
Rapid formation of self-organised Ag nanosheets with high efficiency and selectivity in CO2 electroreduction to CO†
Chong-Yong
Lee
 *a,
Yong
Zhao
a,
Caiyun
Wang
a,
David R. G.
Mitchell
b and
Gordon G.
Wallace
*a
*a,
Yong
Zhao
a,
Caiyun
Wang
a,
David R. G.
Mitchell
b and
Gordon G.
Wallace
*a
aARC Centre of Excellence for Electromaterials Science, Intelligent Polymer Research Institute, AIIM, University of Wollongong, Innovation Campus, Wollongong, NSW 2500, Australia. E-mail: cylee@uow.edu.au; gwallace@uow.edu.au
bElectron Microscopy Centre, AIIM, University of Wollongong, Innovation Campus, Wollongong, NSW 2500, Australia
First published on 6th April 2017
Abstract
Rapid and scalable production of electrocatalysts with high conversion efficiency and product selectivity are essential for practical application of electrochemical CO2 reduction. Here we report highly efficient and selective free-standing silver (Ag) nanosheet-based electrocatalysts that were produced in less than 10 min via an electrochemical oxidative–reductive approach. The hierarchical structures of Ag nanosheets provide an enhanced surface area and favourable gas transport/diffusion. The interconnected nanosize Ag (∼45 ± 10 nm) sheets produce among the best performance for aqueous CO2 to CO reduction for this class of materials. The conversion selectivity was approximately 95% at an overpotential as low as 0.29 V. This rational experimental design strategy may inspire efforts towards developing rapidly synthesized and efficient CO2 reduction electrocatalysts.
Combustion of fossil fuels is one of the major sources of anthropogenic CO2. Reducing CO2 emission levels into the atmosphere is among the key scientific challenges of the age.1 CO2 capture and storage through geological sequestration is costly, requires considerable infrastructure and may be prone to leakage and increased seismicity risks.2 In contrast, direct molecular conversion of CO2 means it becomes a feedstock for manufacturing useful products.3 Such products, including carbon-based fuels, can be obtained by electrochemical CO2 reduction. The electrical power required to drive this conversion process can be obtained from renewable energy sources, hence offering a sustainable approach. However, the success of this technology depends significantly on the development of CO2 reduction electrocatalysts which are not only highly efficient, selective and stable, but can also be rapidly prepared and scaled to meet the industrial demand for cost-effective production. Among the approaches, electrochemical anodisation offers a simple and low cost, yet powerful route towards synthesizing electrocatalysts of self-organising nanostructured morphology that readily formed on the conductive substrates.4,5 The efficiency in processing speed, the ease of up-scaling, and the cost–benefit aspects have promoted industrial applications of anodisation technology since 1920s.5,6 These technological features highlight the merits of employing the electrochemical anodisation method towards synthesizing new CO2 reduction electrocatalysts.
Metallic Ag surfaces show relatively high selectivity towards CO2 reduction to CO, displaying discrimination towards H2 evolution activity. Ag is less expensive and more abundant than Au which is known to be the best metallic CO2 reduction electrocatalyst.7 Furthermore, the intrinsic properties of Ag-based catalysts can be tailored to improve the performance of CO2 conversion to CO. Among the strategies used for this improvement were formation of nanoporous Ag catalysts by de-alloying an Ag–Al precursor,8 anodising Ag in electrolytes containing NaNO3,9 immobilising Ag nanoparticles on carbon supports,10 by reducing AgO to oxide-derived metallic Ag,11 and mesostructuring of Ag electrodes.12 In particular, the presence of halides such as Cl− and Br− in the solution is found to enhance the selectivity of CO formation in the electrocatalytic CO2 reduction.13 The Cl− residue in Ag nanocoral structures formed via anodisation is proposed to effectively inhibit the side reaction of H2 evolution, hence enhancing the electrocatalytic performance for CO2 reduction to CO.14,15
However, the approaches used to form catalysts for CO2 reduction on conductive support materials to date require lengthy processing times, typically many hours (see Table S1† for comparison). Here, we report rapid and template-less formation of a halide (chloride) derived porous self-organised Ag nanosheet-based electrocatalyst with high efficiency and selectivity for CO2 to CO conversion. Ag nanosheets with a beneficial hierarchical structure were produced in less than 10 min (Fig. 1a). In fact, the AgCl nanosheet precursors with a tunable layer thickness can be readily formed within 60 s, followed by an electroreduction to metallic porous Ag nanosheets. To the best of our knowledge, this facile and scalable approach offers, to date, the shortest fabrication time for producing electrocatalysts directly onto conductive materials yet with high CO2 to CO conversion performance.
 | ||
| Fig. 1 (a) Schematic representation of the process to fabricate Ag nanosheets on a Ag foil, (b) schematic illustration of the interdependency of the chloride concentration ([Cl−]) and anodisation potential (U) towards the formation of Ag nanosheet arrays. The brown contour represents the high density of nanosheet arrays achieved using the optimal conditions, (c) the transformation from microchannels (left-hand image) to nanosheets (right-hand image) results from optimizing the anodisation conditions. SEM images of: (d) as prepared AgCl with 90 s anodisation, (e) the Ag nanosheet formed upon electroreduction at −2.0 V (vs. Ag/AgCl 3 M NaCl) for 5 min. (f and g) Higher magnification images showing the nanosheets and the interconnected nanoparticles. | ||
Previous studies reported the anodic formation of AgCl with barrier and microchannel morphologies.14–18 Herein we devise a strategy to form, for the first time, self-organised nanosheets of AgCl. We identify two key parameters influencing the morphologies of the anodised layers: the chloride concentration and anodisation potential. The formation of AgCl films via oxidation of silver in chloride containing solution can be expressed as:16–18
| Ag + Cl− = AgCl + e− | (1) |
And the half cell potential follows a Nernstian behaviour:
E = −0.015 − 0.059![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log[Cl−] (VSCE) log[Cl−] (VSCE) | (2) |
As schematically illustrated in Fig. 1b, the chloride concentration and anodisation potential have a profound influence on the film morphology, changing the structure from barrier/microchannels to nanosheets (see Fig. 1c). The formation of microchannels results from the coalescence of voids between neighbouring AgCl grains.17 The optimum processing region for achieving the nanoporous sheet microstructure (Fig. 1b – brown domain) shows how a high applied potential and high chloride concentration are required to achieve this. Processing conditions within the light grey domain result in a lower density of nanosheets.
With anodisation performed at 1.0 V, for 90 s, in 0.5 M NaCl, microchannels of AgCl were formed with only a small number of AgCl sheets evident (Fig. S1†). Increasing the chloride concentration to 1.0 M resulted in a slight increase in the density of nanosheets. An extended anodisation for 300 s in 1.0 M NaCl did not promote the formation of AgCl nanosheets. The layer thickness did increase and this resulted in a cracked surface (Fig. S1c†). At 2.0 M (Fig. S1e†) and 3.0 M (Fig. 1d) NaCl, significant arrays of Ag nanosheets appeared. This result indicates that a high chloride concentration is required to form self-organised AgCl nanosheets.
As shown in eqn (2) and Fig. 1b, the anodisation potential critically influences the formation of the nanosheets. In the electrolyte containing 3.0 M NaCl, at 0.1 V, interconnected nanoparticles with nanochannels were formed, whereas increasing the potential to 0.3 V successfully promoted the formation of nanosheets. Increasing the potential to 0.6 V and above resulted in the formation of a significant number of nanosheets (Fig. S2†). Fig. 1d shows significant arrays of nanosheets formed at 1.0 V. We postulate that a sufficiently high anodic potential provides an essential kinetic driving force to rapidly oxidise Ag atoms forming a high local concentration of Ag+, which would readily react with high [Cl−] in a short diffusion distance. This may favour the morphological changes.
We have selected an optimal condition of 3.0 M NaCl and 1.0 V for the anodisation time–thickness dependent study (see Fig. S3 and S4,†2b and c1–c3). The current–time profile in Fig. 2a shows a rapid decay of current during the initial 30 s, indicating the rapid formation of a nanochannel AgCl layer (see Fig. S4†). This layer has a sub-micron, equiaxed grain size and is densely covered, suggesting widespread nucleation and growth of AgCl across the surface of the foil, during the initial phase of the reaction. This is followed by a gradual decline in the current and a transition to the growth of a larger structure with sheet morphology with lateral dimensions of up to ∼14 μm in size (Fig. S5a†). Cross-sectional SEM data (Fig. 2c1–c3 and S5b†) further indicate that the growth of AgCl nanosheets involves two stages: the first stage is the formation of nanochannels; the second stage involves the growth of nanosheets, which only occurs under suitable anodisation conditions. This observation is in agreement with a growth model in which the AgCl layer grows by an outward diffusion of Ag+.17,19 Ag+ acts as the main charge carrier, where new material is produced at the AgCl|bulk-solution interface. The surface roughness formed at the edges of the nanosheets may result in concentration of the electric field. This field then encourages ionic migration to these regions at high [Cl−] and U. Thereafter the nanosheets grow outward at their edges, with their aspect ratio increasing. The initial fine grained product is buried by the outwardly growing film.
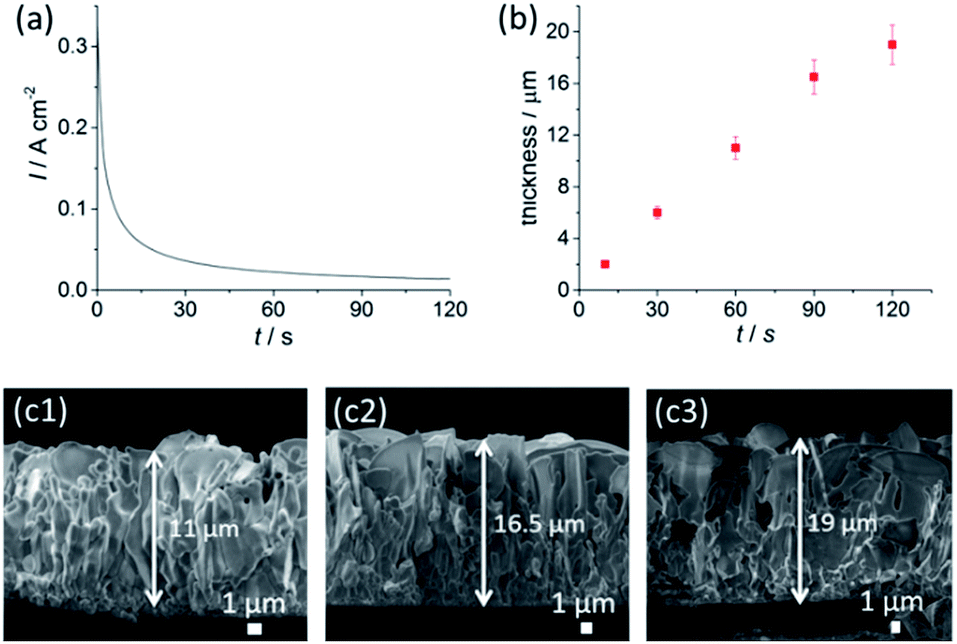 | ||
| Fig. 2 (a) Current–time profile upon anodisation of Ag foil at 1.0 V in 3 M NaCl solution. (b) The thickness dependence of Ag nanosheets as a function of anodisation time. SEM images of anodisation performed at (c1) 60 s, (c2) 90 s and (c3) 120 s, respectively. | ||
An apparent nanosheet morphology appeared within just 60 s under the conditions used. This fast formation indicates that the saturation of Ag+ (U effect) and Cl− (NaCl effect) in addition to a highly conductive NaCl electrolyte promotes instantaneous formation of nanosheets with an excellent coverage across the Ag foil. This feature, in addition to the very short anodisation time, greatly promotes the scalability of the anodised AgCl layer, with ease of up-scaling of AgCl growth on a large area of Ag foil. Extending the anodisation time to 120 s gradually increases the AgCl layer thickness to 19 ± 2 μm.
The free-standing AgCl nanosheet can be readily reduced by cathodic reduction at −2.0 V vs. Ag/AgCl (in 0.5 M NaHCO3 under constant CO2 purging) for 5 min, resulting in the formation of Ag nanosheets with interconnected Ag particles (Fig. 1a, e–g). The particle size of Ag was found to depend upon the rate of nucleation, where a higher cathodic current density resulted in a smaller average particle size.17 The Ag nanoparticles formed under the aforementioned synthesis conditions were found to be 50 ± 10 nm in diameter with an interconnected nanocoral structure. Importantly, the nanosheet structure remained and this unique hierarchical architecture provides not only an enhanced surface area but also allows efficient transport of reactants and products to/from the active sites of Ag for electrocatalytic application. The electrochemical surface area measurement (Fig. S6†) indicates that the relative area of the Ag nanosheet-based electrode is about 17 times larger than that of the polycrystalline Ag foil.
To gain the compositional, crystal and structural information, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and high resolution scanning transmission electron microscopy (STEM) analyses were performed on the Ag foil, as-formed AgCl and reduced Ag layers. XRD data (Fig. 3a) confirm the formation of the as-anodised AgCl, with peak positions being in full agreement with the reference file for AgCl (JCPDS no. 31-1238). The reduced Ag nanosheet shows a typical diffraction pattern of metallic Ag (JCPDS no. 65-2871) indicating successful conversion of AgCl to Ag. This is further confirmed by the shift of Ag 3d XPS peaks after AgCl electroreduction (Fig. 3b). Some residual Cl− is observed in the reduced Ag nanosheets (Fig. 3c) suggesting strong adsorption of Cl− on the Ag surface. High resolution STEM bright field images (Fig. 3d) reveal that the Ag nanosheets are composed of interconnected crystalline Ag nanoparticles with a mean diameter of 45 ± 10 nm, consistent with the SEM data. The nanosheets occur as single layer sheets with a thickness of typically ∼50 nm or as a thicker bi-layer sheet (Fig. S7†). Measurement of high resolution STEM images indicates a lattice fringe spacing of 0.23 nm corresponding to the Ag (111) spacing (Fig. 3d, bottom right). Importantly, large numbers of twins were observed between and within individual grains (Fig. 3d, bottom left, see Fig. S7† for larger images). Recent studies by Kanan and Chorkendorff et al. implicate grain boundaries as active sites for CO2 reduction.20 Therefore, the hierarchical Ag nanosheets with a high density of grain boundaries would be beneficial for the electrocatalytic CO2 reduction.
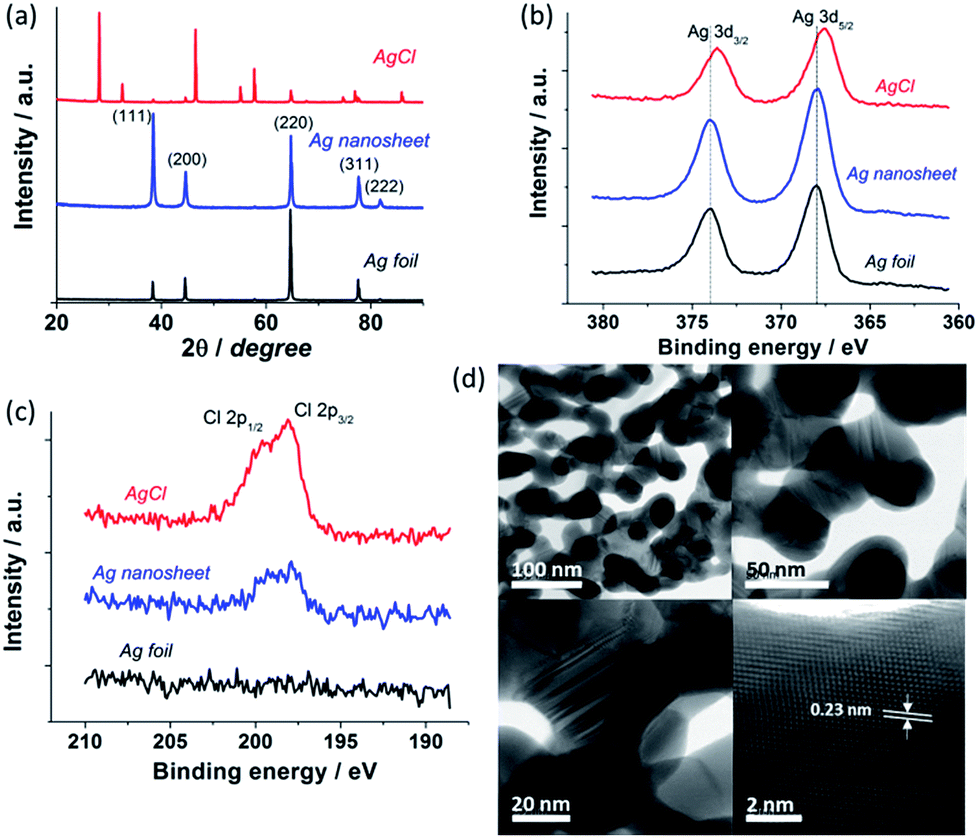 | ||
| Fig. 3 XRD patterns (a) and XPS (b and c) of Ag foil, AgCl, and reduced Ag nanosheets. (d) Representative high resolution STEM bright field images of the reduced Ag nanosheet. | ||
We evaluated CO2 electrocatalytic reduction performance of the polycrystalline Ag foil in comparison to the reduced Ag nanosheets. The reduction was performed in CO2 saturated 0.5 M NaHCO3 electrolyte (pH 7.2) at ambient temperature and pressure. Fig. 4a shows that the geometrical current density obtained with the Ag nanosheet-based electrode was significantly higher than that for the Ag foil. At −0.6 V, the current density obtained with the Ag nanosheet-based electrode was 37 times (vs. 17 times increased in the surface area) larger than that for the polycrystalline Ag, indicating that the intrinsic activity of the Ag nanosheets contributes to the enhanced electrocatalytic performance. Fig. 4b shows the CO conversion faradaic efficiency in the range of 95% up to an overpotential of 0.29 V. At an overpotential of 0.24 V, a faradaic efficiency of ∼70% was achieved. These interconnected nanosize Ag nanosheets therefore produce among the best performances for aqueous CO2 to CO reduction. A current efficiency of 95% was achieved at an overpotential as low as 0.29 V. The high intrinsic activity and ease of reactant access to the porous layer resulting from the nanosheet structure may contribute to this performance. Fig. 4c shows the Tafel slope of 59 mV dec−1 for nanosheets, indicating fast reaction kinetics for the Ag nanosheet-based electrode.8 The Ag nanosheet electrode shows good stability (Fig. S8†) with the retention of ∼70% current density, and a ∼80% CO conversion faradaic efficiency after 16 hours of electrolysis; and the nanosheet structure remained intact.
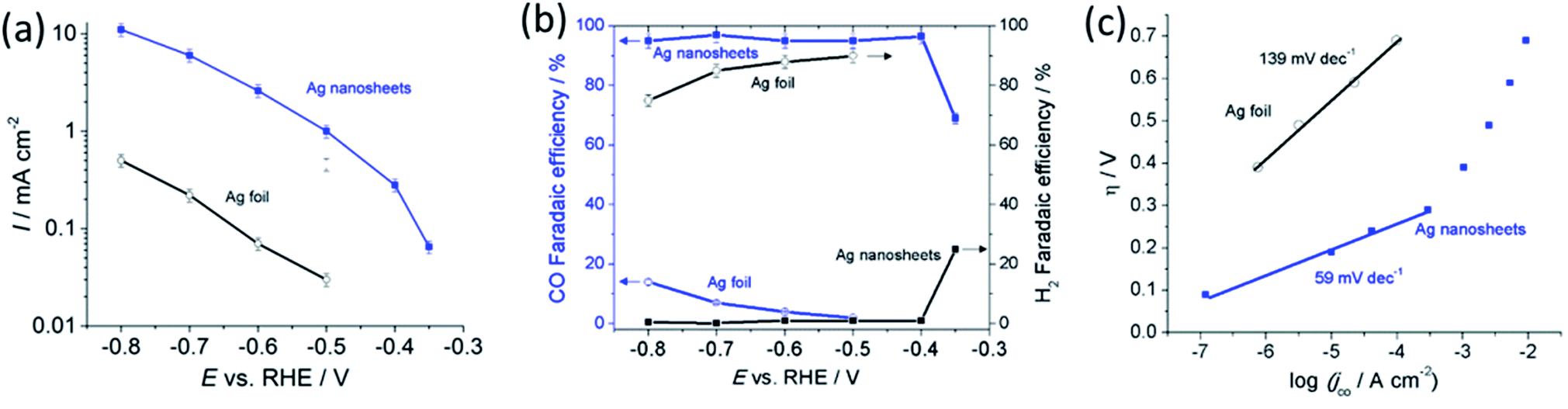 | ||
Fig. 4 Current density (a), CO and H2 faradaic efficiency (b), and Tafel plots of the CO partial current density (c) for polycrystalline Ag foil (black,  ) and Ag nanosheets (blue, ) and Ag nanosheets (blue,  ). The Ag nanosheets were obtained from anodisation at 1.0 V, in 3.0 M NaCl for 90 s, subsequently reduced at −2.0 V, in 0.5 M NaHCO3 for 5 min. ). The Ag nanosheets were obtained from anodisation at 1.0 V, in 3.0 M NaCl for 90 s, subsequently reduced at −2.0 V, in 0.5 M NaHCO3 for 5 min. | ||
To further demonstrate the advantages of the hierarchical structure of Ag nanosheets, we decorated this structure with Au by using a simple electroless galvanic replacement.21 Upon immersion in aqueous 0.5 M HAuCl4 for 60 s, Au decorated Ag nanosheets were formed, exhibiting high catalytic currents from ∼−0.4 V vs. RHE (Fig. S9†). At −0.6 V, the catalytic current improved about 40%, increasing from −1.6 mA cm−2 to −2.7 mA cm−2, while only a marginal difference was observed on the Au-modified nominally flat Ag nanocorals (see Fig. S10†). The faradaic efficiency for CO formation on the Au-modified Ag nanosheets was over 90%. We postulate this enhancement is due to the larger accessibility of Au onto the exposed Ag surface in Ag nanosheets to form Ag–Au in comparison to the Ag nanocorals.
Conclusions
In summary, we show for the first time a fast, efficient and scalable way to form free-standing halide-derived Ag nanosheets by using a simple electrochemical oxidation–reduction approach. These Ag nanosheets, with their high surface area and open structure, are amenable to surface modifications, which opens up the possibility for further enhancement in the electrocatalytic CO2 reduction performance. The residual Cl− and a significant number of grain boundaries may contribute to the highly efficient conversion of CO2 to CO achieved by this material. However, further investigation is required to fully understand the mechanistic aspects of the CO2 reduction process on these novel hierarchical Ag nanosheets. This hierarchical Ag nanostructure may be also useful for other catalytic and sensing applications. This work may stimulate further efforts towards rapidly synthesizing scalable, efficient and selective electrocatalysts for CO2 reduction that may take us closer to the realisation of technologies for practical application.Acknowledgements
This work was supported by the University of Wollongong's Vice Chancellor Research Fellowship (to CYL) and Australian Laureate Fellowship scheme FL 110100196 (to GGW). Funding from the Australian Research Council Centre of Excellence Scheme (CE 140100012) is gratefully acknowledged. The authors would also like to thank the Australian National Fabrication Facility-Materials Node (ANFF) and UOW Electron Microscopy Centre for equipment use.Notes and references
- (a) J. A. Turner, Science, 1999, 285, 687 CrossRef CAS PubMed; (b) M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981 CrossRef CAS PubMed; (c) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- (a) E. J. W. Alexandra and B. Klass, Emory Law J., 2008, 58, 103 Search PubMed; (b) J. J. C. Phelps, J. C. Blackford, J. T. Holt and J. A. Polton, Int. J. Greenhouse Gas Control, 2015, 38, 210 CrossRef CAS.
- (a) B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, Catal. Today, 2016, 270, 19 CrossRef CAS; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (c) W. l. Christina, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef PubMed; (d) T. E. Rosser, C. D. Windle and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 7388 CrossRef CAS PubMed.
- (a) K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385 CrossRef CAS PubMed; (b) P. Roy, S. Berger and P. Schmuki, Angew. Chem., 2011, 123, 2956 ( Angew. Chem., Int. Ed. , 2011 , 50 , 2904 ) CrossRef; (c) C.-Y. Lee, K. Lee and P. Schmuki, Angew. Chem., Int. Ed., 2013, 52, 2077 CrossRef CAS PubMed; (d) S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048 CrossRef CAS; (e) C.-Y. Lee, L. Wang, Y. Kado, M. S. Killian and P. Schmuki, ChemSusChem, 2014, 7, 934 CrossRef CAS PubMed; (f) C.-Y. Lee, Z. Su, K. Lee, H. Tsuchiya and P. Schmuki, Chem. Commun., 2014, 50, 7067 RSC.
- C.-Y. Lee and P. Schmuki, in Advances in Electrochemical Sciences and Engineering Book Series, ed. R. C. Alkire, P. N. Bartlett and J. Lipkowski, Wiley-VCH, Weinheim, Germany, 2015, vol. 15, ch. 5 Search PubMed.
- S. So, K. Lee and P. Schmuki, J. Am. Chem. Soc., 2012, 134, 11316 CrossRef CAS PubMed.
- (a) Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, USA, 2008, vol. 42, ch. 3 Search PubMed; (b) N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 Search PubMed.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Chem. Commun., 2015, 51, 17704 RSC.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844 CrossRef CAS PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., 2016, 128, 9900 CrossRef.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136 CrossRef CAS.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349 CrossRef CAS.
- L. Zhang, Z. Wang, N. Mehio, X. Jin and S. Dai, ChemSusChem, 2016, 9, 428 CrossRef CAS PubMed.
- (a) T. R. Beck and D. E. Rice, J. Electrochem. Soc., 1984, 131, 89 CrossRef CAS; (b) V. I. Birss and C. K. Smith, Electrochim. Acta, 1987, 32, 259 CrossRef CAS; (c) G. T. Burstein and R. D. K. Misra, Electrochim. Acta, 1983, 28, 371 CrossRef CAS; (d) B. M. Jovic, V. D. Jovic and D. M. Draazic, J. Electroanal. Chem., 1995, 399, 197 CrossRef.
- X. Jin, J. Lu, P. Liu and H. Tong, J. Electroanal. Chem., 2003, 542, 85 CrossRef CAS.
- H. Ha and J. Payer, Electrochim. Acta, 2011, 56, 2781 CrossRef CAS PubMed.
- G. W. D. Briggs and H. R. Thirsk, Trans. Faraday Soc., 1952, 48, 1171 RSC.
- (a) X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606 CrossRef CAS PubMed; (b) A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808 CrossRef CAS PubMed.
- Y. Wang, H. Chen, S. Dong and E. Wang, J. Chem. Phys., 2006, 125, 47710 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00069c |
| This journal is © The Royal Society of Chemistry 2017 |