
Conducting polymer nanofiber-supported Pt alloys: unprecedented materials for methanol oxidation with enhanced electrocatalytic performance and stability†
Srabanti
Ghosh
 *a,
Susmita
Bera
a,
Sandip
Bysakh
b and
Rajendra N.
Basu
*a
*a,
Susmita
Bera
a,
Sandip
Bysakh
b and
Rajendra N.
Basu
*a
aFuel Cell and Battery Division, CSIR – Central Glass and Ceramic Research Institute, 196, Raja S. C. Mullick Road, Kolkata-700032, India. E-mail: ghosh.srabanti@mail.com; rnbasu@cgcri.res.in
bMaterials Characterization Division, CSIR – Central Glass and Ceramic Research Institute, 196, Raja S. C. Mullick Road, Kolkata-700032, India
First published on 26th April 2017
Abstract
Conducting polymer nanostructures can be utilized as catalyst supports as alternatives to traditional carbon support materials, which is promising for fuel cell applications in the near future. We synthesized Pt nanoparticles (NPs) and Pt NPs-based multimetallic alloys on polypyrrole (Ppy) nanofibers by a facile and greener approach via radiolysis without using any reducing agents. Compared with the Pt NPs, the binary (Pt66Pd34/Ppy) and ternary (Pt24Pd26Au50/Ppy) electrocatalysts demonstrate superior catalytic activities for methanol oxidation in alkaline medium as well as better tolerance to intermediate poisoning. Most importantly, the catalytic activity of Pt24Pd26Au50/Ppy significantly improved up to 12.5 A per mg Pt, which is ∼15 times higher than that of commercial Pt/C (0.85 A per mg Pt). This can be attributed to the high number of intrinsic active sites, including Pt–Pd–Au heterojunctions, and cooperative action of the three metals in the alloy composition as well as close contact with the polymer nanofibers. Moreover, the chronoamperometric curves confirm the better stability of the ternary alloy catalysts compared to binary and commercial carbon-supported catalysts. The effective dispersion of the NPs within the polymer nanofibers can improve the catalytic activity. Long-term stability of the catalysts may be achieved via facilitating access of methanol molecules to the catalytic sites and preventing agglomeration of the Pt NPs. Hence, these polymer-supported Pt nanoalloys have promising applications as anode electrocatalysts in direct methanol fuel cells (DMFCs).
1. Introduction
Direct methanol fuel cells (DMFCs) are among the most common direct alcohol fuel cells (DAFCs). Methanol is used as a fuel because it has a higher energy density of 6.1 kW h kg−1 as well as facile transportation and convenient storage in comparison to hydrogen; thus, DMFCs are suitable as power sources for portable applications, such as electronic devices.1–4 During the operation of DMFCs, methanol can easily pass through the Nafion membrane and be oxidized to carbon dioxide at the anode in the presence of a suitable electrocatalyst.5,6 The benchmark Pt catalysts demonstrate high electrocatalytic activities for the methanol oxidation reaction (MOR); however, poor kinetics at the anodic side and parasitic methanol reactions at the cathode associated with methanol crossover hinder the commercialization of DMFCs.7–9 The most significant challenge for DMFCs is the development of active, stable and cost effective electrocatalysts. In order to improve power density and efficiency, much effort has been made to develop active catalysts, such as Pt based alloys,10–12 core shells13,14 and heterostructures.15 MOR activity is enhanced by the presence of second or third metals that form core shells or alloy structures, such as Pd@Pt or AuPd@Pt.16–20 In this regard, Pt-based catalysts doped with low cost transition metals, such as Ni@Pt,21,22 and heterojunction nanohybrid23 bimetallic or multi-metallic alloy-based catalysts24–27 also effectively minimize the loading of Pt metal and remarkably enhance the efficiency and stability of electrocatalysts. Moreover, the formation of alloys with Pt catalysts reduces the Pt–Pt bond distances; consequently, the number of effective surface sites increases.28 The enhanced electrooxidation of alcohol results from the altered electronic properties of the Pt surface, where the alloy composition has more Pt d-band vacancies in comparison to Pt/C.29 Furthermore, Pt-based catalysts are highly active in acidic medium; consequently, CO-poisoning and corrosion remain great challenges for large scale applications.30,31 In contrast, alkaline medium provides the considerable advantage of faster MOR kinetics, which enables the use of Pd noble metal as an efficient electrocatalyst as an alternative to Pt. Additionally, Pd-based catalysts reduce methanol crossover and the risks of electrode damage; they also lower the adsorption of ions on the active catalytic surface, which in turn improves the reaction kinetics.32 However, the MOR activity of pure Pd is remarkably low in alkaline medium in comparison to that of platinum. Hence, combining both metals in an alloy generates active electrocatalysts at relatively lower potentials with enhanced CO tolerance and increased stability for MOR.On the other hand, low durability of a catalyst is associated with dissolution of the metal catalyst along with carbon corrosion of the supporting material; this consequently decreases the effective electroactive surface area as well as the mass transfer.33 To date, Pt nanoparticles (NPs) have been immobilized on a variety of carbon-based supports, such as mesoporous carbon,34 carbon nanofibers,35 carbon nanotubes,36,37 carbon aerogels and xerogels,38 and polyoxometalates;39 this may result in increased surface area, superior electron transfer and reduced risk of nanoparticle agglomeration via improved dispersion. Particularly, the strong interaction between the catalyst and carbon support plays a significant role in the oxidation reaction. For example, graphene-supported Pt electrocatalysts have been reported to be highly active.40,41 Due to the hydrophobic nature of the graphene or carbon nanotube support, further functionalization is needed; this may decrease the strength of the interaction between the carbon support and the metal catalyst.
On the other hand, conducting polymers (CPs) such as poly(3,4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI), and polypyrrole (Ppy) have been also employed as carbon supports with unique π-conjugated structures, high electrical conductivities, and exceptional environmental stabilities.42–44 A series of CP-supported Pt NPs-based electrocatalysts have been developed for electrocatalytic oxidation of methanol. For example, Pt/poly(3,4-ethylenedioxythiophene) nanocomposites have demonstrated high electrocatalytic activity toward MOR.45 Additionally, Pt–Ru and Pt–Sn binary catalysts supported on PANI have been reported for MOR.46 Salavagione et al. explored the preparation of Pt NPs on polyaniline using short-chain alcohols and poly(N-vinyl-2-pyrrolidone) (PVP) as reducing agents for MOR.47 Metal oxide (hydrous molybdenum oxide)-modified platinum NPs electrodeposited onto poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonic acid) film also demonstrated high electrocatalytic activity for MOR with decreased CO poisoning.48 Shi et al. developed a multistep process for the electrochemical deposition of platinum NPs on polyaniline-functionalized carbon nanotubes; the as-prepared Pt/CNT–PANI hybrids exhibited excellent electrochemical activity towards MOR.49 Chen et al. fabricated a Pt/C@PANI core–shell electrocatalyst for the oxygen reduction reaction with enhanced durability and activity.50 Recently, Xu et al. also developed multilayered Pt/CeO2/PANI and ZnO/Pt/CeO2/PANI hybrid hollow nanorod arrays; these catalysts provide short diffusion paths for electroactive species and demonstrate elevated electrocatalytic activity toward MOR.51 It is important to note that conducting polymers with 1D-nanostructures are of special interest as electrocatalyst supports. Extensive effort has been made to obtain anisotropic 1D polymer nanostructures using rack-etch polycarbonate (PC) or polyester (PE) membranes and anodic aluminum oxide (AAO) membranes as solid templates; however, these approaches require multistep processes, and removal of the template may cause deformation of the polymer structure.52,53 Hence, surfactant-based soft templates have been employed for the synthesis of anisotropic CP nanostructures. In the present work, we demonstrate a radiochemical approach for the synthesis of monometallic (Pt, Pd), bimetallic (PtPd) and trimetallic (PtPdAu) catalysts supported on polypyrrole (Ppy) nanofibers without using any toxic reducing agents. The electrocatalytic activity of these catalysts towards methanol oxidation has been measured using electrochemical methods such as cyclic voltammetry (CV) and chronoamperometry (CA). To investigate the kinetics and reaction pathways of MOR in the presence of Pt-based alloys as catalysts, various experimental parameters, such as temperature, methanol concentration, KOH concentration and scan rate, have been studied in alkali medium.
2. Experimental
2.1 Reagents
Pyrrole, gold(III) acetate, palladium(II) acetylacetonate [Pd(C5H7O2)2], platinum(II) acetylacetonate [Pt(C5H7O2)2], 2-propanol, KOH (97%), cetyltrimethylammonium bromide (CTAB, 98% purity), ammonium peroxydisulfate (NH4)2S2O8, HCl, Nafion® suspension (5 wt%), and acetone were purchased from Sigma-Aldrich. All compounds were used as received. Ultrapure water (Millipore System, 18.2 MΩ cm), methanol and ethanol (≥99% for HPLC, purchased from Sigma-Aldrich) were used as solvents.2.3. Structural characterization
High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) observations were performed using a transmission electron microscope (TEM), Tecnai G2 30ST (FEI Company, USA) fitted with a LaB6 thermionic electron source, operating at an accelerating voltage of 300 kV. The TEM was equipped with an energy-dispersive X-ray spectrometer (EDAX Inc., USA), i.e., an EDS composition analyser, scanning TEM (STEM) imaging capability, and a high-angle annular dark-field (HAADF) imaging detector (Fischione Inc., USA). A few drops of the nanostructures in ethanolic solution were applied on carbon-coated copper grids and dried under a N2 flow. In order to obtain a direct visual evidence of alloying and elucidate the distribution of the individual metallic elements throughout the individual alloy nanoparticles and their nano-clusters, STEM-HAADF imaging and elemental mapping using STEM-EDS were employed. X-ray powder diffractograms (XRD) were recorded in the 2θ range of 10° to 80° at a slow scanning rate of 1° min−1 by an X-ray diffractometer (Philips X'Pert, The Netherlands) with Cu Kα radiation (at 40 kV and 40 mA).For XPS studies, Ppy and Pd, Pt or Au/Ppy, PtPd/Ppy, and PtPdAu/Ppy samples were prepared in pellets, and the analysis was carried out with a PHI 5000 VersaProbe II spectrophotometer (Physical Electronics Inc., USA) using a monochromatized Al Kα (∼1486.6 eV) X-ray beam with a size of ∼100 μm. Prior to XPS analysis, the sample surfaces were sputtered with a 2 kV rastered Ar+ ion beam for one minute to clean the surface. During the XPS measurements, a dual beam charge neutralization system was operated in order to neutralize the generated static charges on the sample surface. Charge correction was performed using the C 1s spectrum as a standard with a value of 284.5 eV. The recorded high resolution photoelectron spectra were resolved into their respective Gaussian fits after removal of the background intensity.
