
Mechanosynthesis of pure phase mixed-cation MAxFA1−xPbI3 hybrid perovskites: photovoltaic performance and electrochemical properties†
D.
Prochowicz‡
 *ab,
P.
Yadav‡
a,
M.
Saliba
a,
M.
Saski
b,
S. M.
Zakeeruddin
a,
J.
Lewiński
*bc and
M.
Grätzel
*a
*ab,
P.
Yadav‡
a,
M.
Saliba
a,
M.
Saski
b,
S. M.
Zakeeruddin
a,
J.
Lewiński
*bc and
M.
Grätzel
*a
aLaboratory of Photonics and Interfaces, Institute of Chemical Sciences and Engineering, School of Basic Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: dprochowicz@ichf.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
cFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
First published on 12th April 2017
Abstract
We report a facile mechanochemical route for the stabilization of a structurally stable α-FAPbI3 perovskite by fine and controllable stoichiometry modification in mixed-cation (MA)x(FA)1−xPbI3 perovskites. Moreover, the mechanoperovskite with a composition of (MA)0.25(FA)0.75PbI3 was used for solar cell fabrication showing a power conversion efficiency (PCE) of 14.98% with a high short circuit current (JSC) density at 23.7 mA cm−2.
Hybrid organic–inorganic lead halide perovskite solar cells (PSCs) have gained increasing attention due to their easy fabrication and a certified high power conversion efficiency of 22.1%.1–3 One of the first studied perovskite materials for solar cells was the simple single-cation, single-halide methylammonium lead triiodide (MAPbI3), because it could be easily fabricated via a vast array of processing methods using only MAI and PbI2 as precursor components.4–6 However, as the field has evolved, it was found that MAPbI3 suffers from a poor thermal and moisture stability,7,8 as well as hysteretic current–voltage characteristics.9,10 Then, the formamidinium lead iodide (FAPbI3) perovskite was shown to have improved thermal stability and a beneficial lower bandgap red-shifted towards the single junction Shockley–Queisser optimum.11 However, the photoactive α-FAPbI3 “black phase” is thermodynamically stable only above 160 °C and converts to an undesirable δ-FAPbI3 “yellow phase” at room temperature.12–14
This conversion is driven by the presence of large FA+ that distorts the perovskite lattice.13 An important stabilization strategy to relax the distorted lattice relies on cation mixing by adding the smaller MA cation to FA-based perovskites, which results in the formation of a perovskite material formulated as (MA)x(FA)1−xPbI3.15–18 Substituting as little as 15% FA+ with MA+ was already sufficient to stabilize the black phase of FAPbI3.19 Mixed-cation (MA)x(FA)1−xPbI3 thin films were usually fabricated by a two-step deposition process15,16 (where the inorganic layer was deposited first followed by the organic cation) or a cation exchange interdiffusion method of an already formed single-cation perovskite.20,21 In addition, the mixed-cation perovskites exhibit favourable properties in terms of structural stability translating into a photoactive black phase at room temperature and consequently into photovoltaic performance. However, it is difficult to control the processing parameters during the thin film formation process and thus leads to poor repeatability.
While wet methods are commonly used for the synthesis of hybrid perovskites,22 recently we have developed for the first time a highly efficient, simple and reproducible method for the preparation of MAPbI3 induced by mechanical forces.23 Remarkably, the mechanochemically synthesized polycrystalline MAPbI3 powder was used to fabricate solar cells devices via a one-step deposition method and gave superior photovoltaic performance as compared to standard solution processed synthesis. Very recently, mechanosynthesis has been applied to the preparation of other three types of hybrid iodide perovskites (FAPbI3, Gua2PbI4, and GuaPbI3)24 and bromide perovskites (CsPbBr3, MAPbBr3, and FAPbBr3);25 however, the mechanosynthesis of phase pure mixed-cation perovskites has not yet been reported. Following our research interest in mechanochemistry,26–29 herein we report for the first time stabilization of the α-FAPbI3 perovskite at room temperature achieved by mechanochemistry through fine and controllable stoichiometry modification in mixed-cation (MA)x(FA)1−xPbI3 perovskites. The newly synthesized material was used for fabrication of solar cells by a facile single-step antisolvent method and showed an impressive high short circuit current density of 23.7 mA cm−2, a value rarely reached even by the highest performing perovskite solar cells.1–3 In addition, the resultant device gave superior photovoltaic performance compared to a conventional solution processed device.
By applying the solid-state mechanochemical reaction between starting reagents, we determine the appropriate amount of MA+ to stabilize the black phase of FAPbI3. More specifically, the phase structure of FAPbI3 was investigated by varying the molar ratio of MAI/FAI in the reaction with PbI2 using a ball milling procedure (for experimental details see Experimental section in the ESI†). All powders obtained from this mechanosynthesis method were characterized by powder X-ray diffraction (PXRD). First, we performed the ball milling reaction between FAI and PbI2 in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio and the measured PXRD diagram for such a mechanosynthesized material ideally matches with the simulated PXRD pattern of δ-FAPbI3.12,24 Next, we explored the mechanochemical approach to demonstrate the formation of mixed-organic-cation compositional (MA)x(FA)1−xPbI3 perovskites with x = 15, 20, and 25%. As shown in Fig. 1a–c, all compositions exhibit a typical perovskite peak at ∼14°. For x = 15 and 20%, a second peak at 11.61° is present indicating yellow phase δ-FAPbI3. The yellow phase peak disappears for x = 25% leaving only the black cubic perovskite phase α-FAPbI3 (Fig. 1c and d). In addition, in all investigated compositions of (MA)x(FA)1−xPbI3 the diffraction peaks specific to PbI2 (at 12.7 deg) were not observed.
:1 stoichiometric ratio and the measured PXRD diagram for such a mechanosynthesized material ideally matches with the simulated PXRD pattern of δ-FAPbI3.12,24 Next, we explored the mechanochemical approach to demonstrate the formation of mixed-organic-cation compositional (MA)x(FA)1−xPbI3 perovskites with x = 15, 20, and 25%. As shown in Fig. 1a–c, all compositions exhibit a typical perovskite peak at ∼14°. For x = 15 and 20%, a second peak at 11.61° is present indicating yellow phase δ-FAPbI3. The yellow phase peak disappears for x = 25% leaving only the black cubic perovskite phase α-FAPbI3 (Fig. 1c and d). In addition, in all investigated compositions of (MA)x(FA)1−xPbI3 the diffraction peaks specific to PbI2 (at 12.7 deg) were not observed.
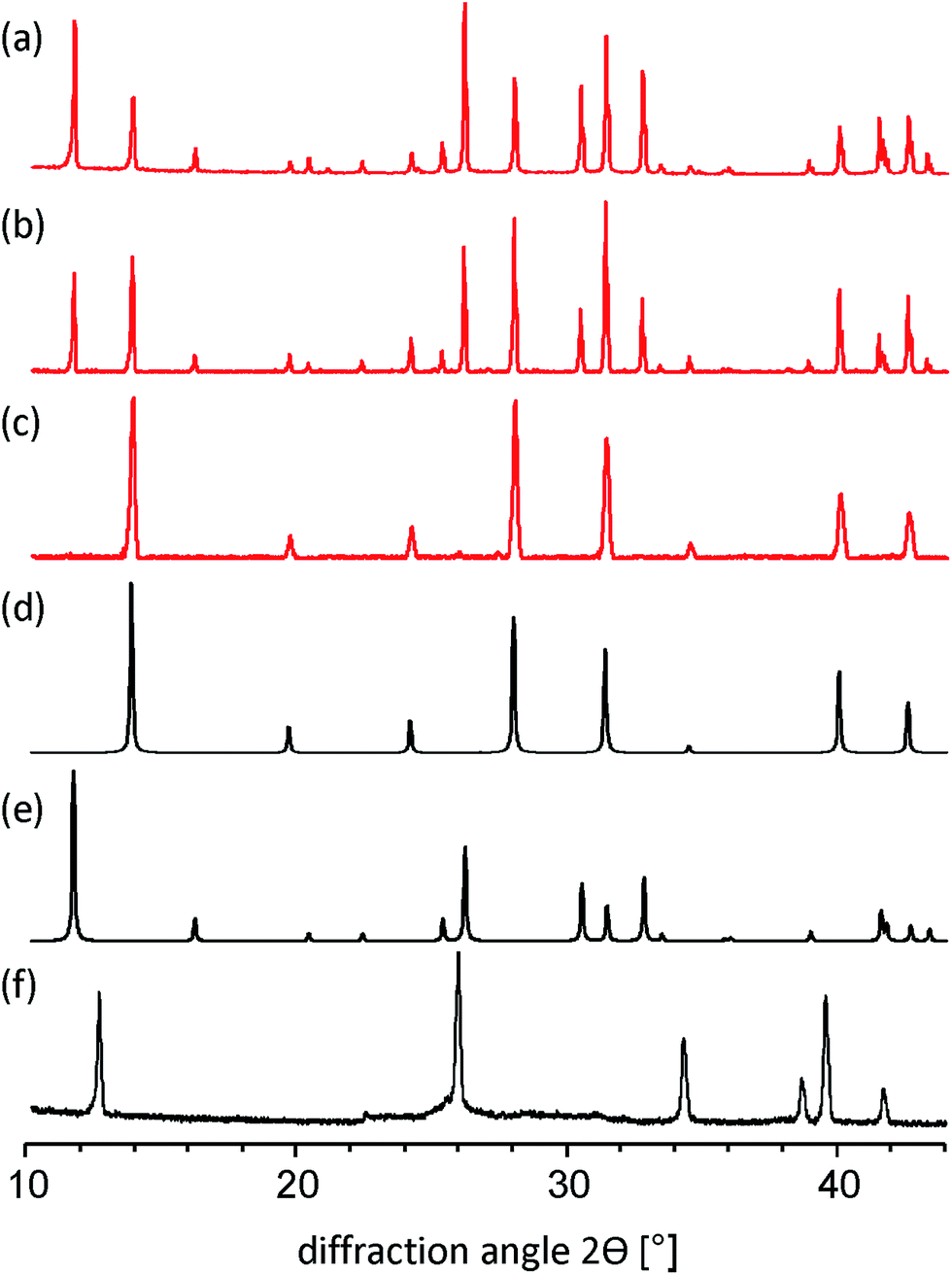 | ||
| Fig. 1 PXRD patterns of: (a) as-ground (MA)0.15(FA)0.85PbI3; (b) as-ground (MA)0.20(FA)0.80PbI3; (c) as-ground (MA)0.25(FA)0.75PbI3; (d) simulated α-FAPbI3 black phase; (e) simulated δ-FAPbI3 yellow phase; (f) PbI2. | ||
In the next step, phase pure mechanoperovskite (MA)0.25(FA)0.75PbI3 was chosen to fabricate solution-processed solid-state photovoltaic devices. To evaluate the quality of the(MA)0.25(FA)0.75PbI3 thin film (using the antisolvent method),30 we first characterized the morphological properties.
The top-view SEM image of the perovskite film clearly shows the compact surface coverage and the particles exhibit average diameters of about 150–350 nm (Fig. 2a). The UV-Vis absorption edge of the compositional (MA)0.25(FA)0.75PbI3 perovskite film is red-shifted from 780 to 830 nm as compared to that of single-cation MAPbI3 (Fig. S1†). We used the thin films for solar cell fabrication using a stack of glass/FTO/bl-TiO2/ms-TiO2-perovskite nanocomposite layer/perovskite upper layer/spiro-OMeTAD/Au. The cross-sectional SEM image of the as-fabricated (MA)0.25(FA)0.75PbI3 perovskite solar cells in Fig. 2b shows a 150 nm thick mesoporous layer and a 200 nm thick capping perovskite layer. Fig. 3 shows the current–density–voltage (J–V) characteristics of the best device measured under AM 1.5G spectrum scanned from forward bias. The device exhibits a short circuit current (JSC) of 23.7 mA cm−2, an open circuit voltage (VOC) of 971 mV, a fill factor (FF) of 65% and a power conversion efficiency (PCE) of 14.98%. Notably, the high JSC at ∼23.76 mA cm−2 is comparable to that of the most efficient perovskite solar cells and deposition methods reported in the literature.13,15 The incident photon-to-electron conversion efficiency spectra and integrated JSC over an AM 1.5G spectrum for an investigated PSC are shown in Fig. S2.† The integrated JSC of 21.8 mA cm−2 from the incident photon-to-current efficiency (IPCE) spectrum is slightly less than the value derived from the corresponding J–V measurement. For comparison of photovoltaic performance, the reference material (MA)0.25(FA)0.75PbI3 was synthesized from the direct solvothermal reaction between PbI2, MAI and FAI in the corresponding ratio (for more details, see the Experimental section). The one-step deposition process provides a device with an open circuit voltage (VOC) of 879 mV, a short-circuit current (JSC) of 18.54 mA cm−2, a fill factor (FF) of 67% and a power conversion efficiency (PCE) of 10.97% (see Fig. S3†). The phase purity of the resultant perovskite films was determined using PXRD analysis. The PXRD of the (MA)0.25(FA)0.75PbI3 film of each device is shown in Fig. S4.† The PXRD spectra of the perovskite thin film prepared from mechanoperovskite particles confirmed the presence of pure phase α-FAPbI3. However, the diffraction pattern of the film prepared from the solution processed synthesis shows two distinct peaks at 11.6° and 13.9° that correspond to the yellow and black FAPbI3 phase, respectively. Thus, the presence of the yellow δ-FAPbI3 phase might be attributed to the detrimental lower photovoltaic performance compared to the PSC fabricated with mechanosynthetically prepared perovskite particles.
 | ||
| Fig. 2 (a) Top view SEM image of the (MA)0.25(FA)0.75PbI3 thin film on FTO/bl-TiO2/ms-TiO2. (b) Cross-sectional SEM image of a device containing (MA)0.25(FA)0.75PbI3. | ||
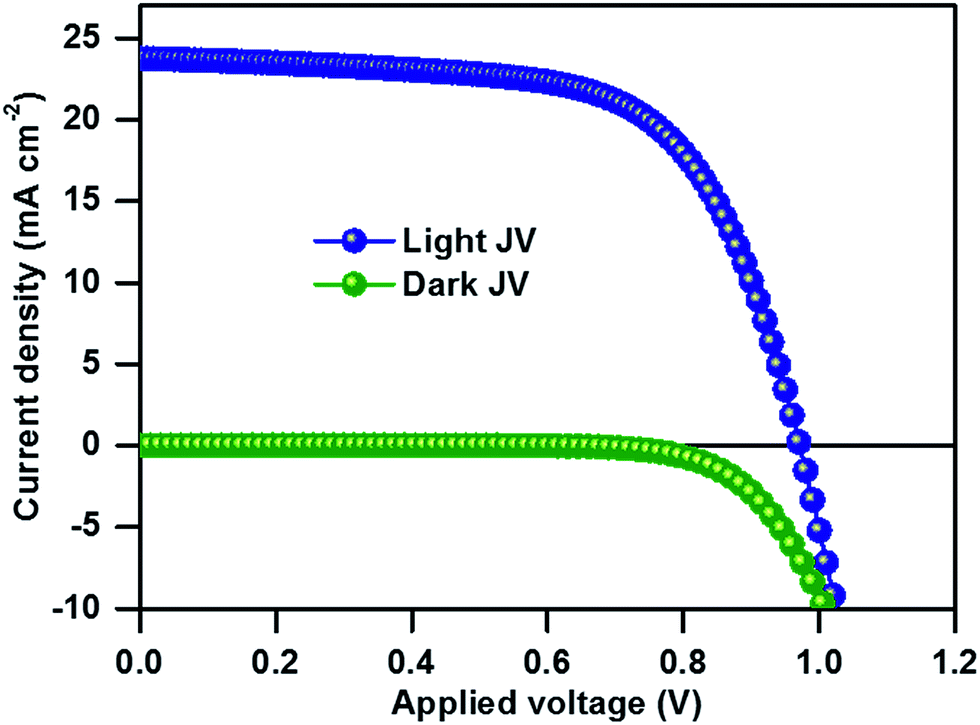 | ||
| Fig. 3 J–V characteristics of the champion device of the (MA)0.25(FA)0.75PbI3 perovskite solar cell recorded under standard illumination. | ||
By using a sweep rate of 50 mV s−1, a moderate hysteresis of 1% in PCE is observed for the solar cell containing the (MA)0.25(FA)0.75PbI3 mechanoperovskite. However, the device PCE is limited by the lower values of FF and VOC. The cross-sectional view of the SEM image shows shunt paths across the interface layer that likely lower the FF and may entail increased recombination that is detrimental for the VOC. Thus, further studies in the optimization of the device architecture are required to improve the PCE.
The key parameters associated with the recombination and transport phenomenon such as the ideality factor (m), series resistance (RS) and reverse saturation current (I0) were also studied. Fig. 4a illustrates the −dV/dJ vs. (JSC − J)−1 plot, calculated from the light J–V characteristics and plotted in the corresponding bias range where the dependence of the net current on the applied potential is least. The device shows a significantly higher value of m ≈ 4.2 attributed to recombination as a dominant loss mechanism. Taking the obtained value of m as an input parameter to the expression dV/dJ = RS + mkT/qI0(exp−qV/mkT), −dV/dJ vs. (exp−qV/mkT) is plotted in Fig. 4b where the linear intercept at the y-axis and the slope depicts RS and J0. The values of RS and J0 were obtained to be 5 Ω cm−2 and 8.8 × 10−6 A, respectively. Along with the higher recombination, a significantly higher value of RS in the present case is also a limiting factor for a lower FF. In general, at VOC, all the photogenerated charges can recombine and it is possible to recalculate it from J0, m and JSC by using the expression: VOC = mkT/qln(JSC/J0). A good correlation between the calculated and measured VOC from J–V characteristics is obtained.
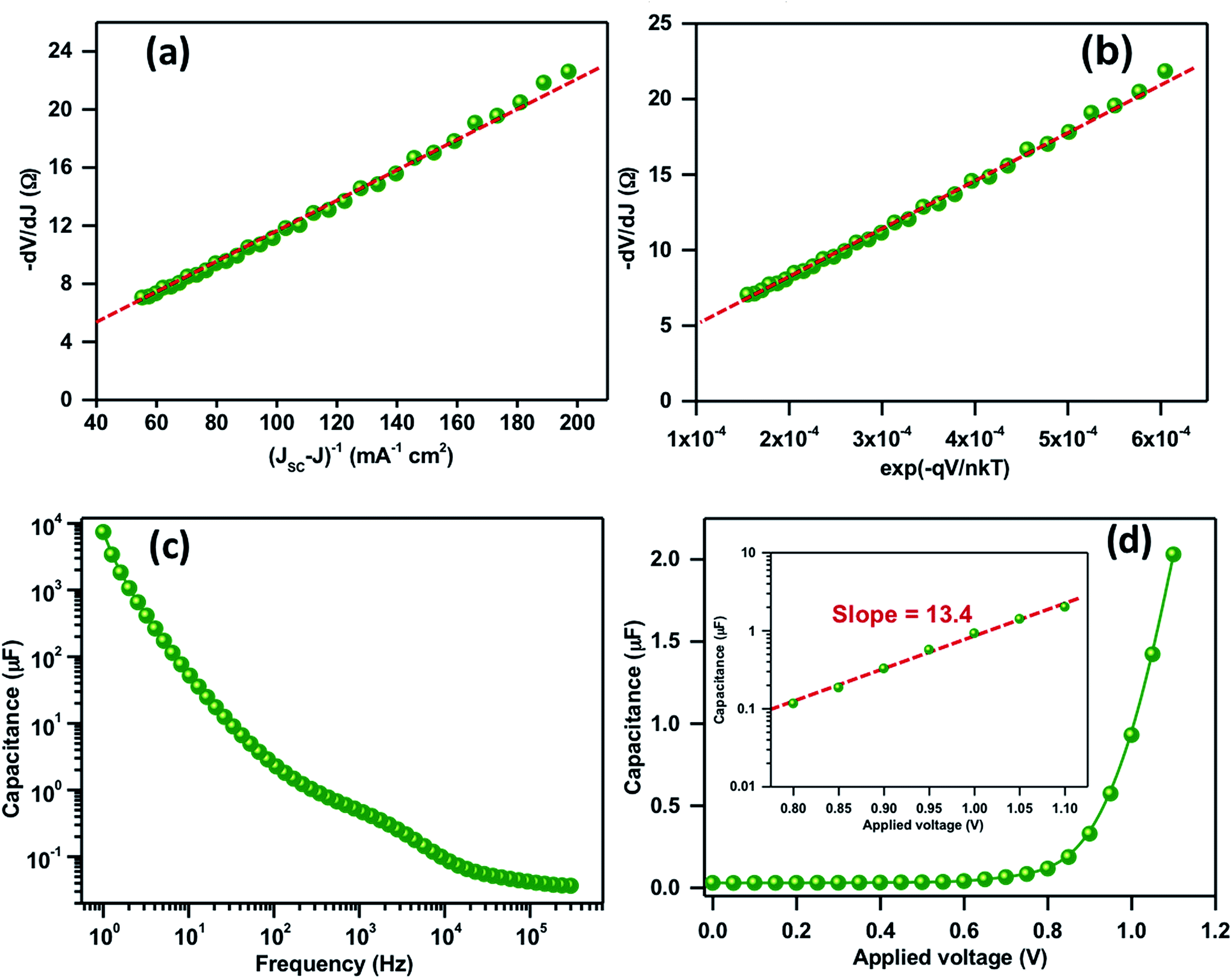 | ||
| Fig. 4 (a) Plot of −dV/dJ versus 1/J curves. (b) Plot of dV/dJ vs. (exp−qV/mkT). The solid lines represent the linear fitting. (c) Frequency-dependent capacitance response of the perovskite solar cell at zero bias. (d) Plot of capacitance vs. applied bias for the perovskite solar cell; the inset shows the semi-logarithmic plot of capacitance in the higher bias with the dashed line showing the linear fit. | ||
Fig. 4c shows the frequency-dependent capacitance response at zero bias where a distinguishable plateau related to the polarization and bulk dielectric process is observed. The high and mid frequency capacitance corresponding to chemical capacitance of TiO2 or di-electric polarization of the absorber material shows the values in the range of 0.5 μF. Most importantly the low frequency capacitance due to charge accumulation at the TiO2/perovskite absorber layer shows a value in the order of 7.4 mF cm−2 at 1 Hz. The obtained capacitance is comparatively higher than the efficient devices but quite similar with the device possessing a similar VOC and efficiency.31–33 In general, the higher value of low frequency capacitance could alter the space-charge and electric field at the interface that leads to an inefficient charge separation and recollection.33Fig. 4d shows the plot of capacitance vs. applied potential at a low frequency. The Fig. 4d inset shows the capacitance vs. applied bias plot in a semi-logarithmic scale in the high potential region. The straight line fit gives a slope of 13.4 that is attributed to a significant accumulation of charges at the TiO2/perovskite absorber layer.32,33
Conclusions
In conclusion, we have demonstrated a mechanochemical strategy for the simple stabilization of a structurally stable α-FAPbI3 perovskite by fine and controllable stoichiometry modification in mixed-cation hybrid (MA)x(FA)1−xPbI3 perovskites. Specifically, we observed that the addition of 25% (molar%) of MAI to the reaction mixture stabilize the black phase of FAPbI3. The newly synthesized perovskite material (MA)0.25(FA)0.75PbI3 was used for solar cell fabrication and showed an impressive high short circuit current density of 23.7 mA cm−2. Moreover, we found that the resultant device gave better photovoltaic performance compared to a conventional solution processed device. We believe that the presented results open up new possibilities for the efficient and sustainable synthesis of hybrid perovskites and, importantly, provide a promising strategy for the highest performing perovskite solar cells. Further studies on the mechanosynthesis of other class of mixed-cation hybrid perovskites and exploration of their photovoltaic performances are in progress.Acknowledgements
D. P. and M. S. acknowledge support from the co-funded Marie Skłodowska Curie fellowship, H2020 Grant agreement no. 707168 and 665667, respectively. M. S, J. L. and M. G. thank funding from the European Union's Horizon 2020 programme, through a FET Open research and innovation action under grant agreement no. 687008. P. Y. gratefully acknowledges financial support from the Swiss Confederation under Swiss Government Scholarship.Notes and references
- X. Li, D. Bi, C. Yi, J. D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- National Renewable Energy Laboratory, Best Research-Cell Efficiencies chart, www.nrel.gov/ncpv/images/efficiency_chart.jpg.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619–2623 Search PubMed.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- T. Baikie, Y. N. Fang, J. M. Kadro, M. Schreyer, F. X. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 RSC.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca and E. Mosconi, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- C. Zhao, B. Chen, X. Qiao, L. Luan, K. Lu and B. Hu, Adv. Energy Mater., 2015, 5, 1500279 CrossRef.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 Search PubMed.
- N. Pellet, P. Gao, G. Gregori, T. Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- F. Ji, L. Wang, S. Pang, P. Gao, H. Xu, G. Xie, J. Zhang and G. Cui, J. Mater. Chem. A, 2016, 4, 14437–14443 RSC.
- Z. Yang, C. C. Chueh, P. W. Liang, M. Crump, F. Lin, Z. Zhu and A. K. Y. Jen, Nano Energy, 2016, 22, 328–337 CrossRef CAS.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 Search PubMed.
- A. Binek, F. C. Hanusch, P. Docampo and T. Bein, J. Phys. Chem. Lett., 2015, 6, 1249–1253 CrossRef CAS PubMed.
- J. Liu, Y. Shirai, X. Yang, Y. Yue, W. Chen, Y. Wu, A. Islam and L. Han, Adv. Mater., 2015, 27, 4918–4923 CrossRef CAS PubMed.
- G. E. Eperon, C. E. Beck and H. J. Snaith, Mater. Horiz., 2016, 3, 63–71 RSC.
- Y. Dang, D. Ju, L. Wang and X. Tao, CrystEngComm, 2016, 18, 4476–4484 RSC.
- D. Prochowicz, M. Franckevicius, A. M. Cieślak, S. M. Zakeeruddin, M. Graetzel and J. Lewiński, J. Mater. Chem. A, 2015, 3, 20772–20777 RSC.
- A. D. Jodlowski, A. Ypez, R. Luque, L. Camacho and G. de Miguel, Angew. Chem., Int. Ed., 2016, 55, 14972–14977 CrossRef CAS.
- A. Jana, M. Mittal, A. Singla and S. Sapra, Chem. Commun., 2017, 53, 3046–3049 RSC.
- D. Prochowicz, K. Sokołowski, I. Justyniak, A. Kornowicz, D. Fairen-Jimenez, T. Friščić and J. Lewiński, Chem. Commun., 2015, 51, 4032 RSC.
- D. Prochowicz, I. Justyniak, A. Kornowicz, T. Kaczorowski, Z. Kaszkur and J. Lewiński, Chem.–Eur. J., 2012, 18, 7367 CrossRef CAS PubMed.
- P. Krupiński, A. Kornowicz, K. Sokołowski, A. M. Cieślak and J. Lewiński, Chem.–Eur. J., 2016, 22, 7817–7823 CrossRef PubMed.
- J. Lewiński, M. Dutkiewicz, M. Lesiuk, W. Śliwiński, K. Zelga, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2010, 49, 8266–8269 CrossRef PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 9898–9903 CrossRef CAS PubMed.
- H.-S. Kim, I. Mora-Sero, V. Gonzalez-Pedro, F. Fabregat-Santiago, E. J. Juarez-Perez, N.-G. Park and J. Bisquert, Nat. Commun., 2013, 4, 2242 Search PubMed.
- H. S. Kim and N. G. Park, J. Phys. Chem. Lett., 2014, 5, 2927–2934 CrossRef CAS PubMed.
- L. Contreras, J. Idígoras, A. Todinova, M. Salado, S. Kazim, S. Ahmad and J. A. Anta, Phys. Chem. Chem. Phys., 2016, 18, 31033–31042 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00094d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |