
Current understanding of chemical degradation mechanisms of perfluorosulfonic acid membranes and their mitigation strategies: a review
M.
Zatoń
,
J.
Rozière
and
D. J.
Jones
*
ICGM – Aggregates, Interfaces and Materials for Energy, UMR 5253, Université de Montpellier 2, Place Eugène Bataillon, 34095 Montpellier, France. E-mail: Deborah.Jones@umontpellier.fr
First published on 5th April 2017
Abstract
This article provides a comprehensive and up-to-date perspective of the understanding of perfluorosulfonic acid (PFSA) fuel cell membrane degradation phenomena, reviews key concepts for the mitigation of membrane degradation, appraises the effectiveness of these strategies by discussing their benefits and drawbacks, and identifies remaining challenges and research priorities for fuel cell membranes with increased longevity. Identification of stressors and improved understanding of how scavenging reactions proceed are essential for accelerated development of new fuel cell membrane materials with antioxidant properties and enhanced durability. Fuel cells convert the chemical energy of a fuel such as hydrogen into electricity and heat and have the potential to deliver environmental and economic benefits across various sectors, including transportation, power generation, industrial equipment, military power, and consumer electronics. The development of Proton Exchange Membrane Fuel Cell (PEMFC) membranes with increased durability is of crucial importance since it directly impacts fuel cell system lifetime and, therefore, large scale implementation of fuel cell technology. The PFSA proton conducting polymer (ionomer) at the heart of the membrane electrode assembly of a PEMFC is subject to chemical degradation as a result of the attack on the polymer chains by reactive oxygen species generated electrochemically in the fuel cell. For the first time, this article reviews the literature both on mechanisms by which this chemical degradation of the ionomer membrane occurs and the sites on the polymer susceptible to free radical attack, as well as how this learning and increased understanding have been used to build and develop successful mitigation strategies designed to annihilate the harmful effect of oxidative species on membrane integrity.
1. Introduction
Fossil fuels are currently the main source of energy. The growing concern for the shrinking availability of the world's oil and gas supplies and the detrimental impact on the environment associated with the use of such sources of energy have motivated the search for alternative, clean energy technologies. Fuel Cells (FC) are devices that convert the chemical energy stored in fuels into electricity via electrochemical reactions. Since fuel cells can generate electricity continuously with high efficiency and low or zero pollution emission at the point of use as long as the fuel and oxidant are supplied, they represent a promising substitute to heat and electricity generation from fossil fuels, and can be used for a wide range of applications from micro power sources to multi – MW power plants. There are many types of fuel cells that can be classified according to the ion transport medium and/or to the operation temperature. Regarding the nature of the electrolyte, an acid or an alkaline fuel cell can be distinguished. According to the operation temperature, fuel cells may be grouped as low (up to 100 °C), intermediate (up to 600 °C) and high temperature (up to 1000 °C). This review is solely focused on Low Temperature Proton Exchange Membrane Fuel Cells (LTPEMFC). PEMFC are known to have interesting properties such as high power density, easy scale-up and fast start-up capability. These features make the device an excellent candidate for automotive and small stationary applications and portable electronics. However the direction of PEMFC development currently poised to make a real impact on emissions reduction is in transportation applications. Prototype Fuel Cell Electric Vehicles (FCEVs) have already been presented by most automakers and were commercialized in 2015 Toyota, and in 2016 by Hyundai and Honda. Furthermore PEM fuel cells are already used in certain stationary, portable and transport applications in most developed countries.1–4 A lot of research is being done in this area due to the economic and environmental benefits that such systems can offer. However there are still challenges that are hindering the global applications of fuel cells. The main issue is the practical long-term operation of the cell components.5–7 There are several losses that limit the long-term stability and the performance of these devices.8 Those losses are due to the degradation of the bipolar plates, the gas diffusion layer, the catalysts and the membranes.9The loss of the active surface area of the catalyst layer, which results in the decrease of the performance of the fuel cell, is caused by platinum oxidation, dissolution and corrosion of the carbon material, which can lead to aggregation of Pt particles and their detachment from the support.10 The high potential delivered by the system as well as the oxidative environment favour the growth of Pt particles. Platinum dissolution is not only accelerated by potential or load cycling but as well as the elevated temperatures used in these devices.11–13 Migration of the dissolved ionic Pt and further precipitation in the membrane can be in certain conditions considered as one of the factors contributing to membrane degradation.14,15 Oxidation of the carbon support is initiated by high-electrochemical potentials caused either by fuel/air boundary passing over the active area during the start-up/shut-down processes or local hydrogen starvation.16 Carbon corrosion results in the thinning of the electrode16 and growth of the platinum particles due to detachment and agglomeration.17–19 GDL degradation is related to loss of hydrophobicity of the macro and microporous layers due to the oxidation of the carbon and the decomposition of the PTFE component. These microstructural changes affect the gas transport and therefore lower the cell performance.20
Degradation of the membrane is one of the major reasons that can lower the durability of a fuel cell. Membrane decomposition can be induced by many factors for instance mechanical degradation which is an effect of non-uniform contact pressure or fatigue from stress during shrinkage and swelling of the membrane during humidity cycling. Reduction of the membrane mechanical strength results in the formation of pinholes and cracks, which can further contribute to the failure of the MEA.21 Another factor affecting the stability of the membrane is the contaminant ions originated from corrosion of the different cell components.22 Released cations such as Fe and Ni catalyse the decomposition of hydrogen peroxide, which is the main source of free radicals. Moreover the counter ions formed tend to associate with the sulfonic acid sites of the polymer, which causes partial drying of the membrane and results in loss of conductivity.23,24 Chemical degradation crucially affects membrane durability. Chemical decomposition is initiated by free radicals, highly reactive oxygen species, which are formed during the chemical and electrochemical reaction of crossover gases. Those species attack the polymer structure causing an “unzipping” reaction on the main chain and the scissions of the side chain, which finally result in thinning of the membrane.
All these damaging effects often occur simultaneously, thereby creating a very complex mechanism of the overall fuel cell degradation.25 Since amelioration of durability is crucial for technology development, understanding the fundamental degradation mechanisms of the fuel cell components is primordial.
Excellent articles by Borup et al.20 and by de Bruijn et al.26 reviewed the literature on degradation and durability of fuel cell components up to 2008 while that of Wu et al.9 additionally discussed possible mitigation approaches. In 2014 the article Dubau et al. provided an updated overview of degradation of fuel cell components.27 None of these reviews was focussed on membrane degradation phenomena. Other authors summarized and classify accelerated stress test (AST) protocols,28,29 aiming to provide practical tools for the study of fuel cell durability. The article from Rodgers et al. reviewed different accelerated stress test and lifetime tests and correlated them to membrane decay and fuel cell lifetime.30 Ishimoto et al. published a survey of literature on the mechanism of chemical degradation of PFSA membranes from an atomistic point of view.31 Despite the significant number of publications on the topics of PFSA membrane degradation and mitigation strategies to reduce this degradation that have been published over the past decade, there is no article providing a comprehensive and detailed review of the field.
The goal here is to review the substantial literature on how increased understanding of factors that lead to the degradation of PFSA membranes has led to the development of mitigation strategies and improvement of membrane and MEA lifetime, in particular since the ground-breaking work introducing effective radical scavengers by Endoh et al. at Asahi Glass Company and by Coms et al. at General Motors.
2. PFSA polymer types
The fuel cell electrolyte must have high proton conductivity in order to maximize the efficiency. It should also be mechanically and chemically robust during fuel cell operation to ensure a long lifetime and finally needs to be impermeable to the direct transfer of reactant gases and electronically insulating. Although many types of FC membranes have been developed over the past two decades32–35 the commonly used proton exchange membranes are perfluorosulfonic acid (PFSA) membranes. The first widely used PFSA membrane is Nafion® developed by DuPont.36 The synthesis of Nafion® is based on the copolymerisation of tetrafluoroethylene (TFE) with a perfluorinated vinyl ether co-monomer and sulfonyl acid fluoride. The product obtained from the copolymerisation is hydrolysed with NaOH and then converted to the acid form. The Nafion® polymer structure comprises two well defined components: the hydrophobic backbone (polytetrafluoroethylene – PTFE) and the hydrophilic side chain terminated with sulfonic acid groups. The PTFE part provides mechanical strength and relatively high stability in very harsh chemical environments, while the conduction properties are due to the presence of hydrated sulfonic acid groups.37 Commercially available polymers with equivalent structure are Flemion® (trade mark of Asahi Glass Company), Aciplex® (produced by Asahi Kasei) and Fumion® F (developed by FuMA-Tech). This type of composition is now known as the long-side-chain (LSC) structure; correspondingly ionomers with a shorter pendant side chain are referred as to as short-side-chain (SSC) ionomers. The chemical structures of PFSAs with different side chain lengths are presented in Fig. 1. | ||
| Fig. 1 The chemical structure of LSC – Nafion® structure (a) and SSC perfluorosulfonic ionomer membranes (b) and (c). Reprinted from Polymer Electrolyte Membrane and Direct Methanol Fuel Cell Technology, vol. 1, D. J. Jones, Polymer Electrolyte Membrane and Direct Methanol Fuel Cell Technology, 27–56.38 Copyright (2012), with permission from Elsevier. | ||
In the SSC polymer structures there is no –O–CF2CF(CF3)– group. Furthermore, the length of the perfluoro vinylether side chain varies between 2 and 4 carbons for the two commercially available SSC ionomers i.e. Aquivion® and 3M™ respectively. Pioneering studies on the synthesis of a SSC polymer date back to 1981 at Dow.39 Early investigations of Dow SSC membranes demonstrated excellent balance between their stability and transport properties.40–42 However a complex and expensive synthesis route impeded their wide commercialisation. More recently, a low cost synthesis route developed by Solvay Solexis resulted in new benchmark Hyflon® Ion membrane,43 renamed in 2010 as the Aquivion® membrane. The parameter that reflects the transport properties of the PFSA membrane and its mechanical integrity, within a given class of PFSA types, is the equivalent weight (EW). EW is defined as dry mass of the polymer in the acid form in grams per mole of exchangeable groups. Another important feature of PFSA membranes in relation to the EW is the ion exchange capacity (IEC). The relationship between these parameters is expressed by: 1000/EW. The IEC of the membrane can be measured experimentally by acid/base titration. Differences in side chain length leads to differences in the molecular weight of the polymer repeat unit and translates into specific properties such as higher degree of crystallinity of SSC membranes44 compared to LSC Nafion® for a given polymer equivalent weight, and higher glass transition temperature (Tg).45 Higher ion exchange capacity46 SSC polymers can be used to higher temperature due to these properties.47–49 In general PFSA membranes possesses good mechanical properties, high proton conductivity and electronic resistance and low gas permeability. All these properties are necessary in order to have an efficient barrier for gas separation and to withstand strong oxidative and reductive environments and severe mechanical stresses imparted by hydration–dehydration cycles compounded by the specific electrochemical environment of the fuel cell. Such harsh conditions nevertheless initiate decomposition or degradation of the ionomer or membrane, of which three main types of membrane degradation may be distinguished: chemical, mechanical and thermal.24
3. Mechanical and thermal degradation of PFSA membranes
Mechanical degradation is an effect of the mechanical forces affecting the integrity of the membrane. Long term fuel cell testing shows a time dependent deformation – creep and micro-crack fracture of Nafion®. These phenomena are related to the repeated stress applied to the membrane between bipolar plates and strain from local changes of the relative humidity. The most important factors inducing micro and macro defects of the membrane are: relative humidity cycling,36,50,51 transients at open circuit voltage (OCV),52 temperature cycling51 and potential cycling. Some of those stressors have a bigger impact on the polymer mechanical durability than others, for example cycling of the temperature was reported to have less influence on membrane breakdown than RH cycling50,53 probably due to the higher strain oscillation amplitude.54 Repeated swelling and shrinking of the membrane causes the gradual reduction of the membrane strength that finally results in membrane dimensional changes, poor interface contact between membrane and electrode,55–57 and defects such as pinhole formation.57,58 Pinhole formation, due to the local stress applied to the membrane is one of the main reasons for the failure of PFSA membranes in PEM fuel cells.59 Accelerated gas crossover of hydrogen and oxygen to the opposite side of the electrolyte through pinholes causes combustion reaction on the catalyst surface and generates local hot spots. The use of high temperature and relative humidity increase gas crossover.60–62 Membrane hydration is a very important factor affecting mechanical and thermal properties of PFSA membranes.31,63,64 Kundu et al. reported that the transition temperatures of Nafion® decrease with increasing membrane hydration.65 Majsztrik et al. described that high hydration of Nafion® at low temperature plasticises its structure,66 whereas at elevated temperature opposite behaviour is observed.67 This implies that at elevated temperature for a highly hydrated membrane, mechanical creep occurs much faster than for a dry polymer. Recently Luo et al. reported significantly higher permeability of degraded Nafion NR211 following 48 h exposure to Fenton reagent compared to the pristine membrane.68 Increased water content and proton mobility, despite lower IEC and proton conductivity of degraded membranes, were explained by the authors as resulting from enhanced water sorption of a damaged polymer membrane structure. Water remaining in non-ionic cavities formed through degradation does not contribute to ion conduction but can cause hygrothermal stress and fracture development.69 Also Shi et al. interrogate heterogeneity in permeability of ex situ aged Nafion membranes.70 Venkatesan reported high water uptake of degraded catalyst coated membrane (CCM) subjected to combined mechanical/chemical accelerated stress test.71 Authors demonstrated that voids regions in severely degraded material, depleted of F and C are susceptible to micro crack formation. PEM fuel cells require to be operated over a wide working temperature range. Therefore it is important to study the stability of PFSA membranes at sub-freezing temperature. The existence of three72,73 or even four different “states” of water in Nafion® termed non-freezing water, bound freezing water and free water has been reported.74 The chemically bound water does not freeze down to −120 °C, whereas it is probable that the water which is non-bonded to the polymer chain freezes below 0 °C.73 Phase transformation as well as difference in densities of water and ice can cause the degradation of PFSA membranes.75 However McDonald et al. reported that after 385 cycles in the temperature range from −40 °C to 80 °C no catastrophic failure was observed.76 On the other hand, Plazanet et al. observed ice formation outside of the membrane with simultaneous increase of concentration of hydronium ions in membrane.77 This phenomenon was related to desorption of water upon cooling and contraction of the membrane. In such a scenario, the formation of ice or frost at the membrane electrode interface is detrimental for the catalyst layer structure. The impact of mechanical degradation can be significantly decreased by optimizing the design of the fuel cell components i.e. by choosing appropriate materials and operating conditions.78 On the other hand, chemical degradation appears more complex and difficult to mitigate as the process has a larger impact on the integrity of the whole polymer.794. Chemical degradation of PFSA
Chemical degradation of the PFSA membrane is the major issue considered in this paper and it will therefore be discussed starting from the structure of the membrane at the microscopic level. The mechanism of membrane degradation at the meso and macroscopic levels is also fully described followed by the classification of free radicals generated under fuel cell operating conditions. Finally a summary of mitigation strategies that have been proposed is discussed at the end of this review.4.1 Radical attack on polymer structure
Chemical degradation of PFSA membranes involves radical-induced decomposition of the polymer structure. The process of chemical degradation is not yet fully understood, however the damage caused to the membrane and its properties are evident, for instance a decrease in the ion exchange capacity followed by conductivity losses, as well as fluoride emission and subsequent membrane thinning (subsection 3.2). The current generally accepted mechanism for chemical degradation of perfluorosulfonic acid polymers is by radical attack on the polymer main chain and side chain.80–85 Reactive oxygen species (ROS) can be generated directly via chemical or electrochemical reaction of crossover gases over the electrocatalyst surface,86–88 however their short lifetime limits to a large extent their diffusion length.80 Another pathway for radical formation occurs through homolysis of hydrogen peroxide (H2O2).23,89 H2O2 itself is not a strong enough oxidant to damage the polymer structure substantially.90–92 However, iron or other multivalent metal ions like Cu2+ or Ti3+, originated from corrosion of the cell or stack materials or humidifiers, catalyse the decomposition of H2O2 to produce hydroxyl radicals as shown in eqn (1).93,94 Pozio et al. performed experiments where stainless steel plates were replaced with iron-free aluminium alloy plates.89 The membrane degradation after 1200 h under such conditions was significantly reduced, which clearly shows correlation between fluoride emission rate and trace amounts of transition metal ions. Moreover the amount of iron can be as low as 1 part per 5–25 parts of H2O2 for the reaction to occur for a concentration of hydrogen peroxide of <10–25 mg L−1.95| H2O2 + Mz+ → M(z+1)+ + HO˙ + OH− | (1) |
The hydroxyl radical produced viaeqn (1) can further undergo reaction (eqn (2)) with hydrogen peroxide to form hydroperoxyl radical (more detail in Section 7).
| HO˙ + H2O2 → H2O + HOO˙ | (2) |
| M(z+1)+ + H2O2 → Mz+ + HOO˙ + H+ | (3) |
Furthermore, the hydroperoxyl radical can also be produced by the reaction of transition metal ions with hydrogen peroxide, eqn (3). In situ hold at open circuit voltage (OCV hold testing)96–98 is the accepted means to accelerate membrane chemical degradation, while outside of the fuel cell environment, the Fenton reagent comprising hydrogen peroxide and ferrous ions is frequently used.70,99–102 In both cases accelerated membrane decomposition is due to high concentration of hydroxyl radicals that attack the most vulnerable sites in polymer structure: carboxylic acid end groups; C–S bonds; tertiary carbon atoms and ether groups. These four main mechanisms of radical attack on the polymer structure are discussed. The first, generally referred to as the “unzipping reaction” is a radical reaction occurring in non-so-called “stabilised” PFSA polymers, and starting on weak terminal –COOH groups of the polymer main chain (Fig. 2). Such terminal carboxylic acid groups can also be generated through transformation of non-perfluorinated groups via hydroxyl radical attack.103 As described in eqn (4)–(6) end carboxylic groups react with hydroxyl radicals to produce CO2 and HF, and reform a terminal –COOH group.104 Several propagation steps of this reaction brings about decomposition of the fluorocarbon main chain by consistent loss of CF2 units.105 When the polymer unzipping reaches a junction with a side chain, cleavage of the overall side chain groups generates perfluoro(3-oxa-5-methyl)pentane-1-sulfonic-5-carboxylic diacid, HCOO–CF(CF3)–O–CF2CF2–SO3H.105 This species can diffuse out of the membrane or can undergo decomposition by the unzipping reaction, as a carboxylic acid end group is also present in its molecular structure. This mechanism was the first to be identified, following seminal work at General Motors and DuPont, and this greater understanding of what was understood at that time to represent a primary cause of polymer degradation led directly at DuPont (and other producers of PFSAs) to post-fluorination of terminal carboxylic acid groups with the aim of stabilising them against the unzipping reaction.