The metal (M = Pd, Pt, Au, PtPd, PtPdAu) content deposited onto the electrodes was determined by a SpectroCiros Vision inductively coupled plasma atomic-emission spectroscopy (ICP-AES) instrument, Spectro GmbH, Germany (Table 1).
| Metal loaded on Ppy | TGA: actual total metal loading (wt%) | ICP-AES | Intended metal composition (atomic, at%) | |||||
|---|---|---|---|---|---|---|---|---|
| Pt–Pd–Au atomic content (at%) | Actual metal loading (wt%) | |||||||
| Pt | Pd | Au | Pt | Pd | Au | |||
| Pt | 15 | 100 | — | 16 ± 2 | 100 | — | — | |
| Pd | 29 | — | 100 | — | 20 ± 1 | — | 100 | — |
| Au | 34 | — | — | 100 | 26 ± 3 | — | — | 100 |
| PtPd | 37 | 66 | 34 | — | 27 ± 2 | 75 | — | 25 |
| PtPdAu | 48 | 24 | 26 | 50 | 30 ± 2 | 25 | 25 | 50 |
2.4. Electrochemical characterization
Cyclic voltammetry (CV) and chronoamperometry measurements were performed using a galvanostat–potentiostat (PGSTAT302N, Autolab, The Netherlands) with a standard three-electrode electrolytic cell. A Pt foil served as the counter electrode, and Ag/AgCl was used as the reference electrode (all potentials were reported against the Ag/AgCl reference electrode). Prior to performing the measurements, the electrolyte solution was purged with high-purity nitrogen gas for at least 30 min. The glassy carbon electrode was pre-treated using the following process. First, the surface of a glassy carbon electrode was polished with 1.0, 0.3 and 0.05 μm α-alumina powders in sequence, rinsed thoroughly with twice distilled water and placed in a water-filled ultrasonic bath over a 5 min period. After drying in air, the nanostructure catalysts were deposited on the electrodes for further use. The electrode with commercial Pt/C catalyst was prepared as follows: two milligrams of Pt/C catalyst (20% Pt on Vulcan XC-72, JM) and 500 μL of ethanol were mixed to obtain the catalyst slurry. Then, 10 μL of the slurry was uniformly spread onto the surface of the glassy carbon electrode. After drying, 4 μL of Nafion (0.5% wt) solution was applied to cover the surface of the catalyst layer. Working electrodes with as-prepared Pt, Pd, Au/Ppy, PtPd/Ppy, and PtPdAu/Ppy were prepared by the same procedure described above. The cyclic voltammetric experiments were carried out in the potential range from −0.8 V to +0.8 V in 0.1 M KOH containing alkaline electrolyte in the absence and presence of 1 M methanol at a scan rate of 20 mV s−1. The variations in KOH concentration, methanol concentration and temperature were also studied.3. Results and discussion
3.1. Synthesis of polymer-supported catalysts
The synthesis strategy for the polymer-supported metal catalysts is shown in Scheme 1. In the first step, a surfactant-based soft template was used for the synthesis of polypyrrole nanofibers (Scheme 1a). In the presence of a chemical oxidant (ammonium persulfate), cetyltrimethylammonium bromide (CTAB) formed a twin-tailed, highly viscous lamellar structure ((CTA)2S2O8) which acted as a soft template to control the formation of the polymer nanostructure.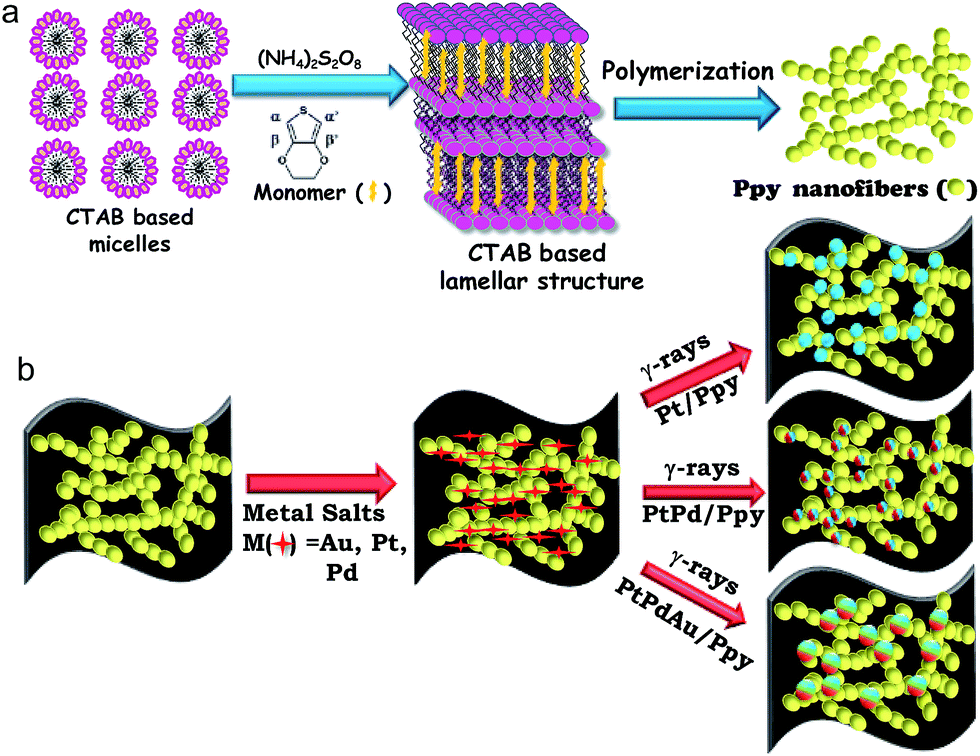 | ||
| Scheme 1 Proposed schematic of the radiolytic synthesis of the nanocomposites. (a) Synthesis of polymer nanofibers using a surfactant-based oxidative template. (b) Deposition of metal nanoparticles on conducting polymer nanofibers by radiolysis. | ||
Au(III) acetate, Pt(II) or Pd(II) acetylacetonate and Ppy nanofibers were used as precursors. Then, Pt(II) or other metal ions were selectively absorbed on the surfaces of the polymer nanostructures due to electrostatic interactions between Pt(II) cations and the surfactant-functionalized polymer nanofibers. Finally, monometallic (Pt, Pd), bimetallic (PtPd) or trimetallic (PtPdAu) NPs were deposited on the polymer nanofibers by radiolysis without using chemical reducing agents or surfactants, as shown in Scheme 1.
Metal ions can be reduced by solvated electrons (esol−), and 2-propanol radicals (CH3)2–C˙OH are produced by solvent radiolysis.55,56
(CH3)2CHOH → esol−; solvated protons ((CH3)2CHOH2+), (CH3)2C˙OH and other radiolytic products.
| (CH3)2CHOH + OH˙(H˙) → (CH3)2C˙OH + H2O | (1) |
| esol− + M+ → M0 | (2) |
| (CH3)2C˙OH + M+ → (CH3)2CO + M0 | (3) |
| PtII + esol−→ PtI | (4) |
| PtII + (CH3)2C˙OH → PtI + (CH3)2CO | (5) |
| 2PtI → Pt0 + PtII | (6) |
| n Pt0 → (Pt)n | (7) |
The radiolytic formation of platinum atoms is followed by association of the atoms with ions and complexes, dimerization reduction reactions, and coalescence of the resulting oligomers, leading to Pt nanostructures.57,58 A similar mechanism is possible for the other metals, viz., Pd or Au nanoparticles. Monometallic clusters are formed initially; further association and reduction reactions gradually build the bimetallic or trimetallic alloyed clusters. The formation of the alloyed structures or the segregation of metals in the core–shell type structures probably depends on the kinetic competition between the irreversible release of metal ions, which are displaced by excess noble metal ions after electron transfer, as well as radiation-induced reduction of both metal ions.
The crystalline structures of the metal polymer composites were explored by XRD. Fig. 1a illustrates typical XRD patterns of the as-prepared nanocomposites, which confirm their face centred cubic (fcc) crystal structures and contain strong and prominent diffraction peaks for each metal. The strong peaks at 2θ values of 39.65, 46.1, 67.5, 81.1, and 86.1 correspond to the (111), (200), (220), (311), and (222) faces of Pt crystal, respectively. In the case of Pt–Pd/Ppy, the major peaks at 39.95°, 46.3°, 67.9° and 82.02° correspond to the (111), (200) (220) and (311) planes, respectively, also of the fcc crystal structure. The angle shift in the diffraction peaks indicates a lattice parameter change due to alloy formation between the metallic phases; this is in agreement with literature reports.59 As Pt, Pd and Au have similar lattice parameters and cubic structures, the diffraction peaks of the resulting PdPt or PtPdAu alloys are similar to those of pure Pt, Pd, and Au. On the other hand, compared to Au/Ppy (37.9°), the (111) peak positions for the trimetallic Pt24Pd26Au50/Ppy (38.3°) nanocomposites are shifted to higher angles, indicating that Pd and Pt are incorporated into the Au fcc structure to form an alloy phase. A characteristic peak for Ppy is centered at ∼27.5°, which can be assigned to the repeat unit of the pyrrole ring; the broad feature of the peak implies an amorphous polymer structure (Fig. S1†). However, the broad peak originating from the Ppy nanofibers was not identified due to the dominating intensity of the diffraction peaks contributed by the metallic NPs present within the metal–polymer nanocomposites.
 | ||
| Fig. 1 (a) XRD patterns for the synthesized Au/Ppy, Pd/Ppy, Pt/Ppy, Pt66Pd34/Ppy, and Pt24Pd26Au50/Ppy nanocomposites. (b) TGA curves of Ppy polymer and the Pd/Ppy, Pt/Ppy, Au/Ppy, Pt66Pd34/Ppy, Pt24Pd26 Au50/Ppy nanocomposites. | ||
The thermal stabilities and metal loadings on the polymer nanostructures were determined by thermogravimetric analysis (TGA). Fig. 1b shows ∼80% weight loss at 186 °C for the Ppy nanofibers, which is due to the removal of low molecular weight oligomers and the thermal decomposition of the polypyrrole backbone. In contrast, the nanocomposites showed a higher decomposition temperature with a high residual mass of Ppy, which indicates metal loading. The residual masses of about ∼19–48% indicate loadings of different metal NPs on the polymer nanofibers, as summarized in Table 1. Hence, the formation of the NPs significantly affects the stability of the polymer structure, which is consistent with a literature report.60
Fig. 2a shows a typical representative transmission electron microscope (TEM) bright-field (BF) image of the Ppy polymer. It can be observed that the polymer has a nanofiber-like morphology, with average diameters of 50 to 65 nm.
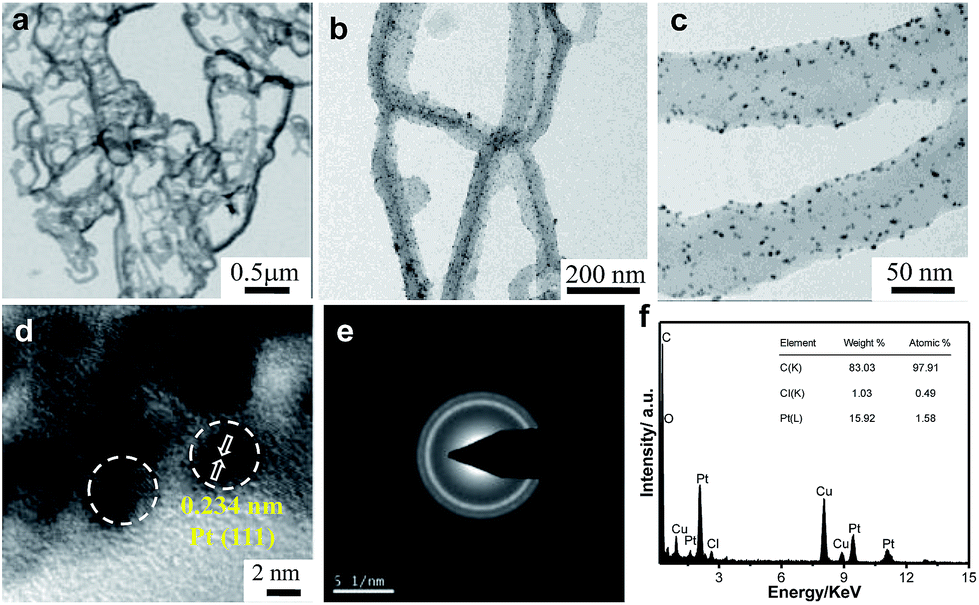 | ||
| Fig. 2 Transmission electron micrographs of (a) Ppy nanofibers and (b and c) Pt/Ppy nanocomposites at two different magnifications. (d) HRTEM image, (e) SAED pattern, (f) EDX spectrum. | ||
The homogeneous distribution of extremely fine Pt NPs supported on the surface of the Ppy nanofibers is evident from the TEM images (Fig. 2b and c). The average particle size of the Pt NPs was found to be 3.5 nm, as measured from the TEM bright-field image. The HRTEM image of Pt/Ppy is shown in Fig. 2d, indicating that the inter-planar distance between the fringes of the Pt NPs is about 0.234 nm; this is consistent with the spacing of (111) planes. Fig. 2e represents a characteristic selected area electron diffraction (SAED) pattern with prominent rings which can be indexed to the fcc structure of the Pt NPs; this is also consistent with the XRD data. The corresponding energy-dispersive X-ray spectrum (EDX) demonstrates the chemical composition of the metal composites with Pt, carbon, and chlorine; this also confirms the formation of the Pt/Ppy nanocomposites (Fig. 2f) with chlorine incorporated within the polymer.
