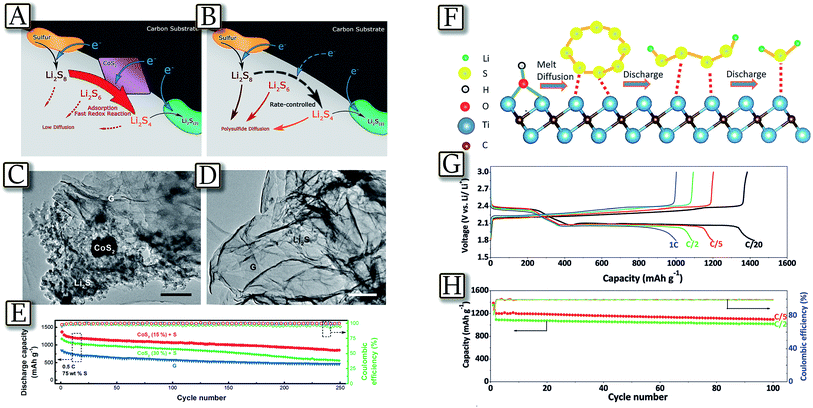 |
| | Fig. 6 Schematic illustration of the discharge process in sulfur cathodes. (A) CoS2 incorporated carbon/sulfur cathode where polysulfide reduction is accelerated and polysulfide diffusion is weakened and (B) pure carbon/sulfur cathode where polysulfide reduction is rate-controlled and polysulfide diffusion is dominated. SEM images of (C) S/CoS2 (15%) + G at the intermediate state and (D) SEM images of S/G at the intermediate state. (E) Electrochemical performance of S/G cathodes with or without CoS2 addition. Reproduced with permission.71 Copyright American Chemical Society 2016. (F) Replacement of the Ti–OH bond on the MXene surface with a S–Ti–C bond on heat treatment or by contact with polysulfides. (G) Voltage profiles of 70S/d-Ti2C at various rates ranging from C/20 to 1C. (H) Cycling performance of 70S/d-Ti2C at C/5 and C/2. Reproduced with permission.74 Copyright Wiley 2015. | |
2.4 Other viable strategies for actually improving the Li/S battery
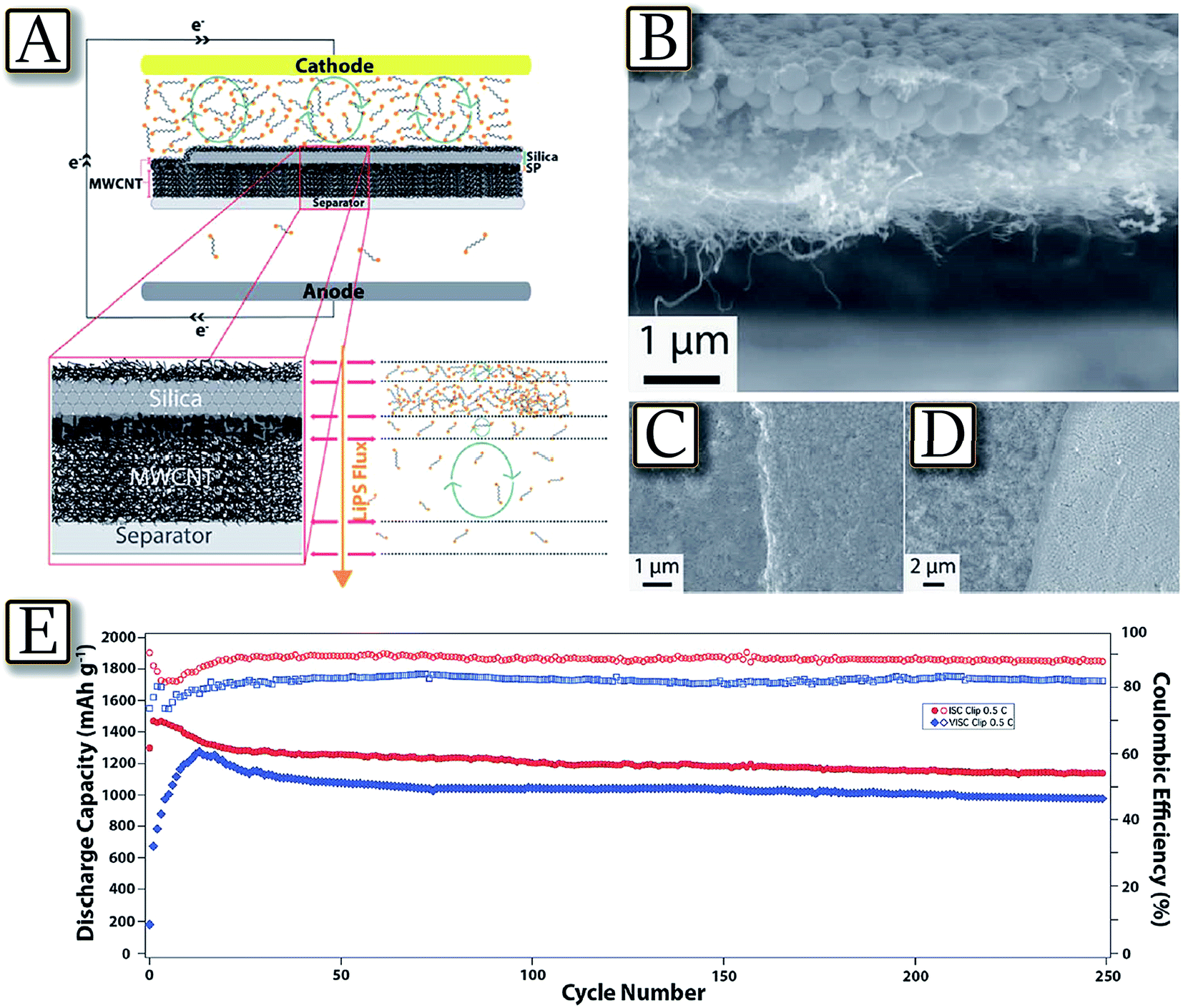
Besides cathode and electrolyte material optimization, which mainly concerns new synthetic pathways for the cathode and chemical additives for the electrolyte,75 several effective approaches are focused on the improvement of the whole cell configuration, simultaneously including electrodes and electrolyte as well as a cell separator considered as concomitant factors for enhancing Li/S cell performance.76 One valid example is represented by the Langmuir–Blodgett technique, which is widely used for preparing coatings of amphiphilic molecules at air/water interfaces with thickness control down to a single molecular layer. Following this approach, multifunctional nanoparticle films of carbon, metal-oxide nanoparticles, polymers, and combinations of these materials may be layer-by-layer deposited using polyolefin separators in lithium sulfur cells to create structured membranes for regulating mass and charge transport.77Fig. 7(A–D)77 illustrate this technique used to create a layered MWCNT/SiO2/MWCNT nanomaterial in a polypropylene membrane (Celgard separator) with a clip-like configuration, for efficiently intercepting and reutilizing dissolved lithium polysulfides in the Li–S battery. The clip coated separator is observed to exhibit largely improved active material utilization, enhanced capacity retention over extended charge/discharge cycling, and attractive high rate capability. Indeed, the cycling test of Fig. 7(E)77 using this cell configuration evidences a capacity of about 1200 mA h g−1 retained for about 250 mA h g−1.
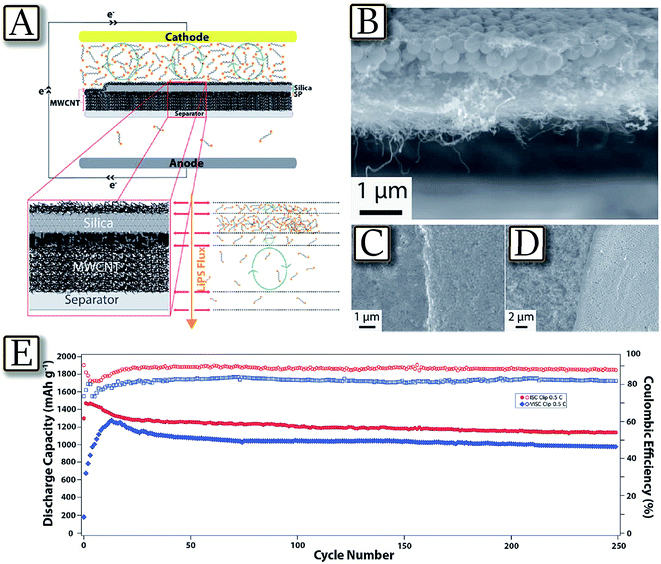 |
| | Fig. 7 Clip configuration coatings on the separator. (A) Clip configuration architecture design and LiPS flux diagram across the separator during the discharge of Li–S cell. (B) Cross-sectional SEM image of the clip coated separator. (C) SEM image of the clip coated separator at the silica-SP boundary layer. (D) SEM image of the clip coated separator at the boundary without final MWCNT coating. (E) Cycling performance of the clip coated separator Li–S cells with ISC and VISC at 0.5C for 250 cycles. Reproduced with permission.77 Copyright Royal Society of Chemistry 2016. | |
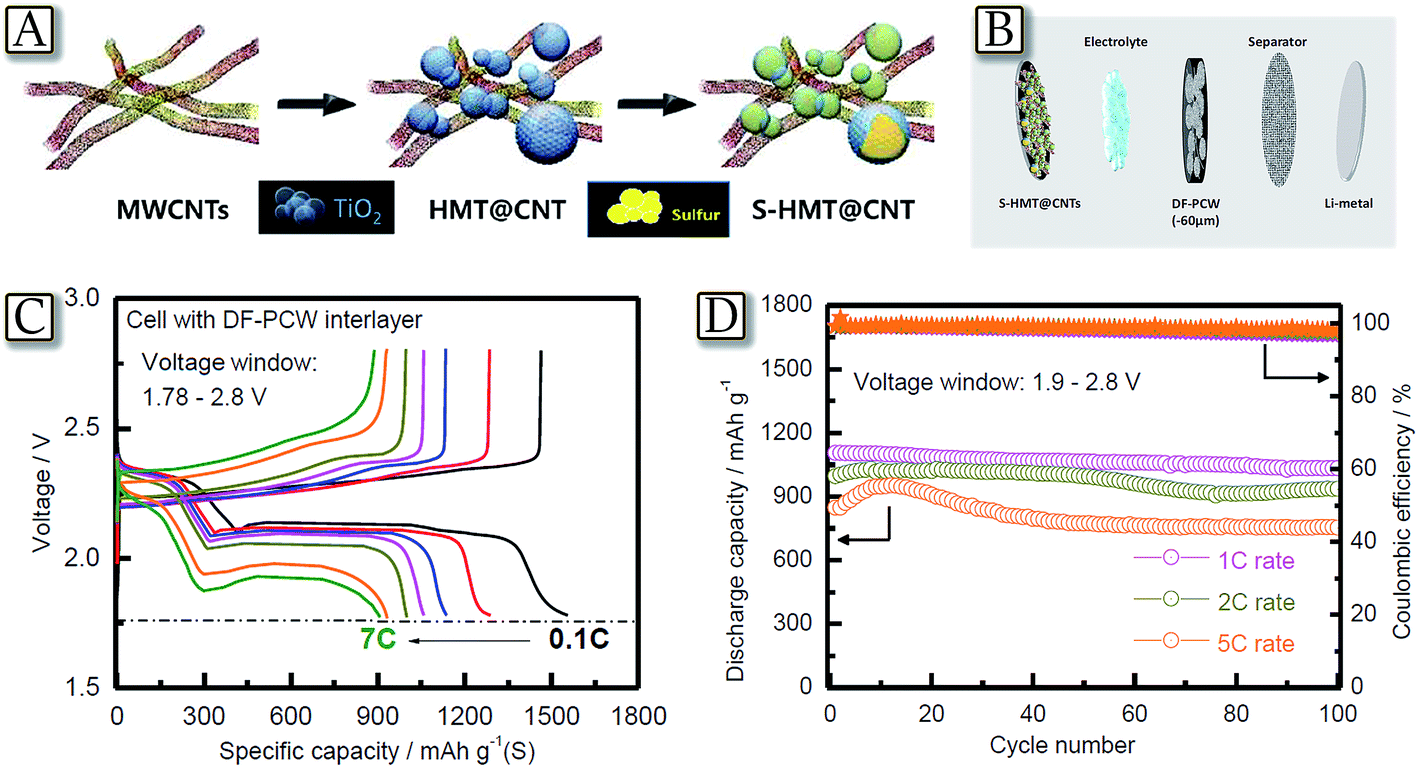
Another example of an inclusive approach taking multiple components is represented by Fig. 8.78 The figure reports a Li/S cell with a cathode consisting of sulfur and hollow-mesoporous titania (HMT) embedded within carbon nanotubes (CNTs), S-HMT@CNT, and a dual functional porous carbon wall interlayer, DF-PCW, obtained by filling the voids in TiO2 spheres with carbon and etching the TiO2 template with a chemical process (Fig. 8(A and B)).78 The DF-PCW interlayer provides a medium for scavenging the lithium polysulfide and suppressing them from diffusing to the anode side when it is inserted between the sulfur cathode and the separator, and improves Li-ion and electron transports. Furthermore, the pores of TiO2 allow high sulfur loadings and accommodation of the volume expansion at the electrode level, while the CNT component constitutes an overlapping network that improves both the electronic conductivity and mechanical strength of the S-HMT@CNT and DF-PCW. These characteristics and the cell design allowed the lithium–sulfur battery to achieve good cycle life, high efficiency, and rate capability extending up to 2C and 5C Fig. 8(C and D).78 Therefore, combinations of multiple and improved components may actually lead to lithium–sulfur cells of high performance, in terms of practical capacity, rate capability, cycle life, and energy and power density, thus allowing the employment of this very appealing system in the next few years.79
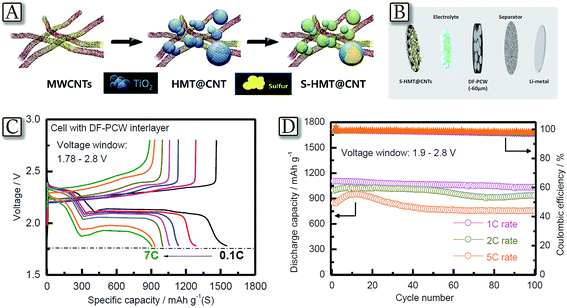 |
| | Fig. 8 (A) Schematic detailing the concept of the containment of sulfur in HMT and CNTs. (B) Schematic representation detailing the cell fabrication process. (C) Voltage profiles of the S-HMT@CNT electrode cycled between 1.78 and 2.8 V under increasing C-rates in cells constructed with a DF-PCW interlayer and (D) discharge capacities and cell efficiencies of the S-HMT@CNT electrode in a cell constructed with a DF-PCW interlayer and cycled between 1.9 and 2.8 V under increasing rates. Reproduced with permission.78 Copyright Wiley 2016. | |
2.5 A highly safe configuration: the lithium-ion sulfur battery
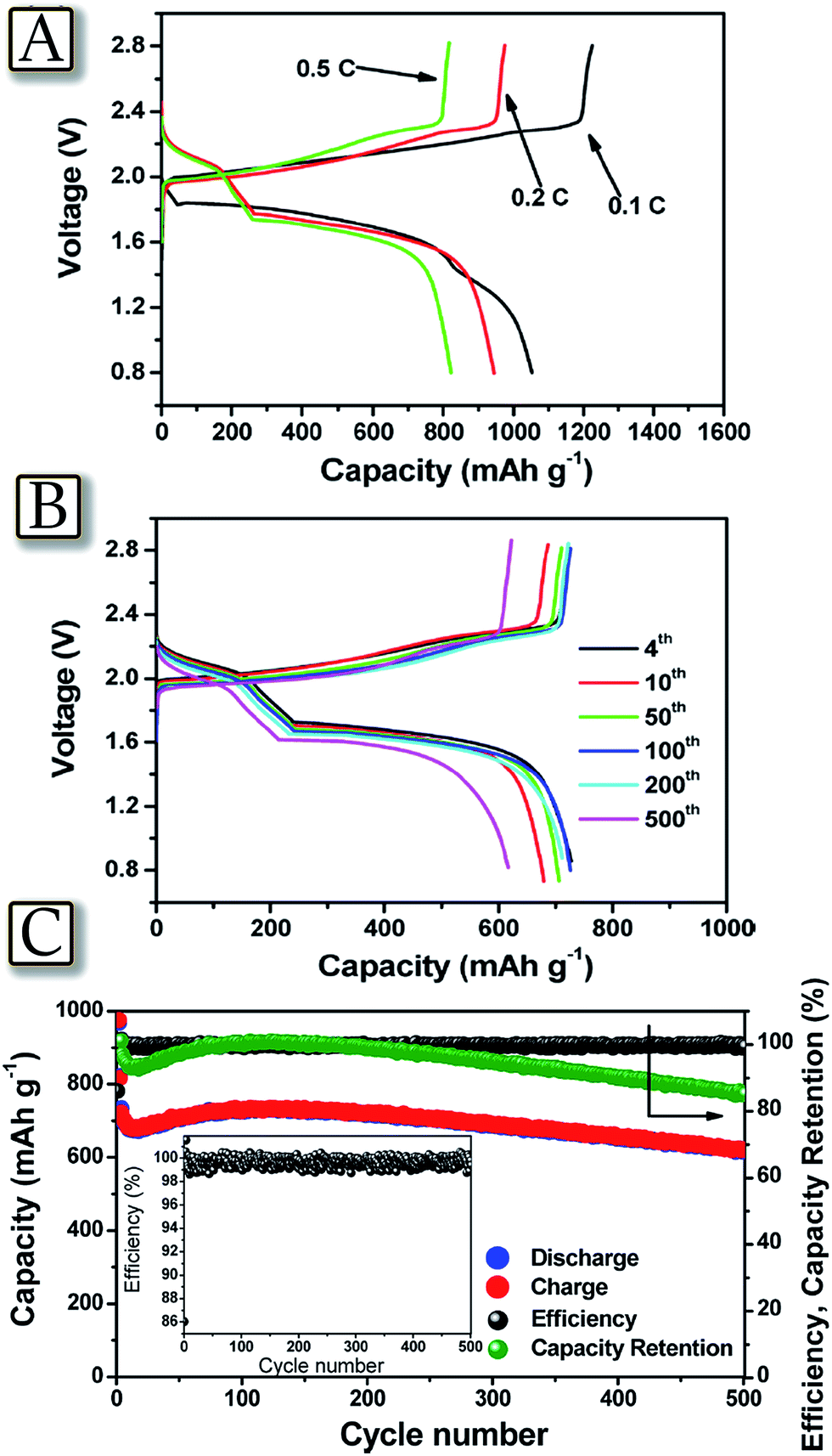
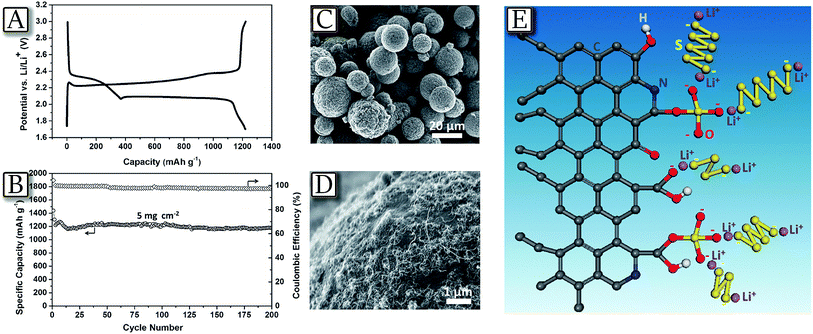
A last step necessary for promoting large scale adoption of the lithium sulfur battery is the increase of battery safety to a level accepted by emerging applications, such as electrified mobility.80 Indeed, lithium–sulfur batteries could become an excellent alternative to lithium-ion batteries, due to their higher energy density and lower production cost.81Fig. 982 represents an example of a highly reliable lithium–sulfur battery with performances comparable to that of lithium-ion batteries, using a reversible dual type sulfur cathode (solid sulfur electrode and polysulfide catholyte) and a lithiated Si/SiOx nanosphere anode.82 The lithium-ion sulfur cell shows high specific capacity, excellent charge–discharge efficiency, and remarkable cycle life. These promising attributes may arise from a synergistic effect of the enhanced electrochemical performance of the newly designed anode and the optimized layout of the cathode. Both the cathodic and anodic half cells demonstrated excellent performances in terms of reversible capacity, cycle life, and cycling efficiency over repeated cycles. The full cell adopting this couple showed a capacity of 750 mA h g−1 at a C-rate of as high as 1C (1675 mA g−1) with an average working voltage of 1.8 V and maintained more than 85% of its initial capacity after 500 cycles. This cell configuration, when coupled with the newly designed electrode materials, represents a significant step toward the realization of commercially available lithium–sulfur battery technology.
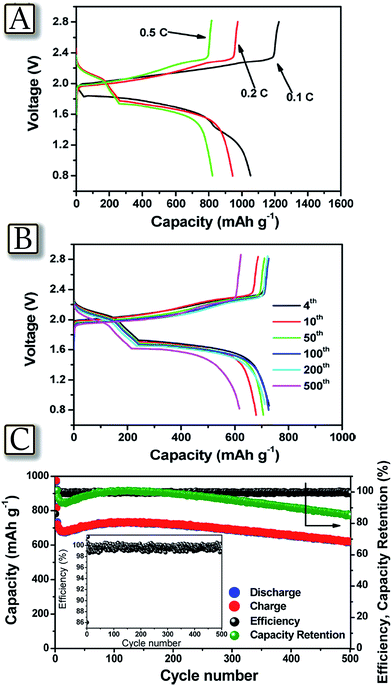 |
| | Fig. 9 (A) Voltage profiles of the full cell adopting the lithiated Si–SiOx nanosphere/DME/DOL (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), Li2S8 (0.05 M), LiTFSI (1 M), LiNO3 (0.4 M)/AC-S structure and including a GDL current collector, cycled at 0.1C (black curve), 0.2C (red curve), and 0.5C (green curve) rates. (B) Voltage profiles and (C) cyclic responses of the lithiated Si–SiOx/DME/DOL (1:1 v/v), Li2S8 (0.05 M), LiTFSI (1 M), and LiNO3 (0.4 M)/AC-S full cell cycled at 1C rate. The upper and lower voltage limits are, respectively, 2.8 and 0.8 V. 1C = 1675 mA h g−1versus overall sulfur weight; temperature, 30 °C. Inset: magnification of coulombic efficiency. Reproduced with permission.82 Copyright American Chemical Society 2015. :1 v/v), Li2S8 (0.05 M), LiTFSI (1 M), LiNO3 (0.4 M)/AC-S structure and including a GDL current collector, cycled at 0.1C (black curve), 0.2C (red curve), and 0.5C (green curve) rates. (B) Voltage profiles and (C) cyclic responses of the lithiated Si–SiOx/DME/DOL (1:1 v/v), Li2S8 (0.05 M), LiTFSI (1 M), and LiNO3 (0.4 M)/AC-S full cell cycled at 1C rate. The upper and lower voltage limits are, respectively, 2.8 and 0.8 V. 1C = 1675 mA h g−1versus overall sulfur weight; temperature, 30 °C. Inset: magnification of coulombic efficiency. Reproduced with permission.82 Copyright American Chemical Society 2015. | |
3 Lithium–oxygen battery
3.1 The electrochemical process
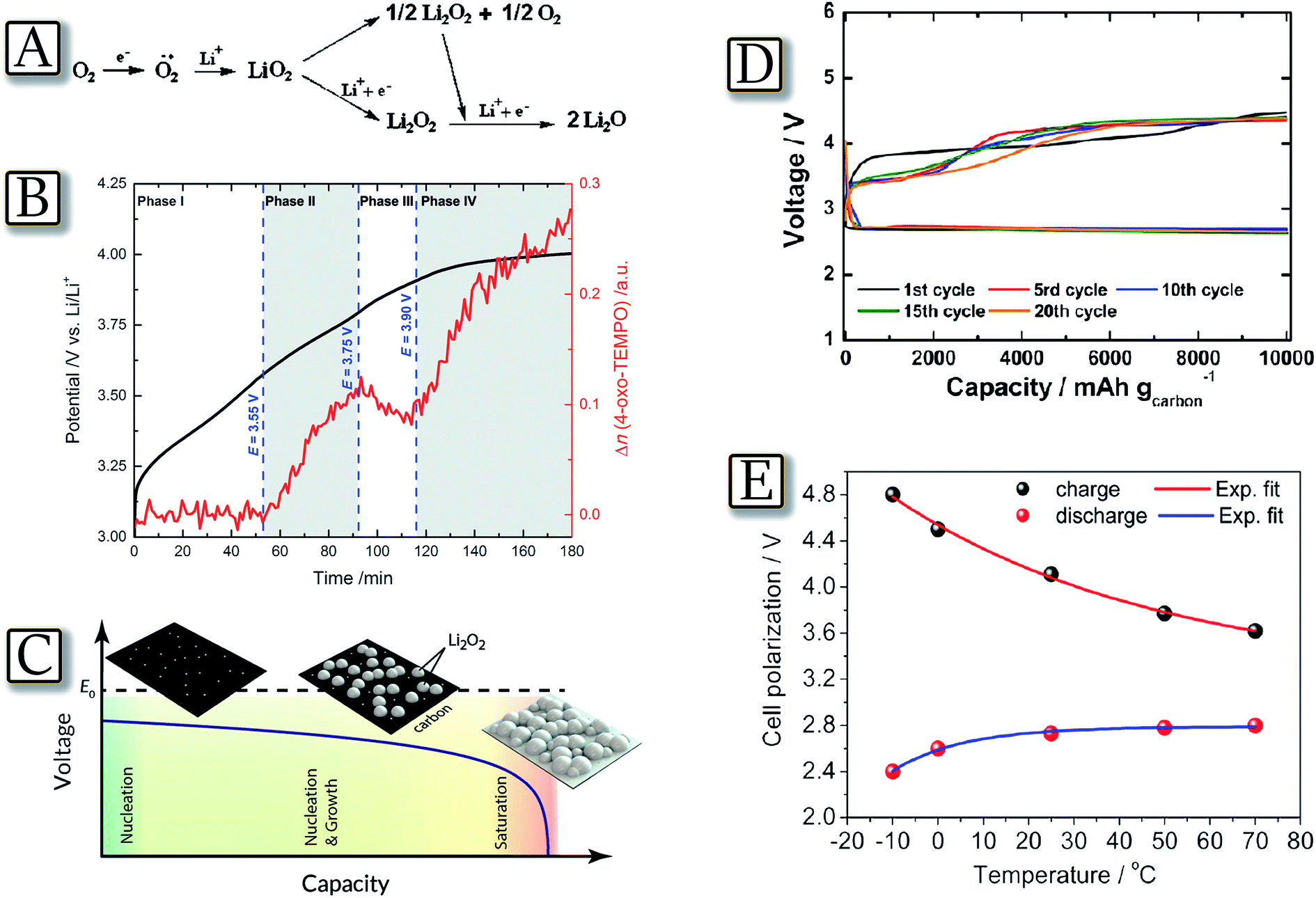
The lithium oxygen electrochemical process evolves at about 3 V through the formation of superoxide radical intermediate species and terminates by formation of lithium peroxide or lithium oxide, leading to a specific capacity higher than 3860 mA h g−1 and, theoretically, to an energy density as high as 11620 W h kg−1.23 However, the formation of nucleophile and highly reactive radicals during the process prevents the utilization of a large fraction of the typical electrolyte solutions, in particular those based on carbonates, i.e., electrophile species which react with the intermediate radicals, decompose and completely hinder the reversibility of the Li/O2 electrochemical process.83Fig. 10(A)84 illustrates the scheme of the process studied in a solvent free, solid-state cell using a poly(ethylene oxide)–lithium triflate (PEO–LiCF3SO3) electrolyte.84 This cell configuration totally avoids side processes of the electrochemical reaction products (particularly of the singlet oxygen) with the polymer electrolyte. The PEO-based electrolyte was chosen as the chemical resistance of the PEO ether linkage is well-documented (e.g. Grignard reagents) and LiCF3SO3 is stable towards nucleophiles, especially under the low current densities. PEO, owing to the tendency of the polymer to arrange into trans–gauche–trans helices to accommodate ions, is far more solvating in the solid state than even its shorter liquid homologues, such as the recently used tetraglyme.22 Therefore, solution and polymers based on ether linkage, such as PEO and glymes, may be considered the most suitable media for application as the electrolyte in the lithium–oxygen cell.85,86 The radicals formed during the lithium–oxygen cell charge process were observed also through a reaction with a spin-trap by time and voltage resolved operando EPR spectroscopy, in a purpose-built spectroelectrochemical cell.87 The study, summarized by Fig. 10(B),87 demonstrates that during the charge process a singlet oxygen (1Dg) is formed upon Li2O2 oxidation at potentials above 3.5 V with a lower limit of approximately 0.5% of the evolved oxygen. The occurrence of highly reactive singlet oxygen might be the long-overlooked missing link in the understanding of the electrolyte degradation and carbon corrosion reactions that occur during the charging of Li/O2 cells. Besides the electrolyte, another factor which may influence the lithium–oxygen cell performance is the morphology of the Li2O2 formed at the electrode surface. The relationship between Li2O2 morphology and the electrochemical performance of the Li/O2 battery is shown in Fig. 10(C)88 by accurately describing and capturing experimental Li/O2 battery discharge curves by a theoretical model in which electrode performance is limited by