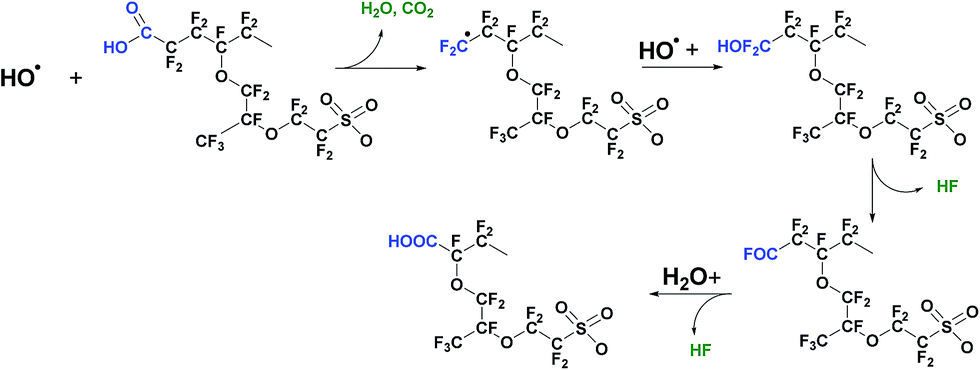 | ||
| Fig. 2 Unzipping mechanism – hydroxyl radical attack on carboxylic acid groups. | ||
4.2 Mechanism I – “unzipping mechanism”
| Rf–CF2COOH + HO˙ → Rf–CF2˙ + CO2 + H2O | (4) |
| Rf–CF2˙ + HO˙ → Rf–CF2OH → Rf–COF + HF | (5) |
| Rf–COF + H2O → Rf–COOH + HF | (6) |
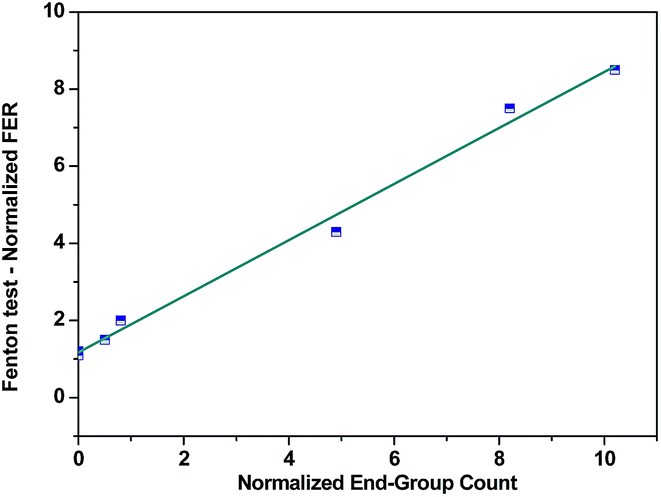 | ||
| Fig. 3 Fluorine emission rates as a function of the content of carboxylic groups in Nafion® structure. Reproduced by permission of The Electrochemical Society from ref. 112. | ||
Other studies demonstrated that Nafion® converted to the Na+, Cs+ or Li+ forms shows two orders of magnitude lower fluoride emission rate compared to the acid form membrane. Such a significant change in the FER values suggest that radicals also attacked the sulfonic acid groups (Fig. 4).91
 | ||
| Fig. 4 Hydroxyl radical attack on C–S bond. | ||
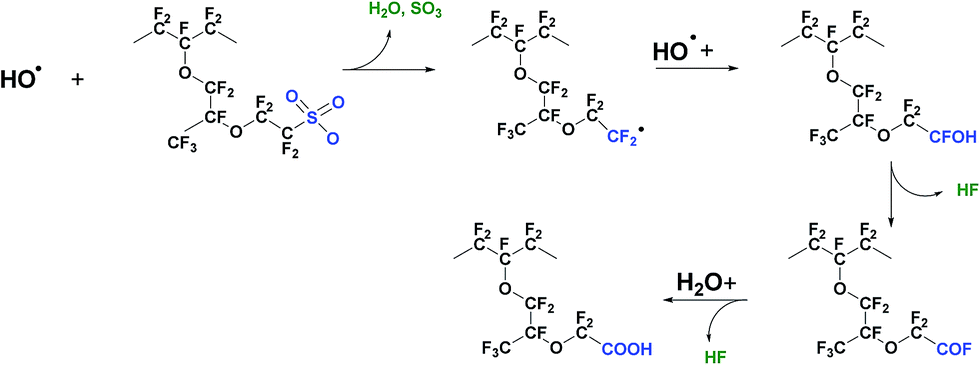
4.3 Mechanism II – attack on C–S bond
A second possible mechanism of membrane degradation is the attack by free radicals on C–S bonds following the reactions shown below:113| Rf–CF2SO3H + HO˙ → Rf–CF2˙ + SO3 + H2O | (7) |
| Rf–CF2˙ + HO˙ → Rf–CF2OH → Rf–COF + HF | (8) |
| Rf–COF + H2O → Rf–COOH + HF | (9) |
The radical attack begins at the end of the side chain on the C–S bond (eqn (7)). After this step the polymer decomposition proceeds with progressive shortening of the side chain and HF emission (eqn (8) and (9)). Eqn (8) and (9) seem to be a repetition of the steps in the unzipping mechanism described in eqn (5) and (6). This conclusion is partially right, the incorporation of a new weak –COOH group accelerates the degradation and therefore the unzipping reaction occurs on both the side and main chains. However as the Rf have different chemical structures, the decomposition products of the side chain have different nature. The detection of –O–CF2–CF2–SO3˙ and –O–CF2–CF2˙ radicals after a UV – induced Fenton test supports the above degradation mechanism.114 This type of membrane decomposition results in the simultaneous loss of weight and conduction properties of the polymer. Recently Kurniawan et al. studied the PFSA degradation mechanism using quantum chemical calculations (QCC) based on density functional theory (DFT).115 Trifluoromethanesulfonate (TFMS) was used as a model of the side chain structure. For these authors, the calculated bond dissociation energies (BDEs) and bond lengths indicate that radical attack on the C–S bond is favourable due to its lower dissociation energy in comparison to the other structural bonds. They further pointed out the behaviour of fluorine atoms as another important factor that can affect the radical attack on the polymer structure, i.e. the electronegative fluorine atoms efficiently shield the carbon backbone from radical reaction. Tokumasu et al. also investigated the BDE of a perfluorosulfonic acid (PFSA) molecule by DFT.116 Similarly to the work of Kurnawian et al. the C–S bond appeared as the weakest bond in the side chain structure of the neutral molecule, thus most susceptible to hydroxyl radical attack. However further analysis showed that the ionisation of the sulfonic group makes this C–S bond stronger while the C–O bond becomes weaker.
From thermochemical and kinetic analysis, Coms deduced further possible route of side chain scissions.117 The mechanism is initiated by sulfonyl radicals, which are formed under dry conditions, where the sulfonic acid groups are not fully ionised. Sulfonyl radicals can be formed directly through hydrogen abstraction from –SO3H by hydroxyl radical or indirectly through reaction with hydrogen peroxide where bissulfonyl peroxide is an intermediate product, which further undergoes homolysis to give sulfonyl radicals (Fig. 5).
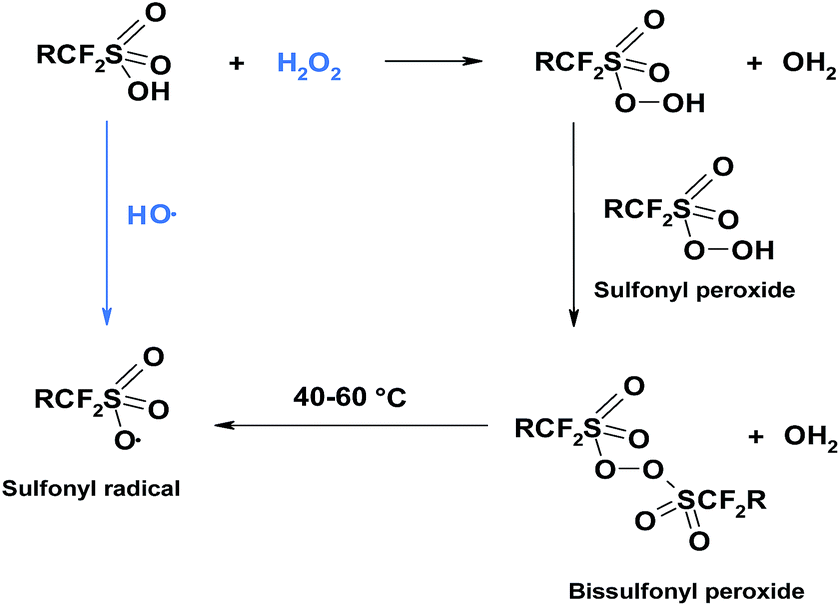 | ||
| Fig. 5 Mechanism of sulfonyl radical formation. | ||
This author pointed out the weakening effect of sulfonyl radicals on the C–S bond. In other words, SO3˙ facilitates the C–S cleavage and formation of the perfluororadical as shown in mechanism II.
4.4 Mechanism III – attack on ether groups
The above discussion brings us to a third mechanism which concerns hydroxyl radical attack on the O–C bond.It was postulated that ether groups next to a C–S bond represent the preferred site for the radical attack in the side chain structure.120,121 This finding might support the above results of Danilczuk et al. where Nafion® ionomer exhibited 20 times lower stability than respective SSC ionomers.108 This is probably due to the absence of OCF2CF(CF3) in the SSC polymer structure. On the other hand, according to studies of Ghassemzadeh et al. the first point of HO˙ attack is located near the sulfonic acid group (α-OCF2) rather than near the main chain (β-OCF2).118,119 After cleaving the α-OCF2 the polymer decomposition continues as shown in Fig. 6(a) and involves the β-OCF2 and CF units. These results are in agreement with theoretical studies of Ishimoto et al.122
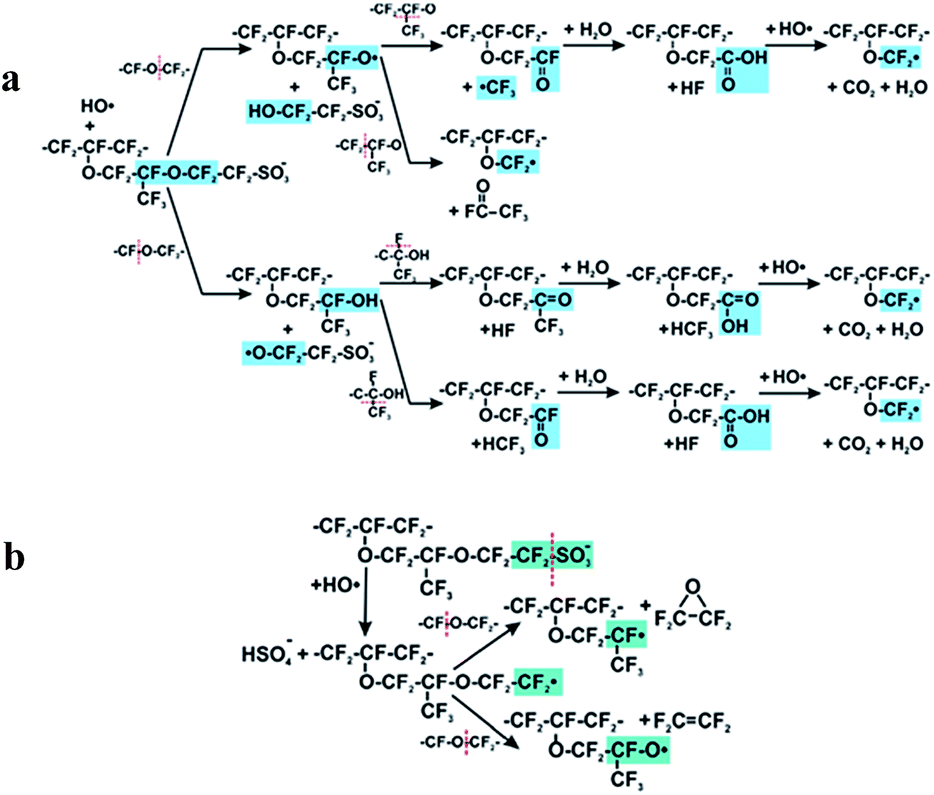 | ||
| Fig. 6 Mechanism of side chain scission by HO˙ radical attack on C–O bond (a) direct attack Reprinted with permission from (J. Am. Chem. Soc., 2013, 135, 15923–15932).118 Copyright (2013) American Chemical Society. (b) Attack as a consequence of bond weakening after C–S bond cleavage. Reprinted with permission from (J. Am. Chem. Soc., 2013, 135, 8181–8184).119 Copyright (2013) American Chemical Society. | ||
4.5 Mechanism IV – attack on the tertiary carbon
A further potential site for radical attack leading to PFSA polymer degradation can occur at the tertiary carbon according to the reaction given in Fig. 7.117 Two tertiary carbons are present in the Nafion® structure; both are adjacent to ether bonds. Radical attack on the CF of the side chain initiates side chain decomposition, whereas attack on the main chain CF results in chain scission. This mode of degradation is characterised by relatively low fluoride emission but with an important increase in COOH groups that can then be degraded by the unzipping mechanism. The vapour phase Fenton test, OCV hold test and low power demand fuel cell test were reported to accelerate Nafion® degradation according to this mechanism.109 Tertiary carbons atoms are assumed to be most fragile to radical attack among the fluorocarbons, due to different thermodynamic stabilities.117 Strong dependence of the C–F bond dissociation energies on abstraction of the fluorine atoms, gives the respective order: tertiary > secondary > primary, with tertiary carbon as the least stable position.116,117 | ||
| Fig. 7 Mechanism of chain scission by radical attack at the tertiary carbon. Reprinted with permission from (J. Am. Chem. Soc., 2013, 135, 15923–15932)118. Copyright (2013) American Chemical Society. | ||
Schiraldi remarked that thermodynamic assumptions should be re-evaluated in relation to the complex morphology of Nafion®, which is likely to increase the kinetic factor in the degradation process.111 According to studies using model compounds, in ex situ accelerated degradation tests a carboxylic acid terminated compound demonstrates 300 times faster generation of fluoride than the corresponding sulfonic acid terminated compound.111 However in their studies on Nafion® degradation by applying in depth profiling by micro FTIR, Danilczuk et al. recently proposed mechanism III as a degradation pathway of membrane decomposition.123 The micro FTIR spectra of Nafion® 115 membrane cross section were recorded after OCV hold testing for 180 h at 90 °C and 30% RH. Two bands C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H characteristic of the degraded membrane appeared at the same profile depths which indicated their generation through a single mechanism, which is fluorine atom abstraction from tertiary carbon atom by H˙. CO and C–H bands were observed at depths of 22 μm and 82 μm from the cathode. The presence of the bands at 22 μm from the cathode was associated with the location of Pt particles precipitated in the membrane, however larger damage was observed near the anode interface at 82 μm where these bands showed highest intensity.
O and C–H characteristic of the degraded membrane appeared at the same profile depths which indicated their generation through a single mechanism, which is fluorine atom abstraction from tertiary carbon atom by H˙. CO and C–H bands were observed at depths of 22 μm and 82 μm from the cathode. The presence of the bands at 22 μm from the cathode was associated with the location of Pt particles precipitated in the membrane, however larger damage was observed near the anode interface at 82 μm where these bands showed highest intensity.
4.6 Summary of radical attack on PFSA type polymers
A graphical summary of degradation mechanisms proposed in the literature is provided in Fig. 8. | ||
| Fig. 8 Summary of the mechanisms of radical attack on the Nafion® polymer structure. | ||
The “unzipping” mechanism, in this work labelled as mechanism I, proposed by Curtin et al.104 and Healy et al.124 is the most frequently cited origin of membrane degradation. However it should be emphasised that the main role of the unzipping mechanism in chemical decomposition of PFSA ionomers changed with the evolution in the ionomer post-fluorination process. A new generation of chemically post-treated PFSA membranes with reduced number of COOH groups show considerably improved durability.108,109 Moreover this improvement significantly increases with elimination of the –OCF2CF(CF3) fragment from the polymer structure.108 This behaviour clearly shows the contribution of radical attack on the side chain to overall membrane decomposition. It can be concluded that all the mechanisms discussed above most probably play a role in the chemical degradation of the membrane during fuel cell operation, where the working conditions determine the contribution of each of them. Very likely, a membrane weakened by one type of radical attack is more susceptible to undergo a second type of radical reaction.119 According to recent reports, hydroxyl radical attack on the susceptible bond in the side chain initiates “unzipping” along the side chain110,117,125 and then main chain structure126,127 as displayed in Fig. 9. Therefore the term “unzipping” cannot be removed in the discourse of degradation; however, it must be approached with greater attention.
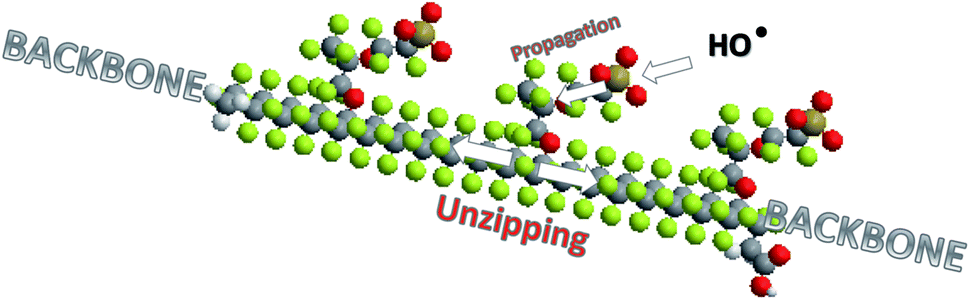 | ||
| Fig. 9 Degradation of the Nafion® structure by radical attack and unzipping of the side and main chains. | ||
Finally two possible sites of the radical attack in the side chain; C–S bond110,113,117,128 and α-OCF2 unit118,119 are suggested in the literature. There is no agreement concerning the dominance of the radical attack on one of these sites however in both scenarios the whole moiety OCF2CF2SO3− is lost. According to Ishimoto et al. the impact of the relative humidity on attack of hydroxyl radical on either ether or sulfonic sides has a great importance.129 Based on DFT calculations these authors concluded that HO˙ attack the ether group under high humidity conditions whereas sulfonic acid groups are more susceptible to radical attack at low relative humidity.
5. Hydrogen peroxide formation in membrane-electrode assemblies (MEA)
The understanding of the different possibilities for radical attack at weakest points in the polymer chain is essential in order to decrease membrane decomposition by chemically stabilising the vulnerable groups in the polymer structure. However it is also important to identify the fragile points of the complete membrane electrode assembly (MEA) with regard to chemical degradation on a macroscopic level. For that reason many researchers have attempted to localise the formation of highly oxidative species in an operating fuel cell. As stated above, it is believed that the main source of hydroxyl radicals is hydrogen peroxide, which is generated in the fuel cell and which has been observed in the membrane,87 exhaust water from the anode and cathode sides and in cathode outlet gas.60,130 The concentration of the H2O2 depends largely on the operating conditions as well as on the membrane thickness. During in situ experiments 8–10 μg cm−3 of hydrogen peroxide was determined in the membrane after 2 h of operation of the fuel cell at 65 °C with H2/O2 as reactants.131 H2O2 can be formed within an operating fuel cell at the cathode side by direct generation during the oxygen reduction reaction (ORR) when the potential is below 0.696 V.| O2 + 2H+ + 2e− → H2O2 | (10) |
| O2 + 4H+ + 4e− → 2H2O | (11) |
Formation of H2O2 at the anode side was first described by LaConti et al.132 who proposed that crossover oxygen can react with hydrogen which was chemisorbed on the Pt catalyst as displayed in eqn (12)–(14).133,134 Another mechanism is the chemical combination of crossover gas,135 where reaction on platinum at the anode, cathode or present in the membrane following degradation of the electrode catalyst layer are all possible.60,86,136
| H2 → 2H˙ | (12) |
| H˙ + O2 → HOO˙ | (13) |
| HOO˙ + H+ + e− → H2O2 | (14) |
Regardless of the exact location of hydrogen peroxide formation, the platinum catalyst is a constant factor in that process. Pt or Pt alloy is considered as being the best catalyst for the hydrogen oxidation reaction and the cathodic oxygen reduction reaction for PEMFC application.137 However, besides the four-electron pathway for ORR leading to production of water as displayed in eqn (11), a partial two-electron reduction reaction to H2O2 also occurs (eqn (10)). Many experiments were developed to simulate hydrogen peroxide generation on Pt particles.138–140Ex situ evaluation of this mechanism using a rotating ring disc electrode (RRDE) demonstrates an increase in H2O2 formation with decreasing electrochemical surface area (ECSA) of the catalyst.138,141 ECSA corresponds to the catalyst active fraction for the electrode reactions and its decrease is due to platinum nanoparticles agglomeration, dissolution or inaccessibility. Therefore the elevated formation of H2O2 was explained by the lower probability of further reduction reaction of hydrogen peroxide re-adsorbed on the sparsely distributed active sites of the Pt particles. Moreover faster decrease in ECSA and further increase of the H2O2 fraction was reported for catalysts with low Pt loading.138,141,142 Adsorption of anions such as Cl−, Br− or of CO on the catalyst surface was reported to accelerate H2O2 formation.139,143 This is in agreement with DFT calculations where the generation of H2O2 and free radicals is expected to occur when the ORR is blocked by an adsorbed species. Four-electron ORR is kinetically promoted for oxygen molecules adsorbed at two Pt sites, in which case any hydrogen peroxide or hydroxyl radical generation is energetically unfavourable. However if O2 bonds with only one Pt site, and accessibility to other active sites is hindered by other adsorbed species, both H2O2 and reactive oxygen species can be easily generated.144,145
H2O2 generation during PEMFC operation can be affected by additional factors impossible to mimic ex situ. One such factor that needs to be considered regarding H2O2/radical formation and degradation mechanism is the relation of the gas crossover to membrane thickness and gas partial pressure.146,147 High oxygen partial pressure leads to high oxygen crossover through the membrane, which elevates the hydrogen peroxide generation at the anode catalyst according to LaConti. This finding was confirmed experimentally by many research groups,60,113,148,149 which have reported negligible membrane degradation in the absence of O2 crossover. The influence of membrane thickness on gas crossover was investigated by Chen et al., who studied different membranes with various thicknesses ranging from 150 to 280 μm and revealed that with the increase in the membrane thickness the concentration of H2O2 in the membrane decreases.131 Also relative humidity plays an important role in the formation of hydrogen peroxide: Sethuraman et al., described a decrease of the concentration of H2O2 in high water activity conditions (i.e. high humidity) in studies of the oxygen reduction reaction on the RRDE.150 Chen et al. reported an increase of H2O2 emission with decrease of relative humidity on both anode and cathode side, which he explained by a low membrane water content and an increase of gas partial pressure that led to higher gas crossover.131 Moreover higher concentration of H2O2 was observed on the anode site. For those reasons low humidity conditions are commonly used to accelerate the polymer degradation rate.78
Decomposition of hydrogen peroxide is one of the possible mechanisms for radical formation in an operating fuel cell. Taking into account only this route to generate reactive oxygen species (ROS), already three different possible areas of chemical attack need to be considered: anode side, cathode side and membrane bulk. Studies carried out in this area show conflicting conclusions. Some researchers propose propagation of the chemical attack from the anode side towards the membrane to the cathode side.151 Others observed that the predominant degradation is at the anode103,123,152–154 or the cathode side,89,98,155,156 while others, did not find any significant difference between anode or cathode membrane–electrode interfaces in term of chemical degradation.157 It is very difficult to distinguish the degradation phenomena as those arising from mechanical and chemical processes respectively.25 To facilitate the analysis of areas of membrane degradation, a bilayer catalyst coated membrane method was proposed.98,156 With such a configuration, post-test analysis could be performed on the detachable membrane and the MEA centre could be easily identified. This method, resulting in membrane thickness much higher than usually applied, can be replaced by using commercially available thin reinforced PFSA membranes such as Nafion XL or GORE-SELECT. The middle of those membranes is clearly determined by the ePTFE reinforcement. Still the difficulty in obtaining answers to the questions on where exactly the chemical attack occurred, and which mechanism dominates in PEM membrane failure, is due to the morphology of the Nafion® membrane, the short life time and the low concentration of the radical species, but also, more importantly, due to the different conditions applied for accelerating the degradation process. Accelerated stress tests (AST) are commonly used in order to avoid thousands of hours of in situ life test to determine membrane durability; however that usually introduces some incertitude as to the relevancy of the methods employed. As an example, open circuit voltage (OCV) hold test is, currently, the most applied in situ stress test to assess the chemical stability and provide better understanding of fuel cell failure mechanisms. It was reported that OCV hold accelerates to greater or lesser extent, depending on the conditions used, such parameters as ECSA losses, voltage decay and gas crossover in comparison to long duration operation.158 The degradation rates calculated from accelerated stress tests tend to be higher than those obtained from a lifetime test. Moreover the H2O2 and concentration profile of the ferrous (Fe2+) at OCV condition also differ from those in normal working mode.159 At OCV, hydrogen peroxide production at the cathode side can be neglected due to high cathode potential and lack of faradic current.80
6. Gas crossover and membrane degradation
Gas crossover is an inevitable process in an operating PEMFC as typically applied electrolyte membranes have a thickness of 20–50 μm. Greater thickness could decrease the permeation of the reactants across the membrane,87,131 however it simultaneously increases the membrane area specific resistance. It should be mentioned that besides the fact of the inevitability of gas crossover, this process is much more pronounced under open circuit conditions as there is no consumption of H2 and O2 through the electrochemical reactions.60 The key role of gas crossover in membrane degradation was demonstrated in series of ex situ experiments. Independently, using different experimental set-ups and methods of evaluation of membrane degradation, several researchers drew the conclusion that the polymer decomposition occurs only when O2, H2 and Pt coexist.113,135,148 That initiated further studies on hydrogen crossover investigated in an operating fuel cell. Inaba et al., in their work on membrane degradation at OCV, demonstrated the incontestable increase of hydrogen crossover with simultaneous drop of OCV and increase of fluoride emission.60 It was pointed out that measurement of the hydrogen permeation across the electrolyte might be a reliable indicator of membrane degradation.60,160 Other authors have investigated the influence of cell temperature, humidification (refer to Fig. 10) and gas pressure on hydrogen crossover. Clearly the H2 crossover current density increases with increasing temperature (from 40 to 80 °C), relative humidity (here from 40 to 80%) and (not included on Fig. 8) gas pressure. This finding was further confirmed by Baik et al. in similar range of temperature and relative humidity.161 Moreover a particular design of the bipolar plate allowed the measurement of the hydrogen crossover rate over a local area. The local variation of crossover (higher H2 crossover was measured near the inlet of the gas than near the outlet) was attributed to the partial pressure gradient of H2.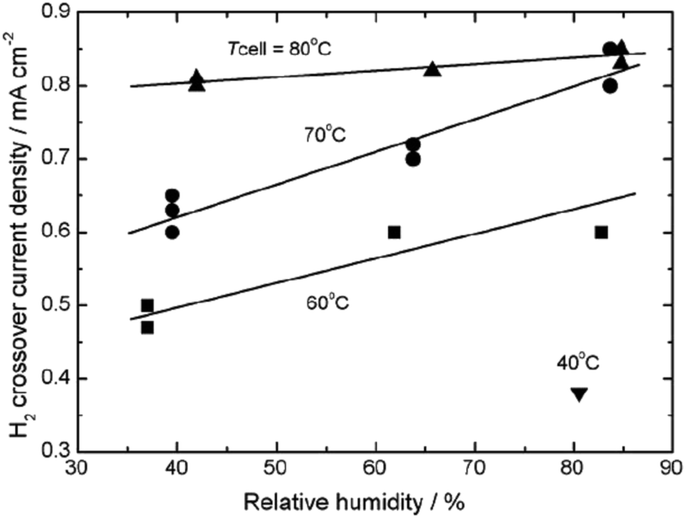 | ||
| Fig. 10 Effect of temperature and relative humidity on H2 crossover current density at atmospheric pressure. Reprinted from (vol. 51, M. Inaba et al., Gas crossover and membrane degradation in polymer electrolyte fuel cells, 5746–5753).60 Copyright (2006), with permission from Elsevier. | ||
As stated above, high temperature162 and gas pressure lead to elevated crossover rates and accelerate membrane degradation. For this reason, those parameters are typically applied in AST. However the influence of relative humidity on the mechanism of membrane chemical decomposition is much more complex. According to the previous discussion, H2 permeation increases with increasing relative humidity. This observation was interpreted using a multi-structural model of Nafion®, comprising an ion cluster region, an interfacial region and a fluorocarbon phase.163 Based on the assumption that H2 permeates through flexible, amorphous interfacial fluorocarbon regions,164 it was proposed that water absorbed by the membrane increases the size of the ion clusters, plasticizes the interfacial regions60 and enlarges the free volume165 where gas permeability can occur. Keeping in mind the relationship between H2 crossover and membrane degradation, the observation that low relative humidity166 or humidity cycling167 accelerates the polymer decomposition to larger extent than high relative humidity is rather surprising. This discrepancy clearly shows that for the mechanism of radical formation and membrane degradation, the degree of humidification itself is one of the critical factors. Inaba et al. argued that it might be related to a higher concentration of hydrogen peroxide under low RH,60 no experimental confirmation was provided. Some more light on this issue is given by research performed recently by Prabhakaran et al.168 These authors investigated formation of the reactive oxygen species (ROS) in an operating fuel cell using in situ fluorescence spectroscopy. Again a clear correlation between generation of ROS and PFSA membrane degradation was demonstrated under different fuel cell operating conditions. ROS formation was found to be enhanced at lower relative humidity (50% > 75% > 95%), elevated cell temperature (100 °C > 80 °C > 60 °C > 40 °C) and finally at higher cathode potential (0.8 V > 0.6 V > 0.4 V). An excellent graphical summary of these results is shown in Fig. 11, which plots the fluoride emission flux vs. the in situ radical formation rate for all conditions investigated.