The size distribution of pure Pd NPs estimated from the TEM bright-field image (considering representative number >300 isolated particles), and the average diameter (D) of the Pd NPs is 4.7 nm, as shown in Fig. S2a and b.† The HRTEM image shows the characteristic lattice fringes of the Pd NPs in the surrounding Ppy matrix (Fig. S2c†). The inter-planar distance in the lattice fringes was measured to be 0.22 nm, which corresponds to the (111) plane of metallic Pd. Fig. 2d† demonstrate the formation of relatively large Au nanocrystals on the Ppy nanofibers. The average particle size of the Au NPs is ∼20 nm. HRTEM images of the Au nanocrystals on the polymer show a lattice fringe of 0.235 nm, which corresponds to the (111) plane of the Au NPs (Fig. S2f†).
Furthermore, a series of bimetallic nanocomposites, Pt66Pd34/Ppy, Pt49Pd51/Ppy, and Pt25Pd75/Ppy, and a trimetallic nanocomposite, Pt24Pd26Au50/Ppy, were successfully synthesized by the radiolytic technique. Ppy nanofibers with high and uniform coverage by Pt66Pd34/Ppy were also observed, as shown in Fig. 3a and b, and the average particle size of Pt66Pd34/Ppy anchored on the Ppy nanofibers was found to be 3.1 nm. The HRTEM image of the Pt66Pd34/Ppy nanocomposites (Fig. 3c) also shows high crystallinity. TEM and HRTEM images of the other bimetallic composites, Pt49Pd51/Ppy and Pt25Pd75/Ppy, also indicate the homogeneous distribution and crystallinity of the Pt49Pd51 and Pt25Pd75 NPs on the polymer nanofibers, as shown in Fig. S3a–f.†Fig. 3d and e illustrate well-dispersed Pt24Pd26Au50 NPs on the surfaces of the polymers, with average particle sizes in the range of 10 to 24 nm. Further, the HRTEM images suggest good crystallinity of the as-prepared Pt24Pd26Au50 NPs on Ppy, as shown in Fig. 3f.
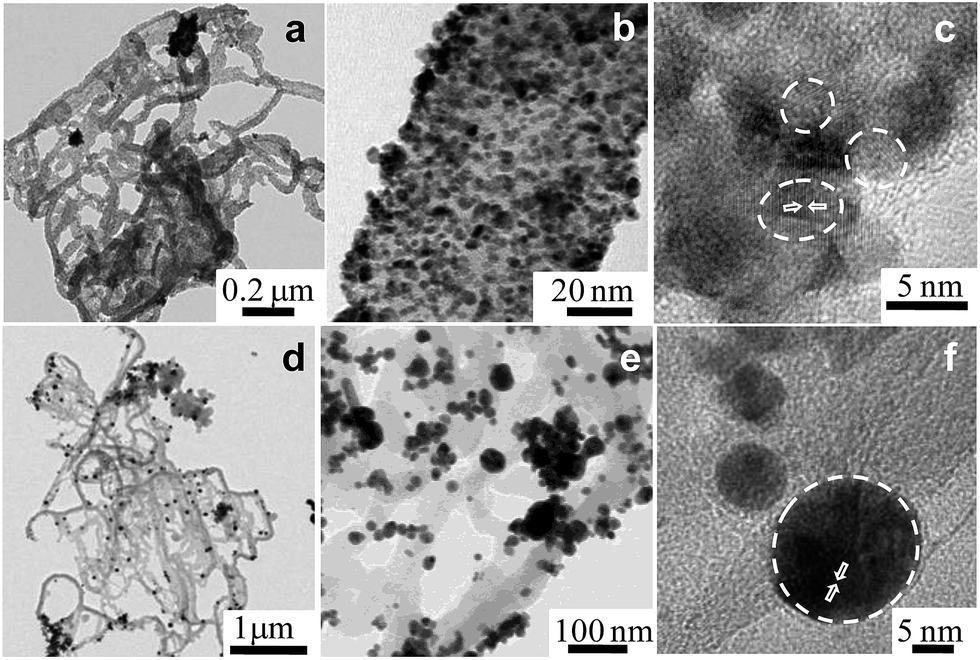 | ||
| Fig. 3 Transmission electron micrographs of (a and b) the Pt66Pd34/Ppy nanocomposite at two different magnifications; (c) HRTEM of Pt66Pd34/Ppy; (d and e) Pt24Pd26Au50/Ppy at two different, higher magnifications; (f) HRTEM of Pt24Pd26 Au50/Ppy. | ||
Fig. 4 and 5 show the results obtained by employing imaging techniques involving scanning transmission electron microscopy (STEM), presenting the high-angle annular dark-field (HAADF) images of the bimetallic and trimetallic alloy nanocomposites together with elemental maps. Fig. 4a and b show STEM-HAADF and corresponding bright field images of the PtPd nanocomposites. Both the HAADF images, showing Z2-contrast, and the BF-TEM image, showing diffraction contrast, reveal extremely fine NPs adhering to the Ppy nanofiber surface. Elemental mapping based on STEM-EDS confirmed that the nanofibers supported nanoparticles composed of Pt–Pd alloys; both Pd and Pt atoms are well distributed (Fig. 4c) among and within the nanoparticles. The similar crystal structures and negligible lattice mismatch between Pd and Pt (0.77%) enable the formation of an alloy structure. Interestingly, the fine NPs are mostly composed of pure Pd or are Pd-rich, while the coarse NPs tend to be rich in Pt with a small amount of dissolved Pd.
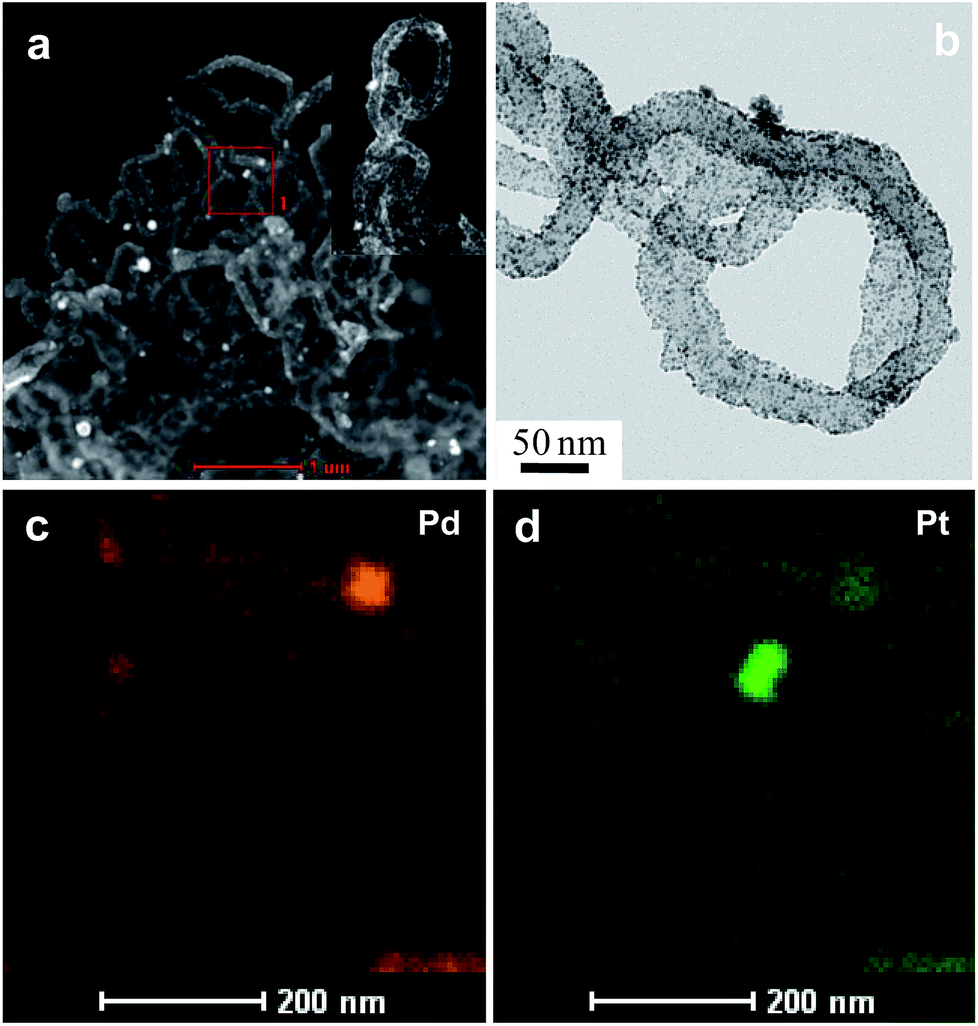 | ||
| Fig. 4 The morphology and structure of Pt66Pd34/Ppy. (a) HAADF-STEM image of the Pt66Pd34/Ppy composite. Inset: high magnification TEM image of the composite. (b) Bright field TEM image of the corresponding area of Pt66Pd34/Ppy. (c and d) HAADF-STEM-EDS mapping images of Pt66Pd34/Ppy. | ||
 | ||
| Fig. 5 The morphology and structure of Pt24Pd26Au50/Ppy. (a) HAADF-STEM image of the Pt24Pd26Au50/Ppy composite. (b–d) HAADF-STEM-EDS mapping images of Pt24Pd26Au50/Ppy. | ||
The TEM bright-field images as well as the STEM-HAADF images of Pt24Pd26Au50/Ppy show the presence of nanoparticles (NPs) of different sizes (ranging from <10 nm to ∼250 nm) adhered to the polymer nanofibers (Fig. 5). The STEM-EDX mapping images revealed that the large NPs (50 to 250 nm) are very rich in Au, with small amounts of both Pd and Pt. The maps also suggest that the Pd NPs are finer (∼10 nm) in general; in the case of small Au-containing NPs, Au formed a shell on the fine Pd–NPs, as indicated by the large sizes in the Au-map and the smaller sizes in the Pd-map for several NPs (indicated by arrowheads). The elemental maps also suggest that Pt forms better alloys with Au than with Pd. Both Pd and Pt tend to retain their very small sizes (i.e., ∼10 nm). The sub-10 nm sizes of the Pd, Pt and Pd–Pt alloy NPs are expected to show enhanced surface activation properties due to their high surface-to-volume ratios.
The elemental composition of the Pt24Pd26Au50/Ppy nanocomposite was further analyzed by energy-disperse X-ray spectroscopy (EDX). A typical EDX spectrum of Pt24Pd26Au50/Ppy indicates the presence of carbon, oxygen, platinum, palladium and gold, as shown in Fig. 6a. Furthermore, X-ray photon spectroscopy (XPS) was carried out to analyze the surface compositions and to predict the oxidation states of the metals in the Pt/Ppy, Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy nanocomposites (Fig. 6b–f).
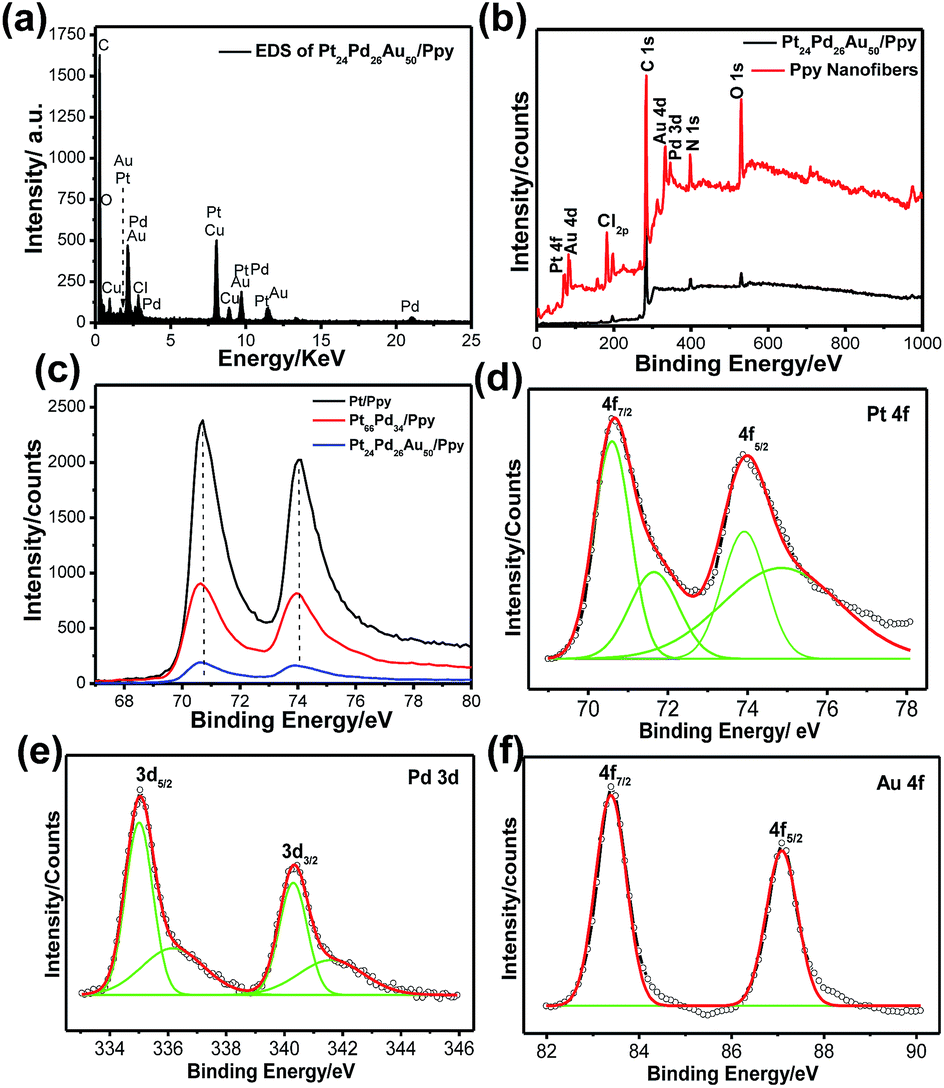 | ||
| Fig. 6 (a) EDS spectrum of Pt24Pd26Au50/Ppy, (b) XPS patterns of the synthesized Ppy, Pt/Ppy and nanocomposites, (c) magnified XPS spectra of Pt 4f in Pt/Ppy, Pt24Pd26Au50/Ppy, and Pt24Pd26Au50/Ppy, (d–f) magnified XPS spectra of (d) Pt 4f, (e) Pd 3d and (f) Au 4f. | ||
Compared with the wide region spectrum of Ppy alone, the signals located at 70.6 and 74.08 eV can be assigned to the binding energies of Pt 4f7/2 and Pt 4f5/2 of metallic Pt0, respectively.61 Other strong XPS signals at 196.5, 285.5, 401.5, and 532 eV are assigned to Cl 2p, C 1s, N 1s, and O 1s, respectively, in the survey spectra of both Ppy and Pt24Pd26Au50/Ppy (Fig. 6b); this indicates that the chemical environment of Cl, C, N, and O originates from PPy.62 The C(1s) core level spectra have two main features; a strong peak at 284.5 eV corresponds to the conjugated carbon atoms in the Ppy chain (Fig. S4a–c†). Additionally, definite positive shifts of ∼0.25 eV in the binding energy of Pt 4f in the spectra of Pt24Pd26Au50/Ppy and Pt66Pd34/Ppy were observed in comparison to that of Pt/Ppy, indicating a change in the electronic states of the Pt atoms (Fig. 6c). Fig. 4e and f shows the XPS spectra of the Pt 4f, Pd 3d and Au 4f regions. The core-level Pd 3d spectra display doublet signals with binding energies of 335.3 and 340.4 eV for Pd 3d5/2 and Pd 3d3/2, respectively; these are comparable to the signals of palladium metal, confirming the Pd signal.63 Another small doublet around 336.7 and 342.1 can be assigned to the Pd 3d3/2 and Pd 3d5/2 peaks of PdO (Fig. 6e).64 The peaks at ∼83.4 and 87.1 eV in the binding energy scale, respectively observed from Au 4f7/2 and Au 4f5/2 spin–orbit doublets, correspond to metallic Au (Au0); these are highly consistent with the peaks observed for the Au NPs (Fig. 6f).65 It is important to note that the lower binding energy of less than 84 ± 0.1 eV indicates the presence of Au metal with higher electron density than typical metallic gold. For the binary composition, both Pt and Pd peaks were obtained, which suggests the formation of Pt66Pd34/Ppy composites (Fig. S4d†). Hence, the XPS data also support the formation of the Pt24Pd26Au50/Ppy nanocomposites.
3.2. Electrochemical activity
Cyclic voltammograms (CVs) of glassy carbon electrodes modified with Pt/Ppy, Pd/ppy, the binary Pt66Pd34/Ppy, Pt51Pd49/Ppy, and Pt25Pd75/Ppy nanocomposites, and the ternary Pt24Pd26Au50/Ppy nanocomposite were measured in argon-saturated 0.1 M KOH at the potential range between −0.8 V and 0.8 V with a scan rate of 20 mV s−1 (Fig. 7a). Pt/Ppy shows two potential peaks at −350 mV, which can be attributed to the adsorption of OH−, and another peak above 190 mV, which may be due to the formation of a Pt oxide layer.| Pt + OH− → Pt–OHads + e− | (8) |
| Pt–OHads + OH− ↔ Pt–O + H2O + e− | (9) |
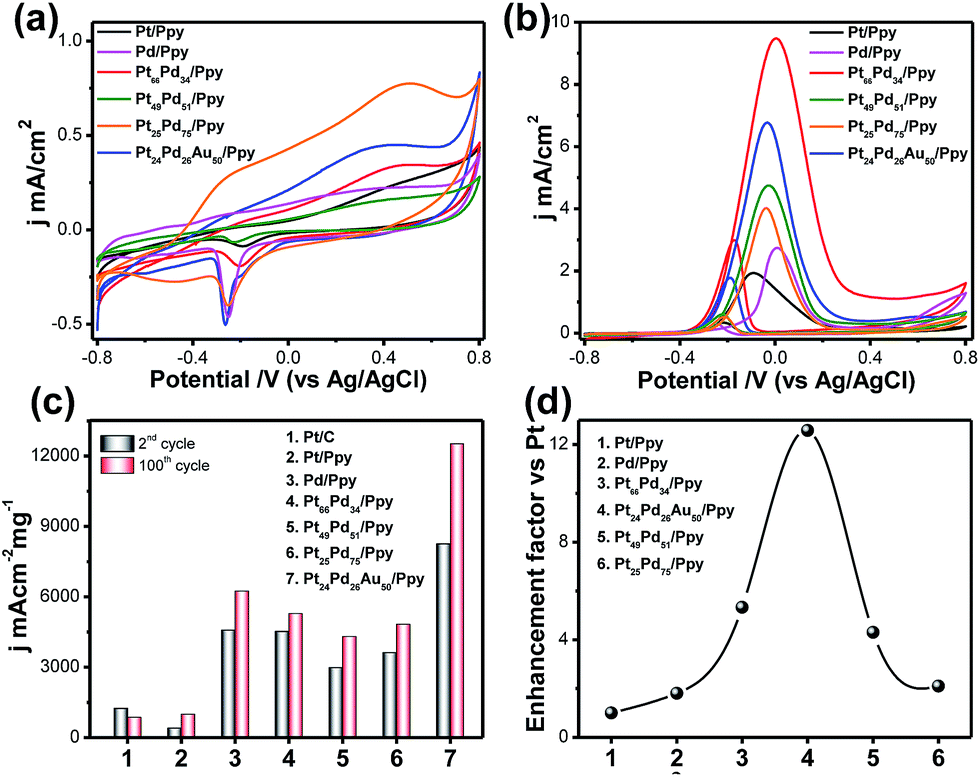 | ||
| Fig. 7 (a) Cyclic voltammograms (in mA cm−2) of Pt/Ppy, Pd/Ppy, Pt66Pd34/Ppy, Pt49Pd51/Ppy, Pt25Pd75/Ppy and Pt24Pd26Au50/Ppy at a scan rate of 20 mV s−1 in 0.1 M KOH. (b) Cyclic voltammetric runs associated with the electrocatalytic oxidation of 1 M methanol by Pt/Ppy (black solid line, 100th cycle), Pd/Ppy (pink solid line, 100th cycle), Pt66Pd34/Ppy (red solid line, 100th cycle), Pt49Pd51/Ppy (green solid line, 100th cycle), Pt25Pd75/Ppy (orange solid line, 100th cycle), and Pt24Pd26Au50/Ppy (blue solid line, 100th cycle) at a scan rate of 20 mV s−1 in 0.1 M KOH. The working electrode was a glassy carbon disk modified with the Pt nanostructures. The reference electrode was Ag/AgCl. (c) Comparison of the mass activity values of the Pt/C, Pd/PPy, Pt49Pd51/Ppy, Pt66Pd34/Ppy, Pt11Pd89/Ppy and Pt24Pd26Au50/Ppy electrodes for methanol oxidation in alkaline medium. (d) Volcano plot corresponding to the mass activities of the Pt nanocomposite-based electrocatalysts. | ||
The binary (Pt66Pd34/Ppy, Pt51Pd49/Ppy, Pt25Pd75/Ppy) and ternary (Pt24Pd26Au50/Ppy) catalysts showed characteristics of both Pd/Ppy and Pt/Ppy; the presence of Pd facilitates the adsorption of OH−. In general, the adsorption of OH− starts at a more negative potential on the Pd/Ppy electrode compared to the Pt/C electrode and overlaps the hydrogen desorption peak.66 A noticeable difference in the potentiodynamic curves for the binary and ternary catalysts is observed from the remarkable negative shifts of the hydrogen adsorption/desorption (Had/Hdes) and oxide formation/reduction peaks compared to monometallic Pd/Ppy and Pt/Ppy; this suggests the formation of an alloy of these metals.67 The peak current density is greater for the alloys than for pure Pt/Ppy and Pd/Ppy, indicating possible transmetalation reactions. The peak for the reduction of PdO to Pd around −0.25 V was utilized to determine the mass normalized electrochemical surface areas (ECSAs) of the synthesized catalysts in alkaline medium.
The oxygen desorption method can be used to evaluate the electrochemically active surface area (ECSA) of Pd by determining the coulombic charge (Q) for the reduction of a palladium oxide (PdO) monolayer. Meanwhile, for the Pt-based catalyst, the ECSA was determined by measuring the charge collected in the hydrogen adsorption/desorption region after double-layer correction (Qref = 210 μC cm−2) for the oxidation of a single layer of hydrogen on a smooth Pt surface. The charges required for reduction of the PdO (QPdO-red) and PtO (QPtO-red) monolayers were assumed to be 405 and 420 μC cm−2, respectively.68,69 It is not easy to measure the ECSAs of bi- and trimetallic alloys; the mean value of the charge required for oxide reduction (410 μC cm−2) was used for the calculation of the ECSAs of the alloyed catalysts. The calculated ECSAs of Pt/Ppy, Pd/Ppy, Pt66Pd34/Ppy, Pt51Pd49/Ppy, Pt25Pd75/Ppy and Pt24Pd26Au50/Ppy are 19.5 m2 g−1, 65.5 m2 g−1, 22.3 m2 g−1, 19.9 m2 g−1, 176 m2 g−1 and 265 m2 g−1, respectively. The ECSA for Pt24Pd26Au50/Ppy (265 m2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g−1) was found to be about 14 times higher than the value obtained for Pt/Ppy (19m2g−1). The binary and ternary alloys demonstrate obviously higher ECSA values, which can be attributed to the highly uniform dispersion of metal NPs and synergistic effects between the metal NPs and polymer nanofibers.
g−1) was found to be about 14 times higher than the value obtained for Pt/Ppy (19m2g−1). The binary and ternary alloys demonstrate obviously higher ECSA values, which can be attributed to the highly uniform dispersion of metal NPs and synergistic effects between the metal NPs and polymer nanofibers.
The electrocatalytic activities of the Pt-based catalysts were examined using cyclic voltammetry in argon-saturated 0.1 M KOH and 1 M methanol at a scan rate of 20 mV s−1. Fig. 7b shows a superposition of the 2nd and 100th cycles of Pt/Ppy (black solid line), Pt66Pd34/Ppy (red solid line) and Pt24Pd26Au50/Ppy (blue solid line); the bimetallic and trimetallic catalysts show lower onset potentials for the methanol oxidation reaction (MOR) and large anodic peak current densities compared to Pt/Ppy and Pd/Ppy. The MOR efficiency is higher for the Pt66Pd34/Ppy composite (9.52 mA cm−2) in terms of current density than for Pt24Pd26Au50/Ppy (6.76 mA cm−2). However, Fig. 7c illustrates that in terms of mass activity, Pt24Pd26Au50/Ppy shows superior performance towards the electrooxidation of methanol. The enhanced catalytic activity is associated with the alloying of Pt with Pd, which can control poisoning effects, and enhancement of the reactivity by Au; this is consistent with earlier reports.70,71Fig. 7d shows that the efficiency follows the order Pt24Pd26Au50/Ppy (12-fold) > Pt66Pd34/Ppy (4-fold) > Pt49Pd51/Ppy ≅ Pt25Pd75/(2-fold) > Pd/Ppy (1-fold) > Pt/Ppy (chosen as reference). Considering d-band theory, when metals such as gold or platinum are combined with the structure of Pd, the d-band of Pd may be upshifted; this, in turn, affects the electron affinities of the Pt atoms in the alloys, while the presence of Pd enhances the ability of the catalyst to adsorb oxygenated species. As a result, more hydroxyls may be adsorbed at the catalyst surface, and more active sites are released on the Pt surface in the alloys compared to commercial Pt/C electrocatalyst.72 Furthermore, strong interactions between the metal NPs and π-conjugated carbons can be expected, which in turn enhances the dispersity and availability of metal catalysts at the catalytic sites. The negative shift of the onset potential (Eonset) underscores the increased kinetics of methanol oxidation. Upon cycling, the electro oxidation potential of MOR at Pt66Pd34/Ppy negatively shifts from ca. 0.22 V to 0.256 V. This observation indicates a relatively significant change in the electro-catalytic activity of the electrode material toward methanol oxidation during cycling. The important parameters of the cyclic voltammograms are tabulated below.
It is clear that during potential cycling of the Pt66Pd34/Ppy electrode material, the forward peak current density (jf) increased, whereas the backward peak current density (jb) remained the same and the ratio of jf/jb changed from 3.29 to 3.19 (Table 2). This suggests a decreasing trend of catalytic poisoning due to surface rearrangements and removal of carbonaceous species that cannot be oxidized in the forward scan. It is also clear that for bimetallic and trimetallic compositions, the forward anodic peak currents of 9.52 mA cm−2 and 6.76 mA cm−2, respectively, are greater than those of the monometallic catalysts, 1.96 mA cm−2 and 2.74 mA cm−2 for Pt/Ppy and Pd/Ppy, respectively. The current density of the Pt66Pd34/Ppy nanocomposite is ∼4.8 times higher than that of Pt/Ppy, which suggests that the bimetallic catalysts demonstrated significant catalytic activity towards MOR. Fig. 7b indicates that the forward peak current densities for the pure material and various alloy materials varies in the order Pt24Pd26Au50/Ppy > Pt66Pd34/Ppy > Pt49Pd51/Ppy > Pt/Ppy > Pt11Pd89/Ppy. For further investigation, we chose the Pt66Pd34/Ppy nanocomposite as a representative catalyst due to its high current density and better performance in MOR.
| Material | Cycle | j f (mA cm−2) | j b (mA cm−2) | j f/jb | E onset (mV vs. Hg/HgO) |
|---|---|---|---|---|---|
| Pt/Ppy | 2nd | 0.78 | 0.108 | 7.22 | −230 |
| 100th | 1.96 | 0.40 | 4.9 | −265 | |
| Pt66Pd34/Ppy | 2nd | 8.14 | 2.47 | 3.29 | −222 |
| 100th | 9.52 | 2.98 | 3.19 | −256 | |
| Pt49Pd51/Ppy | 2nd | 3.25 | 0.456 | 2.44 | −204 |
| 100th | 4.74 | 0.594 | 7.97 | −215 | |
| Pt25Pd75/Ppy | 2nd | 3.62 | 0.469 | 7.71 | −220 |
| 100th | 4.03 | 0.627 | 7.70 | −200 | |
| Pt24Pd26Au50/Ppy | 2nd | 4.46 | 1.43 | 3.11 | −225 |
| 100th | 6.76 | 1.73 | 3.9 | −227 | |
| Pd/Ppy | 2nd | 2.05 | 0.115 | 17.43 | −90 |
| 100th | 2.74 | 0.27 | 10.12 | −81 |
Fig. 8a shows cyclic voltammograms for methanol oxidation at different methanol concentrations, ranging from 0.1 mol L−1 to 2 mol L−1, for the Pt66Pd34/Ppy electrode. The significant anodic peak current density for both the forward and reverse scans accelerates to 11.08 mA cm−2 (at 2 mol L−1, blue solid line) from 4.4 mA cm−2 (at 0.1 mol L−1, black solid line); this is associated with the coverage of adsorbed methoxy groups at the catalyst surface with increasing methanol concentration. This increase in the coverage of adsorbed methoxy groups yields an increase in the oxidation current for MOR. Fig. 8b demonstrates the plot of log current vs. log concentration of methanol at a specific potential; an overall reaction order of 0.83 is estimated for MOR.
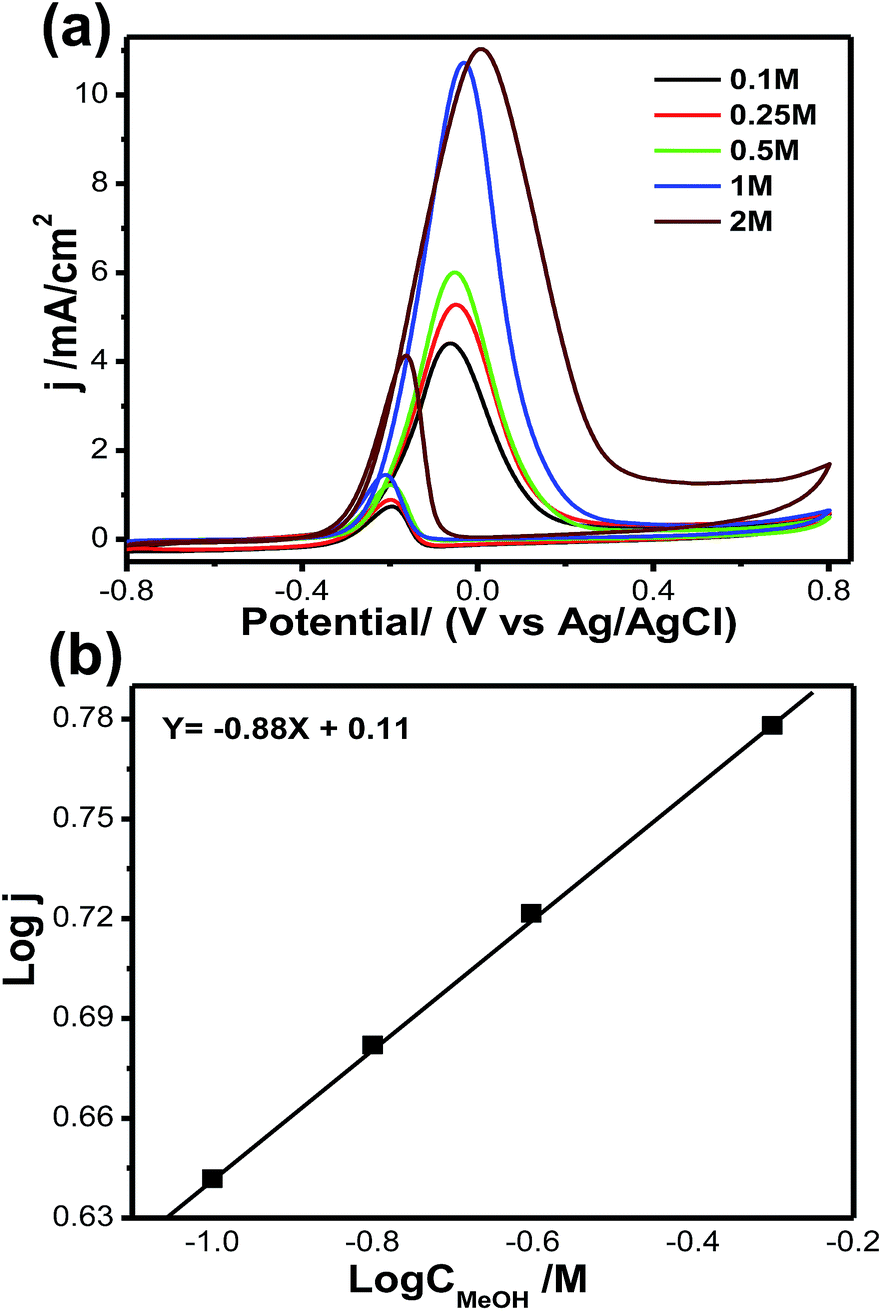 | ||
| Fig. 8 (a) Cyclic voltammograms of methanol oxidation at different methanol concentrations: 0.10 to 2.00 mol L−1. (b) Double logarithm graph of peak current vs. methanol concentration. | ||
Fig. 9a and b illustrate the effects of KOH concentration on methanol oxidation on the Pt66Pd34/Ppy electrode in 1 M methanol at different KOH concentrations. Although the natures of all the CVs are the same, significant enhancement in the forward current was observed with increasing KOH concentration. The oxidation current jf increased by a factor of ca. 3.8 when the KOH concentration was increased from 0.1 M to 1 M; this indicates faster reaction kinetics due to reduced unreacted methoxy groups. It was also observed that the onset potential for each reaction shifted towards a negative potential from −0.22 V (0.1 M, black solid line) to −0.32 V (1 M, blue solid line), indicating that the reaction starts at a lower potential and accelerates towards MOR. The onset potential increases almost linearly with KOH concentration, as shown in the inset of Fig. 9a. Fig. 9b shows the jfvs. KOH concentration plot; the current density changes linearly with KOH concentration due to the progressive oxidation of methoxy groups on the Pt catalyst. We also examined the scan rate variation; a change of current density is observed as the scan rate increases from 10 to 100 mV s−1, as shown in Fig. S5a.† This change may occur due to fast oxidation of methoxy groups on the Pt surface; a linear change in forward current density with respect to scan rate was obtained. It can be clearly observed that the forward peak current density increased gradually, which confirms that the kinetics of the overall oxidation process is mainly controlled by the diffusion process (Fig. S5b†).
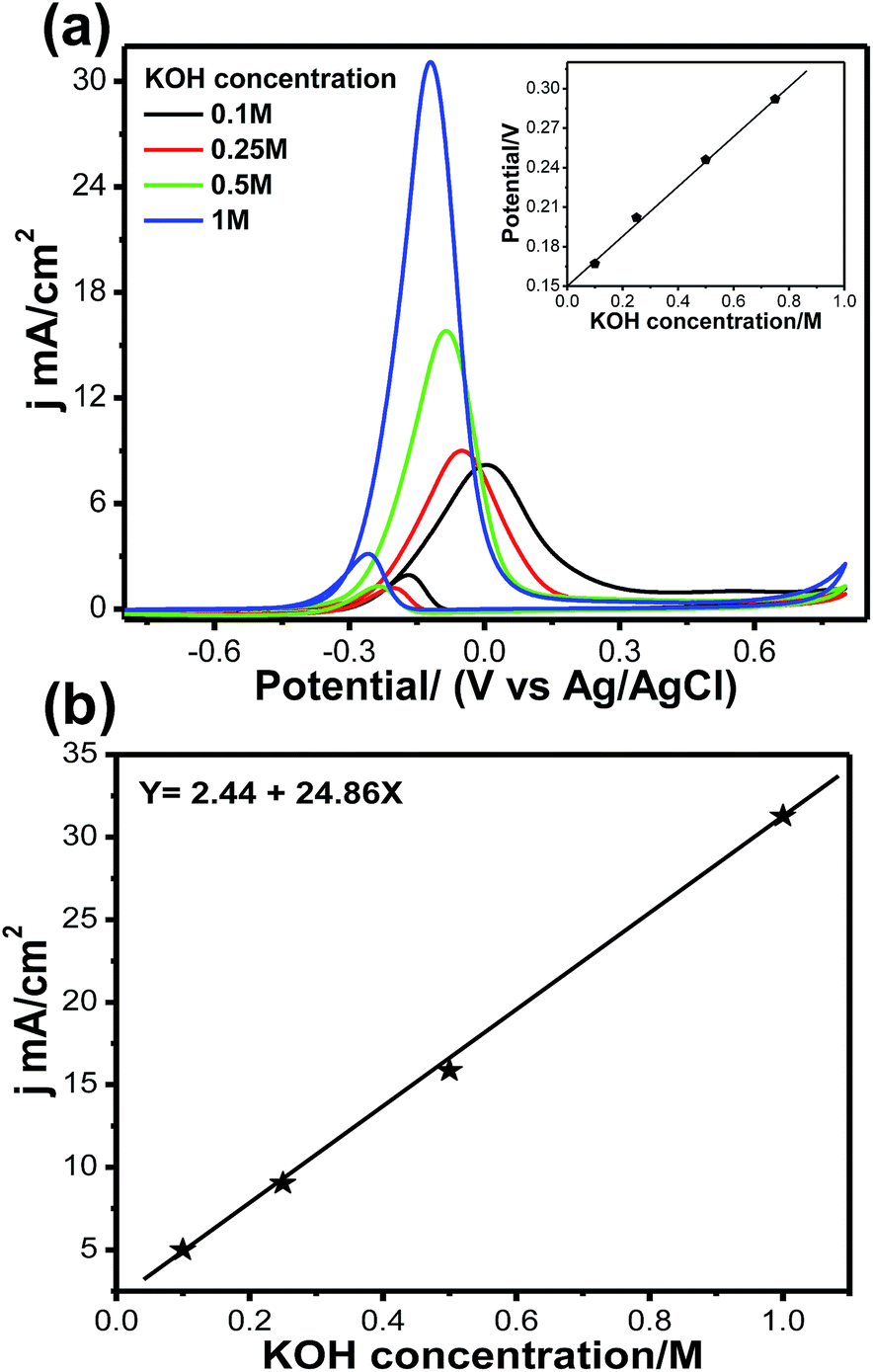 | ||
| Fig. 9 (a) Cyclic voltammetry curves for MOR on Pt66Pd34/Ppy at different KOH concentrations. Inset: variation of onset potential at different KOH concentrations. (b) Variation of anodic peak current density for the electrooxidation of methanol at different KOH concentrations. | ||
It is important to note that the mass of Ppy plays an important role in the electrochemical performance of the catalysts. The initial concentration of polymer (0.1, 0.25, 0.5, 1, 0.5, 2 mg ml−1) was varied during the deposition of metal by radiolysis. Fig. S6† illustrates the Ppy mass dependency of the Pt catalysts on current density obtained at −0.05 V from the forward scan during electrooxidation of methanol. The results suggest that with increasing polymer loading, the current density for methanol oxidation increases linearly up to 10.7 mA cm−2 at a fixed Pt loading of 0.007 mg cm−2. As the polymer loading increased further, the current density reached a saturation value of ∼10.1 mA cm−2; in fact, it slightly decreased in the presence of 2 mg ml−1 polymer solution. Hence, for the present study, the mass of the polymer was fixed at 1 mg ml−1.
Fig. 10a shows CVs for methanol electrooxidation at different temperatures ranging from 20 °C to 60 °C. With increasing temperature from 20 °C to 60 °C, the forward oxidation current increased by a factor of 38 (Fig. 10b). It may be assumed that CO coverage on Pt66Pd34/Ppy decreases with increasing temperature and that methanol adsorption also increases, resulting in a higher methanol oxidation rate. In the reverse scan, progressive reduction occurs; therefore, the backward current density (jb) increases. Thus, as the temperature increases, the potential difference between jf and jb decreases from 2.5 to 1.4 from 30 °C to 50 °C, which suggests decreased catalytic poisoning (Table S1†).
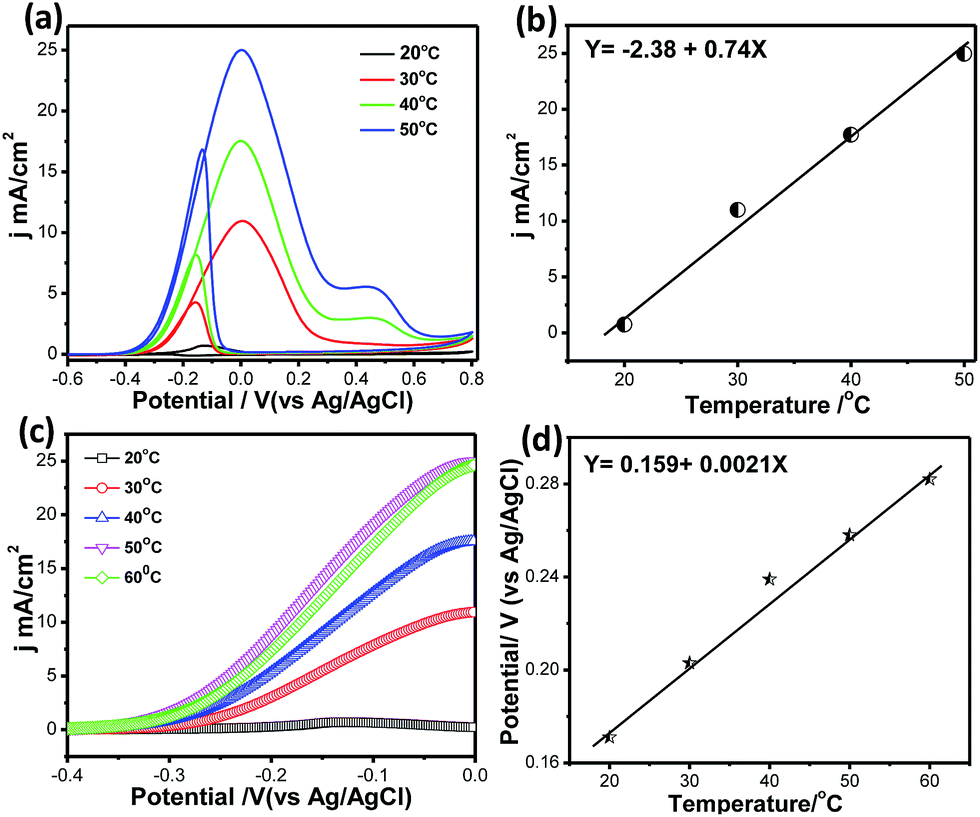 | ||
| Fig. 10 (a) Cyclic voltammetry curves for MOR on Pt66Pd34/Ppy at different temperatures (20 °C to 60 °C). (b) Variation of the current density at different temperatures. (c) Magnified portion of CVs to show the variations in the anodic peak current density. (d) Onset potentials for the electrooxidation of methanol on Pt66Pd34/Ppy catalysts at different temperatures. | ||
Because the backward peak is related to the reduction of adsorbed OH, the vertical increase indicates that reduction of adsorbed OH occurs very rapidly at high temperature on the surface of the catalysts. After a certain temperature, the peak current density of the forward scan attains a steady value because unoxidized methanolic residues are not available on the surface during the forward sweep. In addition, the onset potential of methanol oxidation shows temperature dependence. Magnified portions of the CV curves at different temperatures clearly display the change in the onset potential, as shown in Fig. 10c. With increasing temperature, Eonset values from −0.19 V to −0.29 V were obtained; this suggests the dissociation of carbonaceous species, thereby allowing the reaction to proceed at lower potentials while improving the catalytic effects. The potential of the forward peak shifts negatively with temperature, which may be due to the activated H2O decomposition reaction on the surface of Pt/C (Fig. 10d). This indicates that temperature has a significant role in methanol oxidation, leading to faster reaction rates and reducing the unoxidised residue. The activation energy calculated from Fig. 8d using the Van't Hoff equation is ∼41 kJ mol−1.
To compare the kinetics of the monometallic catalysts with bimetallic and trimetallic alloys, Tafel plots were considered using potentiodynamic pseudo-steady state polarization at a low scan rate of 1 mV s−1 while keeping the other conditions the same, following the equation:
| η = a + blogi | (10) |
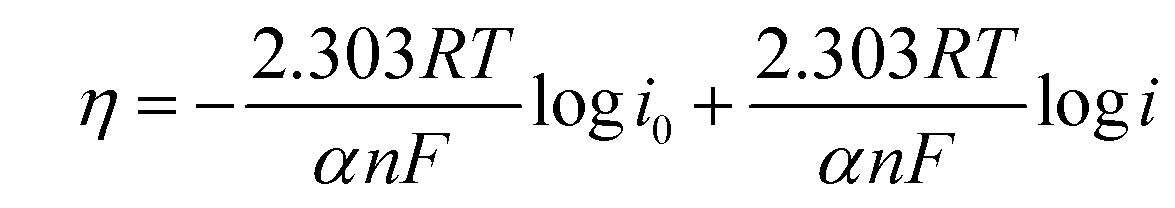 | (11) |
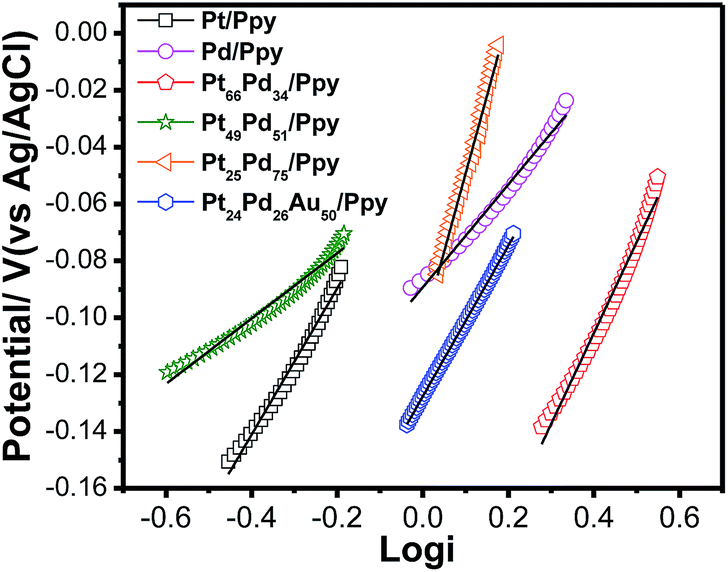 | ||
| Fig. 11 Tafel plots for the electrooxidation of methanol on the polymer nanocomposites based electrodes at a scan rate of 1 mV s−1. | ||
The exchange current density is ca. 0.33 mA for Pt24Pd26Au50/Ppy and ca. 0.19 mA for Pt66Pd34/Ppy. The calculated charge transfer coefficients (α) of the methanol oxidation reaction for the as-prepared composites are summarized in Table 3. The charge transfer coefficient for Pt24Pd26Au50/Ppy is higher than that of Pt66Pd34/Ppy, which demonstrates that the trimetallic alloy and conducting polymer support enhanced the catalytic performance during MOR.
| Electrode | Intercept V | Slope V dec−1 | R 2 value | I 0 mA cm−2 | Α |
|---|---|---|---|---|---|
| Pt/Ppy | −0.18 | 0.266 | 0.972 | 0.21 | 0.114 |
| Pd/Ppy | −0.178 | 0.36 | 0.984 | 0.16 | 0.165 |
| Pt66Pd34/Ppy | −0.233 | 0.32 | 0.984 | 0.19 | 0.186 |
| Pt51Pd49/Ppy | −0.19 | 0.33 | 0.977 | 0.26 | 0.18 |
| Pt25Pd75/Ppy | −0.4 | 0.516 | 0.994 | 0.16 | 0.115 |
| Pt24Pd26Au50/Ppy | −0.127 | 0.265 | 0.999 | 0.33 | 0.225 |
Fig. 12a shows the chronoamperometric (CA) measurements for Pt/C (brown solid line), Pd/Ppy (pink solid line), Pt/Ppy (black solid line), Pt66Pd34/Ppy (red solid line), Pt51Pd49/Ppy (green solid line), Pt25Pd75/Ppy (orange solid line), and Pt24Pd26Au50/Ppy (blue solid line) at a fixed potential (−0.1 V) to evaluate their long term stabilities. The chronoamperometric curve shows the time response current density decay of all the catalysts and commercial Pt/C for methanol electrooxidation. For all the catalysts, the current density decays gradually up to 500 seconds and then attains a steady state thereafter, finally reaching a zero value; however, for Pt66Pd34/Ppy, the current density decayed up to 1500 seconds and then attained a steady state. Pt/Ppy and Pt/C are rapidly deactivated and the current continues to decrease, while the other electrode materials reach steady-state currents. In fact, for the Pt/C catalyst, the current decreased to almost zero; however, for Pt24Pd26Au50/Ppy, the current decay reached a steady-state current after roughly 10 min. This clearly indicates that the bimetallic and trimetallic nanocomposites form stable films on glassy carbon electrode and are more stable electrocatalysts compared to the monometallic nanocomposites for methanol oxidation. However, gradual current decay occurs due to the formation of intermediate carbonaceous species, such as CO-like species, and their coverage on the surface active sites can hinder the adsorption of fresh fuel on the surface of the catalysts. The nonzero current density values after 3600 seconds for the Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy electrocatalysts is possibly due to the close contact between the metal NPs and the polymer nanofibers, resulting in strong interactions which may improve the mass transfer rates.
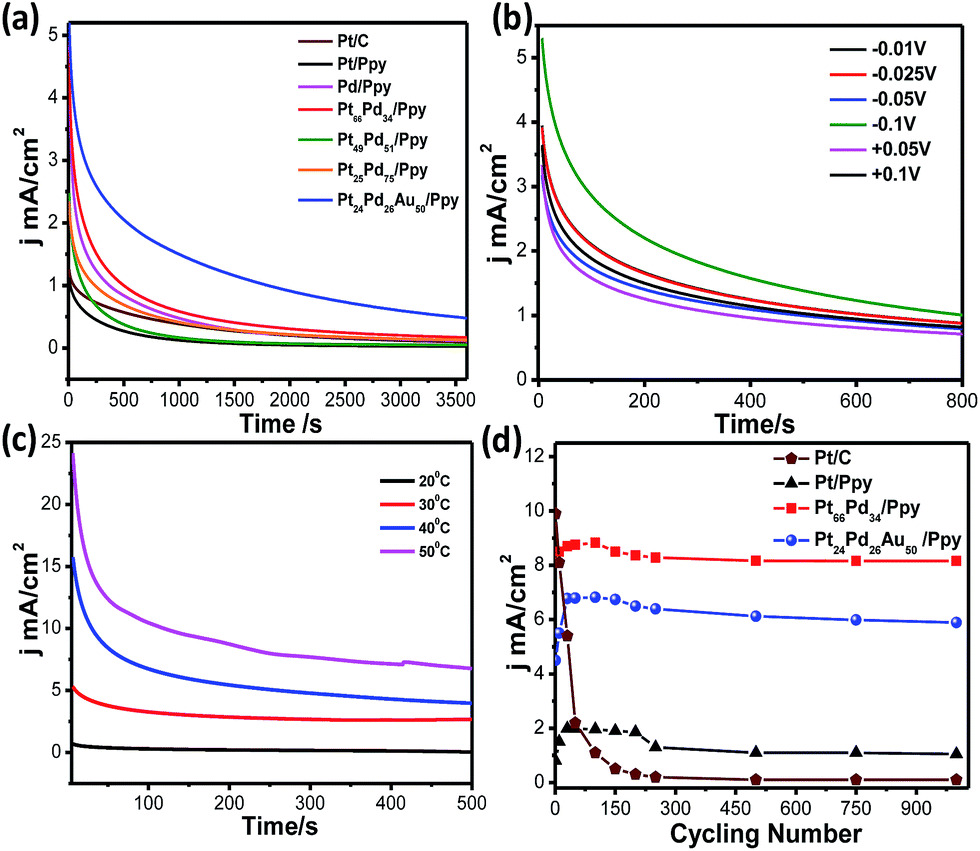 | ||
| Fig. 12 (a) Chronoamperometric curves for methanol oxidation at constant potential of −0.1 V vs. Ag/AgCl on Pt/C, Pd/Ppy, Pt/Ppy, Pt66Pd34/Ppy, Pt49Pd51/Ppy, Pt11Pd89/Ppy and Pt24Pd26 Au50/Ppy/Ppy electrodes. (b) Chronoamperometric curves for methanol oxidation at different potentials. (c) Chronoamperometric curves for methanol oxidation at a constant potential of −0.1 V vs. Ag/AgCl at different temperatures. (d) Long term cycling studies of Pt/C, Pt/Ppy, Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy electrodes in a solution of 0.1 M KOH and 1 M methanol at a scan rate of 20 mV s−1. | ||
The long term stability of a catalyst can be evaluated from CA curves. The calculated loss rates from the CA curves of Pt/Ppy, Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy are 0.113% s−1, 0.018% s−1, and 0.0085% s−1, respectively, demonstrating that Pt66Pd34/Ppy is more stable than the other catalysts. The slow decay rate with time and the lower decay of current density after long cycles reflects favourable adsorption–desorption kinetics on the catalyst surface and confirms greater tolerance to methanol; thus, conducting polymer-supported Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy can be reasonably identified as the best composites for the methanol oxidation reaction in alkaline media. Fig. 12b shows the chronoamperometric curves obtained at different potentials for Pt66Pd34/Ppy catalysts in the oxidation of methanol. In the first several minutes, all the catalysts exhibited a pronounced current decay, which could be caused by the accumulation of poisonous intermediates. Fig. 12c shows the chronoamperometric curves obtained at different temperatures for the Pt66Pd34/Ppy catalysts during the electrooxidation of methanol. With increasing temperature, the stability of the catalyst was enhanced due to the improved kinetics. To compare the stabilities of electrodes using Pt/C, Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy, cyclic voltammetry experiments were performed under similar conditions up to 1000 cycles. It is evident that the conducting polymer-supported bi and tri-metallic nanocomposites are highly active catalysts for MOR in alkali medium up to 1000 potential cycles, as shown in Fig. 12d. There is almost no change in the current densities of the electrode materials based on polymer nanocomposites examined after 1000 potential cycles during MOR. For commercial Pt/C, the current density began to decrease as early as the 2nd cycle, and 98% decay was observed after 1000 cycles. The decays of current density for the MOR were 5.7% on the Pt66Pd34/Ppy electrocatalyst and 12% on the Pt24Pd26Au50/Ppy catalyst, which are significantly lower than that of Pt/Ppy (43%) after 1000 cycles. This indicates better tolerance of Pt66Pd34/Ppy and Pt24Pd26Au50/Ppy electrocatalysts to CO or carbonaceous intermediates formed during the MOR. Hence, radiolytic synthesis of bimetallic and trimetallic alloy-based polymer nanocomposites is suitable for direct alcohol fuel cell applications.
The use of Ppy nanofibers as a support can effectively increase the dispersion of metal catalysts, which in turn enhances the oxidation of organic molecules on the Pt surface and simultaneous adsorption of hydroxyl species on Pd; this produces synergic effects in improving the kinetics of methanol oxidation in Pt66Pd34/Ppy bimetallic catalysts, while the addition of gold increases the reactivity of the Pt24Pd26Au50/Ppy system. The large specific surface area as well as the anisotropic morphology of Ppy nanofibers can facilitate the transportation of electroactive species; also, the small, well dispersed Pt NPs on the nanofibers may enhance the availability of the Pt catalyst and can efficiently prevent Ostwald ripening and aggregation of Pt during methanol oxidation. On the other hand, metal NPs supported on a conductive polymer surface have enhanced structural stability and provide a direct path for electron transport, resulting in excellent electrical contact with the current collectors. However, supported catalysts are among the most difficult to study because of their complexity; in order to fully understand the catalytic potential of a bimetallic system, surface science investigations must be performed to collect atomic level information on the structure, composition, adsorption and desorption, surface chemical bonding, and catalytic activity and selectivity of the catalyst. This approach helps us to envisage the surface dynamics and molecular transformation pathways on the surface sites.
In order to shed light on the path of anodic oxidation of methanol, cyclic voltammetric studies were carried out using Pt/Ppy and Pt66Pd34/Ppy electrodes immersed in 1 M KOH with methanol, formaldehyde and sodium formate fuels (100 mM) in the potential range of −0.8 V to +0.8 V at a scan rate of 20 mV s−1. Fig. 13a demonstrates that the respective peak current densities in mA cm−2 of the Pt66Pd34/Ppy electrode follow the order HCOONa (4.7) < HCHO (7.3) < CH3OH (8.2), representing the current-providing capability of each fuel. The potential of the most intense forward peak in the CV of the HCOONa fuel has a low potential and may follow an easier pathway of oxidation compared to other fuels, such as HCHO and CH3OH, on the Pt66Pd34/Ppy electrode. Fig. 13a also illustrates that the kinetics of the oxidation of formate is affected by poisonous intermediates, as evident from the large anodic peak during the reverse sweep. A large peak appears in the lower potential region for the Pt66Pd34/Ppy electrode due to the formation of carbonate as formate is also oxidized to carbonate.20 In contrast, CH3OH has a low potential and a more favourable oxidation pathway on the Pt/Ppy electrode. Fig. 13b displays that the respective peak current densities in mA cm−2 of the Pt/Ppy electrode follow the order CH3OH (0.5) < HCOONa (2.0), where oxidation of HCHO did not occur on the catalyst surface. The oxidation of formate is more favourable on the Pt/Ppy electrode than on the Pt66Pd34/Ppy alloy electrode.
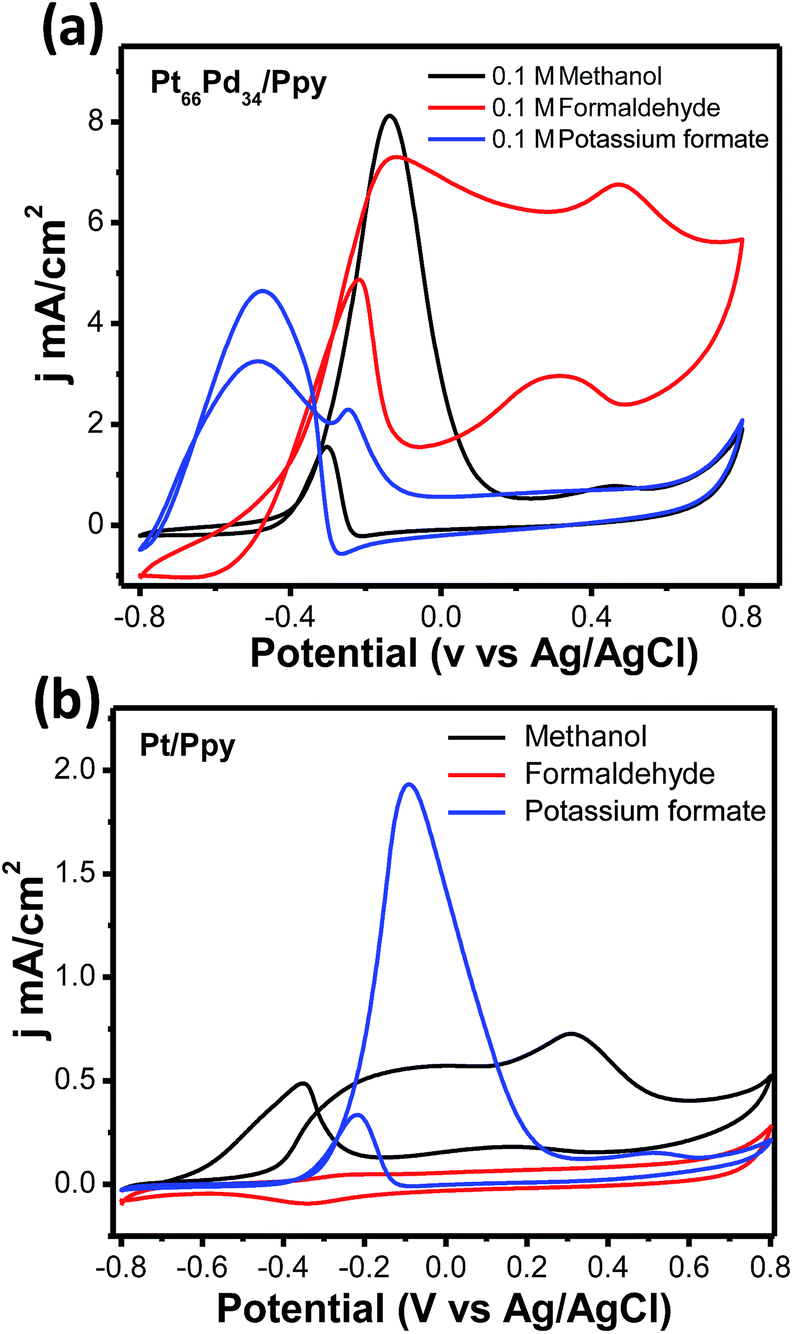 | ||
| Fig. 13 (a) Cyclic voltammetric profiles for the Pt66Pd34/Ppy catalyst in 0.1 M aqueous KOH containing 100 mM of MeOH, HCHO, or HCOONa as fuel at a scan rate of 20 mV s−1. (b) Cyclic voltammetric profiles for the Pt/Ppy electrode in 0.1 M aqueous KOH containing 100 mM of MeOH, HCHO, or HCOONa as fuel at a scan rate of 20 mV s−1. | ||
4. Conclusions
In conclusion, a facile strategy has been developed to synthesize conducting polymer-supported Pt-based monometallic and multimetallic nanocomposites using a radiolytic technique without any chemical reducing agents. When noble metals are incorporated into conducting polymer nanofibers, the synergetic effects of the redox-rich properties of the polymer support and the high catalytic activity of the metal nanoparticles with unique structures are suitable for the electrooxidation of methanol. The bimetallic composition shows a pronounced impact on MOR because of the uniform dispersion of Pt66Pd34 NPs on the conducting nanofibers. Moreover, the trimetallic composition demonstrated superior mass activity and stability for MOR. Most importantly, due to the strong interaction between the NPs and the polymer nanofibers, the stability of electrodes is significantly enhanced for methanol oxidation. The mass activities of the binary and ternary alloy catalysts were successfully increased, and the higher negative onset potential reveals faster kinetics. Additionally, the Pt loading in the bimetallic and trimetallic catalysts was significantly lower, which lowers the effective cost of the catalyst. Hence, polypyrrole nanofiber-supported metal NPs are promising anode catalysts for low temperature fuel cell applications. With further modifications, the polymer nanofiber-supported catalysts may also be useful in a wide range of other catalytic applications, such as the oxygen reduction reaction and dehydrogenation of organic molecules.Acknowledgements
The authors acknowledge Director, CSIR-CGCRI for his kind permission to publish the work. One of the authors (SG) is thankful to the Council of Scientific & Industrial Research (CSIR), India, for providing a CSIR-Senior Research Associate (Scientists' Pool Scheme) award. The authors also wish to thank Dr Biswanath Kundu, Bioceramics and Coating Division, CSIR –CGCRI for his help in carrying out the radiolysis of the samples.Notes and References
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J. M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- S. P. S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 Search PubMed.
- X. Li and A. Faghri, J. Power Sources, 2013, 226, 223–240 CrossRef CAS.
- N. Radenahmad, A. Afif, I. Perta, S. M. H. Rahman, S. G. Eriksson and A. K. Azad, Renewable Sustainable Energy Rev., 2016, 57, 1347–1358 CrossRef CAS.
- Y. Liu, S. F. Zhao, S. X. Guo, A. M. Bond, J. Zhang, G. Zhu, C. L. Hill and Y. V. Geletii, J. Am. Chem. Soc., 2016, 138, 2617–2628 CrossRef CAS PubMed.
- Z. Yang and N. Nakashima, Sci. Rep., 2015, 5, 12236 CrossRef CAS PubMed.
- Z. Wen, J. Liu and J. Li, Adv. Mater., 2008, 20, 743–747 CrossRef CAS.
- D. Chen, F. Ye, H. Liu and J. Yang, Sci. Rep., 2016, 6, 24600 CrossRef CAS PubMed.
- D. He, L. Zhang, D. He, G. Zhou, Y. Lin, Z. Deng, X. Hong, Y. Wu, C. Chen and Y. Li, Nat. Commun., 2016, 7, 12362 CrossRef CAS PubMed.
- Y. Wang, K. Yin, J. Zhang, C. Si, X. Chen, L. Lv, W. Ma, H. Gao and Z. Zhang, J. Mater. Chem. A, 2016, 4, 14657–14668 RSC.
- M. K. Min, J. Cho, K. Cho and H. Kim, Electrochim. Acta, 2000, 45, 4211–4217 CrossRef CAS.
- X. Liu, G. Xu, Y. Chen, T. Lu, Y. Tang and W. Xing, Sci. Rep., 2015, 5, 7619 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasché and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 3273–3291 CrossRef CAS.
- J. Wu, Y. Hou and S. Gao, Nano Res., 2011, 4, 836–848 CrossRef CAS.
- N. B. Devi, F. Kadirgun, J. Beyhan and T. Atilan, Int. J. Hydrogen Energy, 2009, 34, 4312–4320 CrossRef.
- S. Sakong and A. Groß, ACS Catal., 2016, 6, 5575–5586 CrossRef CAS.
- W. Zhou, Z. Zhou, S. Song, W. Li, G. Sun, P. Tsiakaras and Q. Xin, Appl. Catal., B, 2003, 46, 273–285 CrossRef CAS.
- S. Ghosh, H. Remita, P. Kar, S. Choudhury, S. Sardar, P. Beaunier, P. S. Roy, S. K. Bhattacharya and S. K. Pal, J. Mater. Chem., A, 2015, 3, 9517–9527 RSC.
- S. Roy Chowdhury, S. Ghosh and S. K. Bhattachrya, Electrochim. Acta, 2017, 225, 310–321 CrossRef CAS.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao, M. Zeng, J. Zhong, C. Jin, Y. Li, S. T. Lee and H. Dai, Nat. Commun., 2015, 6, 10035 CrossRef CAS PubMed.
- A. Dutta and J. Ouyang, ACS Catal., 2015, 5, 1371–1380 CrossRef CAS.
- C. Y. Su, Y. C. Hsueh, C. C. Kei, C. T. Lin and T. P. Perng, J. Phys. Chem. C, 2013, 117, 11610–11618 CrossRef CAS.
- A. Alshammari, V. N. Kalevaru and A. Martin, Catal., 2016, 6, 97–121 CrossRef.
- Q. Lu, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao, H. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6, 6567 CrossRef CAS PubMed.
- S. Ghosh, Y. Holade, H. Remita, K. Servat, P. Beaunier, A. Hagège, B. Kokoh and T. W. Napporn, Electrochim. Acta, 2016, 212, 864–875 CrossRef CAS.
- X. Huang, Y. Li, Y. Li, H. Zhou, X. Duan and Y. Huang, Nano Lett., 2012, 12, 4265 CrossRef CAS PubMed.
- M. K. Min, J. Cho, K. Cho and H. Kim, Electrochim. Acta, 2000, 45, 4211–4334 CrossRef CAS.
- Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- K. I. Ozoemena, RSC Adv., 2016, 6, 89523–89550 RSC.
- C. Lamy, A. Lima, V. Lerhum, F. Delim, C. Cautenceau and J. M. Legar, J. Power Sources, 2002, 105, 283–287 CrossRef CAS.
- R. Ramachandran, S. M. Chen, G. P. G. kumar, P. Gajendran and A. Xavier, Int. J. Electrochem. Sci., 2016, 11, 1247–1270 CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- J. C. Calderón, M. R. Ráfales, M. J. N. Monge, J. I. Pardo, R. Moliner and M. J. Lázaro, Nanomaterials, 2016, 6, 187–197 CrossRef PubMed.
- L. Tao, S. Dou, Z. Ma and S. Wang, Electrochim. Acta, 2015, 157, 46–53 CrossRef CAS.
- J. Kim, J. S. Jang, D. H. Peck, B. Lee, S. H. Yoon and D. H. Jung, Nanomaterials, 2016, 6, 148–159 CrossRef PubMed.
- C. Alegre, M. E. Gálvez, R. Moliner and M. J. Lázaro, Catalysts, 2015, 5, 392–405 CrossRef CAS.
- D. Pan, J. Chen, W. Tao, L. Nie and S. Yao, Langmuir, 2006, 22, 5872–5876 CrossRef CAS PubMed.
- R. Krishna, D. M. Fernandes, J. Ventura, C. Freire and E. Titus, Int. J. Hydrogen Energy, 2016, 41, 11811–11822 CrossRef CAS.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- S. Ghosh, K. A. Natalie, L. Ramos, S. Remita, A. Dazzi, A. D. Besseau, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- S. Ghosh, M. Thandavarayan and R. N. Basu, Nanoscale, 2016, 8, 6921–6947 RSC.
- S. Ghosh, L. Ramos, S. Remita, A. Dazzi, A. D. Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, New J. Chem., 2015, 39, 8311–8320 RSC.
- S. Patra and N. Munichandraiah, Langmuir, 2009, 25, 1732–1738 CrossRef CAS PubMed.
- C. T. Hable and M. S. Wrighton, Langmuir, 1993, 9, 3284–3290 CrossRef CAS.
- H. J. Salavagione, C. Sanchís and E. Morallón, J. Phys. Chem. C, 2007, 111, 12454–12460 CrossRef CAS.
- C. W. Kuo, C. Sivakumar and T. C. Wen, J. Power Sources, 2008, 185, 807–814 CrossRef CAS.
- L. Shi, R. Liang and J. Qiu, J. Mater. Chem., 2012, 22, 17196–17203 RSC.
- S. Chen, Z. D. Wei, X. Qi, L. Dong, Y.-G. Guo, L. J. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- H. Xu, A. L. Wang, Y. X. Tong and G. R. Li, ACS Catal., 2016, 6, 5198–5206 CrossRef CAS.
- C. W. Kuo, C. Sivakumar and T. C. Wen, J. Power Sources, 2008, 185, 807–814 CrossRef CAS.
- Y. Ma, S. Jiang, G. Jian, H. Tao, L. Yu, X. Wang, J. Zhu, Z. Hu and Y. Chen, Energy Environ. Sci., 2009, 2, 224–229 Search PubMed.
- Z. Liu, X. Zhang, S. Poyraz, S. P. Surwade and S. K. Manohar, J. Am. Chem. Soc., 2010, 132, 13158–13159 CrossRef CAS PubMed.
- J. Belloni, M. Mostafavi, H. Remita, J. L. Marignier and M. O. Delcourt, New J. Chem., 1998, 1239–1255 RSC.
- J. W. T. Spinks and R. J. Woods, Water and Inorganic Aqueous Systems, in, An introduction to Radiation Chemistry, 3rd edn, Wiley Interscience, New York, 1990, pp. 255–283 Search PubMed.
- J. H. Baxendale and P. Wardman, J. Chem. Soc., 1973, 169, 584–594 Search PubMed.
- N. Getoff, A. Ritter, F. Schworer and P. Bayer, Radiat. Phys. Chem., 1993, 41, 797–801 CrossRef CAS.
- Y. Liu, S. Liu, Z. Che, S. Zhao, X. Sheng, M. Hana and J. Bao, J. Mater. Chem., A, 2016, 4, 16690–16697 RSC.
- Y. Min, M. Akbulut, K. kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS PubMed.
- S. Basu, H. Paul, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, J. Catal., 2005, 229, 298–302 CrossRef CAS.
- H. Xu, L. X. Ding, C. L. Liang, Y. X. Tong and G. R. Li, NPG Asia Mater., 2013, 5, 69–79 CrossRef.
- N. Maity, P. R. Rajamohanan, S. Ganapathy, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, J. Phys. Chem. C, 2008, 112, 9428–9433 CrossRef CAS.
- L. S. Kibis, A. I. Titkov, A. I. Stadnichenko, S. V. Koscheev and A. I. Boronin, Appl. Surf. Sci., 2009, 255, 9248–9254 CrossRef CAS.
- A. A. Melvin, K. Illath, T. Das, T. Raja, S. Bhattacharyya and C. S. Gopinath, Nanoscale, 2015, 7, 13477–13486 RSC.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- S. S. Mahapatra and J. Datta, Int. J. Electrochem., 2011, 2011, 563495–563511 Search PubMed.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- G. F. Álvarez, M. Mamlouk and K. Scott, Int. J. Electrochem., 2011, 2011, 684535–684547 Search PubMed.
- R. Doherty, P. Krafft, J. M. Methivier, C. Casale, S. Remita, H. Louis and C. Thomas, J. Catal., 2012, 287, 102–113 CrossRef CAS.
- S. T. Nguyen, H. M. Law, H. T. Nguyen, N. Kristian, S. Wang, S. H. Chan and X. Wang, Appl. Catal., B, 2009, 91, 507–515 CrossRef CAS.
- S. Mukerjee, S. Srinivasan and M. P. Soriaga, Electrochem. Soc. Interface, 1995, 142, 1409–1422 CrossRef CAS.
- Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS PubMed.
- T. Huang, S. Mao, G. Zhou, Z. Zhang, Z. Wen, X. Huang, S. Cib and J. Chen, Nanoscale, 2015, 7, 1301–1307 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information, XRD, TEM and HRTEM images, XPS and CV patterns. See DOI: 10.1039/c7se00126f |
| This journal is © The Royal Society of Chemistry 2017 |