the nucleation and growth of discrete Li2O2 nanostructures in the cathode.88 The study, using a combination of the atomistic nucleation theory and Kolmogorov's phase transformation theory, shows that the characteristic sharp voltage-drop at the end of the discharge curve is caused by an increased kinetic over-potential due to Li2O2 covering up the available surface area for LiO2 intermediate formation. This view is consistent with the current understanding of the Li2O2 formation mechanism as determined by experiments. Preventing excessive surface nucleation is therefore highlighted as a core strategy for increasing Li/O2 battery capacity. Therefore, the amount and morphology of Li2O2 species should be carefully monitored, e.g., by time-controlling the capacity of the lithium oxygen cell as indeed reported by Fig. 10(D).89 The figure shows that a lithium oxygen cell can reversibly operate in a tetraethylene glycol dimethyl ether–lithium triflate (TEGDME–LiCF3SO3) electrolyte by limiting the capacity to 10000 mA h g−1 as referred to the carbon at the electrode.89 Further correlations may be found by taking into account the lithium oxygen cell polarization, Li2O2 morphology, crystallinity and the temperature, in particular considering the effects of the temperature on oxygen solubility and electrolyte viscosity.90 An example of possible correlation is given by Fig. 10(E)91 which reports a Li/TEGDME–LiCF3SO3/O2 cell operating at temperature ranging from −10 to 70 °C.91 The study reveals that the electrochemical process rate decreases at low temperature due to a limited diffusion of the lithium ions from the electrolyte to the electrode interface, while it enhances at high temperature due to the decreased electrolyte viscosity and consequent increased oxygen mobility. The study shows that the temperature also influences the crystallinity and morphology of lithium peroxide, namely of the product formed during cell discharge.91
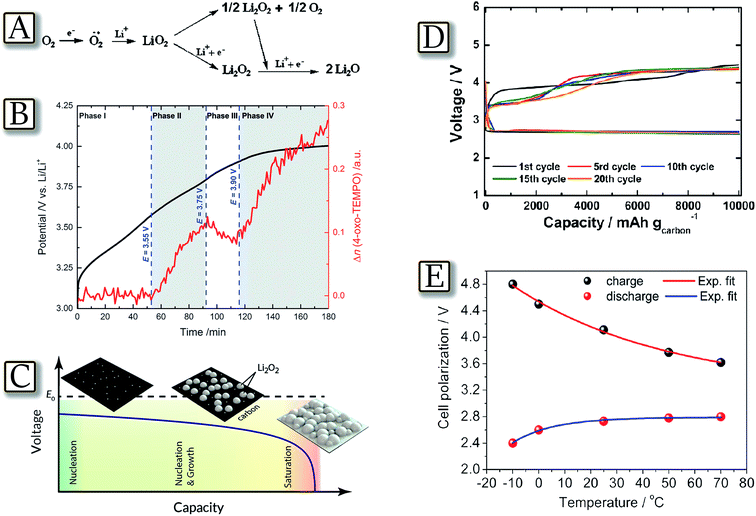 |
| | Fig. 10 (A) Scheme of the possible oxygen-molecule reduction in aprotic systems. Reproduced with permission.84 Copyright Wiley 2011. (B) Voltage profile and EPR signal amplitude for 4-oxo-TEMPO formed during the charging (i = 60 mA gC−1) of Li2O2 with 0.5 M LiTFSI in diglyme containing 0.1 M 4-oxo-TEMP as a spin trap. Reproduced with permission.87 Copyright Wiley 2016. (C) Cathode surface coverage scheme compared with cell voltage as a function of capacity discharged. Reproduced with permission.88 Copyright American Chemical Society 2015. (D) Discharge–charge cycles of a lithium–oxygen cell in TEGDME–LiCF3SO3 at room temperature. Galvanostatic, time-controlled mode. Current: 500 mA gcarbon−1 (0.5 mA cm−2 current, 1 mg cm−2 carbon loading). Time: 20 h. Cycling capacity: 10000 mA h gcarbon−1. Reproduced with permission.89 Copyright American Chemical Society 2012. (E) Cell voltage vs. temperature trend of the lithium oxygen battery in TEGDME–LiCF3SO3, capacity limited to 1000 mA h g−1 carbon. Current: 100 mA gcarbon−1. Reproduced with permission.91 Copyright American Chemical Society 2013. | |
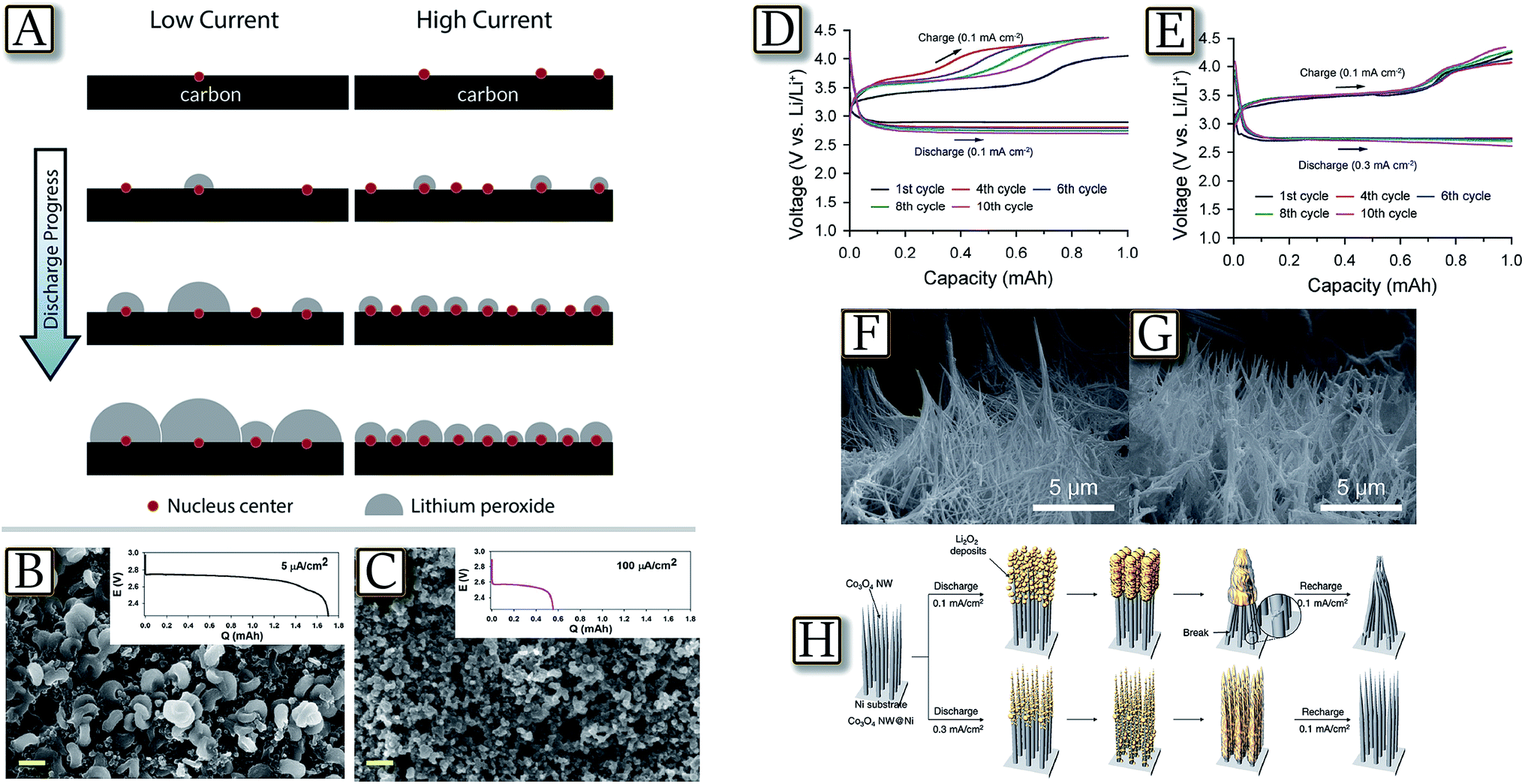
3.2 More light on morphological aspect: Li2O2 deposition
An important relationship between Li2O2 morphology and the electrochemical conditions of Li/O2 operation is demonstrated by theoretical calculation as summarized in Fig. 11(A).88 The figure reveals that increasing discharge current in a Li/O2 cell leads to an increase of the ratio of nucleation centers on which the Li2O2 starts depositing. At a fixed amount of deposited Li2O2, this phenomenon causes a larger number of nucleation sites at higher current and, therefore, smaller particle size.88 This aspect is experimentally confirmed by diffraction, FESEM and STEM study in Fig. 11(B and C).92 The measurements show that lower current densities favor aggregation of lithium peroxide nano-crystallites nucleated via solution dismutase on the surface of the electrode, whereas fast rates deposit quasi-amorphous thin films. The latter provide a lower overpotential on charge due to their nature and close contact with the conductive electrode surface, albeit at the expense of lower discharge capacity. This result indicates that the higher active surface area (of non-carbonaceous materials) and improved electronic conductivity of the oxygen electrode will enable better rate capabilities and support peroxide film formation. In line with the results reported above, Fig. 11(D–H)93 demonstrate that the use of a carbon free and binder-free cathode for Li/O2 batteries based on vertically grown Co3O4 nanowire (NW) arrays on Ni-foam provides high capacity with a reduced overvoltage.93 Interestingly, the figure shows that the charge voltage profile changes with the discharge rate, which is associated with the variation in the phase and local distribution of the discharge products. At a low discharge rate, a morphology resembling the pointed tip of a brush is observed, thus indicating that the crystalline discharge products are concentrated on the skin of the cathode and deform the NW array. At a high discharge rate, a more uniform distribution of the quasi-amorphous discharge products is favored, resulting in a relatively stable voltage profile with cycling. This is demonstrated by the FE-SEM image of a Co3O4 NW@Ni-foam air cathode after charging at discharge–charge rates of 0.1/0.1 mA cm−2 and 0.3/0.1 mA cm−2, and by the corresponding schematic illustration.93 These findings suggest the importance of the electrode structure and discharge product distribution during the design of carbon- and binder-free cathodes.