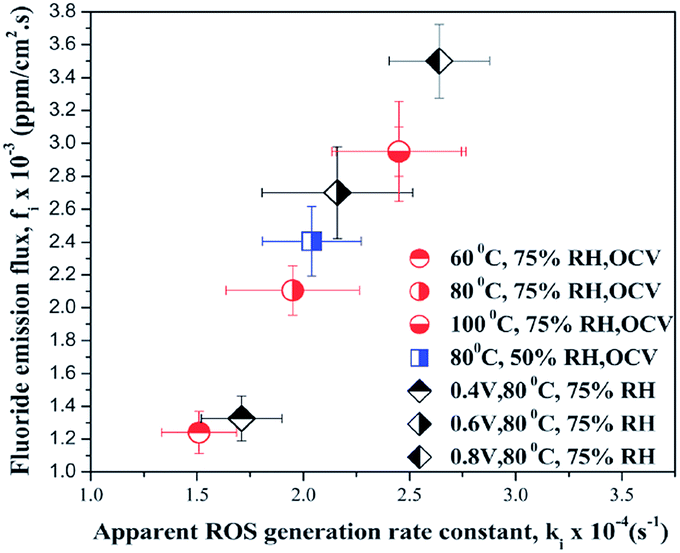 | ||
| Fig. 11 Correlation between in situ reactive oxygen species ROS formation rate and membrane degradation expressed as fluoride emission rate. Reproduced from ref. 168 with permission of the PCCP Owner Societies. | ||
This experiment validates the assumption of a high concentration of reactive oxygen species at low relative humidity and confirms the role of the high temperature and cathode potential as stressors for membrane degradation.
The mechanism of the membrane decomposition depends largely on the experimental conditions; however it always results in scission or cross-linking133 of polymer chains. Recent publications in this field show that attention is increasingly paid to chemical analysis of exhaust gas and water, since this analysis can provide information about the presence of three general groups of species considered from the perspective of membrane degradation. The first group consists of products of membrane decomposition such as fluoride ion,169,170 sulfate ion or polymer fragments.109,113,124,170 The second group are pollutants171,172 which act as degradation catalyst. Contaminants in a fuel cell set up originate from gas or water supplied to the system, from the cell hardware or the alloy catalysts. Gaseous impurities such as HCOOH, CO, NO2, SO2, H2S, Cl2 can cause blocking of the catalyst active sites, affect the ORR mechanism or accelerate Pt dissolution.173,174 Hydrated cations such as Fe2+, Fe3+, Cu2+ or Al3+ accelerate membrane degradation and cause performance losses.175,176 Also certain degradation compounds such as: in case of Nafion – perfluoro(2-methyl-3-oxa-5-sulfonic pentanoic) acid and 3M membranes – perfluoro(4-sulfonic butanoic) acid may migrate and adsorb into the catalyst layer leading to a loss of ECSA and ORR activity.177 The last group of species consists of free radicals, which are initiators but often also products of chemical degradation or of the decomposition of hydrogen peroxide.
7. Influence of Pt band on membrane degradation
The above discussion concludes that there are different contributions of the cathode and anode catalyst to the overall chemical degradation, however recently more attention has been focused on the influence of platinum in the membrane (PTIM) on the acceleration/diminution of its chemical degradation.136 One of the first reports concerning Pt band formation in PFSA membrane appears in 2005, when Ferreira observed the existence of a Pt band in the membrane after 2000 h of OCV hold test,12 and further studies revealed that a Pt band can be formed already after the first 50 h of OCV hold.136 The Pt catalyst is unstable in the acid environment of the fuel cell at high voltage and can dissolve at the cathode due to electrochemical oxidation to Ptz+ ions178,179 as displayed in eqn (15):180| Pt → Pt2+ + 2e− | (15) |
Then Pt ions migrate into the membrane due to the concentration gradient and undergo reduction in the presence of the crossover hydrogen according to the eqn (16).
| Pt2+ + H2 → Pt0 + 2H+ | (16) |
Pt atoms formed on that way (Pt0) can further diffuse and agglomerate in the membrane in order to minimise their high surface energy forming Pt particles.180
| (Pt)n + Pt0 → (Pt)n+1 | (17) |
Pt ions can also precipitate electrochemically at low potentials.
| (Pt)n + Pt2+ → (Pt)n+12+ | (18) |
| (Pt)n+12+ + H2 → (Pt)n+1 + 2H+ | (19) |
Such dynamic oxidation/dissolution and reduction/deposition processes results in Pt band formation. The Pt particle shape, size and distribution is strongly influenced by the composition and partial pressure of the crossover oxygen and hydrogen gases.10,180 Kim et al. studied the location of the Pt band in a Nafion® membrane with 25 μm thickness and a ePTFE reinforcement incorporated at the centre.181 A Pt band was observed between 1 and 10 μm from the cathode membrane interface, where oxygen is typically depleted. These authors reported a greater deposition of Pt nanoparticles in the polymer structure under OCV hold test conditions than under constant current operation. Similarly OCV hold was reported to accelerate polymer degradation. It was proposed that the presence and location of the platinum band might correlate with the formation of highly reactive oxygen species.182 Hatanaka et al. carried out studies with pristine Nafion® and with a membrane where a Pt-band had been intentionally introduced.183 After the OCV hold testing, the FER of the membrane containing platinum was clearly higher than that of the pristine membrane. Ohma et al. employed micro-Raman spectroscopy to investigate the degradation profile of in situ aged membranes and found enhanced degradation of the polymer around the Pt band.14,15 Also post-mortem SEM study of an aged MEA revealed membrane thinning at the cathode site in presence of PTIM and conductive AFM measurements showed electronic connections across the membrane.184 These results were interpreted as formation of electronic short-circuits due to accumulation of Pt particles. Studies of Liu and co-workers revealed an increased degradation when a membrane exposed to H2O2 solution was Pt catalyst coated in comparison to a bare membrane in an H2O2 flow cell experiment.135 Hydrogen peroxide formed at the electrode surface can potentially decompose to hydroxyl radicals on the active Pt sites,186 and free radicals can also be directly generated from the crossover gases on the platinum particles precipitated in the membrane (Fig. 12).137,187 Evidence for such a mechanism was provided by the work of Kim et al. where formation of carbon radicals on the Pt particles was confirmed by using electron spin resonance.187 Ohguri also demonstrated the formation of HO˙ in the presence of dispersed Pt particles83 however it is necessary to keep in mind that metallic platinum is a less active Fenton catalyst than iron and therefore direct formation of HO˙ radical is much less effective.188
 | ||
| Fig. 12 Formation of free radicals on Pt and the Pt dissolution mechanism. Reprinted from (vol. 195, D. Zhao, B. L. Yi, H. M. Zhang and M. Liu, The effect of platinum in a Nafion® membrane on the durability of the membrane under fuel cell conditions, 4606–4612).185 Copyright (2010), with permission from Elsevier. | ||
Pt(II) species present within the membrane can lead to the generation of free radicals136 and can affect the membrane physical properties due to the interaction with the perfluorosulfonic groups resulting in loss of conductivity and membrane stiffening.189 The lowest fluoride emission rate153 and no Pt band formation was observed for MEAs with air supplied to the cathode;14,190 on the contrary to induce PTIM formation in the membrane pure oxygen is commonly used as the reactant gas.156 All those findings confirm the destructive effect of the Pt particles deposited in the membrane on its degradation.
On the other hand, some studies show no correlation between the presence of the PTIM and membrane degradation152 or even a decrease of PFSA ionomer degradation (lower fluoride emission rate) of Pt-containing membranes in comparison to pristine polymer, due to the scavenging of hydrogen peroxide and hydroxyl radicals by Pt particles.148,152,191 Macauley et al. reported enhanced stability of a field-operated MEA with a PTIM compared to a freshly prepared MEA. The pristine membrane showed higher OCV decay and fluoride emission rate after 10 hours of AST cycle as well as a decrease in IEC after exposure to Fenton's reagent.191 This group developed an Accelerated Membrane Durability Test (AMDT) protocol to mimic membrane degradation during heavy duty fuel cell operation.192 Once again PTIM prolonged MEA life-time under these AMDT conditions. Peron et al. suggested that this discrepancy arises from coexistence in the membrane of different Pt species such as Pt(0), Pt(II) or Pt(IV). While metallic platinum might act as radical quencher, Pt(II) catalyses the hydrogen peroxide decomposition and radical formation. Later Gummalla et al. explained these nonlinear trends by two competitive reactions which can occur on the Pt surface: production and deactivation of free radicals.186 In that context such parameters like: the Pt particle size,186 shape,193 loading and spacing186,194,195 (see Fig. 13) define its potential radical scavenging or generating properties.
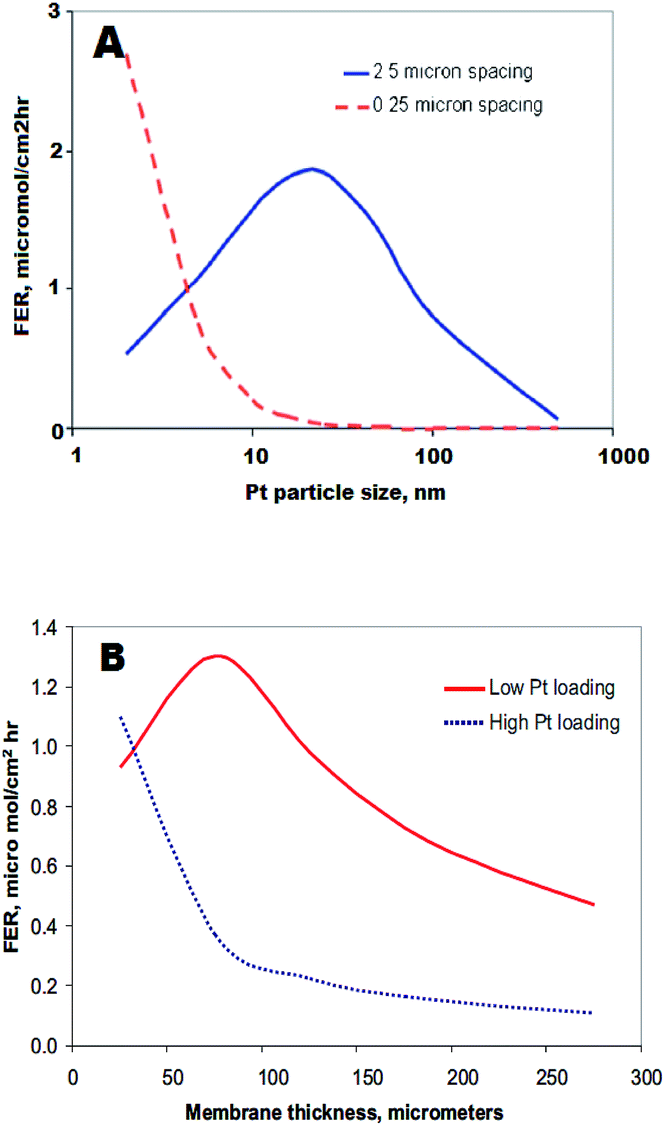 | ||
| Fig. 13 The effect of membrane thickness and Pt particle size on FER for different platinum loadings (B) and spacing (A) in the membrane. Reproduced by permission of The Electrochemical Society from ref. 186. | ||
Furthermore those parameters combined with temperature, level of hydration or concentration of crossover gases appear as crucial key factors impacting polymer decomposition. The proposed model finds confirmation in recently performed studies, on the degradation of a Pt-containing membrane.195 Rodgers et al. studied the effect of precipitated platinum with concentrations of 0, 10, 30 and 50 mol% on membrane degradation. The highest FER with similar magnitude to that observed with Pt/C catalyst coated on the polymer, was found for a membrane containing 10 mol% Pt. In contrast, samples containing 0, 30 and 50 mol% Pt gave very low FER. It was proposed that a dense distribution of Pt particles in the two last membrane samples, enabled the deactivation of the generated radicals prior to the polymer decomposition.196 Finally, non-platinum-group-metal ORR catalysts are being increasingly investigated as an option for fuel cell application. However, catalysts with Fe or Co and N species anchored on carbon substrates (Fe/Co–N–C) have to be considered in the context of PFSA membrane degradation. Fe/Co–N–C catalysts have low stability197 and suffer from dissolution/leaching of (Fenton active) metal, oxidative attack of hydrogen peroxide and protonation of active sites on neighbouring N species.198 All three mechanisms not only cause a strong decrease in catalyst activity, but also threaten the integrity of PFSA membranes.199 Surprisingly little work has been carried out on membrane degradation in the presence of non-noble metal catalysts, and well designed in situ accelerated stress testing is required to develop proper understanding.
8. Radical types, reactivity and lifetime
After examining the degradation mechanisms in the context of the polymer structure susceptible to radical attack, as well as H2O2 formation area and conditions, in this section we will consider the radical types and their generation in a fuel cell in more detail, up to this point the membrane degradation was described mainly as hydroxyl radical HO˙ attack on polymer structure. However in a working fuel cell H˙ and HOO˙ radicals are present in concentrations much higher than those of HO˙ which, once generated from hydrogen peroxide, is further consumed to produce HOO˙ or scavenged by crossover gases. An excellent summary of reactions likely taking place in a fuel cell (Table 1) was given by Gubler et al.81| Reaction | Rate constant (M−1 s−1) | |
|---|---|---|
| 1 | HO˙ + RfCF2SO3H → unzipping | — |
| 2 | HO˙ + RfCF2COOH → unzipping | <106 |
| 3 | H2O2 → 2HO˙ | 1.2 × 10−7 |
| 4 | HO˙ + H2O2 → HOO˙ + H2O | 2.7 × 107 |
| 5 | HOO˙ + H2O2 → HO˙ + H2O + O2 | ≤1 (reasonable upper limit value) |
| 6 | HO˙ + H2 → H˙ + H2O | 4.3 × 107 |
| 7 | H˙ + O2 → HOO˙ | 1.2 × 1010 |
| 8 | 2HOO˙ → H2O2 + O2 | 8.6 × 105 |
| 9 | Fe2+ + H2O2 + H+ → Fe3+ + HO˙ + H2O | 63 |
| 10 | Fe2+ + HO˙ + H+ → Fe3+ + H2O | 2.3 × 108 |
| 11 | Fe2+ + HOO˙ + H+ → Fe3+ + H2O2 | 1.2 × 106 |
| 12 | Fe3+ + HOO˙ → Fe2+ + O2 + H+ | 2 × 104 |
| 13 | Fe3+ + H2O2 → Fe2+ + HOO˙ + H+ | 4 × 10−5 |
This inventory of relevant mechanisms helped to create simulation of possible radical attack. Simulation based on the reaction (2) represents the chemical attack of the hydroxyl radical on the terminal –COOH groups, i.e. the “unzipping mechanism” discussed in the previous section. In their studies of the kinetics of membrane degradation, Gubler et al. did not take into consideration the radical attack on the side chain (cited previously as mechanisms II and III and represented in the table by reaction (1)) due to the lack of information on the kinetics of these reactions in the literature. Two sets of reactions used by Gubler et al. in their studies can be distinguished with hydrogen peroxide as the precursor for the radical intermediates.
Reactions (3)–(8) describe the homolytic dissociation of H2O2 (reaction (3)) and consumption of free radicals originating from H2O2 in the absence of iron catalyst. Reaction (3) occurs with very low rate constant approximately 1.2 × 10−7 M−1 s−1. As displayed in eqn (4), HO˙ reacts with hydrogen peroxide, to form HOO˙, while reaction (5) follows a similar pathway but in this case HOO˙ is the reactant for the generation of hydroxyl radicals. Furthermore the reaction of H˙ and HO˙ radicals with crossover gases in a fuel cell follows the eqn (6) and (7). Finally hydrogen peroxide can be regenerated as described by eqn (8) due to HOO˙ disproportionation. However in real working fuel cell conditions, reactions (9)–(13) are the most relevant, since H2O2 decomposition is catalysed by Fe ion impurities. Reaction (9) is widely known as the Fenton reaction and it is the primary source of hydroxyl radicals, where Fe2+ is the reducing agent regenerated after the reaction of Fe3+ with H2O2 and HOO˙. The rate constant of reaction (9) (here 63 M−1 s−1) is much higher that previously cited in the homolytic dissociation of hydrogen peroxide, which further means higher rate of HO˙ generation.