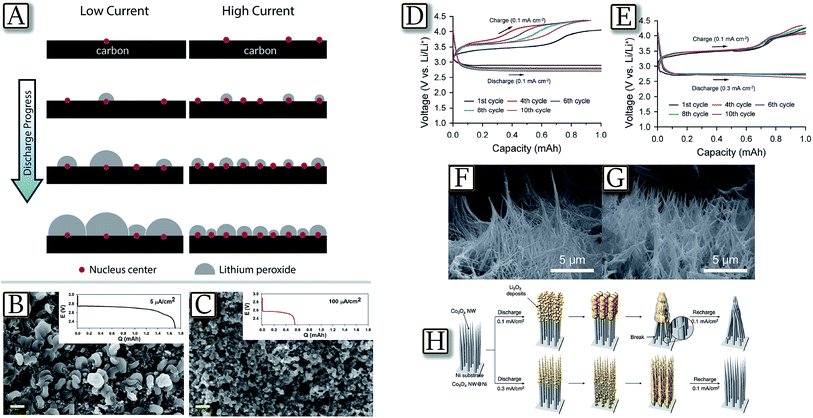 |
| | Fig. 11 (A) Illustration of the model, showing the effect of current density on the nucleation rate and the final amount of Li2O2 deposited. Reproduced with permission.88 Copyright American Chemical Society 2015. FESEM images at a magnification of 20000× after full discharge at (B) 5 μA cm−2 (C) 100 μA cm−2, with the corresponding discharge curves. Scale bar = 400 nm. Reproduced with permission.92 Copyright Royal Society of Chemistry 2013. The discharge–charge voltage profiles of the Co3O4 NW@Ni-foam air cathode at the first, fourth, sixth, eighth, and tenth cycle for (D) the cell at a discharge–charge rate of 0.1/0.1 mA cm−2 and for (E) the cell at a discharge–charge rate of 0.3/0.1 mA cm−2, FE-SEM image of a Co3O4 NW@Ni-foam air cathode after charging at discharge–charge rates of (F) 0.1/0.1 mA cm−2 and (G) 0.3/0.1 mA cm−2. (H) A schematic illustration of the structural evolution of the Co3O4 NW array with low and high discharge rates. Reproduced with permission.93 Copyright Royal Society of Chemistry 2014. | |
3.3 Catalysts for the Li/O2 electrochemical reaction
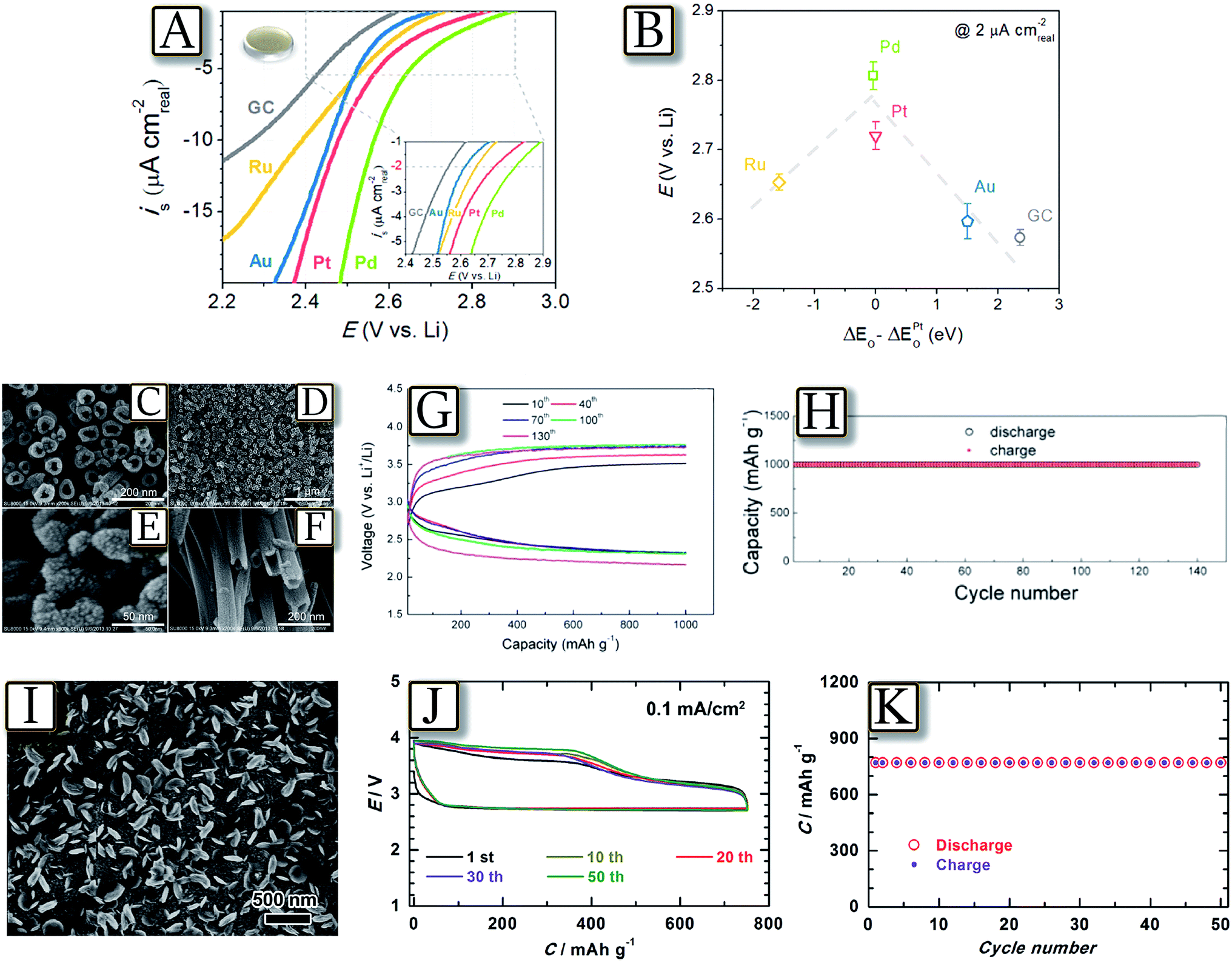
The lithium oxygen cell, due to a three-side interface, including gas/solid (oxygen/gas diffusion layer) and solid/liquid (electrode/electrolyte) interface, exhibits some similarity to the hydrogen fuel cell based on a Nafion membrane.94 Fuel cells require a noble metal catalyst at the electrode side in order to enable the hydrogen/oxygen electrochemical process;95–97 this similarity attracted attention to this class of catalyst for application in lithium oxygen cells.98,99Fig. 12(A and B)100 report the intrinsic oxygen reduction reaction (ORR) activity of polycrystalline palladium, platinum, ruthenium, gold, and glassy carbon surfaces in 0.1 M LiClO4 1,2-dimethoxyethane via rotating disk electrode measurements.100 The ORR activity of these surfaces primarily correlates with oxygen adsorption energy, forming a “volcano-type” trend. Accordingly, the ORR activity trend was found in the order of Pd > Pt > Ru ≈ Au > GC on bulk surfaces. However, most of the noble metal catalysts show a specific activity, i.e., during the oxygen reduction reaction (ORR) or oxygen evolution reaction (OER), while a more adequate catalyst suitable for both the processes should be ad hoc designed by combining various noble metals.101 These difficulties, as well as problems associated with the high cost of a noble metal catalyst, have driven increasing interest on new classes of materials, including a combination of noble and non-noble metals, metal oxides and other materials.102 An example of recent development is represented by Fig. 12(C–H)103 which reports a TiO2 nanotube array growing on Ti foam which is used as the catalyst support to construct carbon and binder free oxygen diffusion electrodes.103 After being coated with Pt nanoparticles by a cool sputtering approach, the TiO2 nanotube arrays are used as cathodes of the Li/O2 battery employing a tetraethylene glycol dimethyl ether electrolyte (TEGDME)-based electrolyte. Benefiting from the stability of TiO2 in the discharge/charge processes, the Li/O2 battery revealed enhanced cyclability at high current densities (for instance, more than 140 cycles at 1 or 5 A g−1), within wide discharge/charge voltage windows (for instance, 1.5–4.5 V). X-ray photoelectron spectra and scanning electron microscopy images of the cathodes after cycling at 5 A g−1 150 times indicated that the TiO2 nanotubes can remain stable in the long term cycle test, while 1H nuclear magnetic resonance analysis revealed that the TEGDME-based electrolyte suffers no degradation. Another alternative catalyst is represented by Fig. 12(I–K)104 which reports Sb-doped tin oxide (STO, ca. 6 nm in size) supported Ru nanoparticles (Ru/STO), applied as a carbon-free cathode in a lithium–oxygen battery delivering a specific capacity of 750 mA h g−1 with low discharge/charge over-potentials.104 The relevant performance of the Li/O2 battery with Ru/STO has been attributed to the improved conductivity and interfacial contact between the STO nanoparticles, and the enhanced ORR and OER catalytic activities related to the addition of Ru nanoparticles.
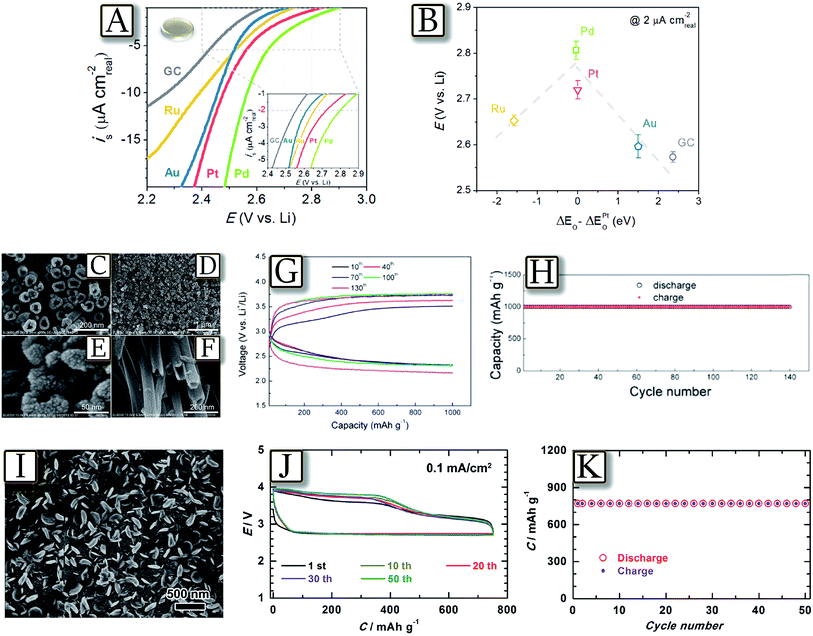 |
| | Fig. 12 (A) Background- and IR-corrected specific ORR polarization curves of polycrystalline Pd, Pt, Ru, Au, and GC surfaces in O2-saturated 0.1 M LiClO4 in DME at 100 rpm and 20 mV s−1. (B) ORR potentials at 2 μA cm2 as a function of calculated oxygen adsorption energy, ΔEO (per oxygen atom relative to an atom in the gas phase). The oxygen adsorption energy on GC is estimated from the oxygen adsorption energy on graphite. Error bars represent standard deviations of at least three independent measurements. Reproduced with permission.100 Copyright American Chemical Society 2011. SEM images of Pt modified TiO2 nanotube arrays prepared by anodic oxidation of Ti foam and subsequent cool sputtering: top views (C–E), side view (F). The electrochemical characteristics of the Li–O2 battery with Pt/TNT as a cathode: (G) discharge/charge curves of the 10th, 40th, 70th, 100th, and 130th cycles at 1 A g(Pt)−1 and (H) cyclability at a current density of 1 A g(Pt)−1 with a limited capacity of 1000 mA h g(Pt)−1. Reproduced with permission.103 Copyright American Chemical Society 2014. (I) SEM images of the cathode Ru/STO at the discharge state, (J) 50 discharge–charge cycles of the Li–O2 Ru/STO battery in 0.5 M LiClO4–TEGDME at 0.1 mA cm−2 with a capacity cut-off value of 750 mA h g−1 and (K) the discharge and charge capacity against the cycle number in. Reproduced with permission.104 Copyright Wiley 2014. | |
3.4 Enhanced carbons and catalyst-modified carbons: high performance lithium oxygen battery
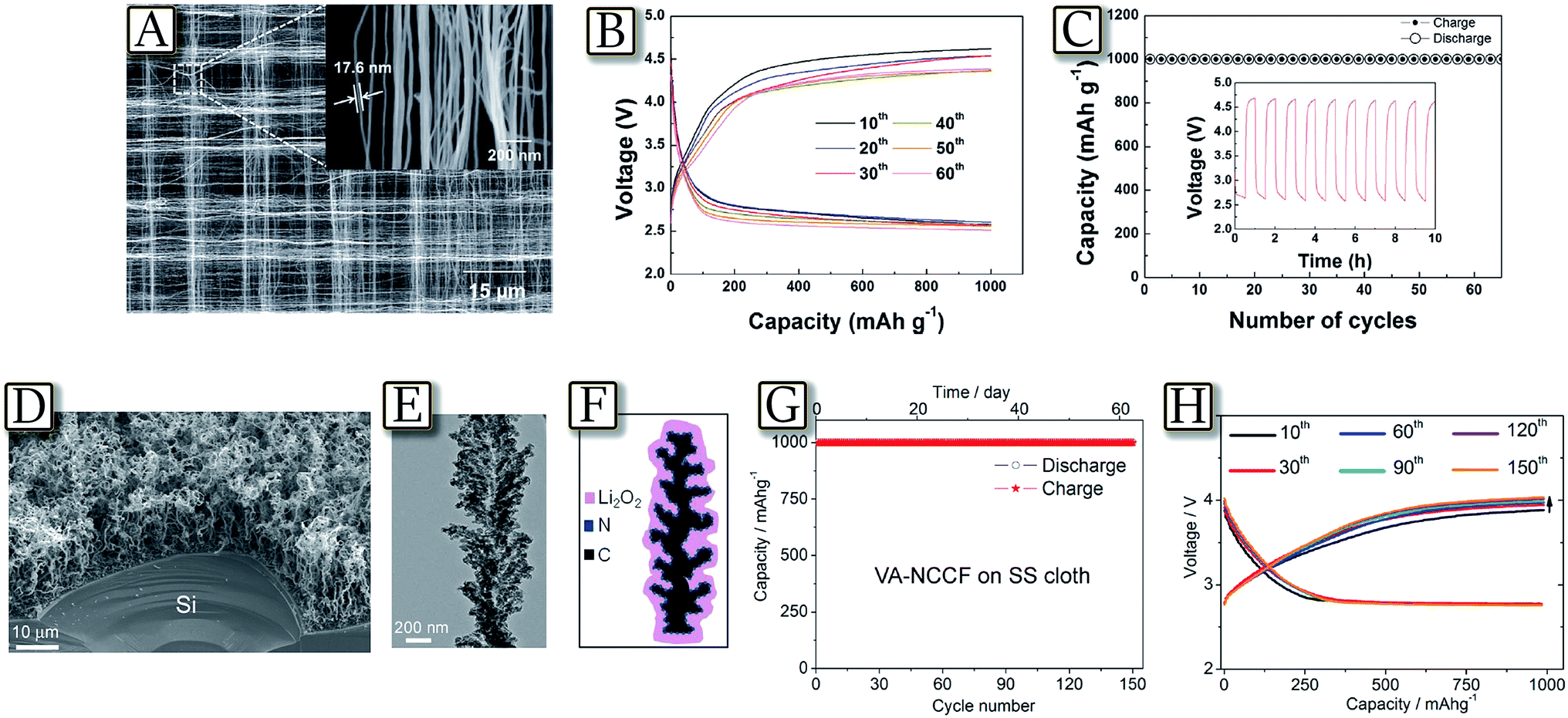