Indeed the simulation studies of the radical attack performed by Gubler showed higher rate of the ionomer attack under ex situ, Fenton reagent conditions, in comparison to that observed in an OCV hold test.81 It is most likely due to a higher concentration of hydroxyl radicals. Moreover, based on the kinetic model the theoretically estimated fluoride emission rate after Fenton aging was in good agreement with FER reported in the literature.100,150,200 However a similar FER estimation for in situ accelerated tests were 2–3 orders of magnitude lower than the literature values.91,188 The differences between the in situ measurement and the kinetic simulation might be due to the absence in the provided model of reaction (1), which describes the side chain decomposition. Moreover two other factors were not included in Gubler's model i.e. the influence of the direct formation of free radicals on Pt nanoparticles in the membrane and the influence of the electrode surface on membrane degradation. A large difference between simulated and experimental values of FER on OCV hold indicates the substantial role of these factors on the chemical decomposition of the ionomer membrane in the MEA.
Another important issue is the oxidative strength of free radicals. The most cited radical with regard to chemical degradation is the hydroxyl radical HO˙. This is due to the ranking of the reactivity of the oxidative species, where the hydroxyl radical takes a first position: HO˙ > H˙ > HOO˙ > H2O2.80 High oxidising power of HO˙ is reflected in higher reduction potential of HO˙ in comparison to the hydroperoxyl radical and hydrogen peroxide (Table 2).
| Half-cell reaction | Standard electrode potential (V) | |
|---|---|---|
| 1 | HO˙ + H+ + e− → H2O | 2.59 |
| 2 | H˙ + H+ + e− → H2 | 2.32 (ref. 80) |
| 3 | HOO˙ + H+ + e− → H2O2 | 1.48 |
| 4 | H2O2 + 2H+ + 2e− → 2H2O | 1.74 |
Similarly, the difference in the rate of hydrogen abstraction can be estimated based on the experimental bond strength.117 As the O–H bond in water is very strong, the hydroxyl radical has strong thermodynamic driving force (497.9 kJ mol−1) to abstract the hydrogen and form an H2O molecule. In contrast it is very unlikely that hydrogen peroxide or HOO˙ will cause hydrogen abstraction with direct detrimental effect on the PFSA membrane. The main role of those species is therefore as a source of much more reactive HO˙.
9. Impact of operating conditions on PFSA membrane chemical degradation
The evaluation of membrane durability in an operating fuel cell is challenging since many factors including operation temperature, pressure, humidity, operating voltage and current, start-up/shut-down conditions, the composition and design of other cell components and the effect of contaminants have to be considered. The synergetic effect of all these factors in operating fuel cell make identification of the local phenomena leading to membrane chemical decomposition quite complex. Test protocols have been designed to limit certain phenomena and exacerbate others, and furthermore harmonized methods provide the basis for comparing results and facilitate assessment of technology status. In real operation these conditions are rarely separated and performance losses can arise from pinhole formation201 following mechanical/chemical membrane degradation cycling, while RH fluctuations, temperature and loading cycling and start-up, shut-down can all cause membrane dimensional change, and lead to plastic deformation, cracks and pinhole formation. Following pinhole formation, high hydrogen crossover causes accelerated radical formation and further polymer decomposition. The degradation pathway continues until the membrane, thinned and weakened by radical attack membrane, can no longer withstand mechanical stress when, regardless of the decomposition mode by chemical attack or shear forces, the consequence is a MEA failure (Scheme 1).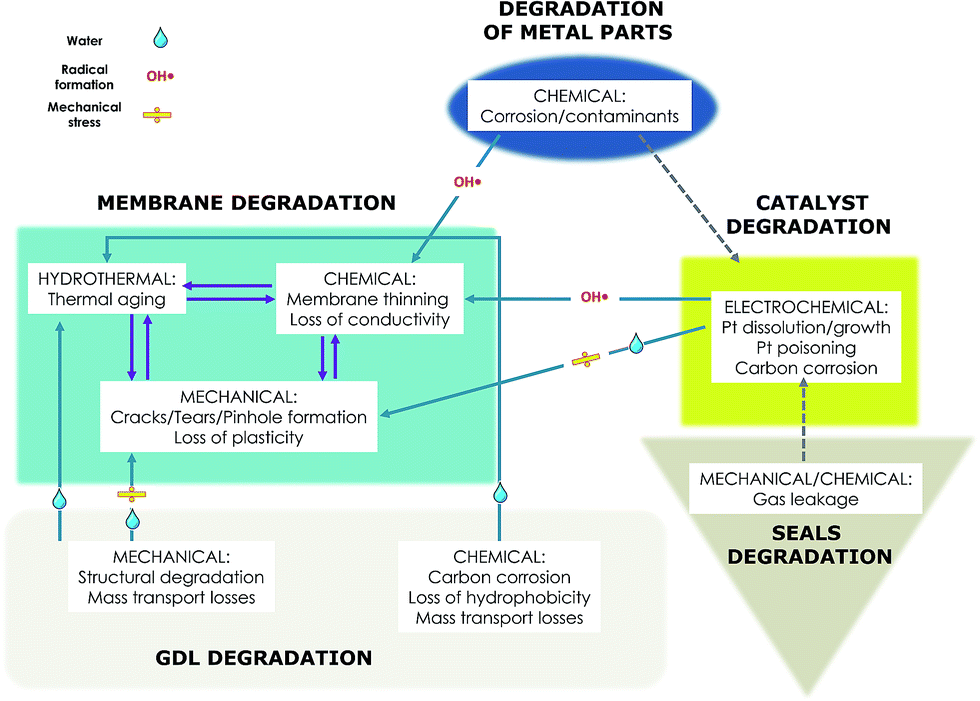 | ||
| Scheme 1 Factors impacting PFSA fuel cell membrane degradation. | ||
10. Mitigation of membrane degradation
The foregoing discussion shows that degradation of PFSA membranes is currently considered to involve complex interconnected mechanical and chemical mechanisms. Due to the complexity of these factors contributing to the overall degradation, many different approaches have been developed to minimise polymer cleavage processes. The main mitigation strategies proposed in the literature include development of new reinforced or composite membranes, stabilisation of most vulnerable PFSA polymer groups, incorporation of a hydrogen peroxide decomposition catalyst or radical quencher in the membrane/electrode, development of new electrode/GDL material.10.1 Development of new reinforced or composite membranes
Preparation of a reinforced material was proposed to improve the durability of the PFSA polymer.107,202–205 In general two widely investigated routes to achieve this goal can be distinguished: chemical modification of the polymer structure and physical reinforcement.203 Chemical modifications include membrane annealing206–212 uniaxial stretching213,214 and chemical cross linking of the polymer,203,215 these were used to reduce swelling of the membrane and improve mechanical durability. Annealing of PFSA polymers leads to more efficient chain packing and higher polymer crystallinity. Thermal treatment is extremely important, especially for a solvent cast membrane, as the annealing procedure improves intrinsic properties of the polymer and makes them similar to those of an extruded membrane. Treatment by uniaxial stretching has been demonstrated to effectively increase Young's modulus, reduce area swelling and slightly increase proton conductivity. Other chemical modifications such as polymer cross linking can be carried out by conversion of the sulfonic acid side chain however such method leads to inevitably high conductivity losses and therefore was not widely studied. Incorporation in the polymer structure of new cross-linkable functional groups appears as the more appropriate method and was performed with sulfonyl fluoride and sulfonamide216,217 or sulfonyl fluoride and 3-aminopropyltriethoxysilane (APrTEOS) as reactants. A different method based on pendant alkyl bromide group, which can be cross linked by thermal treatment or electron beam irradiation has also been investigated.218 In general new cross linked materials show reduced dimensional swelling and higher mechanical stability in comparison to non-reinforced PFSA membranes. However a similar effect can be achieved using physical reinforcements, in which the membrane preparation is further simplified. To fabricate a reinforced composite membrane, a highly mechanically stable organic or inorganic matrix is impregnated or otherwise associated with the PFSA dispersion. A large spectrum of membrane reinforcements has been developed, including expanded porous PTFE sheets204,219–221 or fibrils as well as nanofibre mats prepared by electrospinning of polymers such as poly(vinylidene fluoride) (PVDF),222,223 poly(vinyl alcohol) (PVA),223,224 polybenzimidazole (PBI),225 poly(phenyl sulfone), UHMWPE (ultrahigh molecular weight polyethylene)226 or nanofiber network silicon carbide SiC.227 Among these, low cost expanded PTFE (ePTFE) supports have been particularly successful. ePTFE reinforced membranes are usually prepared by soak or spray methods.228 The ionomer fills the pores of the thin ePTFE film and forms a layer of ionomer on either side of PTFE mat. The advantages of using PTFE as the membrane support include the reduction of membrane cost228 and improved dimensional stability and durability of composite membrane229 when subjected to mechanical accelerated tests such as freeze/thaw or, humidity cycling230,231 as PTFE suppresses swelling of PFSA electrolyte within the porous matrix. However, PTFE/PFSA membranes demonstrate higher proton transport resistivity than the respective non modified membranes.228 This negative effect can be minimized by applying ultrathin PTFE mats with high porosity,232,233 or by hydrophilic treatment of hydrophobic PTFE structure.230 Such treatment improves the wettability and adhesion of more hydrophilic polymer which in turn allows good interfacial compatibility. However according to Kundu et al. the ePTFE reinforced PFSA membranes undergo accelerated chemical degradation at low relative humidities 20–50%.166 Thus increased mechanical durability of the membrane might extend lifetime but not suppress chemical failure. Inorganic fillers resulting in nanocomposite membranes like Nafion® – SiO2,234–238 Nafion® – TiO2,236,238–242, Nafion® – SnO2,243–246 Nafion® – ZrO2 (ref. 236, 238, 247 and 248) or Nafion® – ZrP248–251 deserved special attention. Incorporation of inorganic particles can not only enhance the mechanical properties of the PFSA membranes, but also increase the water retention, which allows higher performance at elevated operating temperatures and low relative humidity.249 Furthermore inorganic particles can be successfully applied as a hydrogen peroxide decomposition catalyst or radical scavenger, as will be discussed in the later section of this review. PFSA-inorganic nanoparticles composite membranes can be prepared either from a dispersion of oxide particles in the ionomer solution prior to membrane casting or by in situ generation of inorganic particles within the membrane/ionomer through a sol–gel-like process. The properties of such prepared composite membranes depend to large extent on the type of the polymer, the particle size, their dispersion through the membrane and any preferential orientation, in the case of inorganic particles with large aspect ratio.25210.2 Stabilisation of most vulnerable PFSA polymer groups – development of SSC PFSA membrane
Stabilisation of the most vulnerable groups of the PFSA polymer is another method to mitigate membrane degradation. It was discussed previously in connection with different variants of chemical decomposition and should be considered in relation to the mechanism of free radical attacks on the polymer structure. Nafion® with chemically stabilised carboxylic acid end groups – the most susceptible point in polymer – shows reduced but not eliminated mass loss. Lower fluoride emission of chemically stabilised Nafion® is due to limited participation of the unzipping mechanism in the overall decomposition reaction, however weight decrease is still noticeable and it is related to the continuously occurring side chain degradation with tertiary carbon, ether bonded carbon and/or sulfonic acid groups as the points of initial attack as these are the most vulnerable points. Although the development of a perfluorinated ionomer with a shorter pendant side chain than Nafion®, the so-called short side chain (SSC) ionomer pre-dates understanding of membrane degradation, the short side structure is expected to bring several advantages regarding membrane durability. Elimination from the side chain structure of the tertiary carbon (–CF) and one of the ether bonded carbon atoms (–O–CF2) decreases the susceptibility of the SSC membrane to radical attack, a finding that was confirmed in several studies.100,108,149 More recently attention has been drawn to low EW multi-acid side chain proton exchange membranes developed by 3M.253–256 In these new materials high number of tetrafluoroethylene units in the backbone structure has been combined with side chains which carry more than one acid site. 3M perfluoro-imide acid (PFIA) membranes have high acid content while preserving excellent mechanical stability when compared to similar EW PFSA membranes.253 Although fundamental studies regarding mechanical, conductive properties of 3M PFIA membranes have been carried out no reports have been published so far on PFIA durability. Only conclusion which might be drawn today is that both the SSC (including PFIA) and long side chain (LSC) membranes have a sulfonic acid as functional ion transporting groups, therefore they both can suffer the decomposition which begins under certain conditions at the C–S bonds. To mitigate degradation of the side chain, incorporation of a free radical scavenger as a chemical trap for highly reactive oxygen species was proposed.10.3 Incorporation into membrane/electrode hydrogen peroxide decomposition catalyst (HPDC) or radical quencher
In this manner we come to the highly effective approach to minimize the chemical degradation, which involves incorporation, into the membrane electrode assembly, of species with peroxide decomposition or radical scavenging properties. This concept has been investigated already before 2005. Convincing results in this area have led to the increased interest of many researchers and several patents.257–262 Examples of hydrogen peroxide decomposition catalysts (HPDC) applied for mitigation of membrane degradation are: metal oxides like TiO2,239,263–265 MnO2 (ref. 266) and ZrO2,200 heteropoly acids,267–270 carbon–tungsten oxide (C/WO3) or carbon–phosphotungstic acid (Pt/C-PTA).271,272 Metal particles such as Ce,108,273–277 Mn,274,276,277 Pd, Ag, Au or Pt,278 metal oxides CeO2,279–289 MnO2,290,291 SnO2 (ref. 292 and 293) and terephthalic acid (TPA)294 also demonstrate ability to mitigate chemical degradation.The main role of HPDC is to lower the H2O2 concentration and indirectly decrease the formation of free radicals. In contrast introduction of a regenerative radical scavenger into MEA components causes direct reduction of the concentration of already created highly oxidative species. For these reasons two potential areas for HPDC/radical scavenger incorporation may be distinguished: the polymer membrane and the electrode. In the first case, preparation of a composite membrane was accomplished by mixing the HPDC/radical scavenger nanoparticles or chemical compounds with the ionomer suspension prior to formation of the membrane. In this approach, hydrogen peroxide can be neutralised only after its diffusion into the polymer membrane. In the work of Xiao et al., zirconia particles were prepared by a hydrothermal method and incorporated in the Nafion® membrane. The resulting ZrO2 composite membrane was examined under OCV hold (80 °C; 50% RH; H2/O2) and Fenton's test conditions. Accelerated stress tests show reduced FER (by up to an order of magnitude) and thinning, relative to native Nafion®.200 As zirconium oxide enhances the membrane durability (due to its radical scavenging ability) and water retention at high temperature and low relative humidity, development of composite ZrO2/PFSA membranes is an interesting approach to mitigate mechanical and chemical degradation. A similar concept to reduce both mechanical and chemical degradation was investigated by Patil et al. by using a TiO2/Nafion® composite membrane.265 A titania “quasi-network” in the polymer structure was developed by in situ sol–gel polymerisation of titanium isopropoxide. The EW of the modified membrane remains unaltered, however the hydration capacity decreased in relation to the pristine Nafion®. The authors postulated that the incorporation of small (nano size) and evenly dispersed particles, placed along the gas diffusion pathways, potentially increase tortuosity and reduce fuel crossover. Additionally, if the titania particles form a slightly interconnected network, due to the presence of weak metal oxide bonds, they can constitute a mechanical membrane reinforcement.252 Indeed the modification provides significant dimensional membrane stability (higher modulus and lower creep) and minimised voltage loss at OCV hold test.239 It should be noted that use of ZrO2 and TiO2 as additives for PFSA membranes was carried out with the aim of improving their water-retention and mechanical properties, and the radical scavenging effect of those compounds was discovered later. Similarly heteropolyacid/PFSA membranes were initially developed to maintain proton conductivity of membrane at elevated temperature and low relative humidity conditions.295–298
Haugen et al., investigated the durability of 3M membrane modified by the addition of various heteropolyacids (HPAs) as HPDC.267 Of these, λ-H3P2W18O62, H6P2W21O71 and H4SiW12O40 significantly reduced the amount of fluorine released under accelerated stress test conditions. Incorporation of the HPAs enhanced performance of the MEA in a narrow concentration range of HPA. The efficacy of the mitigation properties of metal nanoparticles such as Au, Ag, Pt and Pd in Nafion® was studied by Trogadas et al.278 The idea of using those elements as radical scavengers was first provided by their known activities in biology as antioxidant species. Composite membranes were prepared by casting dispersions with 3 wt% metals loading. Such a metal content lowers the conductivity of Nafion® due to dissolution of the metal particles above a certain potential i.e. >0.4 V vs. SHE for Ag and >0.8 V vs. SHE for Pd, and the formation of ionic species that exchange with protons. Platinum particles were an exception, where in situ water production on Pt surface compensates metal dissolution and ion exchange with the sulfonic acid groups. Regarding the radical scavenging properties of the incorporated metals, the addition of Ag, Pt, Pd and Au nanoparticles decreased the fluoride emission rate by 35%, 60%, 80% and 90% respectively. To minimise the impact of metal dissolution on membrane conductivity or further performance, metal particles were supported on silica, SiO2. Indeed, the conductivity of the composite membrane with SiO2-supported metal particles increased significantly and reached values similar to the reference Nafion®. The impact of silica incorporation was especially visible for SiO2/Pt Nafion® membrane. However a lower concentration and simultaneously larger size of the supported metal nanoparticles brought considerable FER increase. α-Tocopherol (α-TOH) is other natural antioxidant (vitamin E component) which, when embedded into Nafion® prevented membrane degradation and performance losses.299 α-TOH acts as a trap for hydroxyl and peroxyl radicals once oxidized, α-TOH can be further reduced by crossover hydrogen during fuel cell operation. The reversibility of α-TO˙/α-TOH system assures its high efficiency as a radical scavenger. However the long-term stability of α-TOH incorporated into electrolyte is unknown. Recently Zhu et al. reported a new method to increase the chemical stability of PFSA membrane by the incorporation of an organic radical scavenger – terephthalic acid (TPA).294,300Fig. 14 displays a possible mechanism of the reaction of TPA with a hydroxyl radical. The generated intermediate, hydroxycyclohexadienyl radical can either undergo a disproportionation reaction to form hydroxylated terephthalate and regenerate TPA, or can be catalysed by an Fe3+ ion to form the hydroxylated terephthalate.
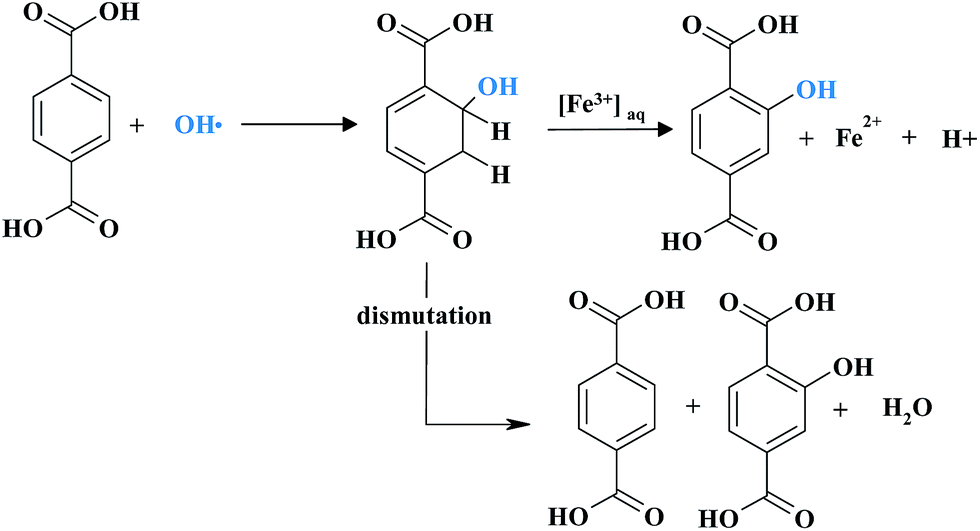 | ||
| Fig. 14 Mechanism of HO˙ radical trapping process by TPA proposed by Zhu. Reprinted from (vol. 432, Y. Zhu, S. Pei, J. Tang, H. Li, L. Wang, W. Z. Yuan and Y. Zhang, Enhanced chemical durability of perfluorosulfonic acid membranes through incorporation of terephthalic acid as radical scavenger, 66–72).294 Copyright (2013), with permission from Elsevier. | ||
A composite TPA/PFSA membrane aged under ex situ Fenton test conditions shows higher durability than pristine PFSA membrane, which indicates HO˙ scavenging ability. However these promising results need to be further confirmed in situ in a fuel cell in order to verify the performance, stability and impact on other MEA components of this newly developed approach.
The second strategy involving the addition of peroxide decomposition catalysts or radical scavengers in the catalyst layers has the advantage that the H2O2 can be decomposed already at the catalyst surface. Typically a catalyst ink includes: carbon support, metal catalyst and ionomer. The ionomer can comprise an HPDC associated with anion groups in the ionomer salt form or suspended as particles in the catalyst ink. Trogadas et al. validated the second approach by preparing various hybrid catalysts such as C/WO3, C-PTA271 or Pt/C–MnO2,266 which were studied for their efficacy of hydrogen peroxide decomposition and mitigation of membrane degradation. RRDE measurements showed significant reduction in the concentration of H2O2 produced during ORR on these catalysts. Also in situ OCV hold tests of the MEAs with HPDC additives revealed decrease of the rate of membrane degradation which was reflected in lower fluoride emission rate. The best results in H2O2 decomposition were given by the incorporation of MnO2 particles with mixed valence state whereby they can undergo relatively easy reversible redox reaction with H2O2, moreover the free hydroxyl radicals – HO˙ can be scavenged at the same time. On the other hand, the performance of an MEA containing an MnO2 hybrid catalyst was inadequate, due to low stability of MnO2 in acidic media at high potentials. Recently developed MnO2 nanotubes and nanowires in small content did not affect the available catalyst area.290 The performance of manganese oxide doped catalyst layer measured at 100% RH was similar to that with a conventional catalyst material. Interestingly for lower RH values, incorporation of 1D MnO2 decreased the sensitivity of the fuel cell to variation in hydration level. Moreover, OCV hold degradation tests have shown much lower OCV decay with an MnO2 composite catalyst in comparison to commercial material especially if incorporated on the anode side (see Table 3) Tacconi et al. developed a Pt/C–TiO2 composite catalyst by using photocatalytic synthesis,263 and found that the incorporation of TiO2 in the catalyst layer brings significant increase to the electrode and membrane durability with comparable performance of a Pt/C–TiO2 composite for oxygen reduction reaction to that with a commercial catalyst. Finally, Brooker et al.268 incorporated adsorbed HPAs on high surface area carbon at the membrane–electrode interface. After OCV hold testing, a cell with a sub-layer enriched with phosphotungstic acid (PW12O43−) showed decreased OCV loss and fluoride release of around 50% over that of the non-HPA sub-layer cell. However the inclusion of the carbon sub-layers significantly increased the electrode ohmic and diffusion losses as well as OCV losses in comparison to the cell with no sub-layer. To overcome these problems Chen et al. impregnated catalyst layers of CCM with phosphotungstic acid (PTA).272 Composite PTA-CCM exhibited better performance (especially at high current density) and stability than a standard non modified CCM. The power density of PTA-CCM decreased by 14% in comparison to 33% of non-modified CCM after 100 h aging, and the authors related these results to scavenging properties of PTA and low degradation of ionomer in catalyst layer.
| REF | Form/location | Content | Accelerated durability test | |
|---|---|---|---|---|
| Ex situ ageing | In situ ageing | |||
| 271 | Hybrid catalyst | 15 wt% | RRDE | OCV: 90 °C 50% RH O2/H2 |
| C/WO3 | 30–40% reduction in the amount of H2O2 produced for 15 wt% C/WO3 and C/PTA versus Pt/C. | FER: (ppm) | ||
| C/PTA | Anode: | |||
| C – 1.3 | ||||
| C/WO3 – 0.5 | ||||
| C/PTA–0.7 | ||||
| Cathode: | ||||
| C – 0.55 | ||||
| C/WO3 – 0.38 | ||||
| C/PTA–0.36 | ||||
| 266 | Hybrid catalyst | 5 wt% | RRDE | OCV: 90 °C 50% RH Air/H2 |
| FER: (ppm) | ||||
| CATHODE | 50–60% reduction in the amount of H2O2 produced for 5 wt% Pt/C/MnO2 versus Pt/C. | Anode: | ||
| Pt/C/MnO2 | Pt/C−1.1 | |||
| Pt/C/MnO2−0.22 | ||||
| Cathode: | ||||
| Pt/C−0.84 | ||||
| Pt/C/MnO2−0.59 | ||||
| 290 | Hybrid catalyst | – | OCV: 60 °C 75% RH Air/H2 decay: (μV min−1) | |
| CATHODE/ ANODE | Pt/C−51.2 | |||
| Pt/C/MnO2 | Cathode : Pt/C/MnO2 − nanowire − 39.4 | |||
| Cathode : Pt/C/MnO2 − nanotubes − 38.8 | ||||
| Anode : Pt/C/MnO2 − nanowire − 17 | ||||
| Anode : Pt/C/MnO2 − nanotubes − 18.6 | ||||
| 283 | Hybrid catalyst | 20 wt% | – | OCV: 90 °C 30% RH O2/H2 decay: (mV) |
| CATHODE | Pt/C − 0.72 | |||
| Pt/C/CeO2 | Cathode : Pt/C/MnO2 − 0.079 | |||
| 326 | Hybrid catalyst | TiO2 | – | FER: (μmol cm−2 h−1) |
| CATHODE | 0 wt% | ETEK (Pt 5 wt%) − 0.03 | ||
| Pt/C/TiO2 | 5 wt% | SIDCAT (Pt 5 wt%) − 0.3 | ||
| 5 wt% | SIDCAT (Pt 10 wt%) − 0.05 | |||
| 0 wt% | TKK (Pt 50 wt%) − 0.75 | |||
| 5 wt% | SIDCAT (Pt 50 wt%) − 0.06 | |||
| 10 wt% | SIDCAT (Pt 50 wt%) − 0.02 | |||
| 294 | Composite membrane | 0.5 wt% | Fenton test F− (%) | – |
| TPA/PFSA − 12% (48h) | ||||
| TPA/PFSA | PFSA − 14% (48h) | |||
| TPA/PFSA − 21% (120h) | ||||
| PFSA − 29% (120h) | ||||
| 278 | Nanocomposite membrane | 3 wt% | – | OCV: 90 °C 30% RH O2/H2 |
| FER:(μmol cm−2 h−1) | ||||
| Ag | Nafion − 0.41 | |||
| Pt | Nafion/Ag − 0.275 | |||
| Pd | Nafion/Pt − 0.18 | |||
| Au | Nafion/Au − 0.05 | |||
| Nafion/Pd − 0.08 | ||||
| 280 | Nanocomposite membrane | – | – | OCV: 90 °C 30% RH O2/H2 |
| – | FER: (μmol cm−2 h−1) | |||
| CeO2/Nafion® | CeO2: | Anode: | ||
| 0 wt% | Nafion® − 0.2 | |||
| 0, 5 wt% | Nafion®/CeO2 − 0.007 | |||
| 1 wt% | Nafion®/CeO2 − 0.006 | |||
| 1 wt% | Nafion®/CeO2(nc) − 0.005 (nc – non commercial) | |||
| 3 wt% | Nafion®/CeO2 − 0.008 | |||
| – | Cathode: | |||
| 0 wt% | Nafion® − 0.08 | |||
| 0,5 wt% | Nafion®/CeO2 − 0.006 | |||
| 1 wt% | Nafion®/CeO2 − 0.005 | |||
| 1 wt% | Nafion®/CeO2(nc) − 0.003 (nc – non commercial) | |||
| 3 wt% | Nafion®/CeO2 − 0.007 | |||
| 286 | Nanocomposite membrane | 1 wt% | – | OCV: 90 °C 30% RH Air/H2 FER: (μmol) |
| PFSA − 3800 | ||||
| CeO2/Nafion® | Ceria(nc) − 340 (nc – non commercial) | |||
| Ceria(com.) − 33 (com commercial) | ||||
| 287 | Nanocomposite membrane | CeO2: | Fenton test F− (μmol g−1) | – |
| 0 wt% | PFSA: 2600(LF) 2600(GF) | |||
| CeO2/Nafion® | 0.5 wt% | Ceria(nc): 500(LF) 1100(GF) | ||
| 1 wt% | Ceria(nc): 400(LF) 800(GF) | |||
| 2 wt% | Ceria(nc): 200(LF) 250(GF) | |||
| 0.5 wt% | Ceria(com.): 400(LF) 1600(GF) | |||
| 1 wt% | Ceria(com.): 350(LF) 700(GF) | |||
| 2 wt% | Ceria(com.): 100(LF) 350(GF) | |||
| (LF) – Fenton reaction in solution phase | ||||
| (GF) – Fenton reaction in gas phase | ||||
| 282 | Nanocomposite membrane | CeO2: | Fenton test F− (mg h−1) | OCV: 95 °C RH cycling Air/H2 decay: (mV s−1) |
| 0 wt% | Nafion − 55.78 | Nafion − 11.7 × 10−4 | ||
| CeO2/Nafion® | 1 wt% | Ceria(self assembled) − 43.05 | – | |
| 3 wt% | Ceria(self assembled) − 8.67 | – | ||
| 5 wt% | Ceria(self assembled) − 6.01 | Ceria(self assembled) − 1.13 × 10−4 | ||
| 10 wt% | Ceria(self assembled) − 4.47 | – | ||
| 5 wt% | Ceria(sol–gel) − 11.64 | Ceria(sol–gel) − 5.78 × 10−4 | ||
| 281 | Nanocomposite membrane | – | – | OCV: 90 °C 30% RH O2/H2 |
| Pt: | FER: (μmol cm−2 h−1) | |||
| Pt/CeO2/Nafion® | 0 wt% | Nafion® − 0.6 | ||
| – | Nafion®/CeO2 − 0.01 | |||
| 0.5 wt% | Nafion®/Pt/CeO2 − 0.007 | |||
| 1 wt% | Nafion®/Pt/CeO2 − 0.007 | |||
| 2 wt% | Nafion®/Pt/CeO2 − 0.007 | |||
| – | Nafion®/MnO2 − 0.008 | |||
| 327 | Nanocomposite membrane | 1 wt% | Fenton test F− (μmol/gh) | OCV: 80 °C 50% RH O2/H2 |
| Nafion®: 2.25 | FER: (μmol cm−2 h−1) | |||
| CsxH3−xPW12O40/CeO2 | Nafion®/CeO2: 0.6 | Anode: | ||
| H3−xPW12O40/CeO2: 0.5 | Nafion® − 0.6 | |||
| Nafion®/CeO2 − 0.4 | ||||
| H3−xPW12O40/CeO2 − 0.025 | ||||
| Cathode: | ||||
| Nafion® − 0.8 | ||||
| Nafion®/CeO2 − 0.5 | ||||
| H3−xPW12O40/CeO2 − 0.1 | ||||
| 264 | Nanocomposite membrane | 20 wt% | – | OCV: 90 °C 30% RH O2/H2 FER: (μg cm−2 h−1) |
| Anode: | ||||
| TiO2/Nafion® | Nafion® − 3.681 | |||
| TiO2/Nafion® − 0.354 | ||||
| Cathode: | ||||
| Nafion® − 4.98 | ||||
| TiO2/Nafion® − 0.1755 | ||||
| 265 | Nanocomposite membrane | 20 wt% | – | OCV: 100 °C 25% RH O2/H2 FER: (μg cm−2 h−1) |
| Anode: | ||||
| TiO2/Nafion® | Nafion® − 1.43 | |||
| TiO2/Nafion® − 0.07 | ||||
| Cathode: | ||||
| Nafion® − 1.46 | ||||
| TiO2/Nafion® − 0.12 | ||||
| 200 | Nanocomposite membrane | 3 wt% | Fenton test | OCV: 80 °C 50% RH O2/H2 |
| FER (μmol g−1 h−1) | FER: (μmol cm−2 h−1) | |||
| ZrO2/Nafion® | Anode: | |||
| Nafion® < 85 | Nafion® − 0.6 | |||
| ZrO2/Nafion® < 75 | ZrO2/Nafion® − 0.06 | |||
| Cathode: | ||||
| Nafion® − 0.8 | ||||
| ZrO2/Nafion® − 0.02 | ||||
| 291 | Nanocomposite membrane | 3 wt% | Fenton test | OCV: 90 °C 50% RH Air/H2 |
| FER (μmol g−1 h−1) | FER: (μmol cm−2 h−1) | |||
| MnO2/SiO2–SO3H | Anode: | |||
| Nafion® − 0.8 | Nafion® − 0.8 | |||
| MnO2/Nafion® − 0.04 | MnO2/Nafion® − 0.04 | |||
| MnO2/SiO2/Nafion® − 0.05 | MnO2/SiO2/Nafion® − 0.05 | |||
| Cathode: | ||||
| Nafion® − 1.1 | ||||
| MnO2/Nafion® − 0.2 | ||||
| MnO2/SiO2/Nafion® − 0.08 | ||||
| 279 | CeO2 free particles in fenton solution | 1 mM, 10 μM | Fenton test – EPR STUDIES | – |
| Higher radical scavenging properties of CeO2 with lower (10 μM) concentration | ||||
| 267 | HPAs doped membranes | – | – | OCV: 90 °C Air/H2 FER: (μg per cm2 per day) |
| HP2W18 | 10 wt% | 0.6 | ||
| HP2W21 | 10 wt% | 0.8 | ||
| HSiW | 20 wt% | 0.84 | ||
| 275 | Ce ion exchange membrane | Ce: | – | OCV: 90 °C 30% RH Air/H2 FER: (g cm−2 h−1) |
| 0 wt% | Nafion® − 1 × 10−6 | |||
| 5 wt% | Nafion®/Ce − 1 × 10−9 | |||
| 10 wt% | Nafion®/Ce − 1.6 × 10−10 | |||
| – | – | OCV: cold start conditions FER: (μmol) | ||
| 0 wt% | Nafion(50)− 0.45 (Nafion50 – after 50 OCV cycles) | |||
| 10 wt% | Nafion(50) ≈ 0 | |||
| 276 | Ion exchange membrane | (μmol) | Hydrogen peroxide flow cell experiments | OCV: 95 °C 50% RH O2/H2 FER: (g cm−2 h−1) |
| Mn2+ − anode | 2.2 | Co-doping of Ce into membrane containing 25 ppm of iron eliminates the accelerating impact of Fe catalyzed hydroxyl radical generation. | 2 × 10−7 | |
| Mn2+ − cathode | 2.2 | 3 × 10−7 | ||
| Ce3+ – anode | 2.2 | 5 × 10−8 | ||
| Ce3+ – anode/cathode | 2.8 | 3 × 10−8 | ||
| Ce3+ – membrane | 45 | 3 × 10−8 | ||
| 268 | HPA-carbon membrane-electrode sublayers | mg cm−2 | – | OCV: 90 °C 30% RH Air/H2 FER: (μmol cm−2) |
| PW12O403− – PTA | 0.13 | No sublayer − 40 | ||
| PW11O404− – PTA-1V | No HPA − 48 | |||
| SiW11O405− – STA-1V | PW12O403− − 27 | |||
| PW11O404− − 35 | ||||
| SiW11O405− − 40 | ||||
Finally stabilisation301 or development of new ORR electrocatalysts can also be considered as a mitigation strategy as elimination of potential catalytically active sites for hydrogen peroxide and radical generation is of great importance in increasing membrane durability. Platinum alloys are promising candidates for PEMFC application due to their lower cost and high performance.302 Rodgers et al. carried out studies on the influence of a PtCo/C catalyst on membrane durability.303 They showed that higher stability of the PtCo/C material in comparison to the Pt/C catalyst is translated into lower membrane degradation which results in reduced FER, lower hydrogen crossover and voltage decay. Ramaswamy et al. also reported higher durability of a membrane associated with an electrocatalyst enriched with cobalt using novel segmented cell design, where membrane decay is correlated to losses in its ionic conduction.95 Based on RRDE measurement they concluded that such behaviour is due to lower adsorption of oxygenated species on the Pt in the presence of Co, thereby diminishing H2O2 formation.
Both cerium and manganese in ionic or oxide form have been commonly applied to increase MEA durability by their incorporation into the membrane or electrode. Due to the high effectiveness in mitigation of the membrane degradation and exceptional mechanism of the radical decomposition they deserve special attention. The idea of incorporation in the polymer matrix of cerium and manganese ions by partial ion exchange, was developed by Endoh et al.274,304
The concept proposed by Endoh assumed that when the ionomer is ionically cross-linked with a radical scavenger cation, it can show improved mechanical properties and higher chemical stability (Fig. 15). The in situ durability studies of newly developed material called NPC – New Polymer Composite membrane performed by Endoh under the following fuel cell conditions: OCV hold test, 20% RH and temperature 120 °C, demonstrated excellent stability of the NPC of over 1000 h with FER in range of 2 × 10−8 g cm−2 h−1.
 | ||
| Fig. 15 Incorporation of radical quencher – concept of highly durable PFSA membrane developed by Endoh.274 | ||
Further research of Coms et al. in this area shed more light on the scavenging properties of both Mn and Ce cations.276 Coms et al. concluded that there is no impact of the initial location of the radical quencher on the membrane stability based on in situ degradation test of pristine and cation-containing MEAs, where an ion-exchanged ionomer was used for preparation of the catalyst ink. In other words, doping of the anode or cathode electrode with radical scavenger gave a similar effect on the MEA durability. Coms argued that the hot-pressing procedure promoted cation migration from the catalyst layer to a low concentration area – here the membrane – and that such migration occurred irrespective of which electrode was initially doped. These authors did not find evidence of cation leakage out of the system however. Another important conclusion concerned the higher efficiency of cerium cation in comparison with manganese. Both cations undergo easy redox reaction with HO˙ to form water as displayed in eqn (20) and (21). These reactions have higher rate constants than those of hydrogen abstraction reactions by hydroxyl radicals, which results in the scavenging properties of Ce3+ and Mn2+ ions. However, the studies of Coms et al. demonstrated that for an equal molar basis, cerium was much more effective at reducing FER. The authors explained such behaviour by a higher rate constant of radical reduction reaction by cerium ions k = 3 × 108 M−1 s−1. A similar rate constant of Ce3+ with HO˙ was reported Danilczuk et al.273 based on the competitive kinetics approach.
| HO˙ + Ce3+ + H+ → H2O + Ce4+, k = 3 × 108 M−1 s−1 | (20) |
| HO˙ + Mn2+ + H+ → H2O + Mn3+, k = 4 × 107 M−1 s−1 | (21) |
An excellent overview of all reaction involving Ce and Mn cations was described by Gubler et al. in the framework of their kinetic modelling studies.81 Gubler confirmed the correlation presented by Coms between reaction rate constants of HO˙ with radical quenchers and their scavenging efficiency. As an example, the concentration of 1% of Mn cations with respect to the sulfonic acid groups scavenges 46% of HO˙ whereas the same concentration of Ce cations scavenges 89%. Further studies in this area underline the significant impact of the ratio of the oxidation states of Ce and Mn on their mitigation properties. Only Ce3+ in the Ce3+/Ce4+ redox couple is able to react with hydroxyl radicals, therefore high dynamic equilibrium of Ce3+/Ce4+ is necessary to keep high efficiency of the system. Cerium has advantage to be self-regenerative, especially in an acidic medium. Fast reduction of Ce4+via reaction with H2O2 ensures rapid reactivation of Ce3+. It was reported that Ce4+ can be also reduced by H2, H2O or HOO˙ on the Pt catalyst layer273,276 but, as argued by Gubler, the reaction of Ce4+ with H2O2 is fast enough to convert essentially all cerium ions to Ce3+ form. Similarly to cerium ions, Mn2+ is also effectively restored via reaction with H2O2 or HOO˙.81 However the Mn2+/Mn3+ system is much more complex than that of cerium. Species with different oxidation states such as Mn+; Mn2+; Mn3+; and MnO2+ need to be considered.81,305 Indeed Tanuma et al. provided confirmation for this through their studies where the X-ray absorption spectroscopy (XAFS) was employed to measure a shift in absorption energy in the XANES (X-ray adsorption near edge structure) spectra of Ce and Mn ion exchanged membranes before and after OCV hold.305 No difference was found in the XANES spectra of Ce exchanged membrane before and after accelerated degradation, whereas in the case of the Mn exchanged membrane, a shift in the XANES spectrum to higher energy after OCV hold confirmed presence of Mn atoms with valence state higher than +2.
| HOO˙ + Ce4+ → O2 + Ce3+ + H+ | (22) |
| H2O2 + Ce4+ → HOO˙ + Ce3+ + H+ | (23) |
| HOO˙ + Mn3+ → O2 + Mn2+ + H+ | (24) |
| H2O2 + Mn3+ → MnO2+ + 2H+ | (25) |
Experimental evidence of the Ce3+/Ce4+ redox couple was provided by Danilczuk et al.93 by in situ monitoring of radical formation at the anode and cathode sides of pristine (MEA/H) and Ce ion exchanged (MEA/Ce) Nafion® 117 membrane. The 5,5-dimethylpyrroline-N-oxide (DMPO) was applied as a spin trap.
Both MEAs types examined showed completely different behaviour under OCV hold testing. For baseline MEA/H the dominant adducts were DMPO/OH and DMPO/CCR – carbon centred radicals derived from Nafion®, whereas for MEA/Ce the main adduct detected after membrane aging was DMPO/OOH. The absence of hydroxyl radicals as well as the absence of carbon centred radicals confirmed the scavenging properties of Ce3+. Moreover, the presence of DMPO/OOH indicates reaction of hydrogen peroxide with Ce4+ to restore the Ce3+. Another study of the same research group performed with an ionomer dispersion demonstrated a concentration of OH and CCR adducts lower by a factor of 12 for a dispersion enriched with cerium ions than for a pristine polymer dispersion.108 These studies also indicate scavenging properties of cerium, but also point out the role of membrane morphology in the radical attack as different degradation products were found for membrane and polymer dispersion.
Endoh et al. investigated the influence of the incorporation of Ce3+ ions in PFSA membranes on their degradation under the low humidity subzero conditions of −30 °C.275 After 50 temperature cycles from 70 °C to −30 °C, a 10% Ce3+ doped MEA showed very low 1.6 × 10−10 g cm−2 h−1 FER in contrast to 1 × 10−6 g cm−2 h−1 with pristine Nafion®. This result indicates that Ce3+ ions can mitigate the membrane degradation under repeated cold-starts.
 | ||
| Fig. 16 Mitigation of ionomer degradation as a function of total Ce-ion content (Ce3+ + Ce4+). (a) HO˙ concentration and rate of PFSA ionomer attack, expressed as fluoride emission rate (FER) assuming a membrane thickness of 50 μm, at a carboxylic end-group concentration of [–COOH] = 18 mM. (b) Fraction of HO˙ reacting with the ionomer and Ce3+. Reproduced by permission of The Electrochemical Society from ref. 81. | ||
However besides obvious advantages of the incorporation of Ce or Mn ions in MEAs, there are some disadvantages mainly a decrease of membrane conductivity resulting in fuel cell performance losses. Another important problem is the mobility of the ions. As was reported by Coms et al., cerium ions incorporated in the catalyst layer after hot pressing migrated into the PFSA membrane.276 It can be expected that scavengers can migrate under fuel cell operation conditions from an initially ion exchanged membrane to the electrodes as well, and further be washed out with exhaust water. Decrease of the scavenger concentration can be reflected in the simultaneous decrease of mitigating efficiency, which makes this strategy inappropriate for long-term use. According to Gubler et al. the relation between the membrane degradation rate and ions concentration can be expressed by the Fig. 16.81
Cheng et al. postulated scavenger migration from membrane to electrode layers. He demonstrated accelerated performance degradation after AST for MEAs with Ce and Mn ions added to the membrane.277 This author assumed that this higher performance degradation might be due to increased cathode catalyst layer (CCL) ionic losses. However further studies showed no impact of scavengers on Pt dissolution, agglomeration, carbon corrosion or oxidation.