Several recent studies well demonstrated the crucial role of the carbon, used as the support for the Li/O2 electrochemical process and generally coated on a gas diffusion layer (GDL).105 Carbons have been indicated to control the morphology of the formed products and the kinetics of the OER/ORR reactions, thus determining the lithium–oxygen cell polarization, the rate capability, the delivered capacity and the cycle life.106 Among the interesting examples, Fig. 13(A–C)107 demonstrate that hierarchical porous electrodes comprising well-aligned CNT fibrils significantly enhance the cycle stability and rate capability of the Li/O2 battery.107 The controlled porous framework of these woven CNT electrodes enables effective formation/decomposition of lithium peroxide by providing facile accessibility of oxygen to the inner side of the air electrode and preventing the clogging of pores by the discharge product, even during the deep discharge. The catalyst-free porous morphology of the air electrode allowed a uniform deposition of the discharge products on the individual CNTs and CNT bundles and reduced pore clogging leading to high current density (2 A g−1). Another relevant configuration is reported by Fig. 13(D–H)108 in which an electrode based on a vertically aligned nitrogen-doped coral-like carbon nanofiber (VA-NCCF) array supported by stainless steel cloth is used in a nonaqueous Li/O2 battery, with an energy efficiency as high as 90% and a narrow voltage gap of 0.3 V between discharge/charge plateaus.108 The observed performance has been attributed to the unique electrode morphology, the high catalytic activity of the N-doped carbon and to an optimized oxygen/electron transportation capability, coupled with the microporous stainless steel substrate. The vertically aligned coral-like fiber structure and the highly conductive microporous SS cloth support, with a minimized contact resistance, have provided a large free space for efficient Li2O2 deposition and enhanced the electron/electrolyte/reactant transport. Furthermore, the electrolyte decomposition was minimized due to the low overpotential, and the battery operated for more than 150 cycles with a good reversibility under considerably high specific capacities (up to 1000 mA h g−1) per cycle.108
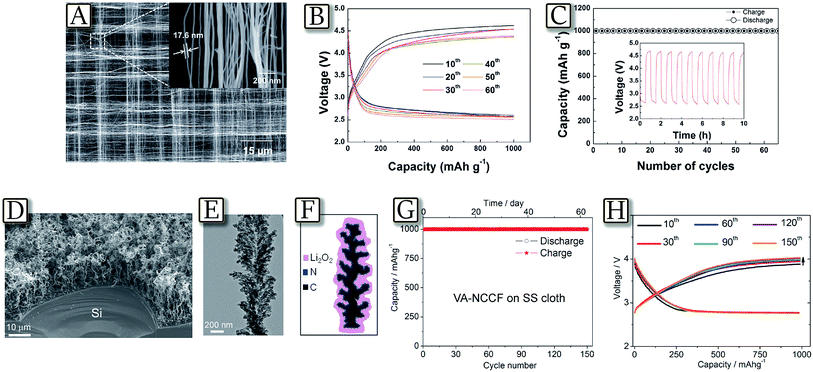 |
| | Fig. 13 (A) SEM images of the CNT fibril at high magnification (inset: large area image of the air electrode), (B) discharge/charge profiles, and (C) cyclability of Li–O2 cells using an air electrode based on a woven CNT at a current rate of 2000 mA g−1 (inset: voltage vs. time graph of the initial 10 cycles). Reproduced with permission.107 Copyright Wiley 2013. (D) SEM image of a VA-NCCF array grown on a piece of Si wafer by CVD. (E) TEM image of an individual VANCCF. (F) The sketch of Li2O2 grown on a coral-like carbon fiber, which has an advantage to tightly hold the Li2O2 deposit by the rugged surface, and (G) cycling performance of the VA-NCCF array on the SS cloth substrate. (H) The representative discharge–charge curves from the 10th to 150th cycle for the VA-NCCF electrode. Current density was 500 mA g−1, and the controlled capacity was 1000 mA h g−1. Reproduced with permission.108 Copyright American Chemical Society 2014. | |
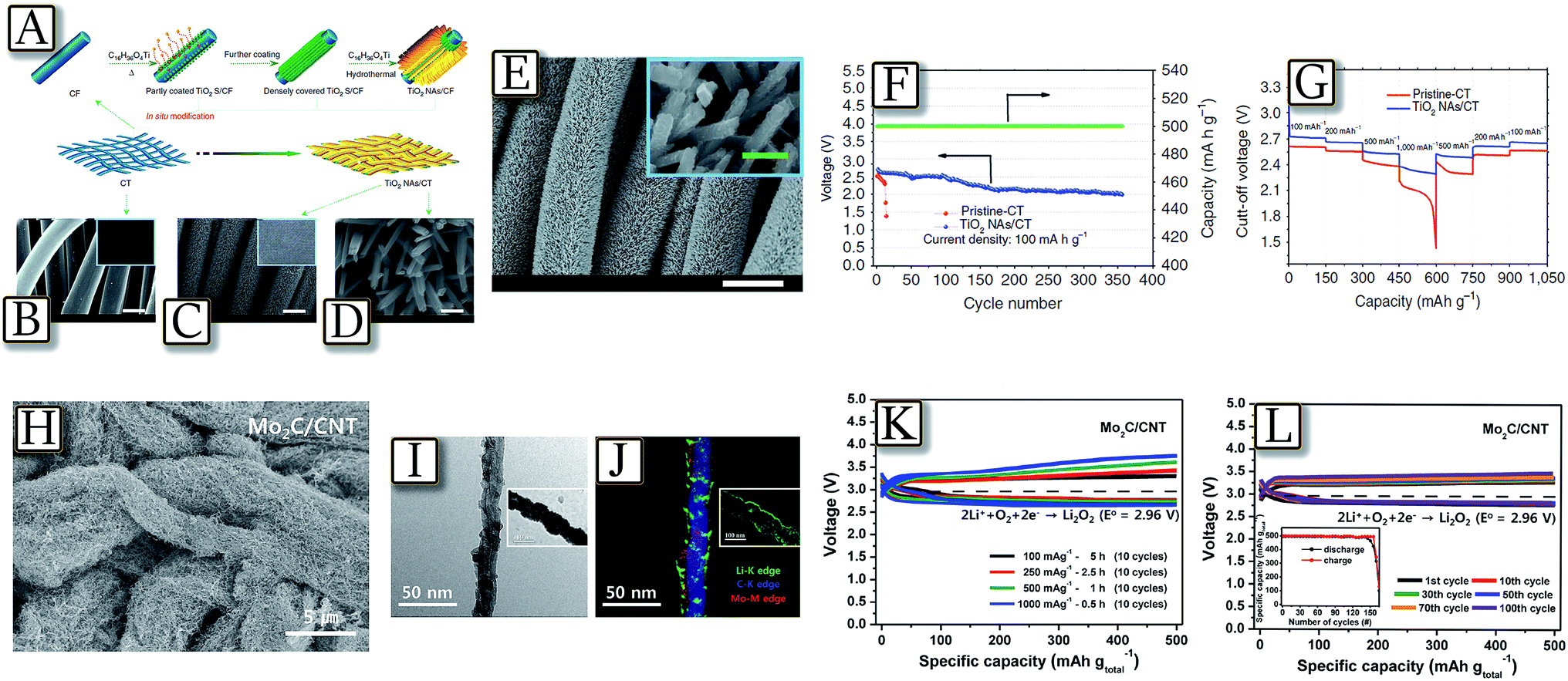
Combination of carbons of enhanced morphology, and new-generation catalysts designed ad hoc for the lithium–oxygen electrochemical process109 has led to batteries of remarkably enhanced performances such as that reported by Fig. 14(A–G).110 The figure shows a Li/O2 battery with technologically relevant electrochemical performances, even under stringent bending/twisting conditions and extended cycle life, fabricated using unique TiO2 nanowire arrays grown onto carbon textiles (NAs/CT) as a free-standing cathode.110 These enhanced characteristics have been attributed to the tailored properties of the TiO2 NAs/CT cathode, which include high mechanical and chemical stability as well as relevant catalytic activity. Remarkably, Fig. 14(H–L)111 present a nanostructured cathode based on molybdenum carbide nanoparticles (Mo2C) dispersed on carbon nanotubes, in a lithium–oxygen battery with energy efficiency increasing up to 88%, and a cycle life of more than 100 cycles.111 Mo2C nanoparticle catalysts are shown to promote the formation of well-dispersed lithium peroxide nano-layers (Li2O2) on the Mo2C/carbon nanotubes with a large contact area during the oxygen reduction reaction (ORR). This Li2O2 structure decomposes at low potential upon the oxygen evolution reaction (OER) by avoiding the energy loss associated with the decomposition of the more common Li2O2 morphology. The Li/O2 battery using the Mo2C/carbon nanotube electrode was cycled with low charge potential (3.25–3.4 V) over 100 cycles.111
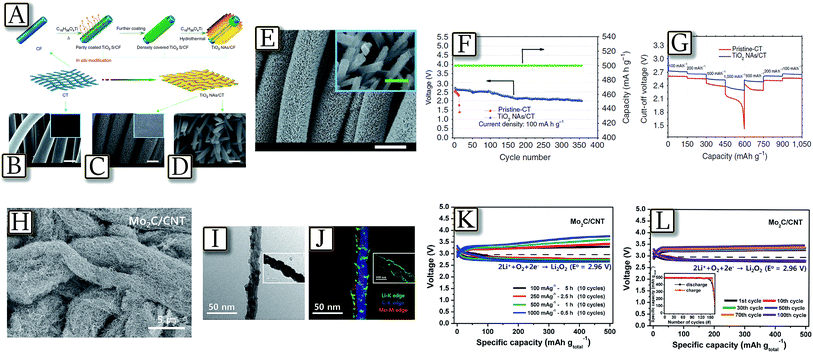 |
| | Fig. 14 Scheme for the fabrication and structure of the TiO2 NAs/CT cathode. (A) Schematic representations for the design and preparation of the TiO2 NAs/CT. (B) Scanning electron microscopy image and photograph (inset) of pristine–CT (scale bar, 10 μm). (C) SEM image and photograph (inset) of the obtained TiO2 NAs/CT cathode (scale bar, 10 μm). (D) Enlarged image of (C) with 500 nm scale bars; (E) SEM images of the discharged TiO2 NAs/CT cathode with a current density of 100 mA g−1 (white scale bar, 10 μm, green scale bars: 500 nm). (F) Voltage versus cycle number on the discharge terminal of the Li–O2 cell with a TiO2 NAs/CT cathode and (G) the rate capability of the Li–O2 cells with the two types of cathodes at different current densities. Reproduced with permission.110 Copyright Nature Publishing Group 2015. (H) SEM images of the Mo2C/carbon nanotube, (I) HR-TEM image and (J) EF-TEM maps of the Mo2C/carbon nanotube electrode after the first discharge (discharge depth is 1000 mA h gtotal−1). A fully discharged Mo2C/carbon nanotube electrode to 2.5 V is also presented in the inset data (length of the scale bar in inset data is 100 nm). (K) The first 10 cycles of Li–O2 batteries with the Mo2C/carbon nanotube electrode at current densities of 100, 200, 500, and 1000 mA gtotal−1 with a discharge capacity of 500 mA h gtotal−1 and (L) cycling performance of the Li–O2 battery with the Mo2C/carbon nanotube electrode at a discharge capacity of 500 mA h gtotal−1 and at a current density of 100 mA h gtotal−1. Reproduced with permission.111 Copyright American Chemical Society 2015. | |
3.5 Alternative low polarization lithium–oxygen batteries: redox mediators and ionic liquids
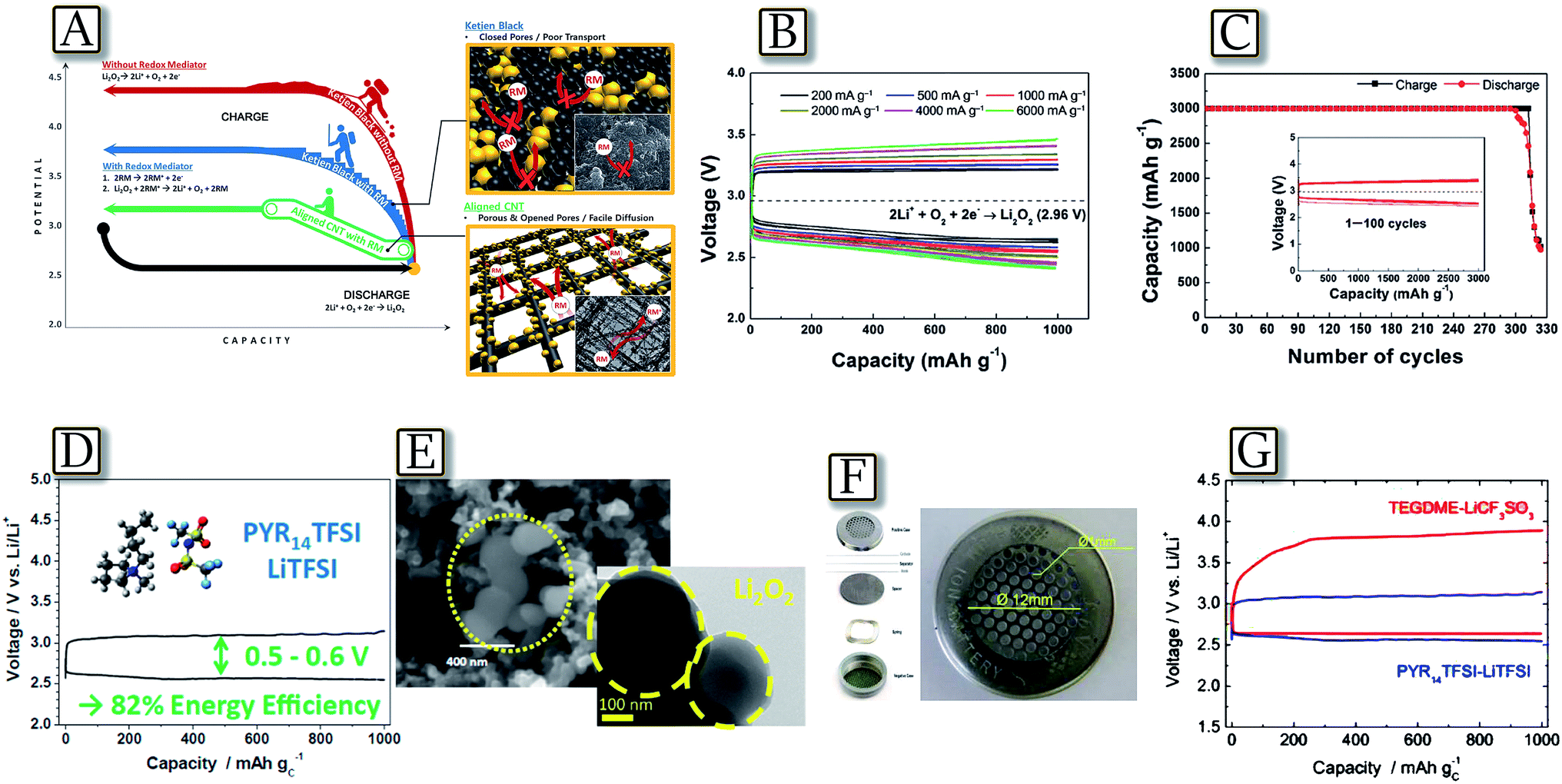
Besides electrode improvements, alternative strategies have been proposed to enhance the lithium–oxygen battery performances, in particular in terms of cell polarization, i.e., energy density and cycle life.112 These novel approaches involved the electrode nature, chemical composition, salts and additives that improved the redox process.113–115Fig. 15(A–C)116 report a suitable lithium–oxygen cell with high reversibility and good energy efficiency using a soluble catalyst combined with a hierarchical nanoporous electrode.116 Through the porous three-dimensional network of the air electrode, not only lithium ions and oxygen but also soluble catalysts can be rapidly transported, enabling ultra-efficient electrode reactions and significantly enhanced catalytic activity. The battery can deliver a high reversible capacity (3000 mA h g−1) up to 300 cycles with reduced polarization (about 0.25 V). The use of a hierarchically aligned porous carbon electrode and a soluble LiI redox mediator provides effectively rapid channels for both the reaction products and the soluble catalyst. The facile diffusion of the soluble catalyst throughout the entire air electrode decreases the polarization during charge, enabling enhanced rechargeability and energy efficiency.116 A further intriguing example is given by the lithium–oxygen battery exploiting a PYR14TFSI–LiTFSI ionic liquid electrolyte, reported in Fig. 15(D–G).117 The study, involving electrochemical impedance spectroscopy, capacity-limited cycling, field emission scanning electron microscopy, high-resolution transmission electron microscopy, and X-ray photoelectron spectroscopy, demonstrates that this Li/O2 cell is characterized by a stable electrode–electrolyte interface and a highly reversible charge–discharge cycling behavior. Most remarkably, the study revealed a charge process (oxygen oxidation reaction) characterized by a very low overvoltage, enhancing the energy efficiency up to 82%, thus, addressing one of the most critical issues preventing the practical application of lithium–oxygen batteries. The favorable performance of the battery and particularly its capability of cycling with a low charging over-potential were directly linked to the transport properties of the IL-electrolyte medium. The discharge (ORR) process in the PYR14TFSI–LiTFSI electrolyte has led to lithium peroxide particles having a size much smaller than that usually obtained in other media and allowed a facile reconversion in the charge (OER) process. Furthermore, the high thermal stability of the IL-based electrolyte was supposed to offer the additional bonus of enhancing the safety of the battery.117
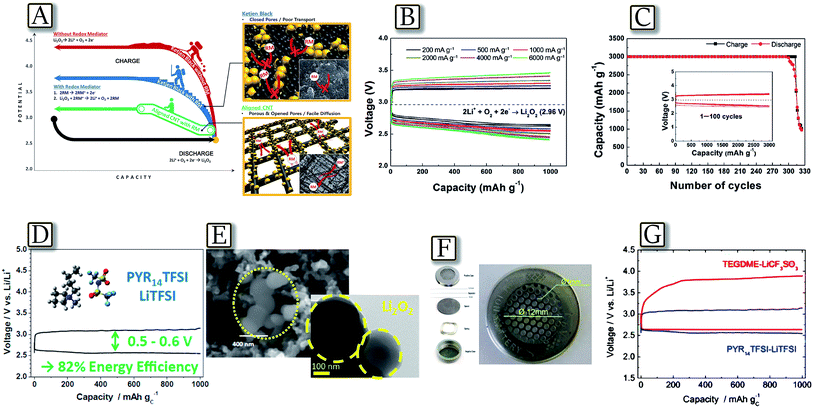 |
| | Fig. 15 (A) Schematic illustration of the role of the redox mediator (RM) in a Li–O2 battery system made using a hierarchical CNT fibril electrode, (B) power capability of CNT fibril electrodes in the presence of a LiI catalyst at current rates from 200 mA g−1 to 6000 mA g−1 and (C) cyclability of the CNT fibril electrodes in the presence of a LiI catalyst at a discharge depth of 3000 mA h g−1. Reproduced with permission.116 Copyright Wiley 2014. (D) Voltage profile of the lithium oxygen cell galvanostatically cycled applying a current of 100 mA g−1 and a capacity limited to 1000 mA h g−1 exploiting PYR14TFSI–LiTFSI as the ionic liquid-based electrolyte, (E) FESEM images of the GDL-SP electrode after discharge and higher magnification. (F) Cell configuration and (G) voltage profile comparison with different electrolytes. Reproduced with permission.117 Copyright American Chemical Society 2014. | |
3.6 A new safe and high energy system: the lithium-ion oxygen battery
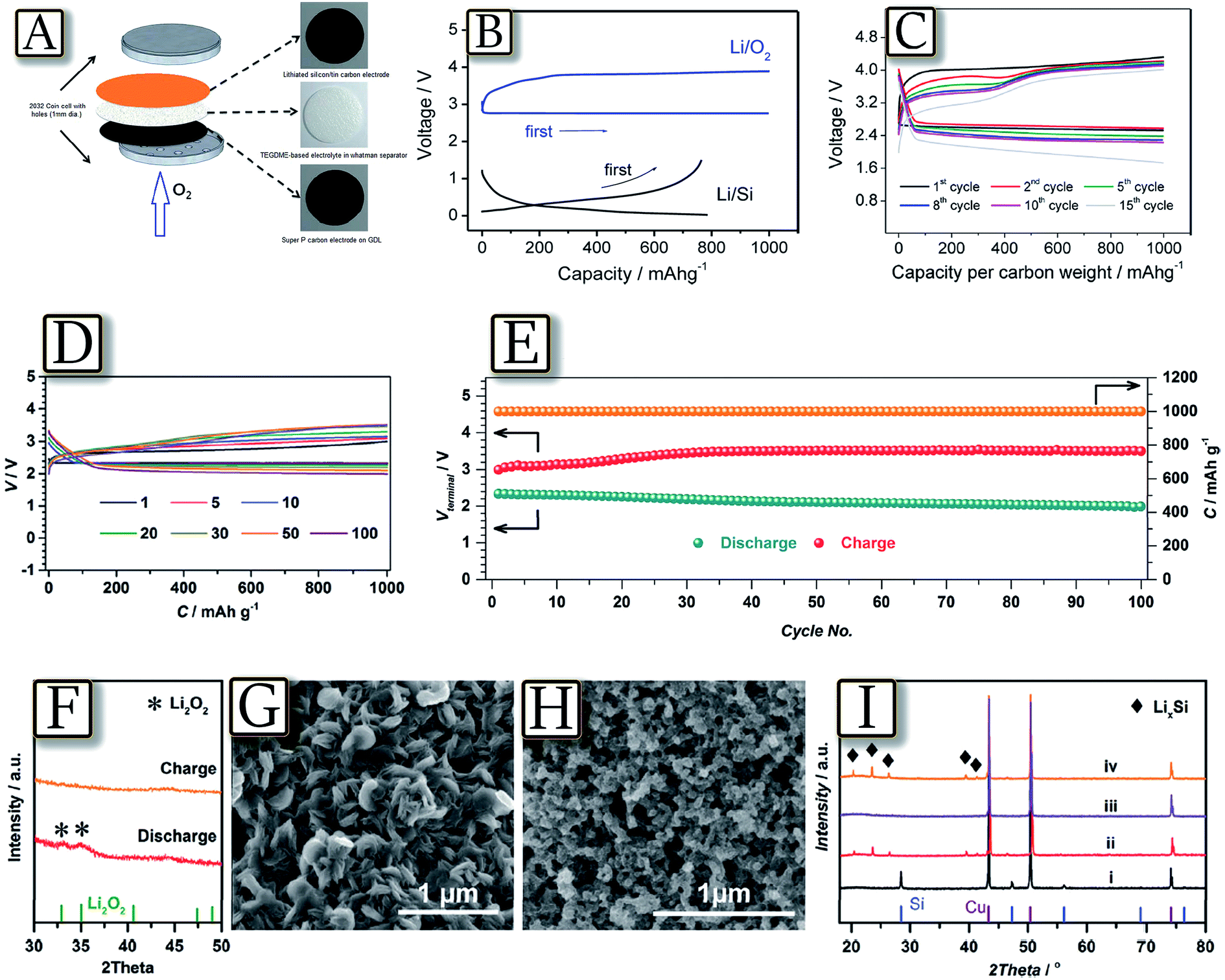
The lithium–oxygen battery is expected to deliver high energy density, well suitable for the growing electric vehicle market.118 However, such a challenging application requires safety targets to which the presence of highly reactive lithium metal may be a serious obstacle.119,120 Attempts to solve this severe issue have been devoted to the use of alternative anodes, characterized by high storage capacity and, at the same time, higher safety content than lithium. Among the suitable materials, lithium alloying compounds, such as tin121 and silicon,122 have been proposed in their nanostructured configuration in carbon matrices, i.e., a configuration almost completely solving the severe issues ascribed to volume variation of the electrode during the electrochemical process and consequent loss of contact and cell performance deterioration.123 Therefore, nanostructured Sn–C and Si–C, in their lithiated configuration,124 successfully served as the lithium-ion reservoir anode in a new configuration of the cell, i.e., the lithium-ion oxygen battery. Fig. 16(A–C)125 report the first attempt of a lithium-ion oxygen battery, where the common lithium metal anode is replaced by a lithiated silicon–carbon composite.125 The results, based on X-ray diffraction and galvanostatic charge–discharge analyses, demonstrate the reversibility of the electrochemical process of the battery that can be promisingly cycled with a rather high specific capacity. The figure shows that the lithium–metal-free/oxygen battery reversibly operates with a discharge voltage of 2.40 V and a capacity of 1000 mA h g−1 referred to the carbon electrode, and shows a profile resulting from the combination of lithium–oxygen and lithium–silicon alloying processes. However, the system reveals a voltage decay by cycling due to the oxygen dissolution within the TEGDME-based electrolyte, permeation and direct reaction with the lithiated anode. The study proposed anode protection by membranes or glassy electrolytes of the anode side as suitable solution to mitigate this issue.125 Another recent example of the efficient lithium-ion oxygen cell based on the silicon anode, achieving 100 discharge/charge cycles with low over-potentials, is proposed by Fig. 16(D–I).126 The composition of the SEI film grown in the glyme-based electrolyte was originally analyzed, and the resulting strong tendency of the unique SEI film to inhibit oxygen crossover was determined as the reason for stable cycling, as also confirmed by the storage experiment. The composition of the optimized SEI film, including organic species, Li2CO3, LiF and specifically, polyfluorocarbon, endowed the Si electrodes with protection against corrosion from O2 crossover side reactions.126 Despite these encouraging results, these alternative systems require longer study, characterization and optimization in order to allow practical application.
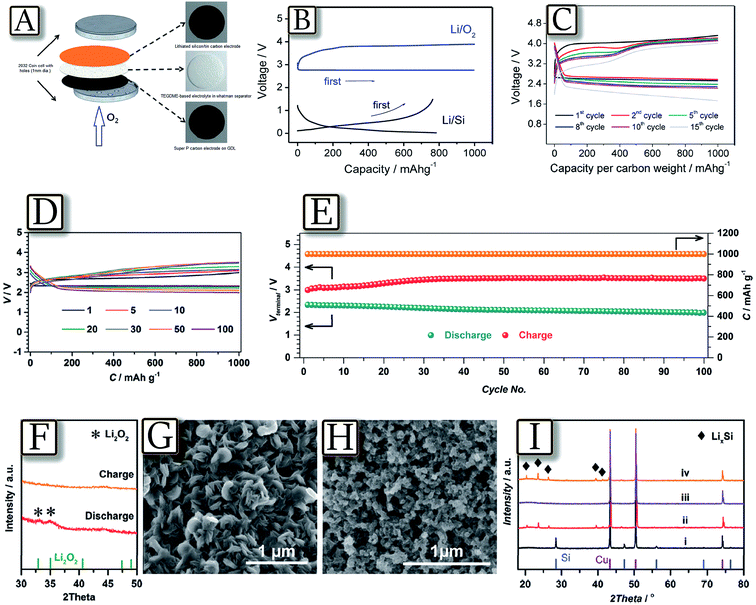 |
| | Fig. 16 (A) Lithium ion cell configuration scheme, the cathode is a large-surface-area Super P carbon that is coated on a gas diffusion layer (GDL), the electrolyte is a solution of LiCF3SO3 in TEGDME (molar ratio of 1:4) adsorbed in a glass fiber separator and the anode is a lithiated silicon–carbon composite. (B) Voltage profiles of the carbon–oxygen electrode (upper part) and the lithiated-silicon–carbon electrode (lower part) in lithium cells galvanostatically cycled at a current of 50 mA g−1. (C) Voltage profiles of the lithiated-silicon/carbon–oxygen cell. Cycling current: 200 mA gcarbon−1. Reproduced with permission.125 Copyright American Chemical Society 2012. (D) The selected discharge–charge profiles of Li ion O2 batteries with F-L-Si anodes. (E) The specific capacities along with discharge and charge terminal voltages against the cycle number of Li ion O2 batteries with F-L-Si anodes. (F–I) Product analysis after discharge and charge of Li ion O2 batteries with F-L-Si anodes. (F) Ex situ XRD analysis of the discharged and charged Ru/KB cathodes. SEM images of the discharged (G) and charged (H) Ru/KB cathodes. (I) Ex situ XRD patterns of the pristine Si electrodes (i), the F-L-Si electrodes (ii), and the F-L-Si anodes in discharged (iii) and recharged (iv) Li-ion O2 batteries. After optimization of the Si anodes, the Li-ion O2 batteries show excellent cycling stability. Reproduced with permission.126 Copyright Royal Society of Chemistry 2016. | |
4 Conclusion
In this review paper, we focused attention on the most recent, high-energy systems based on lithium–sulfur and lithium–oxygen electrochemical processes. Several improvements were observed for both systems during the last few years, mainly concerning the significant increase of the cycle life and the decrease of the cell polarization. The improvements were achieved by controlling the course of the electrochemical reactions which lead to the formation of soluble polysulfide in the lithium sulfur system and reactive intermediate radicals in the lithium oxygen one, as clearly demonstrated by the recent literature. From the Li/S side, the full control of the shuttle reaction by using an electrolyte additive favorable for the formation of a protecting SEI film at the anode side represented the first crucial step.48,50,127–129 Furthermore, composite sulfur electrodes with a large variety of structures and morphologies including carbons, inert materials, doping, nanostructures, and microstructures as well as new kinds of electrolytes effectively avoided polysulfide dissolution and shuttle reactions, i.e., the most severe problems affecting the lithium sulfur cell. Various types of carbons characterized by enhanced architectures, oxides, catalysts and alternative solutions of stable electrolytes allowed a sizeable decrease of the lithium–oxygen reaction over-potential and the remarkable increase of the lithium–oxygen cell cycle life. Certainly, advanced cell configuration, geometry and engineering played a crucial role in determining the progress of these intriguing systems. Among the two batteries, lithium–oxygen appeared the most energetic one; however, the lithium–sulfur battery was considered the most practically advanced, and therefore suitable for short-term application, as indeed demonstrated by the Li/S prototypes already available in the energy storage market.130 Furthermore, efforts suitable for moving from the lithium–oxygen battery to the more practical lithium–air battery may certainly come from the use of molecule-selective membranes at the external cell case in order to trap moisture and CO2 from air, thus allowing an efficient lithium oxygen electrochemical process. Last but not least, the severe safety requirements of the market triggered the study on a novel cell prototype in which the lithium metal was replaced by a Li-ion anode with lower energy, however avoiding risks associated with possible cell damage during operation. These findings open a new exciting scenario for alternative energy storage systems characterized both by high energy content and sustainability.
The lithium sulfur battery is not far from large scale diffusion, since it holds at the present state satisfactory energy, cycle life and rate capability;130 however further efforts aimed toward an additional increase of the battery life and the safety may be necessary. In contrast, the lithium oxygen cell, i.e., the most appealing system in terms of energy, is still an important but emerging system which requires several improvements in the next few years, in particular in terms of reversibility and cycle life, for achieving a satisfactory performance. The two systems are, in our opinion, the new frontiers of sustainable energy storage, suitable for emerging markets such as electric vehicles and renewable energy plants.
Acknowledgements
The work was performed within the collaboration project “Accordo di Collaborazione Quadro 2015” between University of Ferrara (Department of Chemical and Pharmaceutical Sciences) and Sapienza University of Rome (Chemistry Department) and the project “Fondo di Ateneo per la Ricerca Locale FAR 2016”, University of Ferrara. The work at Hunter College was supported by a grant from the U.S. Office of Naval Research.
References
- R. Van Noorden, Nature, 2014, 507, 26–28 CrossRef CAS PubMed.
- H. D. Yoo, E. Markevich, G. Salitra, D. Sharon and D. Aurbach, Mater. Today, 2014, 17, 110–121 CrossRef CAS.
- E. M. Erickson, C. Ghanty and D. Aurbach, J. Phys. Chem. Lett., 2014, 5, 3313–3324 CrossRef CAS PubMed.
- L. Carbone, M. Gobet, J. Peng, M. Devany, B. Scrosati, S. Greenbaum and J. Hassoun, ACS Appl. Mater. Interfaces, 2015, 7, 13859–13865 CAS.
- D. Devaux, R. Bouchet, D. Glé and R. Denoyel, Solid State Ionics, 2012, 227, 119–127 CrossRef CAS.
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed.
- M. Agostini and J. Hassoun, Sci. Rep., 2015, 5, 7591 CrossRef CAS PubMed.
- H. Kim, J. T. Lee, A. Magasinski, K. Zhao, Y. Liu and G. Yushin, Adv. Energy Mater., 2015, 5, 1501306 CrossRef.
- M. Agostini, S. Xiong, A. Matic and J. Hassoun, Chem. Mater., 2015, 27, 4604–4611 CrossRef CAS.
- M. R. Busche, P. Adelhelm, H. Sommer, H. Schneider, K. Leitner and J. Janek, J. Power Sources, 2014, 259, 289–299 CrossRef CAS.
- T. Yamada, S. Ito, R. Omoda, T. Watanabe, Y. Aihara, M. Agostini, U. Ulissi, J. Hassoun and B. Scrosati, J. Electrochem. Soc., 2015, 162, A646–A651 CrossRef CAS.
- J. Zhang, Z. Dong, X. Wang, X. Zhao, J. Tu, Q. Su and G. Du, J. Power Sources, 2014, 270, 1–8 CrossRef CAS.
- F. Wu, J. T. Lee, F. Fan, N. Nitta, H. Kim, T. Zhu and G. Yushin, Adv. Mater., 2015, 27, 5579–5586 CrossRef CAS PubMed.
- J. Zhang, J. Xiang, Z. Dong, Y. Liu, Y. Wu, C. Xu and G. Du, Electrochim. Acta, 2014, 116, 146–151 CrossRef CAS.
- S. Chen, F. Dai, M. L. Gordin and D. Wang, RSC Adv., 2013, 3, 3540–3543 RSC.
- J. Liu, L. Yuan, K. Yuan, Z. Li, Z. Hao, J. Xiang and Y. Huang, Nanoscale, 2016, 8, 13638–13645 RSC.
- M. Hagen, D. Hanselmann, K. Ahlbrecht, R. Maça, D. Gerber and J. Tübke, Adv. Energy Mater., 2015, 5, 1401986 CrossRef.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- S. a. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- B. D. Adams, R. Black, Z. Williams, R. Fernandes, M. Cuisinier, E. J. Berg, P. Novak, G. K. Murphy and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1–11 Search PubMed.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1 CrossRef CAS.
- G. A. Elia, R. Bernhard and J. Hassoun, RSC Adv., 2015, 5, 21360–21365 RSC.
- X. Zhu, T. S. Zhao, Z. Wei, P. Tan and L. An, Energy Environ. Sci., 2015, 1–10 Search PubMed.
- A. Débart, J. Bao, G. Armstrong and P. G. Bruce, J. Power Sources, 2007, 174, 1177–1182 CrossRef.
- R. R. Mitchell, B. M. Gallant, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2952 CAS.
- W. H. Ryu, F. S. Gittleson, M. Schwab, T. Goh and A. D. Taylor, Nano Lett., 2015, 15, 434–441 CrossRef CAS PubMed.
- F. Li, D. M. Tang, Y. Chen, D. Golberg, H. Kitaura, T. Zhang, A. Yamada and H. Zhou, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed.
- Y. Xu, Y. Chen, G. Xu, X. Zhang, Z. Chen, J. Li, L. Huang, K. Amine and S. Sun, Nano Energy, 2016, 28, 63–70 CrossRef CAS.
- F. Wu, Y. Xing, X. Zeng, Y. Yuan, X. Zhang, R. Shahbazian-Yassar, J. Wen, D. J. Miller, L. Li, R. Chen, J. Lu and K. Amine, Adv. Funct. Mater., 2016, 7626–7633 CrossRef CAS.
- G. A. Elia, D. Bresser, J. Reiter, P. Oberhumer, Y.-K. Sun, B. Scrosati, S. Passerini and J. Hassoun, ACS Appl. Mater. Interfaces, 2015, 7, 22638–22643 CAS.
- K. Song, D. A. Agyeman, J. Jung, M. R. Jo, J. Yang and Y. M. Kang, Isr. J. Chem., 2015, 715, 458–471 CrossRef.
- P. Sennu, M. Christy, V. Aravindan, Y. G. Lee, K. S. Nahm and Y. S. Lee, Chem. Mater., 2015, 27, 5726–5735 CrossRef CAS.
- Z. Cui, L. Li, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 7278–7281 CrossRef CAS PubMed.
- C. Barchasz, F. Molton and C. Duboc, Anal. Chem., 2012, 84, 3973–3980 CrossRef CAS PubMed.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Electrochem. Soc., 2012, 159, A421 CrossRef CAS.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41–63 CrossRef CAS.
- J. Xiao, J. Z. Hu, H. Chen, M. Vijayakumar, J. Zheng, H. Pan, E. D. Walter, M. Hu, X. Deng, J. Feng, B. Y. Liaw, M. Gu, Z. D. Deng, D. Lu, S. Xu, C. Wang and J. Liu, Nano Lett., 2015, 15, 3309–3316 CrossRef CAS PubMed.
- Y. Gorlin, M. U. M. Patel, A. Freiberg, Q. He, M. Piana, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A930–A939 CrossRef CAS.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694 CrossRef CAS.
- S. Walu
![[s with combining umlaut]](https://www.rsc.org/images/entities/char_0073_0308.gif) , C. Barchasz, R. Bouchet, J. C. Leprêtre, J. F. Colin, J. F. Martin, E. Elkaïm, C. Baehtz and F. Alloin, Adv. Energy Mater., 2015, 5, 1500165 CrossRef.
, C. Barchasz, R. Bouchet, J. C. Leprêtre, J. F. Colin, J. F. Martin, E. Elkaïm, C. Baehtz and F. Alloin, Adv. Energy Mater., 2015, 5, 1500165 CrossRef.
- S. Waluś, C. Barchasz, J.-F. Colin, J.-F. Martin, E. Elkaïm, J.-C. Leprêtre and F. Alloin, Chem. Commun., 2013, 49, 7899–7901 RSC.
- Q. Wang, J. Zheng, E. Walter, H. Pan, D. Lv, P. Zuo, H. Chen, Z. D. Deng, B. Y. Liaw, X. Yu, X. Yang, J.-G. Zhang, J. Liu and J. Xiao, J. Electrochem. Soc., 2015, 162, A474–A478 CrossRef CAS.
- L. Ji, M. Rao, H. Zheng, L. Zhang, O. Y. Li and W. Duan, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- J. J. Chen, R. M. Yuan, J. M. Feng, Q. Zhang, J. X. Huang, G. Fu, M. Sen Zheng, B. Ren and Q. F. Dong, Chem. Mater., 2015, 27, 2048–2055 CrossRef CAS.
- W. H. Shin, H. M. Jeong, B. G. Kim, J. K. Kang and J. W. Choi, Nano Lett., 2012, 12, 2283–2288 CrossRef CAS PubMed.
- A. Rosenman, R. Elazari, G. Salitra, E. Markevich, D. Aurbach and A. Garsuch, J. Electrochem. Soc., 2015, 162, A470–A473 CrossRef CAS.
- S. Xiong, K. Xie, Y. Diao and X. Hong, J. Power Sources, 2014, 246, 840–845 CrossRef CAS.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y.-M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- S. Xiong, K. Xie, Y. Diao and X. Hong, Electrochim. Acta, 2012, 83, 78–86 CrossRef CAS.
- S. S. Zhang, Electrochim. Acta, 2012, 70, 344–348 CrossRef CAS.
- S.-H. Chung, C.-H. Chang and A. Manthiram, Energy Environ. Sci., 2016, 9, 3188–3200 CAS.
- L. Qie, C. Zu and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502459 CrossRef.
- Y. S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- Z. Yang, Z. Zhu, J. Ma, D. Xiao, X. Kui, Y. Yao, R. Yu, X. Wei, L. Gu, Y.-S. Hu, H. Li and X. Zhang, Adv. Energy Mater., 2016, 1600806 CrossRef.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Adv. Energy Mater., 2016, 6, 1–9 Search PubMed.
- D. Sun, Y. Hwa, Y. Shen, Y. Huang and E. J. Cairns, Nano Energy, 2016, 26, 524–532 CrossRef CAS.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 11, 19 Search PubMed.
- S. Zhang, N. Li, H. Lu, J. Zheng, R. Zang and J. Cao, RSC Adv., 2015, 5, 50983–50988 RSC.
- W. Li, Z. Liang, Z. Lu, H. Yao, Z. W. Seh, K. Yan, G. Zheng and Y. Cui, Adv. Energy Mater., 2015, 5, 1500211 CrossRef.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- J. G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- H. Sohn, M. L. Gordin, T. Xu, S. Chen, D. Lv, J. Song, A. Manivannan and D. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 7596–7606 CAS.
- C. Xu, Y. Wu, X. Zhao, X. Wang, G. Du, J. Zhang and J. Tu, J. Power Sources, 2015, 275, 22–25 CrossRef CAS.
- J. Guo, J. Zhang, F. Jiang, S. Zhao, Q. Su and G. Du, Electrochim. Acta, 2015, 176, 853–860 CrossRef CAS.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 4325–4329 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- P. Zhu, J. Song, D. Lv, D. Wang, C. Jaye, D. A. Fischer, T. Wu and Y. Chen, J. Phys. Chem. C, 2014, 118, 7765–7771 CAS.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS PubMed.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- C. L. Lee and I. D. Kim, Nanoscale, 2015, 7, 10362–10367 RSC.
- L. Chai, J. Wang, H. Wang, L. Zhang, W. Yu and L. Mai, Nano Energy, 2015, 17, 224–232 CrossRef CAS.
- M. S. Kim, L. Ma, S. Choudhury, S. S. Moganty, S. Wei and L. A. Archer, J. Mater. Chem. A, 2016, 4, 14709–14719 CAS.
- J. Y. Hwang, H. M. Kim, S. K. Lee, J. H. Lee, A. Abouimrane, M. A. Khaleel, I. Belharouak, A. Manthiram and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1–7 CrossRef.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- M. Liu, D. Zhou, H. R. Jiang, Y. X. Ren, F. Y. Kang and T. S. Zhao, Nano Energy, 2016, 28, 97–105 CrossRef CAS.
- L. Ma, K. E. Hendrickson, S. Wei and L. A. Archer, Nano Today, 2015, 10, 315–338 CrossRef CAS.
- S. K. Lee, S. M. Oh, E. Park, B. Scrosati, J. Hassoun, M. S. Park, Y. J. Kim, H. Kim, I. Belharouak and Y. K. Sun, Nano Lett., 2015, 15, 2863–2868 CrossRef CAS PubMed.
- J. Lu, Y. Jung Lee, X. Luo, K. Chun Lau, M. Asadi, H.-H. Wang, S. Brombosz, J. Wen, D. Zhai, Z. Chen, D. J. Miller, Y. Sub Jeong, J.-B. Park, Z. Zak Fang, B. Kumar, A. Salehi-Khojin, Y.-K. Sun, L. A. Curtiss and K. Amine, Nature, 2016, 529, 1–7 CrossRef PubMed.
- J. Hassoun, F. Croce, M. Armand and B. Scrosati, Angew. Chem., Int. Ed., 2011, 50, 2999–3002 CrossRef CAS PubMed.
- C. V. Amanchukwu, J. R. Harding, Y. Shao-Horn and P. T. Hammond, Chem. Mater., 2015, 27, 550–561 CrossRef CAS.
- S. Wu, J. Tang, F. Li, X. Liu and H. Zhou, Chem. Commun., 2015, 51, 16860–16863 RSC.
- J. Wandt, P. Jakes, J. Granwehr, H. A. Gasteiger and R. A. Eichel, Angew. Chem., Int. Ed., 2016, 55, 6892–6895 CrossRef CAS PubMed.
- S. Lau and L. A. Archer, Nano Lett., 2015, 15, 5995–6002 CrossRef CAS PubMed.
- H.-G. Jung, H.-S. Kim, J.-B. Park, I.-H. Oh, J. Hassoun, C. S. Yoon, B. Scrosati and Y.-K. Sun, Nano Lett., 2012, 12, 4333–4335 CrossRef CAS PubMed.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518 CAS.
- J. B. Park, J. Hassoun, H. G. Jung, H. S. Kim, C. S. Yoon, I. H. Oh, B. Scrosati and Y. K. Sun, Nano Lett., 2013, 13, 2971–2975 CrossRef CAS PubMed.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1777 CAS.
- H. Lee, Y.-J. Kim, D. J. Lee, J. Song, Y. M. Lee, H.-T. Kim and J.-K. Park, J. Mater. Chem. A, 2014, 2, 11891 CAS.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. Zhang, D. Song, Z. S. Liu, H. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS.
- Y. C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- V. R. Chitturi, M. Ara, W. Fawaz, K. Y. S. Ng and L. M. R. Arava, ACS Catal., 2016, 6, 7088–7097 CrossRef CAS.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- F. S. Gittleson, W.-H. Ryu, M. Schwab, X. Tong and A. D. Taylor, Chem. Commun., 2016, 52, 6605–6608 RSC.
- M. Sevim, C. Francia, J. Amici, S. Vankova, T. Şener and Ö. Metin, J. Alloys Compd., 2016, 683, 231–240 CrossRef CAS.
- Y. C. Lu, H. A. Gasteiger and Y. Shao-Horn, J. Am. Chem. Soc., 2011, 133, 19048–19051 CrossRef CAS PubMed.
- J. Jung, K. Song, Y. Bae, S. Il Choi, M. Park, E. Cho, K. Kang and Y. M. Kang, Nano Energy, 2015, 18, 71–80 CrossRef CAS.
- Y. S. Jeong, J.-B. Park, H.-G. Jung, J. Kim, X. Luo, J. Lu, L. Curtiss, K. Amine, Y.-K. Sun, B. Scrosati and Y. J. Lee, Nano Lett., 2015, 15, 4261–4268 CrossRef CAS PubMed.
- G. Zhao, R. Mo, B. Wang, L. Zhang and K. Sun, Chem. Mater., 2014, 26, 2551–2556 CrossRef CAS.
- F. Li, D. M. Tang, Z. Jian, D. Liu, D. Golberg, A. Yamada and H. Zhou, Adv. Mater., 2014, 26, 4659–4664 CrossRef CAS PubMed.
- D. Zhai, K. C. Lau, H. H. Wang, J. Wen, D. J. Miller, J. Lu, F. Kang, B. Li, W. Yang, J. Gao, E. Indacochea, L. A. Curtiss and K. Amine, Nano Lett., 2015, 15, 1041–1046 CrossRef CAS PubMed.
- Y. Jing and Z. Zhou, ACS Catal., 2015, 5, 4309–4317 CrossRef CAS.
- H. Lim, K. Park, H. Song, E. Y. Jang, H. Gwon, J. Kim, Y. H. Kim, M. D. Lima, R. O. Robles, X. Lepró, R. H. Baughman and K. Kang, Adv. Mater., 2013, 25, 1348–1352 CrossRef CAS PubMed.
- J. Shui, F. Du, C. Xue, Q. Li and L. Dai, ACS Nano, 2014, 8, 3015–3022 CrossRef CAS PubMed.
- M. D. Radin, C. W. Monroe and D. J. Siegel, Chem. Mater., 2015, 27, 839–847 CrossRef CAS.
- Q.-C. Liu, J.-J. Xu, D. Xu and X.-B. Zhang, Nat. Commun., 2015, 6, 7892 CrossRef CAS PubMed.
- W. Kwak, K. C. Lau, C. Shin, K. Amine, L. A. Curtiss and Y. Sun, ACS Nano, 2015, 9, 4129–4137 CrossRef CAS PubMed.
- D. Sun, Y. Shen, W. Zhang, L. Yu, Z. Yi, W. Yin, D. Wang, Y. Huang, J. Wang, D. Wang and J. B. Goodenough, J. Am. Chem. Soc., 2014, 136, 8941–8946 CrossRef CAS PubMed.
- D. Sharon, D. Hirsberg, M. Afri, F. Chesneau, R. Lavi, A. A. Frimer, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2015, 7, 16590–16600 CAS.
- Y. G. Zhu, X. Wang, C. Jia, J. Yang and Q. Wang, ACS Catal., 2016, 6, 6191–6197 CrossRef CAS.
- T. Zhang, K. Liao, P. He and H. Zhou, Energy Environ. Sci., 2015, 9, 1024–1030 Search PubMed.
- H. D. Lim, H. Song, J. Kim, H. Gwon, Y. Bae, K. Y. Park, J. Hong, H. Kim, T. Kim, Y. H. Kim, X. Leprõ, R. Ovalle-Robles, R. H. Baughman and K. Kang, Angew. Chem., Int. Ed., 2014, 53, 3926–3931 CrossRef CAS PubMed.
- G. a. Elia, J. Hassoun, W.-J. Kwak, Y.-K. Sun, B. Scrosati, F. Mueller, D. Bresser, S. Passerini, P. Oberhumer, N. Tsiouvaras and J. Reiter, Nano Lett., 2014, 14, 6572–6577 CrossRef CAS PubMed.
- H. Horie, T. Abe, T. Kinoshita and Y. Shimoida, World Electric Vehicle Journal, 2008, 2, 25–31 Search PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- P. G. Balakrishnan, R. Ramesh and T. Prem Kumar, J. Power Sources, 2006, 155, 401–414 CrossRef CAS.
- J. Hassoun, G. Derrien, S. Panero and B. Scrosati, Adv. Mater., 2008, 20, 3169–3175 CrossRef CAS.
- L.-F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2009, 9, 491–495 CrossRef CAS PubMed.
- J. Hassoun, P. Reale and B. Scrosati, J. Mater. Chem., 2007, 17, 3668 RSC.
- W.-J. Kwak, H.-J. Shin, J. Reiter, N. Tsiouvaras, J. Hassoun, S. Passerini, B. Scrosati and Y.-K. Sun, J. Mater. Chem. A, 2016, 1–5 Search PubMed.
- J. Hassoun, H. G. Jung, D. J. Lee, J. B. Park, K. Amine, Y. K. Sun and B. Scrosati, Nano Lett., 2012, 12, 5775–5779 CrossRef CAS PubMed.
- S. Wu, K. Zhu, J. Tang, K. Liao, S. Bai, J. Yi, Y. Yamauchi, M. Ishida and H. Zhou, Energy Environ. Sci., 2016, 9, 3262–3271 CAS.
- L. Carbone and J. Hassoun, Ionics, 2016, 22, 2341–2346 CrossRef CAS.
- M. Agostini, B. Scrosati and J. Hassoun, Adv. Energy Mater., 2015, 5, 1500481 CrossRef.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef CAS.
-
http://www.sionpower.com/
.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Steve G.
Greenbaum
b and
Jusef
Hassoun
a,
Steve G.
Greenbaum
b and
Jusef
Hassoun