Trogadas and co-workers developed an alternative approach to include cation radical scavengers, namely incorporation of cerium oxide in the membrane.280 Ceria particles, commonly known as a crucial component in three-way catalysts, emerged as an efficient hydrogen peroxide decomposition catalyst.306 A CeO2 – Nafion® composite membrane showed decrease in the FER of greater than an order of magnitude after accelerated degradation testing.280 This observation was attributed to quenching of the highly oxidative species by the non-stoichiometric CeO2. This assumption finds a confirmation in further studies of Prabhakaran et al. with a fluorescent molecular probe in conjunction with in situ fluorescence spectroscopy to investigate the membrane chemical degradation and CeO2 mitigation properties in an operating fuel cell.284,285 This is probably due to the particular electron structure and mixed valence state of CeOx.307–309 It was proposed that scavenging properties of ceria are mediated at oxygen vacancies or defects in the lattice structure.310 Thus the hydroxyl radical can be scavenged by Ce3+ ions, which are further oxidised to Ce4+. Oxidised Ce3+ is regenerated in acidic media through the mechanism displayed in Fig. 17. This regeneration of the Ce3+ form appears as important factor for CeO2 mitigation ability.
 | ||
| Fig. 17 Scavenging mechanism of CeO2 and regeneration of Ce3+ active sites. Reprinted with permission from ref. 284. | ||
To tune the Ce3+/Ce4+ ratio, Trogadas et al. proposed two strategies.309 A first approach was based on particle size control, as the concentration of oxygen vacancies and Ce3+ formation might depend on particle size. The second strategy included doping of ceria particles with Zr4+ cations in order to improve the stability and oxygen storage capacity of the CeO2 microstructure. To investigate the influence of ceria particle size and Zr incorporation, Ce0.25Zr0.75O2 nanoparticles and ceria particles with varying Ce3+ surface concentrations were synthesised incorporated in Nafion, and operated in a fuel cell MEA. After accelerated in situ degradation testing, the MEAs with composite membranes containing CeO2 or Ce0.25Zr0.75O2 demonstrated one order of magnitude lower fluorine emission rate compared to that of an MEA with a pristine membrane. Furthermore, the Ce3+ surface concentration of incorporated particles was reflected in FER values. The general conclusion can be drawn from this study is that a higher Ce3+ content lowers the FER from OCV hold-tested MEAs. Following this concept, Trogadas et al.281 performed another interesting investigation by platinum incorporation in the polymer membrane. Nafion® doped with CeO2 – supported platinum demonstrated over an order of magnitude reduction of fluoride emission rate. This system showed increased scavenging ability of cerium oxide nanoparticles independently of the Pt concentration (0.5–2 wt%). This observation was explained in terms of enhanced rate of Ce4+ to Ce3+ reduction in the presence of Pt. Wang et al. also studied the durability and performance of a Pt/CeO2-Nafion composite membrane.311 This author pointed out the higher performance of the Pt/CeO2-Nafion composite membrane in comparison to NRE-211 membrane at 35% RH that can be due to a self-hydrating function of Pt particles, a concept first introduced by Watanabe.312 Furthermore, the composite material demonstrated stable OCV over 180 h of AST performed at 70 °C/35% RH and H2/O2 as reactants. After the pioneering studies of Trogadas with CeO2 as radical scavenger, several authors investigated membrane durability with CeO2 additives in the MEA components. Similarly to the above described Ce and Mn ion radical scavengers, CeO2 can be incorporated in both the membrane and electrode structure. Wang and co-workers reported the reduction of ionomer degradation through incorporation of CeO2 in the catalyst layer.283 A newly prepared catalyst was applied either on the anode or the cathode side. MEAs containing CeO2, regardless of the initial position of the radical scavenger, showed similar improvement of the membrane durability after in situ OCV hold testing performed at 90 °C and 30% RH in comparison to the baseline MEA. After 200 hours of accelerated testing, no membrane thinning or pinhole development was observed for the MEA with anode or cathode cerium-rich catalyst. The MEA with CeO2 incorporated in the anode demonstrated a slightly lower performance than the baseline MEA. On the other hand, performance of the cell with CeO2 incorporated on the cathode side was slightly higher than that of baseline MEA. The author linked this result to the oxygen storage ability of CeO2. Increased local concentration of oxygen on the catalyst surface might improve the electrocatalytic reaction. In another study, the enhanced performance observed with a catalyst based on self-assembled mesoporous carbon with ceria nanoparticles has been explained by improved water retention of the modified catalyst.313 The idea of using CeOx as an active and durable catalyst support was followed by Lei et al.314 These authors reported lower chemical degradation of Nafion binder in catalyst layers during CV-cycles in the high voltage region when using CeO2 nanocubes–graphene oxide (CeO2–GO) catalyst support. They observed a slightly increased performance of a single cell with CeO2–GO when CeO2 loading was lower than 8 wt%. Other studies based on ex situ electrochemical characterisation revealed that incorporation of a low content (between 2 and 4 wt%) of amorphous CeO2 in a Pt catalyst enhances its tolerance to SO2 and CO.315 It was reported that the presence of nano-scale ceria facilitates the oxygen transfer thus assists CO oxidation and inhibits SO2 oxidation on Pt surfaces.
Weissbach et al. investigated stability of PFSA membranes co-casted with CeO2, ZrO2 and yttria-stabilized zirconia (YSZ) exposed to Fenton reagent.316 It was found that incorporation of CeO2 into the membrane significantly reduced FER, mass loss and loss of sulfonic groups, while PFSA-ZrO2 and PFSA-YSZ membranes did not present improved stability. Nafion – CeO2 nanocomposite membranes prepared through a “self-assembled” route, where ceria nanoparticles were incorporated into the polymer structure through in situ sol–gel process, were studied by Wang et al.282 The composite material demonstrated slightly lower proton conductivity at 100% RH, whereas at relative humidity below 75%, it showed higher values than pristine Nafion®. Both in situ and ex situ accelerated degradation tests revealed superior durability of self-assembled Nafion® – CeO2 membranes in comparison to the non-modified membrane, as well as to Nafion® – CeO2 membranes prepared by recasting from CeOx – Nafion colloidal suspension. D'Urso et al. studied the effect of the presence of silica supported CeOx in an ePTFE reinforced membrane on its chemical stability.221 The authors did not provide any FER or OCV decay measurement to assess the efficiency of their system. However they reported seven times longer lifetime of MEA enriched with CeOx in comparison to the non-modified membrane. Also Pearman et al. performed in situ and ex situ membrane degradation tests on composite materials consisting of porous PTFE supported cerium oxide impregnated with Nafion.286,287 Mechanical reinforcement of the membrane, enhanced with radical scavenging ability of ceria resulted in high durability over 500 h in an OCV hold test. Moreover the influence of the CeO2 incorporation on Pt band formation was reported. It was found that Pt formed larger particles deposited further into the polymer membrane. The author connected this behaviour with the presence of the ceria particles which affected the potential profile through the membrane thus altered the Pt band. Contrary to reports by Trogadas et al., on the influence of size of ceria particles on FER, Pearman et al. did not find evidence for a lower FER for small CeOx in comparison to larger, commercial ceria particles. A recent study of Schlick et al. shed some new light on the subject.317 These authors emphasize that the effectiveness of Ce nanoparticles may depend not only on the initial particle size or oxidation state but also on the extent of particle dispersion and agglomeration.
It might be concluded that ceria is very efficient radical scavenger. However it should be noted that recent studies on MEAs containing either cerium ions or cerium oxide clearly showed migration of cerium ions through the MEA components,287,318–320 which indicates dissolution of ceria in acid medium and creates a new question regarding the stability of ceria during fuel cell operation. Prabhakaran et al. reported loss of the efficiency of ceria already after 7 hours of an accelerated stress test, based on in situ fluorescence spectroscopy studies.284,285 However the authors related this observation to a decreased concentration of Ce3+ reactive centers rather than to dissolution of ceria particles. In order to improve its regenerative abilities, ceria nanoparticles have been doped with nitrogen by annealing in nitrogen-rich atmosphere.321 After treatment the N-tuned CeOx demonstrated high efficiency in radical scavenging over 90 h of testing which was associated with increased number of Ce3+ active clusters after exposure to the N-rich atmosphere. The possibility of ceria dissolution and cerium ion migration through MEA components was not considered.
A detailed analysis of the effect of positioning of ceria in the MEA was performed by Zatoń et al.319,322 The authors used electrospinning to develop a thin protective composite layer of PFSA nanofibers enriched with cerium oxide particles – NFCeOx that was incorporated into the MEA at the desired anode/cathode interface. In the previous reports incorporation of CeOx has been realized through preparation of a ceria enriched membrane281,287 or catalyst layer.318,323–325 The presence of CeOx in the membrane bulk can restrict the transport pathways and increase tortuosity and thus decrease proton conductivity. Moreover such an approach does not allow studies of the impact of ceria particles on membrane degradation when located at respective anode and cathode sides. The incorporation of CeOx in the catalyst layer gives such possibility, however affects the MEA performance as the charge transfer of ceria is rather low. Zaton et al. demonstrated that the lifetime of MEAs with anode or cathode sides protected by NFCeOx was eight times longer relative to an unmitigated MEA and FER and OCV decay were significantly reduced. The authors also reported that additional protective interlayer was more effective when incorporated on the anode side, possibly due to the reductive environment that accelerates regeneration of active Ce3+ scavenging centers, as well as a result of partial dissolution of CeOx at the anode interface and migration of cerium ions through membrane. In contrast, cerium species created after dissolution of CeOx on the cathode side might be easily leached from the MEA.
Table 3 summarises the results obtained after AST on various composite materials with scavenging properties. This provides a clearer view on the state of the art for mitigation of membrane degradation. Moreover the effectiveness of parameters like scavenger loading, nature of the introduced species, or their location on the membrane durability can be compared using the fluoride emission rate.
11. Summary and outlook
The objective was to review and analyse the reaction mechanisms leading to PFSA membrane degradation and failure, and to examine methodologies and means to reduce this degradation in an operating fuel cell. As discussed in Section 3, membrane degradation is not a single isolated reaction and cannot be classified by any known model. It is a complex combination of different factors including intrinsic properties of the material used for MEA preparation and the operating conditions applied. This work has reviewed the factors that can promote membrane degradation and which, once clearly identified, provide the basis for design of materials with enhanced robustness. Beyond that, this review has also provided the first survey of mitigation strategies designed to annihilate the harmful effect of oxidative species on membrane integrity and has comprehensively appraised their effectiveness by discussing the benefits and drawbacks of each strategy. By identifying stressors and through improved understanding of how scavenging reactions proceed can certainly accelerate development of new materials with antioxidant properties and enhanced durability. To meet fuel cell durability and lifetime targets over the widest range of temperature, pressure and relative humidity operating conditions requires membranes that are highly heterogeneous, comprising not only a reinforcing component, or designed architecture at nano- to micro-metric length scales, but also an advanced radical scavenger component and/or hydrogen peroxide decomposition catalyst. The choice of radical scavenger will be dictated by its efficiency and stability in operating conditions. Controlled release of a radical scavenger or hydrogen peroxide decomposition catalysts in response to a chemical signal from a repository within the membrane would represent a paradigm shift by providing the requisite groundwork for the elaboration of self-healing fuel cell membranes. Further development of the other cell components: GDL, catalyst and bipolar plates will also have, of course, significant influence on membrane stability. This is especially salient in the context of emerging non platinum group metals cathode catalysts of which the most active are iron or cobalt based. Chemical stabilisation of PFSA membranes in an MEA with a catalyst based on Fe2+ ions will require the development of radical scavengers of magnified efficacy, to which early consideration should be given in support of a future generation of fuel cell catalysts using earth-abundant metals.Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2010-2013) for the Fuel Cells and Hydrogen Joint Undertaking under grant agreement IMMEDIATE no. 3034.References
- A. J. Martin, A. Hornes, A. Martinez-Arias and L. Daza, Recent advances in fuel cells for transport and stationary applications, Elsevier B.V., Amsterdam, 2013 Search PubMed.
- J. Garche and L. Jorissen, Electrochem. Soc. Interface, 2015, 24, 3–43 CrossRef CAS.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- T. Wilberforce, A. Alaswad, A. Palumbo, M. Dassisti and A. G. Olabi, Int. J. Hydrogen Energy, 2016, 41, 16509–16522 CrossRef CAS.
- W. Schmittinger and A. Vahidi, J. Power Sources, 2008, 180, 1–14 CrossRef CAS.
- V. Andrea, P. d. S. P. Oliveira, E. I. Santiago and M. Linardi, ECS Trans., 2016, 71, 233–238 CrossRef.
- A. de Frank Bruijn and G. J. M. Janssen, PEM Fuel Cell Materials: Costs, Performance and Durability, Springer New York, New York, NY, 2013 Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- W. Bi, G. E. Gray and T. F. Fuller, Electrochem. Solid-State Lett., 2007, 10, B101–B104 CrossRef CAS.
- R. M. Darling and J. P. Meyers, J. Electrochem. Soc., 2003, 150, A1523–A1527 CrossRef CAS.
- P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- K. Yasuda, A. Taniguchi, T. Akita, T. Ioroi and Z. Siroma, Phys. Chem. Chem. Phys., 2006, 8, 746–752 RSC.
- A. Ohma, S. Yamamoto and K. Shinohara, ECS Trans., 2007, 11, 1181–1192 CAS.
- A. Ohma, S. Yamamoto and K. Shinohara, J. Power Sources, 2008, 182, 39–47 CrossRef CAS.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659–2668 CrossRef CAS.
- D. Spernjak, J. D. Fairweather, T. Rockward, R. Mukundan and R. Borup, ECS Trans., 2011, 41, 741–750 CAS.
- Z. Y. Liu, B. K. Brady, R. N. Carter, B. Litteer, M. Budinski, J. K. Hyun and D. A. Muller, J. Electrochem. Soc., 2008, 155, B979–B984 CrossRef CAS.
- R. Borup, J. Meyers and B. Pivovar, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- F. Nandjou, J. P. Poirot-Crouvezier, M. Chandesris, J. F. Blachot, C. Bonnaud and Y. Bultel, J. Power Sources, 2016, 326, 182–192 CrossRef CAS.
- T. Okada, Effect of ionic contaminants, John Wiley & Sons, Ltd, Chichester, UK, 2003 Search PubMed.
- N. Zamel and X. Li, Prog. Energy Combust. Sci., 2011, 37, 292–329 CrossRef CAS.
- A. Collier, H. Wang, X. Zi Yuan, J. Zhang and D. P. Wilkinson, Int. J. Hydrogen Energy, 2006, 31, 1838–1854 CrossRef CAS.
- A. Sadeghi Alavijeh, M. A. Goulet, R. M. H. Khorasany, J. Ghataurah, C. Lim, M. Lauritzen, E. Kjeang, G. G. Wang and R. K. N. D. Rajapakse, Fuel Cells, 2015, 15, 204–213 CrossRef CAS.
- F. A. de Bruijn, V. A. T. Dam and G. J. M. Janssen, Fuel Cells, 2008, 8, 3–22 CrossRef CAS.
- L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J.-C. Perrin, E. Moukheiber, A. ElKaddouri, G. De Moor, C. Bas, L. Flandin and N. Caqué, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560 CrossRef CAS.
- S. Zhang, X. Yuan, H. Wang, W. Merida, H. Zhu, J. Shen, S. Wu and J. Zhang, Int. J. Hydrogen Energy, 2009, 34, 388–404 CrossRef CAS.
- X.-Z. Yuan, H. Li, S. Zhang, J. Martin and H. Wang, J. Power Sources, 2011, 196, 9107–9116 CrossRef CAS.
- M. P. Rodgers, L. J. Bonville, H. R. Kunz, D. K. Slattery and J. M. Fenton, Chem. Rev., 2012, 112, 6075–6103 CrossRef CAS PubMed.
- T. Ishimoto and M. Koyama, Membranes, 2012, 2, 395–414 CrossRef CAS PubMed.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- J. Roziere and D. J. Jones, Annu. Rev. Mater. Res., 2003, 33, 503–555 CrossRef CAS.
- D. J. Jones and J. Rozière, Inorganic/organic composite membranes, John Wiley & Sons, Ltd, Chichester, UK, 2010 Search PubMed.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- C. Wang, G. Duscher and S. J. Paddison, RSC Adv., 2015, 5, 2368–2373 RSC.
- D. J. Jones, 2-Membrane materials and technology for low temperature fuel cells, Woodhead Publishing, Cambridge, 2012 Search PubMed.
- The Dow Chemical Company, EP Pat., EP0041735A1, 1981.
- Y. M. Tsou, M. C. Kimble and R. E. White, J. Electrochem. Soc., 1992, 139, 1913–1917 CrossRef CAS.
- G. A. Eisman, J. Power Sources, 1990, 29, 389–398 CrossRef CAS.
- A. Ghielmi, P. Vaccarono, C. Troglia and V. Arcella, J. Power Sources, 2005, 145, 108–115 CrossRef CAS.
- V. Arcella, C. Troglia and A. Ghielmi, Ind. Eng. Chem. Res., 2005, 44, 7646–7651 CrossRef CAS.
- J. Li, M. Pan and H. Tang, RSC Adv., 2014, 4, 3944–3965 RSC.
- Q. Zhao and J. Benziger, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 915–925 CrossRef CAS.
- X. Luo, S. Holdcroft, A. Mani, Y. Zhang and Z. Shi, Phys. Chem. Chem. Phys., 2011, 13, 18055–18062 RSC.
- A. S. Aricò, A. Di Blasi, G. Brunaccini, F. Sergi, G. Dispenza, L. Andaloro, M. Ferraro, V. Antonucci, P. Asher, S. Buche, D. Fongalland, G. A. Hards, J. D. B. Sharman, A. Bayer, G. Heinz, N. Zandonà, R. Zuber, M. Gebert, M. Corasaniti, A. Ghielmi and D. J. Jones, Fuel Cells, 2010, 10, 1013–1023 CrossRef.
- A. Stassi, I. Gatto, E. Passalacqua, V. Antonucci, A. S. Arico, L. Merlo, C. Oldani and E. Pagano, J. Power Sources, 2011, 196, 8925–8930 CrossRef CAS.
- M. Marrony, D. Beretta, S. Ginocchio, Y. Nedellec, S. Subianto and D. J. Jones, Fuel Cells, 2013, 13, 1146–1154 CrossRef CAS.
- H. Tang, S. Peikang, S. P. Jiang, F. Wang and M. Pan, J. Power Sources, 2007, 170, 85–92 CrossRef CAS.
- R. M. H. Khorasany, E. Kjeang, G. G. Wang and R. K. N. D. Rajapakse, J. Power Sources, 2015, 279, 55–63 CrossRef CAS.
- X. Huang, M. Rodgers, W. Yoon, B. Li and N. Mohajeri, ECS Trans., 2008, 16, 1573–1579 CAS.
- Y. Xiao and C. Cho, Energies, 2014, 7, 6401–6411 CrossRef CAS.
- R. M. H. Khorasany, A. Sadeghi Alavijeh, E. Kjeang, G. G. Wang and R. K. N. D. Rajapakse, J. Power Sources, 2015, 274, 1208–1216 CrossRef.
- N.-I. Kim, Y. Seo, K. B. Kim, N. Lee, J.-H. Lee, I. Song, H. Choi and J.-Y. Park, J. Power Sources, 2014, 253, 90–97 CrossRef CAS.
- R. Lin, F. Xiong, W. C. Tang, L. Técher, J. M. Zhang and J. X. Ma, J. Power Sources, 2014, 260, 150–158 CrossRef CAS.
- L. Dubau, L. Castanheira, M. Chatenet, F. Maillard, J. Dillet, G. Maranzana, S. Abbou, O. Lottin, G. De Moor, A. El Kaddouri, C. Bas, L. Flandin, E. Rossinot and N. Caqué, Int. J. Hydrogen Energy, 2014, 39, 21902–21914 CrossRef CAS.
- Y. Tang, A. M. Karlsson, M. H. Santare, M. Gilbert, S. Cleghorn and W. B. Johnson, Mater. Sci. Eng., A, 2006, 425, 297–304 CrossRef.
- B. Wu, M. A. Parkes, L. de Benedetti, A. J. Marquis, G. J. Offer and N. P. Brandon, J. Appl. Electrochem., 2016, 46, 1157–1162 CrossRef CAS.
- M. Inaba, T. Kinumoto, M. Kiriake, R. Umebayashi, A. Tasaka and Z. Ogumi, Electrochim. Acta, 2006, 51, 5746–5753 CrossRef CAS.
- H. S. Sodaye, P. K. Pujari, A. Goswami and S. B. Manohar, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 771–776 CrossRef CAS.
- H. S. Sodaye, P. K. Pujari, A. Goswami and S. B. Manohar, Radiat. Phys. Chem., 2000, 58, 567–570 CrossRef CAS.
- M. N. Silberstein and M. C. Boyce, J. Power Sources, 2010, 195, 5692–5706 CrossRef CAS.
- M. Ozmaian and R. Naghdabadi, PCCP, 2014, 16, 3173–3186 RSC.
- S. Kundu, L. C. Simon, M. Fowler and S. Grot, Polymer, 2005, 46, 11707–11715 CrossRef CAS.
- P. W. Majsztrik, A. B. Bocarsly and J. B. Benziger, Rev. Sci. Instrum., 2007, 78, 103904 CrossRef CAS PubMed.
- F. Bauer, S. Denneler and M. Willert-Porada, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 786–795 CrossRef CAS.
- X. Luo, L. Ghassemzadeh and S. Holdcroft, Int. J. Hydrogen Energy, 2015, 40, 16714–16723 CrossRef CAS.
- S. v. Venkatesan, C. Lim, E. Rogers, S. Holdcroft and E. Kjeang, Phys. Chem. Chem. Phys., 2015, 17, 13872–13881 RSC.
- W. Shi and L. A. Baker, RSC Adv., 2015, 5, 99284–99290 RSC.
- S. v. Venkatesan, C. Lim, S. Holdcroft and E. Kjeang, J. Electrochem. Soc., 2016, 163, F637–F643 CrossRef CAS.
- Z. Lu, G. Polizos, D. D. Macdonald and E. Manias, J. Electrochem. Soc., 2008, 155, B163–B171 CrossRef CAS.
- Y. S. Kim, L. Dong, M. A. Hickner, T. E. Glass, V. Webb and J. E. McGrath, Macromolecules, 2003, 36, 6281–6285 CrossRef CAS.
- Z. Lu, G. Polizos, E. Manias and D. Macdonald, ECS Trans., 2010, 28, 81–89 CAS.
- F. Teocoli, A. Paolone, O. Palumbo, M. A. Navarra, M. Casciola and A. Donnadio, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1421–1425 CrossRef CAS.
- R. C. McDonald, C. K. Mittelsteadt and E. L. Thompson, Fuel Cells, 2004, 4, 208–213 CrossRef CAS.
- M. Plazanet, F. Sacchetti, C. Petrillo, B. Deme, P. Bartolini and R. Torre, J. Membr. Sci., 2014, 453, 419–424 CrossRef CAS.
- S. D. Knights, K. M. Colbow, J. St-Pierre and D. P. Wilkinson, J. Power Sources, 2004, 127, 127–134 CrossRef CAS.
- F. Wang, H. Tang, M. Pan and D. Li, Int. J. Hydrogen Energy, 2008, 33, 2283–2288 CrossRef CAS.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755–B769 CrossRef CAS.
- L. Gubler and W. H. Koppenol, J. Electrochem. Soc., 2011, 159, B211–B218 CrossRef.
- N. Ohguri, A. Y. Nosaka and Y. Nosaka, Electrochem. Solid-State Lett., 2009, 12, B94–B96 CrossRef CAS.
- N. Ohguri, A. Y. Nosaka and Y. Nosaka, J. Power Sources, 2010, 195, 4647–4652 CrossRef CAS.
- Y. Nosaka, K. Ohtaka, N. Ohguri and A. Y. Nosaka, ECS Trans., 2010, 33, 899–905 CAS.
- Y. Nosaka, K. Ohtaka, N. Ohguri and A. Y. Nosaka, J. Electrochem. Soc., 2011, 158, B430–B433 CrossRef CAS.
- E. Endoh, S. Terazono, H. Widjaja and Y. Takimoto, Electrochem. Solid-State Lett., 2004, 7, A209–A211 CrossRef CAS.
- W. Liu and D. Zuckerbrod, J. Electrochem. Soc., 2005, 152, A1165–A1170 CrossRef CAS.
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Nørskov, A. Nilsson and H. Ogasawara, Nat. Commun., 2013, 4, 2817 Search PubMed.
- A. Pozio, R. F. Silva, F. M. De and L. Giorgi, Electrochim. Acta, 2003, 48, 1543–1549 CrossRef CAS.
- V. O. Mittal, H. Russell Kunz and J. M. Fenton, Electrochem. Solid-State Lett., 2006, 9, A299–A302 CrossRef CAS.
- V. O. Mittal, H. R. Kunz and J. M. Fenton, J. Electrochem. Soc., 2007, 154, B652–B656 CrossRef CAS.
- X. Fang, P. K. Shen, S. Song, V. Stergiopoulos and P. Tsiakaras, Polym. Degrad. Stab., 2009, 94, 1707–1713 CrossRef CAS.
- M. Danilczuk, F. D. Coms and S. Schlick, J. Phys. Chem. B, 2009, 113, 8031–8042 CrossRef CAS PubMed.
- A. Panchenko, H. Dilger, J. Kerres, M. Hein, A. Ullrich, T. Kaz and E. Roduner, PCCP, 2004, 6, 2891–2894 RSC.
- N. Ramaswamy, N. Hakim and S. Mukerjee, Electrochim. Acta, 2008, 53, 3279–3295 CrossRef CAS.
- S. Xiao, H. Zhang, C. Bi, Y. Zhang, Y. Zhang, H. Dai, Z. Mai and X. Li, J. Power Sources, 2010, 195, 5305–5311 CrossRef CAS.
- M. Zhao, W. Shi, B. Wu, W. Liu, J. Liu, D. Xing, Y. Yao, Z. Hou, P. Ming and Z. Zou, Electrochim. Acta, 2015, 153, 254–262 CrossRef CAS.
- S. Zhang, X.-Z. Yuan, R. Hiesgen, K. A. Friedrich, H. Wang, M. Schulze, A. Haug and H. Li, J. Power Sources, 2012, 205, 290–300 CrossRef CAS.
- S. Mu, C. Xu, Q. Yuan, Y. Gao, F. Xu and P. Zhao, J. Appl. Polym. Sci., 2013, 129, 1586–1592 CrossRef CAS.
- L. Ghassemzadeh, K. D. Kreuer, J. Maier and K. Muller, J. Power Sources, 2011, 196, 2490–2497 CrossRef CAS.
- S. Kundu, L. C. Simon and M. W. Fowler, Polym. Degrad. Stab., 2008, 93, 214–224 CrossRef CAS.
- M. Danilczuk, A. Bosnjakovic, M. K. Kadirov and S. Schlick, J. Power Sources, 2007, 172, 78–82 CrossRef CAS.
- A. A. Shah, T. R. Ralph and F. C. Walsh, J. Electrochem. Soc., 2009, 156, B465–B484 CrossRef CAS.
- D. E. Curtin, R. D. Lousenberg, T. J. Henry, P. C. Tangeman and M. E. Tisack, J. Power Sources, 2004, 131, 41–48 CrossRef CAS.
- T. Xie and C. A. Hayden, Polymer, 2007, 48, 5497–5506 CrossRef CAS.
- S. Hommura, K. Kawahara, T. Shimohira and Y. Teraoka, J. Electrochem. Soc., 2008, 155, A29–A33 CrossRef CAS.
- K. E. Schwiebert, K. G. Raiford, G. Escobedo and G. Nagarajan, ECS Trans., 2006, 1, 303–311 CAS.
- M. Danilczuk, A. J. Perkowski and S. Schlick, Macromolecules, 2010, 43, 3352–3358 CrossRef CAS.
- C. Zhou, M. A. Guerra, Z.-M. Qiu, T. A. Zawodzinski and D. A. Schiraldi, Macromolecules, 2007, 40, 8695–8707 CrossRef CAS.
- N. E. Cipollini, ECS Trans., 2007, 11, 1071–1082 CAS.
- D. A. Schiraldi, Polym. Rev., 2006, 46, 315–327 CAS.
- G. Escobedo, K. Raiford, G. S. Nagarajan and K. E. Schwiebert, ECS Trans., 2006, 1, 303–311 Search PubMed.
- L. Ghassemzadeh, K.-D. Kreuer, J. Maier and K. Muller, J. Phys. Chem. C, 2010, 114, 14635–14645 CAS.
- A. Bosnjakovic, M. Kadirov and S. Schlick, Res. Chem. Intermed., 2007, 33, 677–687 CrossRef CAS.
- D. Kurniawan, H. Arai, S. Morita and K. Kitagawa, Microchem. J., 2013, 106, 384–388 CrossRef CAS.
- T. Tokumasu, I. Ogawa, M. Koyama, T. Ishimoto and A. Miyamoto, J. Electrochem. Soc., 2011, 158, B175–B179 CrossRef CAS.
- F. D. Coms, ECS Trans., 2008, 16, 235–255 CAS.
- L. Ghassemzadeh, T. J. Peckham, T. Weissbach, X. Luo and S. Holdcroft, J. Am. Chem. Soc., 2013, 135, 15923–15932 CrossRef CAS PubMed.
- L. Ghassemzadeh and S. Holdcroft, J. Am. Chem. Soc., 2013, 135, 8181–8184 CrossRef CAS PubMed.
- A. M. Dreizler and E. Roduner, Fuel Cells, 2012, 12, 132–140 CrossRef CAS.
- M. Danilczuk, F. D. Coms and S. Schlick, Fuel Cells, 2008, 8, 436–452 CrossRef CAS.
- T. Ishimoto, T. Ogura and M. Koyama, ECS Trans., 2011, 35, 1–6 CAS.
- M. Danilczuk, L. Lancucki, S. Schlick, S. J. Hamrock and G. M. Haugen, ACS Macro Lett., 2012, 1, 280–285 CrossRef CAS.
- J. Healy, C. Hayden, T. Xie, K. Olson, R. Waldo, M. Brundage, H. Gasteiger and J. Abbott, Fuel Cells, 2005, 5, 302–308 CrossRef CAS.
- C. Lim, L. Ghassemzadeh, F. Van Hove, M. Lauritzen, J. Kolodziej, G. G. Wang, S. Holdcroft and E. Kjeang, J. Power Sources, 2014, 257, 102–110 CrossRef CAS.
- K. H. Wong and E. Kjeang, J. Electrochem. Soc., 2014, 161, F823–F832 CrossRef CAS.
- M. Ghelichi, P.-E. A. Melchy and M. H. Eikerling, J. Phys. Chem. B, 2014, 118, 11375–11386 CrossRef CAS PubMed.
- T. H. Yu, Y. Sha, W.-G. Liu, B. V. Merinov, P. Shirvanian and W. A. Goddard, J. Am. Chem. Soc., 2011, 133, 19857–19863 CrossRef CAS PubMed.
- T. Ishimoto, R. Nagumo, T. Ogura, T. Ishihara, B. Kim, A. Miyamoto and M. Koyama, J. Electrochem. Soc., 2010, 157, B1305–B1309 CrossRef CAS.
- K. Teranishi, K. Kawata, S. Tsushima and S. Hirai, Electrochem. Solid-State Lett., 2006, 9, A475–A477 CrossRef CAS.
- C. Chen and T. Fuller, ECS Trans., 2007, 11, 1127–1137 CAS.
- A. B. LaConti, M. Hamdan and R. C. McDonald, in Handbook of Fuel Cells, John Wiley & Sons, Ltd, 2003 Search PubMed.
- J. Qiao, M. Saito, K. Hayamizu and T. Okada, J. Electrochem. Soc., 2006, 153, A967–A974 CrossRef CAS.
- V. A. Sethuraman, J. W. Weidner, A. T. Haug, S. Motupally and L. V. Protsailo, J. Electrochem. Soc., 2007, 155, B50–B57 CrossRef.
- H. Liu, H. A. Gasteiger, A. Laconti and J. Zhang, ECS Trans., 2006, 1, 283–293 CAS.
- J. Peron, Y. Nedellec, D. J. Jones and J. Roziere, J. Power Sources, 2008, 185, 1209–1217 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- K. Ono, Y. Yasuda, K. Sekizawa, N. Takeuchi, T. Yoshida and M. Sudoh, Electrochim. Acta, 2013, 97, 58–65 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- K. Ke, T. Hatanaka and Y. Morimoto, Electrochim. Acta, 2010, 56, 2098–2104 CrossRef.
- M. Inaba, H. Yamada, J. Tokunaga and A. Tasaka, Electrochem. Solid-State Lett., 2004, 7, A474–A476 CrossRef CAS.
- A. Bonakdarpour, T. R. Dahn, R. T. Atanasoski, M. K. Debe and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11, B208–B211 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger, B. N. Grgur and P. N. Ross, J. Electroanal. Chem., 1999, 467, 157–163 CrossRef CAS.
- V. Atrazhev, S. F. Burlatsky, N. E. Cipollini, D. A. Condit and N. Erikhman, ECS Trans., 2006, 1, 239–246 CAS.
- V. Atrazhev, E. Timokhina, S. F. Burlatsky, V. Sultanov, T. Madden and M. Gummalla, ECS Trans., 2008, 6, 69–74 CAS.
- F. N. Buchi, B. Gupta, O. Haas and G. G. Scherer, Electrochim. Acta, 1995, 40, 345–353 CrossRef CAS.
- M. Marrony, R. Barrera, S. Quenet, S. Ginocchio, L. Montelatici and A. Aslanides, J. Power Sources, 2008, 182, 469–475 CrossRef CAS.
- M. Aoki, H. Uchida and M. Watanabe, Electrochem. Commun., 2005, 7, 1434–1438 CrossRef CAS.
- L. Merlo, A. Ghielmi, L. Cirillo, M. Gebert and V. Arcella, J. Power Sources, 2007, 171, 140–147 CrossRef CAS.
- V. A. Sethuraman, J. W. Weidner, A. T. Haug, S. Motupally and L. V. Protsailo, J. Electrochem. Soc., 2008, 155, B50–B57 CrossRef CAS.
- C. Huang, K. Seng Tan, J. Lin and K. Lee Tan, Chem. Phys. Lett., 2003, 371, 80–85 CrossRef CAS.
- E. Endoh, S. Hommura, S. Terazono, H. Widjaja and J. Anzai, ECS Trans., 2007, 11, 1083–1091 CAS.
- M. Bodner, B. Cermenek, M. Rami and V. Hacker, Membranes, 2015, 5, 888 CrossRef CAS PubMed.
- G. De Moor, C. Bas, N. Charvin, J. Dillet, G. Maranzana, O. Lottin, N. Caqué, E. Rossinot and L. Flandin, Int. J. Hydrogen Energy, 2016, 41, 483–496 CrossRef CAS.
- J. Yu, B. Yi, D. Xing, F. Liu, Z. Shao, Y. Fu and H. Zhang, PCCP, 2003, 5, 611–615 RSC.
- W. Yoon and X. Y. Huang, J. Electrochem. Soc., 2010, 157, B599–B606 CrossRef CAS.
- B. Mattsson, H. Ericson, L. M. Torell and F. Sundholm, Electrochim. Acta, 2000, 45, 1405–1408 CrossRef CAS.
- M. P. Rodgers, R. P. Brooker, N. Mohajeri, L. J. Bonville, H. R. Kunz, D. K. Slattery and J. M. Fenton, J. Electrochem. Soc., 2012, 159, F338–F352 CrossRef CAS.
- K. H. Wong and E. Kjeang, ChemSusChem, 2015, 8, 1072–1082 CrossRef CAS PubMed.
- S. A. Vilekar and R. Datta, J. Power Sources, 2010, 195, 2241–2247 CrossRef CAS.
- K. D. Baik, I. M. Kong, B. K. Hong, S. H. Kim and M. S. Kim, Appl. Energy, 2010, 101, 560–566 CrossRef.
- J. Zhang, Y. Tang, C. Song, J. Zhang and H. Wang, J. Power Sources, 2006, 163, 532–537 CrossRef CAS.
- H. L. Yeager and A. Steck, J. Electrochem. Soc., 1981, 128, 1880–1884 CrossRef CAS.
- Z. Ogumi, Z. Takehara and S. Yoshizawa, J. Electrochem. Soc., 1984, 131, 769–773 CrossRef CAS.
- L. Liu, A. Chakma and X. Feng, J. Membr. Sci., 2008, 310, 66–75 CrossRef CAS.
- S. Kundu, M. W. Fowler, L. C. Simon, R. Abouatallah and N. Beydokhti, J. Power Sources, 2010, 195, 7323–7331 CrossRef CAS.
- K. Panha, M. Fowler, X.-Z. Yuan and H. Wang, Appl. Energy, 2012, 93, 90–97 CrossRef CAS.
- V. Prabhakaran, C. G. Arges and V. Ramani, PCCP, 2013, 15, 18965–18972 RSC.
- T. Madden, D. Weiss, N. Cipollini, D. Condit, M. Gummalla, S. Burlatsky and V. Atrazhev, J. Electrochem. Soc., 2009, 156, B657–B662 CrossRef CAS.
- M. Takasaki, Y. Nakagawa, Y. Sakiyama, K. Tanabe, K. Ookubo, N. Sato, T. Minamide, H. Nakayama and M. Hori, J. Electrochem. Soc., 2013, 160, F413–F416 CrossRef CAS.
- K. Matsuoka, S. Sakamoto, K. Nakato, A. Hamada and Y. Itoh, J. Power Sources, 2008, 179, 560–565 CrossRef CAS.
- D. Imamura and E. Yamaguchi, ECS Trans., 2009, 25, 813–819 CAS.
- M. Chen, C. Du, J. Zhang, P. Wang and T. Zhu, J. Power Sources, 2011, 196, 620–626 CrossRef CAS.
- X. Zhang, H. M. Galindo, H. F. Garces, P. Baker, X. Wang, U. Pasaogullari, S. L. Suib and T. Molter, J. Electrochem. Soc., 2010, 157, B409–B414 CrossRef CAS.
- M. Sulek, J. Adams, S. Kaberline, M. Ricketts and J. R. Waldecker, J. Power Sources, 2011, 196, 8967–8972 CrossRef CAS.
- H. Li, K. Tsay, H. Wang, J. Shen, S. Wu, J. Zhang, N. Jia, S. Wessel, R. Abouatallah, N. Joos and J. Schrooten, J. Power Sources, 2010, 195, 8089–8093 CrossRef CAS.
- J. M. Christ, K. C. Neyerlin, H. Wang, R. Richards and H. N. Dinh, J. Electrochem. Soc., 2014, 161, F1481–F1488 CrossRef CAS.
- V. Berejnov, Z. Martin, M. West, S. Kundu, D. Bessarabov, J. Stumper, D. Susac and A. P. Hitchcock, Phys. Chem. Chem. Phys., 2012, 14, 4835–4843 RSC.
- J. Xie, D. L. Wood, K. L. More, P. Atanassov and R. L. Borup, J. Electrochem. Soc., 2005, 152, A1011–A1020 CrossRef.
- S. F. Burlatsky, M. Gummalla, V. V. Atrazhev, D. V. Dmitriev, N. Y. Kuzminyh and N. S. Erikhman, J. Electrochem. Soc., 2011, 158, B322–B330 CrossRef CAS.
- L. Kim, C. G. Chung, Y. W. Sung and J. S. Chung, J. Power Sources, 2008, 183, 524–532 CrossRef CAS.
- J. Peron, D. Jones and J. Roziere, ECS Trans., 2007, 11, 1313–1319 CAS.
- T. Hatanaka, T. Takeshita, H. Murata, N. Hasegawa, T. Asano, M. Kawasumi and Y. Morimoto, ECS Trans., 2008, 16, 1961–1965 CAS.
- S. Helmly, R. Hiesgen, T. Morawietz, X.-Z. Yuan, H. Wang and K. Andreas Friedrich, J. Electrochem. Soc., 2013, 160, F687–F697 CrossRef CAS.
- D. Zhao, B. L. Yi, H. M. Zhang and M. Liu, J. Power Sources, 2010, 195, 4606–4612 CrossRef CAS.
- M. Gummalla, V. V. Atrazhev, D. Condit, N. Cipollini, T. Madden, N. Y. Kuzminyh, D. Weiss and S. F. Burlatsky, J. Electrochem. Soc., 2010, 157, B1542–B1548 CrossRef CAS.
- T. Kim, H. Lee, W. Sim, J. Lee, S. Kim, T. Lim and K. Park, Korean J. Chem. Eng., 2009, 26, 1265–1271 CrossRef CAS.
- H. Liu, J. Zhang, F. Coms, W. Gu, B. Litteer and H. A. Gasteiger, ECS Trans., 2006, 3, 493–505 CAS.
- C. Iojoiu, E. Guilminot, F. Maillard, M. Chatenet, J. Y. Sanchez, E. Claude and E. Rossinot, J. Electrochem. Soc., 2007, 154, B1115–B1120 CrossRef CAS.
- A. Ohma, S. Suga, S. Yamamoto and K. Shinohara, J. Electrochem. Soc., 2007, 154, B757–B760 CrossRef CAS.
- N. Macauley, L. Ghassemzadeh, C. Lim, M. Watson, J. Kolodziej, M. Lauritzen, S. Holdcroft and E. Kjeang, ECS Electrochem. Lett., 2013, 2, F33–F35 CrossRef CAS.
- N. Macauley, A. S. Alavijeh, M. Watson, J. Kolodziej, M. Lauritzen, S. Knights, G. Wang and E. Kjeang, J. Electrochem. Soc., 2015, 162, F98–F107 CrossRef CAS.
- N. Macauley, K. H. Wong, M. Watson and E. Kjeang, J. Power Sources, 2015, 299, 139–148 CrossRef CAS.
- S. Helmly, B. Ohnmacht, P. Gazdzicki, R. Hiesgen, E. Gülzow and K. A. Friedrich, J. Electrochem. Soc., 2014, 161, F1416–F1426 CrossRef CAS.
- S. Helmly, B. Ohnmacht, R. Hiesgen, E. Gluzow and K. A. Friedrich, ECS Trans., 2013, 58, 969–990 CrossRef.
- M. P. Rodgers, B. P. Pearman, L. J. Bonville, D. A. Cullen, N. Mohajeri and D. K. Slattery, J. Electrochem. Soc., 2013, 160, F1123–F1128 CrossRef CAS.
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS.
- D. Banham, S. Ye, K. Pei, J.-i. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- S. Xiao, H. Zhang, C. Bi, Y. Zhang, Y. Ma, X. Li, H. Zhong and Y. Zhang, J. Power Sources, 2010, 195, 8000–8005 CrossRef CAS.
- M. Hu and G. Cao, Int. J. Hydrogen Energy, 2014, 39, 7940–7954 CrossRef CAS.
- H.-C. Chien, L.-D. Tsai, C.-M. Lai, J.-N. Lin, C.-Y. Zhu and F.-C. Chang, J. Power Sources, 2013, 226, 87–93 CrossRef CAS.
- S. Subianto, M. Pica, M. Casciola, P. Cojocaru, L. Merlo, G. Hards and D. J. Jones, J. Power Sources, 2013, 233, 216–230 CrossRef CAS.
- J. A. Kolde, B. Bahar, M. S. Wilson, T. A. Zawodzinski and S. Gottesfeld, Proc.–Electrochem. Soc., 1995, 95–23, 193–201 CAS.
- B. Bahar, C. Cavalca, S. Cleghorn, J. Kolde, D. Lane, M. Murthy and G. Rusch, J. New Mater. Electrochem. Syst., 1999, 2, 179–182 CAS.
- G. Alberti, R. Narducci, M. L. Di Vona and S. Giancola, Fuel Cells, 2013, 13, 42–47 CrossRef CAS.
- G. Alberti, R. Narducci and M. Sganappa, J. Power Sources, 2008, 178, 575–583 CrossRef CAS.
- M. K. Hassan, A. Abukmail and K. A. Mauritz, Eur. Polym. J., 2012, 48, 789–802 CrossRef CAS.
- L. Maldonado, J.-C. Perrin, J. Dillet and O. Lottin, J. Membr. Sci., 2012, 389, 43–56 CrossRef CAS.
- A. Kusoglu, S. Savagatrup, K. T. Clark and A. Z. Weber, Macromolecules, 2012, 45, 7467–7476 CrossRef CAS.
- X. Wang, H. Tang and M. Pan, J. Membr. Sci., 2011, 379, 106–111 CrossRef CAS.
- J. Li, X. Yang, H. Tang and M. Pan, J. Membr. Sci., 2010, 361, 38–42 CrossRef CAS.
- J. Lin, P. H. Wu, R. Wycisk, A. Trivisonno and P. N. Pintauro, J. Power Sources, 2008, 183, 491–497 CrossRef CAS.
- W. Zhang, R. Wycisk, D. L. Kish and P. N. Pintauro, J. Electrochem. Soc., 2014, 161, F770–F777 CrossRef CAS.
- H. Ghassemi, T. Zawodzinski, D. Schiraldi and S. Hamrock, ACS Symp. Ser., 2012, 1096, 201–220 CrossRef CAS.
- Y. M. Zhang, L. Li, J. Tang, B. Bauer, W. Zhang, H. R. Gao, M. Taillades-Jacquin, D. J. Jones, J. Roziere, N. Lebedeva and R. Mallant, ECS Trans., 2009, 25, 1469–1472 CAS.
- N. Uematsu, N. Hoshi, T. Koga and M. Ikeda, J. Fluorine Chem., 2006, 127, 1087–1095 CrossRef CAS.
- L. Sauguet, B. Ameduri and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4566–4578 CrossRef CAS.
- J. Shim, H. Y. Ha, S.-A. Hong and I.-H. Oh, J. Power Sources, 2002, 109, 412–417 CrossRef CAS.
- P. Xiao, J. Li, H. Tang, Z. Wang and M. Pan, J. Membr. Sci., 2013, 442, 65–71 CrossRef CAS.
- C. D'Urso, C. Oldani, V. Baglio, L. Merlo and A. S. Aricò, J. Power Sources, 2014, 272, 753–758 CrossRef.
- Utc Power Corporation, WO Pat., WO2012099582A1, 2012.
- B. S. Pivovar, Y. Wang and E. L. Cussler, J. Membr. Sci., 1999, 154, 155–162 CrossRef CAS.
- N. W. DeLuca and Y. A. Elabd, J. Power Sources, 2006, 163, 386–391 CrossRef CAS.
- Johnson Matthey Fuel Cells Limited, Centre National de la Recherche Scientifique, Université Montpellier 2, WO Pat., WO2016020668A1, 2016.
- M. H. Yildirim, D. Stamatialis and M. Wessling, J. Membr. Sci., 2008, 321, 364–372 CrossRef CAS.
- T.-E. Kim, S. M. Juon, J. H. Park, Y.-G. Shul and K. Y. Cho, Int. J. Hydrogen Energy, 2014, 39, 16474–16485 CrossRef CAS.
- T.-C. Jao, G.-B. Jung, S.-C. Kuo, W.-J. Tzeng and A. Su, Int. J. Hydrogen Energy, 2012, 37, 13623–13630 CrossRef CAS.
- H. Tang, M. Pan, F. Wang, P. K. Shen and S. P. Jiang, J. Phys. Chem. B, 2007, 111, 8684–8690 CrossRef CAS PubMed.
- J. Park, L. Wang, S. G. Advani and A. K. Prasad, J. Electrochem. Soc., 2012, 159, F864–F870 CrossRef CAS.
- H. L. Tang, M. Pan and F. Wang, J. Appl. Polym. Sci., 2008, 109, 2671–2678 CrossRef CAS.
- X. Zhu, H. Zhang, Y. Liang, Y. Zhang, Q. Luo, C. Bi and B. Yi, J. Mater. Chem., 2007, 17, 386–397 RSC.
- T.-C. Jao, G.-B. Jung, S.-T. Ke, P.-H. Chi and S.-H. Chan, Int. J. Energy Res., 2011, 35, 1274–1283 CrossRef CAS.
- J. Yuan, H. Pu and Z. Yang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2647–2655 CrossRef CAS.
- D. Yuan, Z. Liu, S. W. Tay, X. Fan, X. Zhang and C. He, Chem. Commun., 2013, 49, 9639–9641 RSC.
- N. H. Jalani, K. Dunn and R. Datta, Electrochim. Acta, 2005, 51, 553–560 CrossRef CAS.
- C.-C. Ke, X.-J. Li, Q. Shen, S.-G. Qu, Z.-G. Shao and B.-L. Yi, Int. J. Hydrogen Energy, 2011, 36, 3606–3613 CrossRef CAS.
- K. T. Adjemian, R. Dominey, L. Krishnan, H. Ota, P. Majsztrik, T. Zhang, J. Mann, B. Kirby, L. Gatto, M. Velo-Simpson, J. Leahy, S. Srinivasan, J. B. Benziger and A. B. Bocarsly, Chem. Mater., 2006, 18, 2238–2248 CrossRef CAS.
- Y. Patil, S. Kulkarni and K. A. Mauritz, J. Appl. Polym. Sci., 2011, 121, 2344–2353 CrossRef CAS.
- V. Baglio, A. S. Arico, A. D. Blasi, V. Antonucci, P. L. Antonucci, S. Licoccia, E. Traversa and F. S. Fiory, Electrochim. Acta, 2005, 50, 1241–1246 CrossRef CAS.
- H. Uchida, Y. Ueno, H. Hagihara and M. Watanabe, J. Electrochem. Soc., 2003, 150, A57–A62 CrossRef CAS.
- T. Jian-hua, G. Peng-fei, Z. Zhi-yuan, L. Wen-hui and S. Zhong-qiang, Int. J. Hydrogen Energy, 2008, 33, 5686–5690 CrossRef.
- C. F. Nørgaard, U. G. Nielsen and E. M. Skou, Solid State Ionics, 2012, 213, 76–82 CrossRef.
- F. Chen, A. D'Epifanio, B. Mecheri, E. Traversa and S. Licoccia, ECS Trans., 2009, 25, 1935–1941 CAS.
- S. Brutti, R. Scipioni, M. A. Navarra, S. Panero, V. Allodi, M. Giarola and G. Mariotto, Int. J. Nanotechnol., 2014, 11, 882–896 CrossRef CAS.
- R. Scipioni, D. Gazzoli, F. Teocoli, O. Palumbo, A. Paolone, N. Ibris, S. Brutti and M. A. Navarra, Membranes, 2014, 4, 123–142 CrossRef PubMed.
- A. Saccà, I. Gatto, A. Carbone, R. Pedicini and E. Passalacqua, J. Power Sources, 2006, 163, 47–51 CrossRef.
- S. Subianto, A. Donnadio, S. Cavaliere, M. Pica, M. Casciola, D. J. Jones and J. Roziere, J. Mater. Chem. A, 2014, 2, 13359–13365 CAS.
- M. Casciola, P. Cojocaru, A. Donnadio, S. Giancola, L. Merlo, Y. Nedellec, M. Pica and S. Subianto, J. Power Sources, 2014, 262, 407–413 CrossRef CAS.
- G. Alberti and M. Casciola, Membranes for medium temperature PEFC based on Nafion filled with layered metal phosphates and phosphonates, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- M. Casciola, D. Capitani, A. Donnadio, V. Frittella, M. Pica and M. Sganappa, Fuel Cells, 2009, 9, 381–386 CrossRef CAS.
- D. Jones and J. Rozière, Advances in the Development of Inorganic–Organic Membranes for Fuel Cell Applications, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- M. S. Schaberg, J. E. Abulu, G. M. Haugen, M. A. Emery, S. J. O'Conner, P. N. Xiong and S. Hamrock, ECS Trans., 2010, 33, 627–633 CAS.
- J. K. Clark Ii and S. J. Paddison, Electrochim. Acta, 2013, 101, 279–292 CrossRef CAS.
- N. J. Economou, A. M. Barnes, A. J. Wheat, M. S. Schaberg, S. J. Hamrock and S. K. Buratto, J. Phys. Chem. B, 2015, 119, 14280–14287 CrossRef CAS PubMed.
- L. Puskar, E. Ritter, U. Schade, M. Yandrasits, S. J. Hamrock, M. Schaberg and E. F. Aziz, PCCP, 2017, 19, 626–635 RSC.
- J. A. Leistra, N. E. Cipollini, W. R. Schmidt, J. B. Hertzberg, C. H. Paik, T. D. Jarvi, T. W. Patterson and S. Tulyani, US Pat., US20050095355A1, 2005.
- Ballard Power Systems Inc., US Pat., US20050136308A1, 2005.
- 3M Innovative Properties Company, US Pat., US20070099053A1, 2007.
- 3M Innovative Properties Company, US Pat., US20080160380A1, 2008.
- GM Global Technology Operations, Inc., DE Pat., DE102007048872A1, 2008.
- GM Global Technology Operations, Inc., US Pat., US20120122016A1, 2012.
- N. R. de Tacconi, C. R. Chenthamarakshan, K. Rajeshwar, W.-Y. Lin, T. F. Carlson, L. Nikiel, W. A. Wampler, S. Sambandam and V. Ramani, J. Electrochem. Soc., 2008, 155, B1102–B1109 CrossRef CAS.
- Y. Patil and K. A. Mauritz, J. Appl. Polym. Sci., 2009, 113, 3269–3278 CrossRef CAS.
- Y. Patil, S. Sambandam, V. Ramani and K. Mauritz, J. Electrochem. Soc., 2009, 156, B1092–B1098 CrossRef CAS.
- P. Trogadas and V. Ramani, J. Power Sources, 2007, 174, 159–163 CrossRef CAS.
- G. M. Haugen, F. Meng, N. V. Aieta, J. L. Horan, M.-C. Kuo, M. H. Frey, S. J. Hamrock and A. M. Herring, Electrochem. Solid-State Lett., 2007, 10, B51–B55 CrossRef CAS.
- P. R. Brooker, L. J. Bonville and D. K. Slattery, J. Electrochem. Soc., 2013, 160, F75–F80 CrossRef.
- A. M. Herring, H. M. Gregory, M. Fanqin, N. V. Aieta, J. L. Horan, M. H. Frey, S. J. Hamrock and M. C. Kuo, ECS Trans., 2006, 3, 551–559 Search PubMed.
- J. Rajeswari, Z. Ziegler, G. M. Haugen, S. J. Hamrock and A. M. Herring, ECS Trans., 2011, 41, 1561–1565 CAS.
- P. Trogadas and V. Ramani, ECS Trans., 2007, 11, 949–960 CAS.
- G.-Y. Chen, C. Wang, Y.-J. Lei, J. Zhang, Z.-Q. Mao, J.-W. Guo and J.-L. Wang, Int. J. Hydrogen Energy, 2016, 41, 16167–16172 CrossRef CAS.
- M. Danilczuk, S. Schlick and F. D. Coms, Macromolecules, 2009, 42, 8943–8949 CrossRef CAS.
- E. Endoh, Highly durable PFSA membranes, John Wiley & Sons, Ltd, Chichester, UK, 2010 Search PubMed.
- E. Endoh, N. Onoda, Y. Kaneko, Y. Hasegawa, S. Uchiike, Y. Takagi and T. Take, ECS Electrochem. Lett., 2013, 2, F73–F75 CrossRef CAS.
- F. D. Coms, H. Liu and J. E. Owejan, ECS Trans., 2008, 16, 1735–1747 CAS.
- T. T. H. Cheng, S. Wessel and S. Knights, J. Electrochem. Soc., 2013, 160, F27–F33 CrossRef CAS.
- P. Trogadas, J. Parrondo, F. Mijangos and V. Ramani, J. Mater. Chem., 2011, 21, 19381–19388 RSC.
- S. Babu, A. Velez, K. Wozniak, J. Szydlowska and S. Seal, Chem. Phys. Lett., 2007, 442, 405–408 CrossRef CAS.
- P. Trogadas, J. Parrondo and V. Ramani, Electrochem. Solid-State Lett., 2008, 11, B113–B116 CrossRef CAS.
- P. Trogadas, J. Parrondo and V. Ramani, Chem. Commun., 2011, 47, 11549–11551 RSC.
- Z. Wang, H. Tang, H. Zhang, M. Lei, R. Chen, P. Xiao and M. Pan, J. Membr. Sci., 2012, 421–422, 201–210 CrossRef CAS.
- L. Wang, S. G. Advani and A. K. Prasad, Electrochim. Acta, 2013, 109, 775–780 CrossRef CAS.
- V. Prabhakaran, C. G. Arges and V. Ramani, PNAS, 2011, 109, 1029–1034 CrossRef PubMed.
- V. Prabhakaran, C. G. Arges and V. Ramani, ECS Trans., 2011, 41, 1347–1357 CAS.
- B. P. Pearman, N. Mohajeri, R. P. Brooker, M. P. Rodgers, D. K. Slattery, M. D. Hampton, D. A. Cullen and S. Seal, J. Power Sources, 2013, 225, 75–83 CrossRef CAS.
- B. P. Pearman, N. Mohajeri, D. K. Slattery, M. D. Hampton, S. Seal and D. A. Cullen, Polym. Degrad. Stab., 2013, 98, 1766–1772 CrossRef CAS.
- H. F. Xu and X. L. Hou, Int. J. Hydrogen Energy, 2007, 32, 4397–4401 CrossRef CAS.
- A. M. Baker, L. Wang, W. B. Johnson, A. K. Prasad and S. G. Advani, J. Phys. Chem. C, 2014, 118, 26796–26802 CAS.
- M. Lei, T. Z. Yang, W. J. Wang, K. Huang, Y. C. Zhang, R. Zhang, R. Z. Jiao, X. L. Fu, H. J. Yang, Y. G. Wang and W. H. Tang, J. Power Sources, 2013, 230, 96–100 CrossRef CAS.
- D. Zhao, B. L. Yi, H. M. Zhang and H. M. Yu, J. Membr. Sci., 2010, 346, 143–151 CrossRef CAS.
- M. T. Taghizadeh and M. Vatanparast, RSC Adv., 2016, 6, 56819–56826 RSC.
- M. T. Taghizadeh and M. Vatanparast, J. Mater. Sci.: Mater. Electron., 2017, 28, 778–786 CrossRef CAS.
- Y. Zhu, S. Pei, J. Tang, H. Li, L. Wang, W. Z. Yuan and Y. Zhang, J. Membr. Sci., 2013, 432, 66–72 CrossRef CAS.
- F. Meng, N. V. Aieta, S. F. Dec, J. L. Horan, D. Williamson, M. H. Frey, P. Pham, J. A. Turner, M. A. Yandrasits, S. J. Hamrock and A. M. Herring, Electrochim. Acta, 2007, 53, 1372–1378 CrossRef CAS.
- V. Ramani, H. R. Kunz and J. M. Fenton, J. Membr. Sci., 2005, 266, 110–114 CrossRef CAS.
- V. Ramani, H. R. Kunz and J. M. Fenton, Electrochim. Acta, 2005, 50, 1181–1187 CrossRef CAS.
- Y. Zhou, J. Yang, H. Su, J. Zeng, S. P. Jiang and W. A. Goddard, J. Am. Chem. Soc., 2014, 136, 4954–4964 CrossRef CAS PubMed.
- Y. Yao, J. Liu, W. Liu, M. Zhao, B. Wu, J. Gu and Z. Zou, Energy Environ. Sci., 2014, 7, 3362–3370 CAS.
- Y. Zhu, J. Mai, H. Li, J. Tang, W. Z. Yuan and Y. Zhang, Polym. Degrad. Stab., 2014, 107, 106–112 CrossRef CAS.
- P. Parthasarathy and A. V. Virkar, J. Power Sources, 2011, 196, 9204–9212 CrossRef CAS.
- S. Chen, H. A. Gasteiger, K. Hayakawa, T. Tada and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A82–A97 CrossRef CAS.
- M. P. Rodgers, N. Mohajeri, L. J. Bonville and D. K. Slattery, J. Electrochem. Soc., 2012, 159, B564–B569 CrossRef CAS.
- E. Endoh, ECS Trans., 2006, 3, 9–18 CAS.
- T. Tanuma and T. Itoh, J. Power Sources, 2016, 305, 17–21 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- C. Korsvik, S. Patil, S. Seal and W. T. Self, Chem. Commun., 2007, 1056–1058 RSC.
- Y. Xue, Q. Luan, D. Yang, X. Yao and K. Zhou, J. Phys. Chem. C, 2011, 115, 4433–4438 CAS.
- P. Trogadas, J. Parrondo and V. Ramani, ACS Appl. Mater. Interfaces, 2012, 4, 5098–5102 CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- L. Wang, S. G. Advani and A. K. Prasad, ECS Electrochem. Lett., 2014, 3, F30–F32 CrossRef CAS.
- M. Watanabe, H. Uchida, Y. Seki, M. Emori and P. Stonehart, J. Electrochem. Soc., 1996, 143, 3847–3852 CrossRef CAS.
- M. Lei, T. Z. Yang, W. J. Wang, K. Huang, R. Zhang, X. L. Fu, H. J. Yang, Y. G. Wang and W. H. Tang, Int. J. Hydrogen Energy, 2013, 38, 205–211 CrossRef CAS.
- M. Lei, Z. B. Wang, J. S. Li, H. L. Tang, W. J. Liu and Y. G. Wang, Sci. Rep., 2014, 4, 7415 CrossRef CAS PubMed.
- F. Xu, R. Xu and S. Mu, Electrochim. Acta, 2013, 112, 304–309 CrossRef CAS.
- T. Weissbach, T. J. Peckham and S. Holdcroft, J. Membr. Sci., 2016, 498, 94–104 CrossRef CAS.
- S. Schlick, M. Danilczuk, A. R. Drews and R. S. Kukreja, J. Phys. Chem. C, 2016, 120, 6885–6890 CAS.
- S. M. Stewart, D. Spernjak, R. Borup, A. Datye and F. Garzon, ECS Electrochem. Lett., 2014, 3, F19–F22 CrossRef CAS.
- M. Zaton, D. Jones and J. Rozière, ECS Trans., 2014, 61, 15–23 CrossRef CAS.
- A. M. Baker, R. Mukundan, D. Spernjak, E. J. Judge, S. G. Advani, A. K. Prasad and R. L. Borup, J. Electrochem. Soc., 2016, 163, F1023–F1031 CrossRef CAS.
- V. Prabhakaran and V. Ramani, J. Electrochem. Soc., 2014, 161, F1–F9 CrossRef CAS.
- M. Zatoń, J. Roziere and D. Jones, J. Mater. Chem. A, 2017, 5, 5390–5401 Search PubMed.
- L. Pino, A. Vita, M. Cordaro, V. Recupero and M. S. Hegde, Appl. Catal., A, 2003, 243, 135–146 CrossRef CAS.
- D. Banham, S. Ye, T. Cheng, S. Knights, S. M. Stewart, M. Wilson and F. Garzon, J. Electrochem. Soc., 2014, 161, F1075–F1080 CrossRef CAS.
- D. Banham, S. Ye, T. Cheng, S. Knights, S. M. Stewart and F. Garzon, ECS Trans., 2013, 58, 369–380 CrossRef.
- N. R. deTacconi, C. R. Chenthamarakshan, K. Rajeshwar, W.-Y. Lin, T. F. Carlson, L. Nikiel, W. A. Wampler, S. Sambandam and V. Ramani, J. Electrochem. Soc., 2008, 155, B1102–B1109 CrossRef CAS.
- D. Zhao, B. L. Yi, H. M. Zhang, H. M. Yu, L. Wang, Y. W. Ma and D. M. Xing, J. Power Sources, 2009, 190, 301–306 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |