
Organic salt photovoltaics
M.
Bates
a and
Richard R.
Lunt
 *b
*b
aMichigan State University, USA
bMichigan State University, USA. E-mail: rlunt@msu.edu
First published on 24th April 2017
Abstract
In this article we review the development of organic salts integrated in organic photovoltaics (OPVs) and the potential capabilities that organic salts enable in OPV devices. Organic salts are ionic compounds composed of either organic cations, anions, or a combination of both. These organic salt semiconductors can exhibit highly tunable properties with absorption throughout the visible and deep into the near-infrared. These characteristics have enabled their integration into traditional OPV cells, multijunctions, and transparent photovoltaic devices with unique applications for windows and similar surfaces. We discuss the tunable properties of these salts that make them desirable for use in a range of applications and the historical development of salt based OPVs to provide an overview of the progress made in efficiencies and device architectures. Among their most exciting properties, organic salts have the ability to enable high efficiency transparent photovoltaics (TPVs), tunable energetics, and highly efficient ultra-deep infrared harvesting.
 M. Bates | Matthew Bates is a first year Ph.D. student in chemical engineering at Michigan State University working in the Molecular and Organic Excitonics Lab led by Professor Richard R. Lunt. He received his B.S. in chemical engineering from Oregon State University in 2016. |
 Richard R. Lunt | Richard R. Lunt is the Johansen Crosby Endowed Associate Professor at Michigan State University in the Departments of Chemical Engineering & Materials Science and Physics. He earned his B.S. from the University of Delaware in 2004 and his Ph.D. from Princeton University in 2010. He then worked as a post-doctoral researcher at MIT until 2011. His group focuses on understanding and exploiting excitonic photophysics and molecular crystal growth to develop unique thin-film optoelectronic devices. |
Introduction
Organic salts are ionic compounds comprised of a cation and anion with at least one organic molecule in the pair (Fig. 1). The ionic character is similar to that of inorganic salts (e.g. NaCl, KCl etc.) but more closely related to ionic liquids in both composition and ordering. Indeed, while many semiconducting organic salts have extended conjugation to generate more interesting bandgaps, this typically increases melting points past decomposition limits. Common photoactive cations include cyanine dyes of varying conjugation length, polymethines, hemicyanines, ionic polymers, cyanine dyads, pyrylium dyes, chalcogenopyrylium monomethine, azapyrenes, and non-photoactive tetrabutylammonium, among others.1–16 Anions used in organic salt PVs are generally non-photoactive and include halides, perchlorate, phosphates, antimonates, borates, carbonanes, and even photoactive cyanine sulfonates.17,18 Many of these counterions are weakly coordinating anions (WCAs), which is in part why organic salt OPVs, introduced in the 1970s, were not consistently investigated until a greater range of WCAs emerged.19Fig. 2 depicts many of the cationic cyanines and counterions discussed in this review (cations are referenced by number as they are mentioned). Organic salts have been used historically in a wide range of applications including medical imaging, photographic emulsions, chemical sensors, ‘click’ bioconjugation, solar concentrators, and recording media.20–38 Photovoltaic devices utilizing organic salts in the active layer are generally considered small molecule based organic solar cells, however the unique properties and tunability that organic salts provide warrants consideration as a new class of OPV donors and acceptors. Organic salts have many properties that make them suitable materials for OPVs, including strong absorption demonstrated by high extinction coefficients, tunable absorption in the visible and near-infrared (Fig. 3), precisely defined molecular weights, and are solution processable. Furthermore, the very notion of using organic salts conjures the image of table salt dissolving in water, thus implying the potential for a high sensitivity to moisture and a low likelihood for success in electronic applications. However, many of the demonstrated salts to date are sparingly soluble in water and actually show very good application in optoelectronics devices. The widespread use of these salts and their exceptional properties for OPV applications make them exciting materials for further research.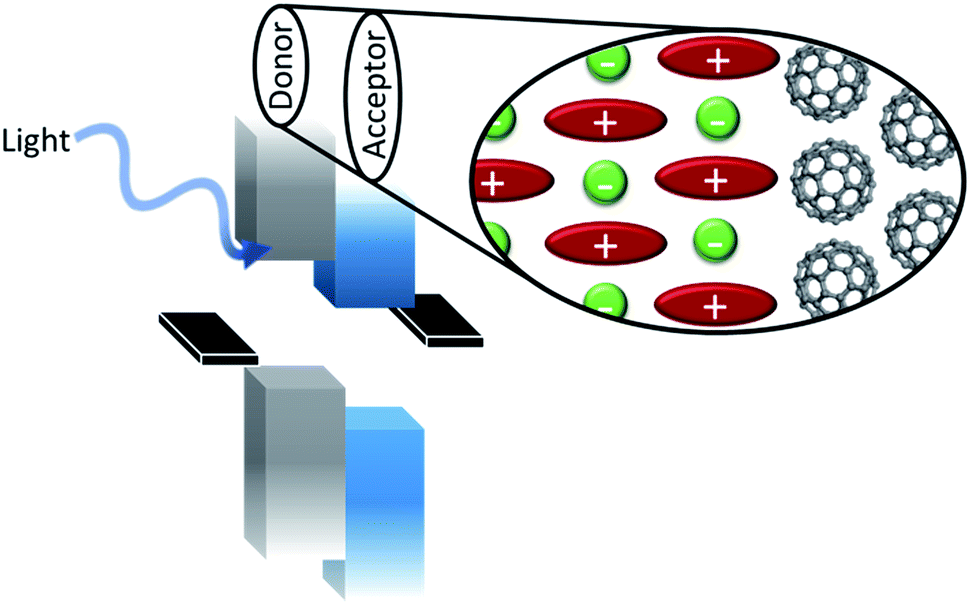 | ||
| Fig. 1 Schematic of an organic salt based PV device showing light absorption by the active layers, one or both of which consist of the organic salt shown in the expanded view. | ||
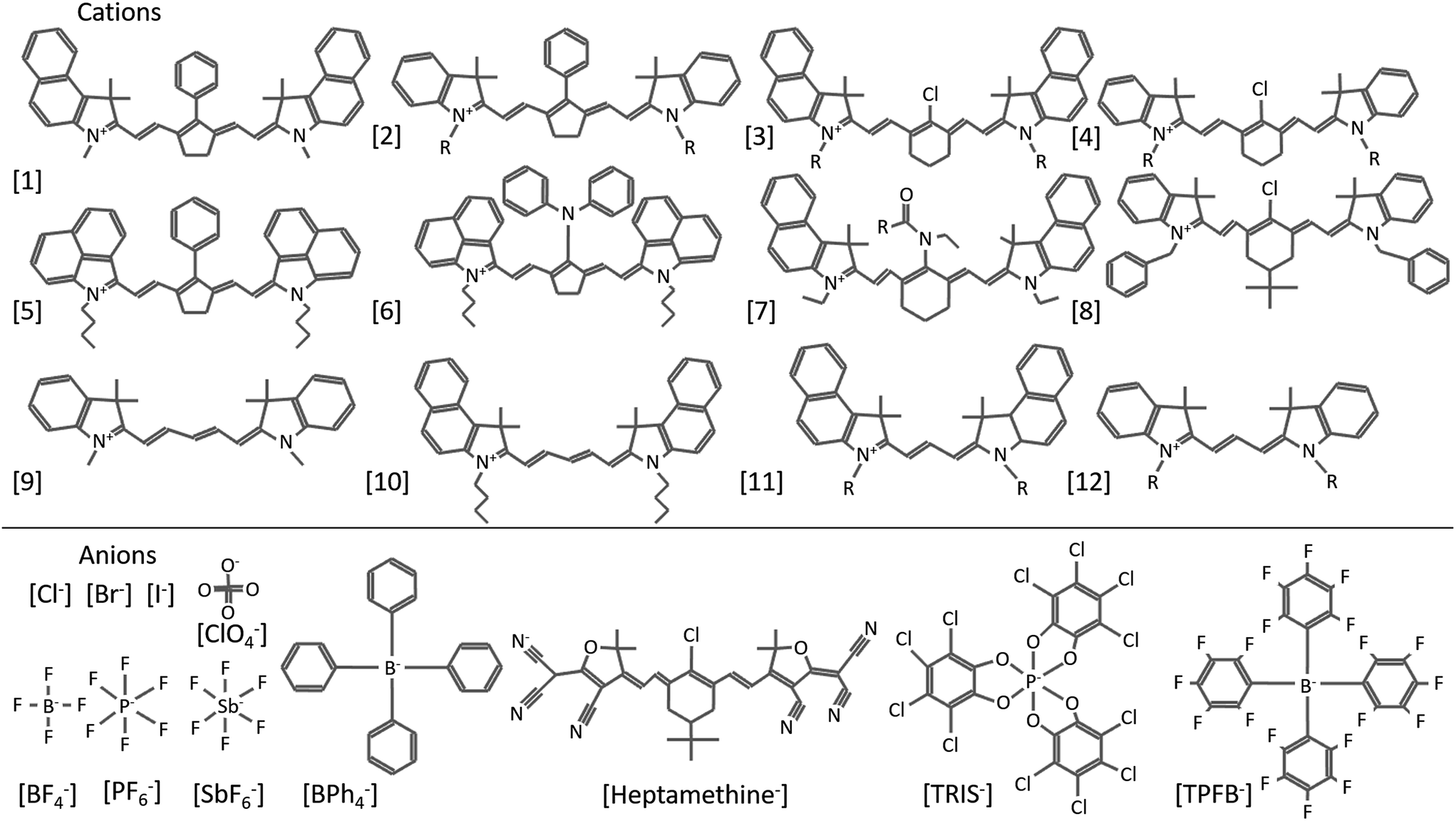 | ||
| Fig. 2 Various cationic cyanine molecules, including heptamethines (1–8), pentamethines (9–10), and trimethines (11–12), and anions used in organic salt based PVs. | ||
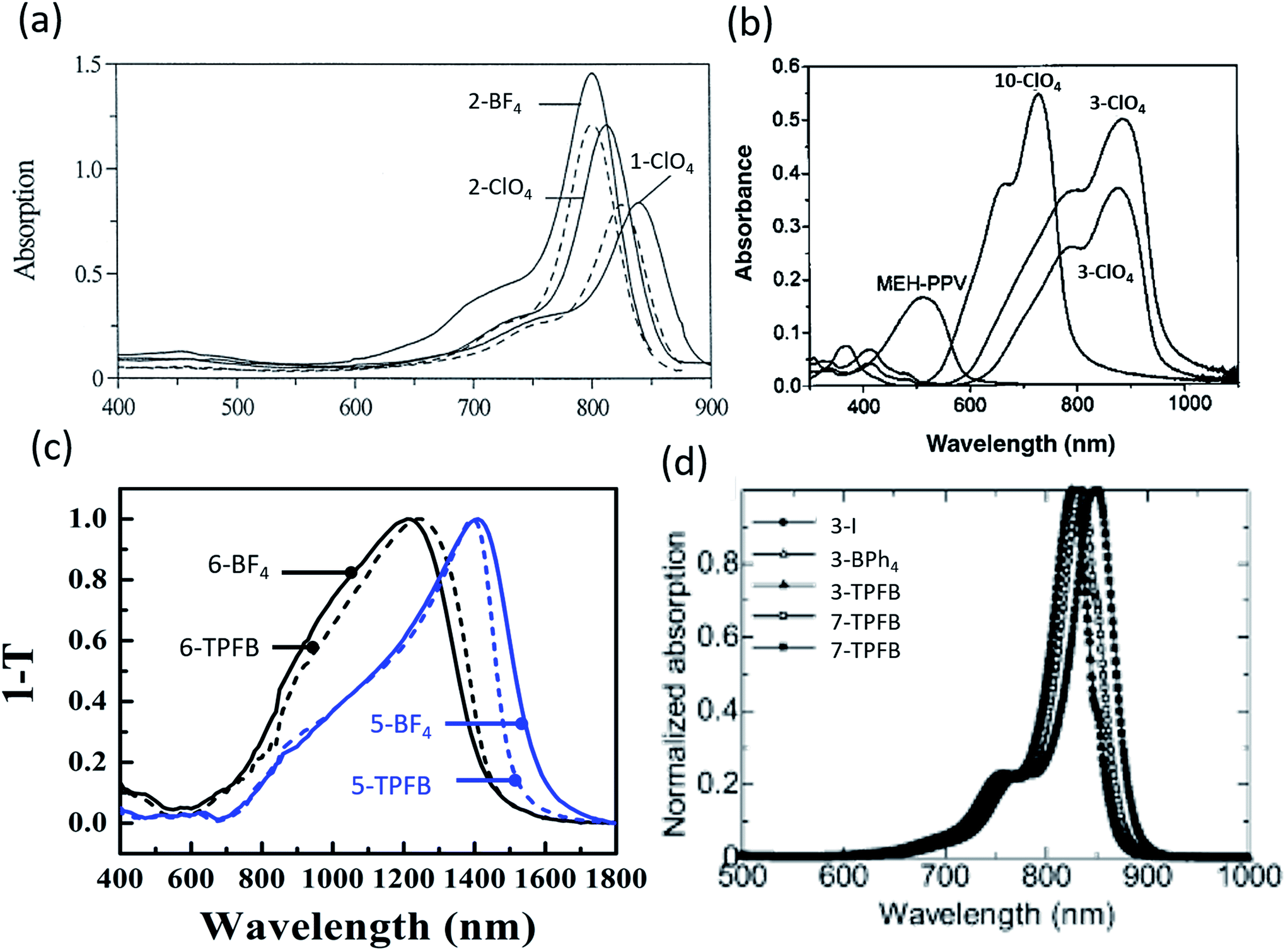 | ||
| Fig. 3 Absorption spectra of (a) organic salts in polyurethane acrylate (solid) and ethanol (dashed),9 (b) organic salt thin films with NIR absorption,50 (c) salt thin films demonstrating deep NIR absorption out to 1600 nm,69 (d) thin films showing NIR absorption.10 | ||
This review is outlined as follows: we first detail the historical integration of organic salts into solar cells before summarizing the properties of weakly coordinating anions. We then review topics unique to organic salt PVs, including the role of the counterion in these devices and ion mobility in the active layer. We then examine detailed photovoltaic processes in organic salt PVs, focusing on exciton diffusion and charge carrier generation, transport, and collection. We conclude by highlighting the capabilities organic salts enable in transparent photovoltaics, as well as the stability of these devices, followed by a summary of important knowledge gaps in this emerging field.
Historical development of organic salt based photovoltaics
We start by reviewing the development of organic salt based organic photovoltaics (OPVs), focusing on device and molecular structure, key photovoltaic parameters, and quantum efficiencies. For context, other single junction small-molecule and polymer based OPVs have been extensively reviewed and have achieved over 9% and 10% efficiencies, respectively, with multijunctions over 11% for polymer and small molecule devices.38–40PCEs for organic salt devices, in contrast, have risen from 0.3% to 3.7%, and parameters such as Jsc, Voc, and FF have similarly increased. External quantum efficiencies (EQEs) have seen massive gains in magnitude and in breadth, reaching as high as 80% and harvesting light in the NIR, and recently extending out as far as 1600 nm.
Ghosh was among the first to propose the use of organic salts as active layers in 1978 in a U.S. patent for photovoltaic devices using an organic layer between two electrodes.41 Ghosh put forth several different organic materials, including cyanines and hemicyanines, even though the organic salt variations of these materials were not reported among the data.
One of the first reports of functioning organic salt PVs came from Stepanova et al., who used pyrylium dye cations with ClO4− counterions as an acceptor material for OPVs in the late 1970s.42 Thin film preparation techniques noted by Whorle et al. in 1991 included vacuum deposition, spin coating, chemical vapor deposition, and plasma polymerization.43 Stepanova's solar cells were fabricated with indium tin oxide (ITO) as the anode followed by vacuum deposited copper phthalocyanine, spin coated organic salt, and an indium cathode. This device demonstrated a power conversion efficiency (PCE) = 0.43%, open circuit voltage (Voc) = 0.45 V, and short circuit current (Jsc) = 1.6 mA cm−2 under 70 mW cm−2 irradiation. Early devices struggled to achieve high PCEs without the metallophthalocyanine donor Simon used.
Although organic salt based OPVs were first used in the 1970s and 1980s, consistent development of such devices did not start in earnest until the early 2000s.44 In 2003, Meng et al. used a trimethine (cation 12)+/ClO4− organic salt as an acceptor and donor layer in OPVs.45 An organic salt donor paired with a fullerene acceptor layer demonstrated efficiency = 0.0038%, with fill factor (FF) = 0.20, Voc = 0.25 V, and Jsc = 0.11 mA cm−2. The band gap of the organic salt was 2.1 eV, an order of magnitude higher than the Voc. The high bandgap salt limits spectral harvesting to photons with equivalent or greater energy, leading to the low Jsc. With this organic salt as the acceptor, paired with a polymer donor layer, the device demonstrated PCE = 0.0082%, Voc = 1.28 V, Jsc = 0.05 mA cm−2, and FF = 0.20. A peak EQE from the organic salt of 4% was demonstrated at 575 nm. Other work showed the use of organic salts as acceptors and donors in bulk heterojunction devices (BHJ) with a thin film of two organic salts on a gold substrate.46 Both organic salts utilized anionic trimethines and exhibited a 2% peak EQE at 650 nm. The reported PCE was 0.05%, with a 0.19 V Voc and 0.007 mA cm−2Jsc.
Despite improvements, early investigations of organic salt PVs suffered from particularly low PCEs. In 2005 Nuesch et al. introduced a PEDOT:PSS interlayer between the ITO anode and trimethine (12)+/ClO4− donor paired with fullerene.47 Devices with PEDOT:PSS had Voc = 0.43 V and Jsc = 0.13 mA cm−2 while those without demonstrated Voc = 0.28 V and 0.1 mA cm−2Jsc. PEDOT:PSS improved the hole extraction into ITO, improving Voc by over 50%. Improving on this initial PEDOT:PSS interfacial layer, Nuesch et al. demonstrated modest PCE = 0.09% at 31 mW cm−2 illumination, which decreased to 0.02% at 310 mW cm−2.48 At higher intensity, the fill factor was notably low at 0.16 (any value less than 0.25 shows inverted curvature in the JV and indicates high resistances). In 2005, Meng et al. synthesized fullerene – organic salt dyads, wherein the trimethine cation of the trimethine+/ClO4− organic salt was covalently bonded to fullerene, paired with an additional fullerene acceptor layer.49 Devices demonstrated 6% peak EQE at 540 nm with PCE = 0.041% at 310 mW cm−2 illumination, a 0.33 V Voc, and 1.2 mA cm−2Jsc. Castro et al. created devices utilizing a polymer/organic salt blend.50 Three organic salts were mixed with the polymer, MEH-PPV, one with a pentamethine (10) cation, the others with heptamethine (3) cations, all with a ClO4− counterion. Fig. 3b shows the absorption spectra for this system. The pentamethine (band gap of 1.5 eV) demonstrated a Voc = 0.63, whereas the heptamethine (band gap of 1.13 eV) showed, surprisingly, a higher Voc = 0.79 V. However, Jsc was small, on the order of 1 μA cm−2 or less for all three devices, which limited the efficiency.
In 2007, Fan et al. investigated the doping of organic salt PVs by ambient air, water, and oxygen, with and without irradiation.51 Fresh devices with a trimethine (12)+/ClO4− salt demonstrated PCE = 0.14%, with Voc = 0.47 V, Jsc = 0.46 mA cm−2, and 0.189 FF. After three hours of exposure to ambient atmosphere and irradiation at 5 mW cm−2 solar cells obtained a 1.2% PCE, 0.73 V Voc, 1.83 mA cm−2Jsc, and 0.274 FF. Doping with humid nitrogen or oxygen without irradiation saw little increase in performance indicating that oxygen exposure paired with irradiation improved devices. Fan et al. followed up that work in 2009 by doping bilayer trimethine (12)+/ClO4− salt (band gap of 1.7 eV) PVs with the nitrosonium salt NOBF4, which has been used previously as an oxidizing agent.52 At full sun irradiation and a concentration of 0.02 wt% NOBF4, a PCE = 2.0% was demonstrated with Voc = 0.72 V, Jsc = 8.3 mA cm−2, and FF = 0.34. The peak EQE of the devices, shown in Fig. 4a, increased to 80% at 580 nm. In achieving such a high surprisingly high EQE, there two possibilities for the mechanism. It is unlikely that the dopant improved the exciton diffusion length or bulk dissociation. Rather, it is more likely that the trimethine salt, already possessing an inherently high exciton diffusion length, showed enhancement from the lowering of a charge collection barrier and/or improved charge collection. Alternatively, it is possible that the nitrosonium dopant promoted C60 diffusion into the trimethine layer, potentially forming a bulk heterojunction structure and increasing the exciton dissociation efficiency. While the mechanism is still unclear, the utilization of PEDOT:PSS and Alq3 interfacial layers, along with NOBF4 as a dopant in trimethine salts resulted in remarkable performance in both PCE and EQE for a planar architecture.
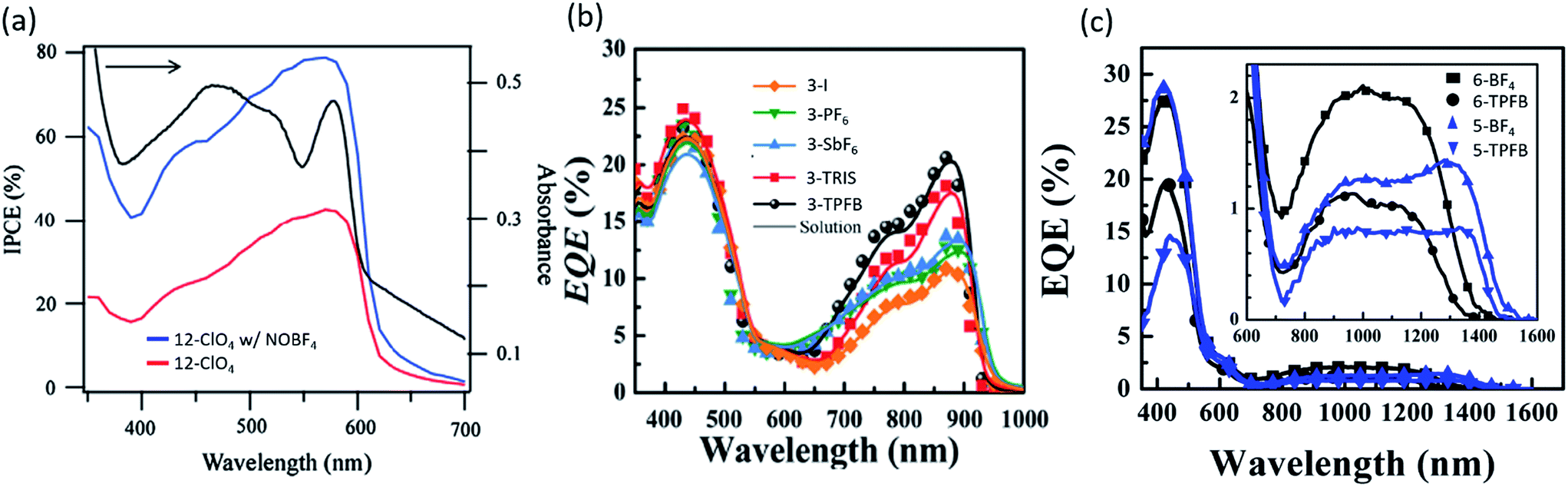 | ||
| Fig. 4 External quantum efficiency (EQE, or IPCE) of organic salt based PVs. (a) Demonstration of 80% peak EQE after addition of dopant NOBF4.52 (b) EQE of various anions paired with the same cation.68 (c) EQE of organic salt devices demonstrating deep NIR (1600 nm) photoresponse.69 | ||
The use of longer conjugated polymethines in OPVs was limited until Bouit et al. used heptamethines in 2009.53 BHJ devices with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 PCBM and heptamethine (8)+/heptamethine− salt layer demonstrated PCE = 0.4%, with Jsc = 3.78 mA cm−2, Voc = 0.37 V, and 11% peak EQE at 785 nm. This is one of the first instances of organic salt OPVs efficiently harvesting NIR light, and although the peak was lower than Fan et al.'s doped devices, it was red shifted over 200 nm. Absorbing NIR photons became a key ability of the organic salts and opened opportunities for deep NIR harvesting and transparent devices.
:2 PCBM and heptamethine (8)+/heptamethine− salt layer demonstrated PCE = 0.4%, with Jsc = 3.78 mA cm−2, Voc = 0.37 V, and 11% peak EQE at 785 nm. This is one of the first instances of organic salt OPVs efficiently harvesting NIR light, and although the peak was lower than Fan et al.'s doped devices, it was red shifted over 200 nm. Absorbing NIR photons became a key ability of the organic salts and opened opportunities for deep NIR harvesting and transparent devices.
In 2010, Fan et al. utilized PANI:DBS instead of PEDOT:PSS as an interfacial layer in trimethine (12)+/PF6− salt (1.8 eV band gap) PVs, demonstrating an EQE above 40% from 400 to 600 nm.54 A PCE = 3% was attained, with FF = 0.61, Jsc = 6.92 mA cm−2, and a 0.72 V Voc. To better understand interfacial layers, Berner et al. investigated the effects of the anode buffer layer, TiOx, smoothness on inverted device performance in 2013, utilizing the organic salt from Fan's work in 2010.55 Sol–gel processed TiOx demonstrated the best performance with PCE = 3.7%, Voc = 0.88 V, Jsc = 6.2 mA cm−2, and FF = 0.678. A TiOx sputtered device achieved PCE = 2.9%, Voc = 0.84 V, Jsc = 5.4 mA cm−2, and FF = 0.63. The sol–gel processed and sputter deposited TiOx results indicate the importance of a smooth, low resistance interfacial layer. Similarly, Malinkiewicz et al. studied the differences between PEDOT:PSS and MoO3 interfacial layers in bilayer polymethine+/anion− organic salt PVs.56 Four salts were used, trimethine (12)+/ClO4−, two other trimethine cations (11) with PF6−, and pentamethine (9)+/PF6−. Organic salts with the PF6− counterion demonstrated improved JV characteristics without the S-shaped curve seen with trimethine+/ClO4− salts that implies a high bulk or interface resistance and which improved the FF. The role of the bulkier counterion PF6− is likely related to ion mobility in the active layer and is discussed in a later section. Replacing PEDOT:PSS with MoO3 as the interfacial layer improved JV characteristics even though the peak EQE from the organic salts decreased from 45% to 30%. The best device utilized a 23 nm layer of trimethine (11)+/PF6− organic salt (1.9 eV band gap) and MoO3, demonstrating a PCE = 2.9% with a Voc = 0.92 V, Jsc = 5.1 mA cm−2, and FF = 0.62. Malinkiewicz also observed changes in device performance based on interfacial layer and counterion selection, demonstrating improved JV characteristics attributed to changes in either material.
Much of the development in organic salt PVs utilized layers of organic salt or a bulk layer of salt paired with fullerene, however, some work has been done in using organic salts as light harvesting dopants in polymer OPVs. In 2010, Yap et al. fabricated MEHPPV:PCBM heterojunction organic solar cells doped with tetrabutylammonium hexafluorophosphate (TBAPF6), an organic salt.57 The organic salt increased the Jsc from 0.54 μA cm−2 to 6.41 μA cm−2, Voc from 0.24 to 0.50 V, and fill factor from 0.16 to 0.18 compared to undoped devices. Due to the low FF and Jsc the PCE was 5.77 × 10−4%. Three years later, Sabri et al. investigated the concentration effect of TBAPF6 doped MEH:PPV devices, demonstrating a 0.15% PCE, 0.53 V Voc, 1.06 mA cm−2Jsc, and 0.27 FF for 20 wt% TBAPF6.58Jsc and Voc increased from 10 to 20 wt% TBAPF6 and decreased at 30 and 40 wt%. Optimization of organic salt doping increased performance but such devices were still an order of magnitude lower in efficiency than organic salt PVs at the time.
Most work with organic salts to this point had focused on relatively small inorganic anions, such as I−, ClO4− and PF6−, but some work had been done with larger counterions, including polymeric counterions. In 2014, Wang et al. fabricated organic solar cells using a sulfoethyl methacrylate/methacrylate anion and trimethine (12) cation for an organic salt.59 A 4.9 nm trimethine (12)+/polymer− layer demonstrated PCE = 0.93% with Voc = 0.63 V, Jsc = 2.3 mA cm−2, and FF = 0.64. The best devices had EQEs between 6 and 10% from 400 to 700 nm. Limited progress has been made working with polymeric anions in the organic salt, although other bulky anions have recently received greater attention.
The anionic polymer discussed in the work of Wang et al. was one of the first organic counterions used and one of the first other than I−, ClO4− and PF6−. A bulkier weakly coordinating organic anion, Δ-tris (tetrachloro-1,2 benzenediolato) phosphate(V) (TRISPHAT− or TRIS−), that was used in several recent reports was introduced by Veron et al. to organic salt bilayer solar cells in 2014.60 Devices with heptamethine (4)+/TRIS− (1.17 eV band gap) salt demonstrated PCE = 2.2%, Voc = 0.63 V, Jsc = 6.4 mA cm−2, and FF = 0.648. Similar devices with PF6− showed PCE = 0.9%, Voc = 0.38 V, Jsc = 3.6 mA cm−2, and FF = 0.54. PF6− devices demonstrated a wider absorption profile paired with decreased current, indicating that a lower fraction of generated excitons diffused to the acceptor/donor interface and separated to create current. The counterion's role in lowering or increasing this charge generation efficiency however was not well understood.
Previous work doping organic salt PVs with oxygen or NOBF4 led to significant improvements in efficiency, and in 2015, Jenatsch et al. utilized a p-type dopant in organic solar cells to investigate the effect of concentration and active layer thickness on device performance.61 A heptamethine (4)+/PF6− salt doped with a Co(III) complex and paired with fullerene was explored. Undoped devices demonstrated a decrease in FF and Jsc as the organic salt thickness increased while Voc initially increased before decreasing for larger thicknesses. Undoped device efficiency increased from 1.20% at 10 nm thickness to 1.23% at 20 nm before declining to 0.95% for 45 nm thick salt layers. Doped devices demonstrated slight gains and losses in FF and Voc, respectively, as the salt thickness increased. Doped PVs had decreased Voc and Jsc relative to undoped devices at the same thickness due to higher recombination losses and exciton quenching, respectively. Undoped devices were more efficient at low thicknesses, although 10 and 20 nm salt layers were the most efficient and doped cells were not fabricated at those thicknesses. Jenatsch' work with doped PVs highlights some of the difficulties of optimizing Voc, Jsc, FF, and EQE via material selection and implementation. While 20 nm undoped organic salt PVs demonstrated the best efficiency, other configurations showed higher Voc or Jsc, indicating that better efficiencies are achievable with the right materials and optimization.
Dye-sensitized solar cells, a subset of organic PVs, have demonstrated efficiencies up to 7.3% with organic salts and peak EQEs at almost 90%.62 Recent progress in utilizing organic salts in DSSCs has been limited and such devices have struggled relative to other DSSCs, such as those utilizing squaraine dyes (8.9% PCE) or a combination of squaraine and other polymethine dyes (7.6%).63,64 Furthermore, all organic salt DSSCs have trailed DSSCs utilizing Ru-complex sensitizers.64 For these reasons, organic salt DSSCs will not be discussed in depth, however, reviews of DSSC progress can be found elsewhere.64–67
While the anion was previously thought to have minimal impact on the organic salt PV, a key discovery was the systematic understanding of the importance and role of the counterion in the organic salt. Suddard et al. were the first to systematically investigate the effect of a range of counterions in organic salt based PVs as shown in Fig. 4b and 5a.68 Device architecture incorporated a heptamethine (3)+/anion− salt paired with fullerene and explored a series of anions including I−, PF6−, SbF6−, TRIS−, and tetrakis(pentafluorophenyl)borate (TPFB−). Devices with TPFB− showed the highest efficiency, demonstrating a 2.0% PCE, 0.71 V Voc, Jsc = 4.7 mA cm−2, and FF = 0.60. Fig. 5a demonstrates the drop from TPFB− to TRIS− devices in Voc from 0.71 to 0.63 V, leading to PCE = 1.7% for 3-TRIS. Short circuit current decreased to 4.4 mA cm−2 and FF increased to 0.62. The EQE peak for TPFB− devices were over 20% at 870 nm, as shown in Fig. 4b, and increased with thickness from 9 nm to 12 nm before declining. The 0.71 V Voc of the TPFB− device was near the predicted excitonic voltage limit for OPVs at that optical excitonic gap, demonstrating the potential of counterions to maximize the open circuit voltage by specifically modulating the frontier orbital level which led to an increase in the junction interface gap (reducing interface recombination). The full implications of the counterion in device performance are discussed in greater detail below.
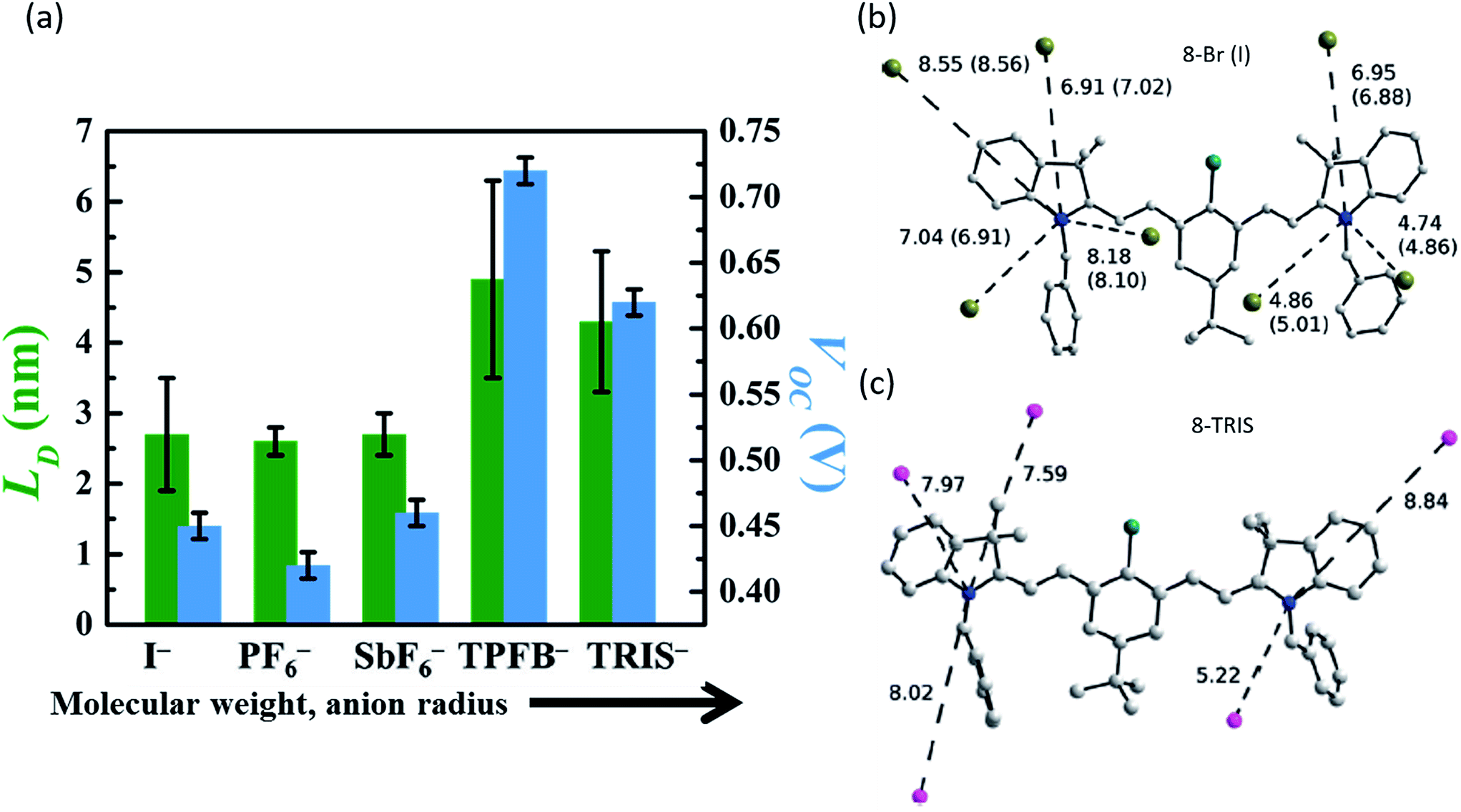 | ||
| Fig. 5 (a) Exciton diffusion length (LD) and Voc of organic salt PVs with cation 3 and a range of anions, demonstrating the variation caused by anion selection.68 Position and distances of the nearest anion neighbours for (b) small counterions I− and Br− and (c) large counterion TRIS− demonstrating bond length alteration due to cation/anion pairing.73 | ||
In 2016, Young et al. used organic salts with benzene and phenylaniline groups anchored to a heptamethine backbone with a benzo[cd]indole end group in OPVs. In that work, they demonstrated the ability to harvest energy from the deep NIR spectrum up to 1600 nm, an unprecedented range for any photoactive organic materials and a result of ultra-low 0.8 eV bandgaps as shown in Fig. 4c.69 Four different combinations of two heptamethine cations (5 and 6) paired with BF4− or TPFB− counterions demonstrated NIR photon harvesting and conversion to current with peak NIR EQEs reaching 2.1% at or past 1000 nm. Device efficiencies were limited by low Voc (max of 0.33 V) and EQEs, but nonetheless demonstrated a route to deep harvesting that is important for the highest efficiency multijunction cells and photodetectors.
Weakly coordinating anions
Many counterions used in organic salt OPVs are classified as WCAs, or non-coordinating anions, and their unique properties are important for understanding why organic salts are such an interesting and exciting class of molecules for OPVs. WCAs are used in ionic liquids, batteries, fuel cells, electrochemistry, and ion catalyzed organic reactions, among other applications.19 WCAs only weakly interact with cations, are unlikely to bind to many metal centers and thus can stabilize cations against oxidation. Some WCAs include BF4−, SbF6−, PF6−, Sb3F16−, B(CF3)4−, B(ArCF3)4−, CHB11Me5Br6−, and B12Cl11NMe3−.70 Common starting materials for WCAs include strong cationic oxidants, Bronsted acids, or metal cations. WCAs have a negative charge spread over many electronegative atoms and often exhibit larger molecular radii, allowing them to weakly interact with and stabilize reactive, unstable, and electrophilic cations.71 Superacids have been demonstrated by WCAs with hydrogen counter ions, and can protonate alkanes to produce carbocations.19,70 WCAs are exciting materials for organic salt based OPVs because of their strong stabilizing properties, allowing for the facile use of a range of cations and imparting additional tunability.Role of the counterion in organic salt photovoltaics
Early work in organic salt PVs assumed the role of the counterion was less important relative to the cation. Several studies investigated counterions and demonstrate that they actually have large effects in such devices. The first evidence for a relevant role of the counterion was found by Demchuk et al., who examined excited state relaxation times for organic salts in different media through time resolved absorption spectroscopy.72 Polar media, such as ethanol, demonstrated no sensitivity to the counterion, however, weakly polar solvents like dichloroethane demonstrated relaxation times ranging from 100 to 50 ps depending on the anion. A clear trend in anion nucleophilicity, size, etc., was not observed. Bouit et al. demonstrated that the anion could impact the structural state (ranging from asymmetric to symmetric) of the cation.73Fig. 5b demonstrates that small anions such as Br− or I− polarized the cation and caused it to take on an asymmetric dipolar structure, whereas bulkier TRIS− ions caused the heptamethine to adopt a symmetric ideal polymethine state as shown in Fig. 5c. Thus it was demonstrated that bond length alteration was impacted by counterion selection, an interesting mechanism with potential for altering optoelectronic properties. Bouit et al. concluded that counterion effects could be important and warranted further investigation.Bulavko et al. examined BHJ polymer solar cells with organic salt dopants and the effect that the anion had on photovoltaic processes.74 Counterions with low oxidation potentials contributed to the process of electron–hole generation, dissociation, and recombination through ion pairs formed by the organic salt wherein photoinduced electron transfer between the anion and cation can occur, forming radical species involved in the generation of current.
Suddard et al. systematically investigated the role of the counterion and demonstrated that anion selection led to widely varied performances in solar cells, with TPFB− achieving Voc = 0.71 V, while devices with I− showed Voc = 0.45 V (Fig. 5a and 6).68 However, Voc improvement was not proportional to anion molecular weight or radius directly but suggested it was tied to the degree of halogenation and the electronegativity of the halogens. Previous work had suggested that anions played no role in the redox potentials measured in solution by cyclic voltammetry.50 However, using photoemission spectroscopy on solid films, Suddard showed that changes in Voc stemmed from the suppression of recombination losses at the interface, a direct result of anion controlled adjustment of the highest molecular orbital by up to 1 eV. Fig. 6a, c, and e show that the energy levels in these salt systems could be finely tuned (∼0.01 eV) by chemically alloying various anions, simultaneously enabling the enhancement of Voc and EQE. This tuning approach can be exploited to optimize energy level alignment for arbitrary donor–acceptor pairings with novel and ultra-low bandgap organic salts, and was extended to several new molecules with Voc near the theoretical limit (for their bandgap) and photoresponse from 950 nm out past 1600 nm.
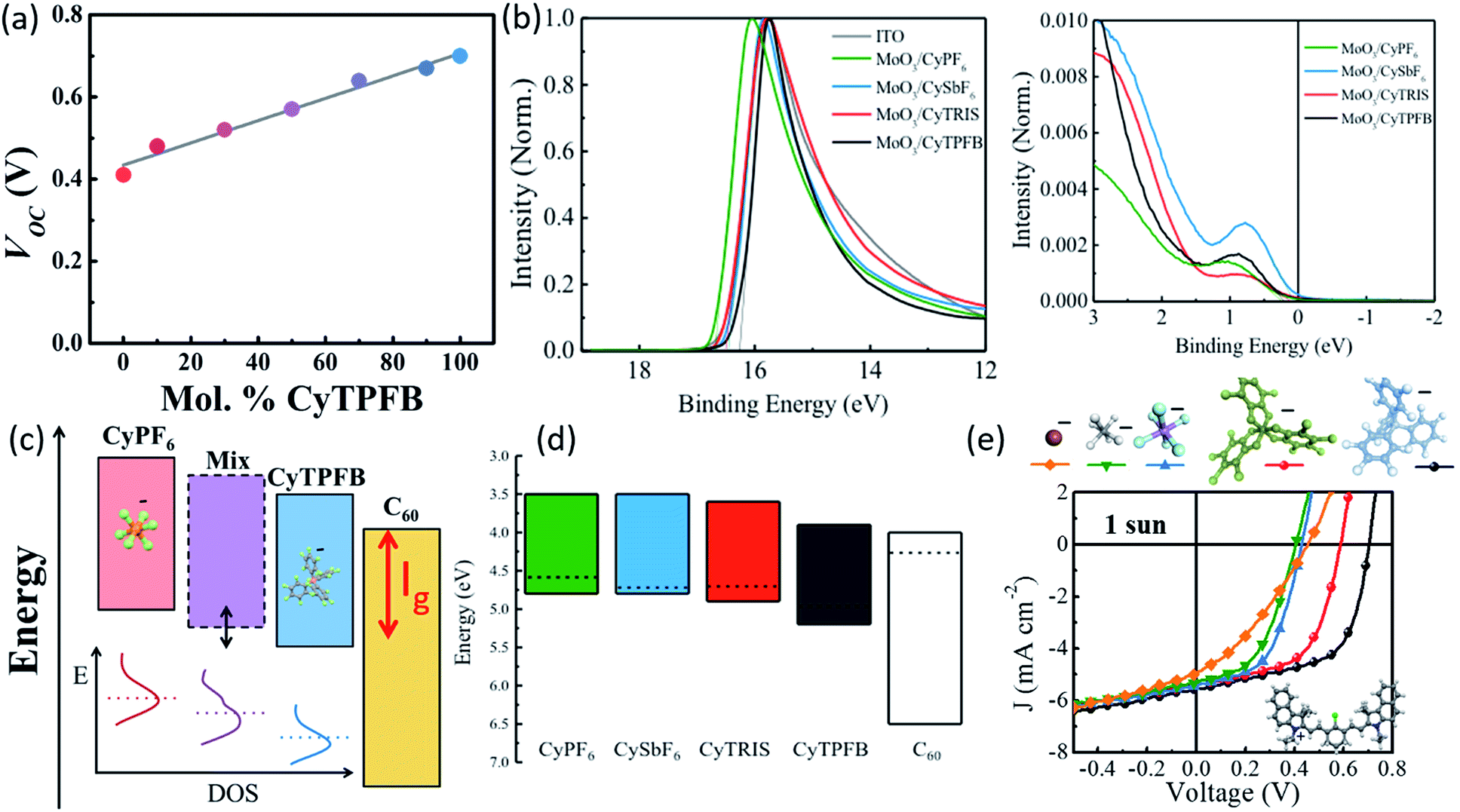 | ||
| Fig. 6 (a) Voc of organic salt thin films with varying compositions of counterions TPFB− and PF6−.68 (b) Ultraviolet photoelectron spectroscopy data for cation paired with different anions.68 (c) Schematic demonstrating improved energy alignment between the organic salt and adjacent C60 layer via counterion selection or blending.68 (d) Schematic showing energy levels for cation 3 paired with various counterions.68 (e) JV curves demonstrating the alteration of Voc for cation 3 paired with various counterions.68 | ||
The counterion clearly plays a significant role in organic salt PV performance. Indeed, organic salts are now a powerful approach to modulate the frontier orbital energy level alignment via anion exchange and blending, demonstrated in Fig. 6c and d, creating an independent chemical tuning parameter to adjust the electronic properties without fundamentally altering the cation bandgap. This tunability can help to bypass the recombination/electron-transfer tradeoff which previously hindered the smallest bandgap molecule-based photovoltaics and limited photoresponse in these materials to <1200 nm and further aid in the optimization for salts throughout the solar spectrum.
Ion mobility
A unique aspect of organic salt PVs is the presence of ions in the active layer, and the mobility of such ions has been an important field of research. Nuesch et al. compared devices with separate cations and anions to those with covalently bonded anions and cations to observe ion mobility effects.48 Absorption profiles were similar for both devices, but the C60 contribution to EQE in the ultra-violet was much larger for mobile counterion devices. Nuesch et al. attributes the greater UV EQE to electric field changes in the fullerene layer, and noted that mobile ions can lead to space charge buildup in the active layer.Benmansour et al. demonstrated that mobile ion generated space charge build up impacts photovoltaic processes.75 Application of a negative bias caused fullerene, normally an acceptor in organic salt PVs, to act as an electron donor due to the migration of Cl− counterions into the fullerene layer, demonstrating the potential of ion mobility to alter organic salt PV processes and performance. Lenes et al. used applied biases to determine the ionic space charge effect on device efficiency, demonstrating that negative biases create space charge that enhances efficiency for trimethine (12)+/PF6− and trimethine (11)+/ClO4− devices (Fig. 7a and b).76 The FF and Jsc were largely unchanged by biasing, whereas Voc decreased for positive bias and increased for negative bias. Positive biasing caused cations to accumulate at the donor/acceptor interface, raising the donor HOMO level and decreasing Voc, while negative biasing moved counterions towards the active layer interface, lowering the donor HOMO level and increasing Voc. Lenes et al. noted that ion mobility could enhance device performance and proposed future work take advantage of this unique ability in organic salt PVs. Similarly, Suddard et al. negatively biased devices out to −1.5 V and demonstrated no change in Voc for heptamethine (3)+/PF6− or heptamethine (3)+/TPFB− salts, as shown in Fig. 7c, suggesting that ion mobility was not a factor in devices with larger conjugated cations paired with bulkier anions. Fig. 7e compares the two salts, where the bulkier salt demonstrates no ion mobility effects.68 Decreased ion mobility is potentially beneficial for stable device performance, although the effect of the larger cation is not yet fully understood.
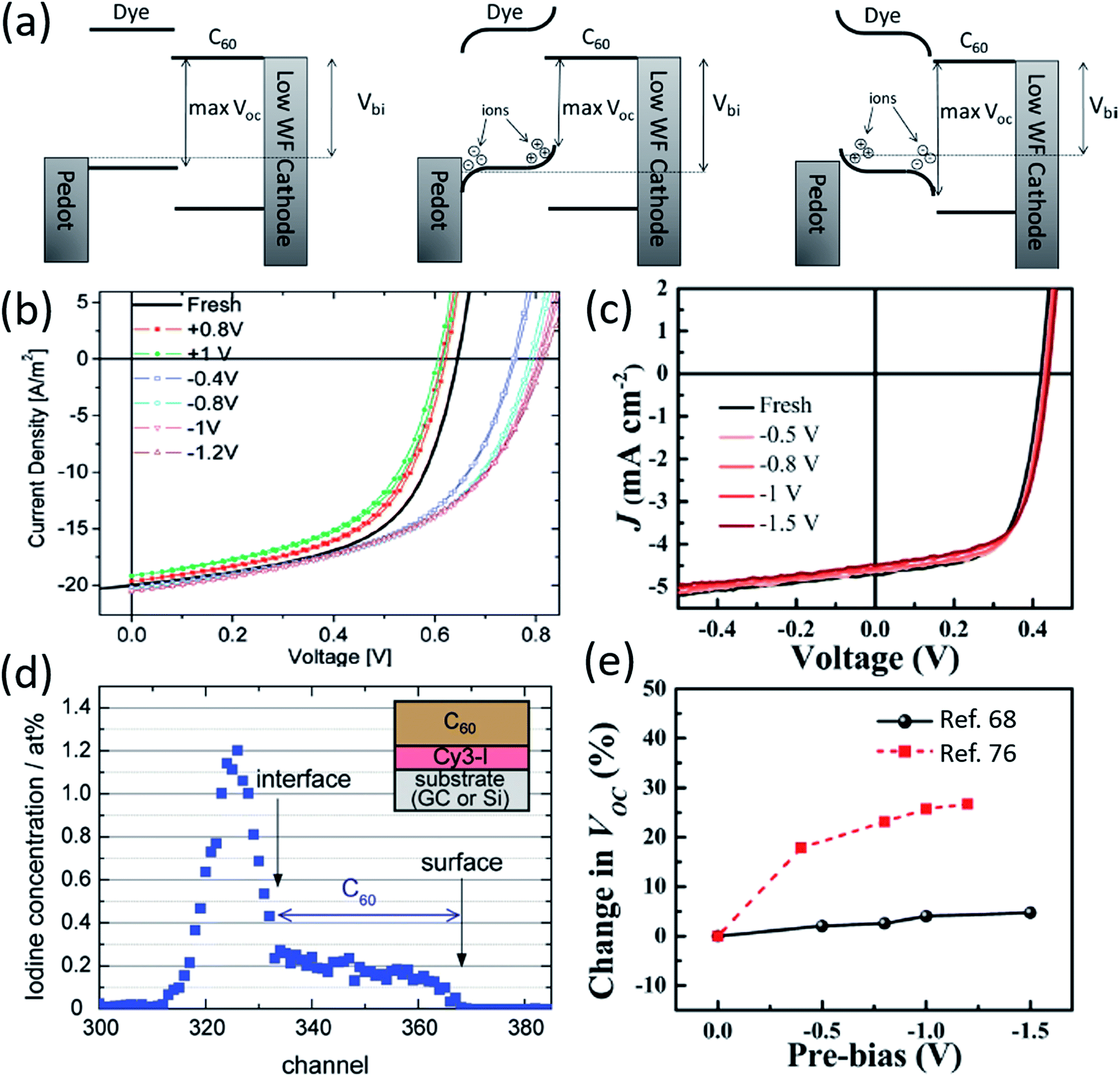 | ||
| Fig. 7 (a) Schematic demonstrating ionic space charge build-up and its impact on Voc for particular cation/anion pairings.76 (b) Data demonstrating the change in Voc for organic salt 11-ClO4 under different biasing conditions, which can create ionic space charge build-up.76 (c) Voc data for 3-PF6 under different biasing conditions, demonstrating no significant ion mobility.68 (d) Rutherford backscattering spectroscopy data for 12-I demonstrating the I− counterion diffusing through the donor layer and C60 acceptor layer.77 (e) Comparison of Voc data under biasing conditions for 11-ClO4 and 3-PF6.68 | ||
Jenatsch et al., provided recent evidence of ion migration through an organic salt PV device and monitored the potential through the fullerene layer as ions migrated.77Fig. 7d shows a profile of the counterion, I−, in regular and inverted structure devices obtained via Rutherford backscattering spectroscopy (RBS). Jenatsch et al. demonstrated significant ion diffusion into the fullerene layer in both structures, although regular architecture showed higher levels of I− in fullerene. A sharp drop off in I− occurs at the donor/acceptor interface. Jenatsch et al. provided the first data directly showing ion migration from the donor layer into the acceptor of an organic salt PV device.
Demonstrating the process and effects of ion migration and space charge buildup are important for understanding one of the fundamental differences between organic salt PVs and other small molecule organic solar cells. Ion migration has most commonly been observed with the smallest anions (e.g. I−), but is also sensitive to the cation structure. The ability to both enhance and suppress such migration may be a critical factor moving forward, and it appears that such control is at hand.
Exciton diffusion and charge carriers in organic salt photovoltaics
The photovoltaic process in bilayer and BHJ organic salt solar cells can be divided into five steps, (1) photon absorption and exciton generation by the active material, (2) exciton diffusion to the donor/acceptor interface, (3) charge transfer of electrons from the donor material and holes from the acceptor, (4) dissociation of electron and hole pairs into free charge carriers, and (5) the transfer of holes and electrons to electrodes and collection of the charge. We discuss these fundamental processes as it is key to optimizing devices and creating high efficiency organic salt PVs.Exciton diffusion
Higgins et al. used near-field scanning optical microscopy (NSOM) to demonstrate that exciton diffusion lengths (LD) were less than 50 nm in pseudoisocyanine organic salts.78 Other groups, including Suddard et al., have since reported exciton diffusion lengths for organic salt PVs on the order of 5 to 10 nm, shown in Fig. 5a, similar to many other small molecule exciton diffusion lengths.68 They demonstrated the LD was dependent on the anion, increasing from 2.5 nm for 3-I, 3-PF6, and 3-SbF6 to 5 nm, for 3-TRIS and 3-TPFB observed alongside a concomitant increase in the optimum salt thickness for the maximum quantum efficiency.68 One possible mechanism for exciton diffusion is via long range Förster transfer, wherein the exciton diffusion length is proportional to R03, the Förster radius and inversely proportional to n2, the index of refraction of the salt at the peak of the overlap integral.79 While increasing the average hopping distance by increasing the anion size, it is possible to counterbalance this effect by the reduction in the non-radiative rate for exciton quenching so that there can be a sensitive optimum.80 A second mechanism is short range (nearest neighbor hopping) Dexter energy transfer, where LD exhibits an exponential decay with separation distance and quickly becomes negligible for non-nearest neighbor interactions. Förster transfer could only explain half of the change in LD due to changes in n, and the lack of photoluminescence (PL) makes Förster transfer a questionable primary exciton diffusion mechanism. Dexter transfer is also improbable due to the larger nearest neighbor distances in heptamethine+/TPFB− or TRIS− compared to I− or PF6− salts, meaning that LD should generally decrease, not increase. Thus, understanding the mechanism of exciton diffusion remains a key unanswered question in organic salts.Given the relatively small exciton diffusion lengths reported so far, the use of bulk heterojunction (BHJ) structures with blended donor–acceptors will be a key approach to avoid exciton diffusion limitations in organic salt photovoltaics.68 Such devices would require shorter exciton diffusion to a donor/interface, increasing the number of excitons that successfully dissociate into charge carriers. Research into BHJ devices with organic salts has been limited, but warrants further investigation to understand how to control phase separation and morphology in these systems.50,53,55–57 It is likely that the thermodynamic processes that drive bulk heterojunction formation will be strongly influenced by both the anion selection and the general ionic character of the organic salts.
Charge injection
The transport and collection of free charge carriers has been improved by the introduction of interlayers and understanding of interfacial interactions. Nuesch et al. introduced a PEDOT layer that limited the injection of electrons from the organic salt donor into ITO, and subsequent generation of photocurrent, via a 1.3 eV energy barrier at that interface.47 Elimination of this loss mechanism improved current generation. Jenatsch et al. observed a hole trapping mechanism at the MoO3/organic salt interface resulting from a thin solvent layer solubilizing part of the salt and lowering charge carrier mobility.81 Trapped hole lifetimes were greater than 200 μs, making them easily distinguishable from free holes. The mechanism was only present when a polar fluorinated alcohol solvent was used, and was absent when a less polar solvent such as chlorobenzene was used. Layer interfaces and trap states for exciton dissociation and free charge carrier collection is an important design consideration for organic salt PVs.Electron transfer
Risse et al. used femtosecond pump-probe experiments to examine the photoinduced electron transfer process, observing a picosecond scale process for cyanine borate organic salt.82 The excited state charge transfer process occurs on the femtosecond timescale when the organic salt is blended with PCBM, typically an acceptor material, and charge is transferred into the PCBM. The kinetics of organic salt to PCBM electron transfer compared to intra-molecular electron transfer are important for understanding BHJ charge transfer mechanisms and rates. Devizis et al. monitored the process of free charge carrier generation and identified exciton dissociation as the rate-limiting step using ultrafast transient Stark shift spectroscopy.83 Bilayer trimethine (12)+/PF6− and fullerene thin films were subjected to an electric field and electromodulated differential absorption (EDA) on a pump-probe spectrometer was used to monitor the generation and transport of free charge carriers. Non-overlapping electroabsorption (EA) spectra of the organic salt and fullerene allowed for independent observation of electron and hole movement in each layer. Fullerene EA bands decreased with time while organic salt EA bands remained constant, indicating that electron motion in fullerene was responsible for decay in the electric field during the nanosecond probing period. Devizis et al. confirmed the orders of magnitude difference of electron mobility in pure C60 (not fullerene as an acceptor in organic salt PVs), and hole mobility in the organic salt reported by Jenatsch et al. Furthermore, Devizis et al. determined the rate limiting step in the photovoltaics process of organic salt PVs to be the escape of the electron from the Coulomb potential at the donor–acceptor interface, which lasts for hundreds of picoseconds, and potentially longer given that the applied electric field likely lowered the barrier for the electron.The exciton binding energy (EBE) is another key parameter as it defines the excess potential required at the interface to promote electron transfer and exciton dissociation. Young et al. used the tunability of the frontier energy levels with anion blending to extract the exciton binding energies of very small bandgap salts.69 By finding the energy level at which there was a sharp EQE cutoff as the HOMO was reduced, the excess energy needed to enable efficient electron transfer was estimated. Binding energies were found to vary significantly both with changes in cation structure and bandgap, where EBE = 0.55 eV for heptamethine (6) with a bandgap of 0.9 eV, and EBE = 0.40 eV for heptamethine (5) with a bandgap of 0.8 eV. The changes correlated with changes in the index of refraction, which were suggestive of changes in the dielectric constant. In this case, the binding energies limited the Voc to roughly half the interface bandgap to maintain efficient dissociation (EQE). Understanding routes to modulate the binding energy, particularly at these very small bandgaps, is a key area of exploration.
Charge carrier mobilities
Jenatsch et al. used photogenerated charge extraction by linearly increasing voltage (photo-CELIV) to determine hole mobility in a trimethine (12)+/PF6− layer.81 A light pulse generated charge carriers in the photovoltaic device, and a voltage ramp extracted the carriers. The delay time between carrier generation and the voltage ramp allows for investigation of the charge carrier recombination kinetics. Conventional methods for determining mobilities requires micron thick layers, such that organic salt based PVs cannot typically be used due to solubility limits. The calculated mobility was attributed to the organic salt because fullerene calculations saw an unreasonable order of magnitude increase in mobility for a similar increase in C60 thickness. The mobility was demonstrated to be 4 × 10−5 cm2 V−1 s−1 and was assigned to hole transport because electron mobility in C60 and PCBM in previously reported work was on the order of 1 cm2 V−1 s−1.84,85Tennakone et al. estimated electron mobilities of 8 × 10−2 cm2 V−1 s−1 in rhodamine B thiocyanate films on a copper thiocyanate surface using the Mott–Gurney equation with measured space-charge saturated current.86 To obtain the space-charge saturated current, the organic salt, rhodamine B thiocyanate was vacuum dried and pressed into a pellet between two steel electrodes with an applied DC current yielding the current density and applied voltage needed for the Mott–Gurney equation. Pitigala et al. utilized the same method to estimate hole mobility in a pentamethine+/SCN− salt at 7.2 × 10−3 cm2 V−1 s−1, two orders of magnitude higher than the mobility Jenatsch et al. reported in organic salt PVs.81,87 From both reports however, the hole mobility was low, and Jenatsch et al. demonstrated the effect of such limited hole mobility in organic salt PVs with active layer thickness dependent performance testing.61 Photovoltaic performance declined with thick (>30 nm) active layers, owing to either exciton diffusion or hole mobility limitations.
Organic salts have also demonstrated usefulness as electron transport layers (ETLs) in organic PVs.88 While organic salt ETLs do not possess the same function as organic salt active layers, this demonstration highlights the capable electron transport properties in such materials and suggests that there are at least modest electron mobilities in organic salts.
To date, however, the mobility of electrons in organic salt PVs have not been well characterized, and few works have reported hole mobility in such devices. Considering the performance of salts in the PVs described above is similar in electrical performance (series resistance, fill factors), hole mobilities are likely to be similar (at least) to other amorphous small molecule semiconductors (∼10−5 to 10−3 cm2 V−1 s−1). Given the impact that the anion can play in the frontier energy levels, work function, charge transfer efficiency, and exciton diffusion length, as shown in Fig. 5 and 6, it is then expected that the anion could play a similarly important and interesting role in the carrier transport properties. Another key factor in the magnitude of electronic properties is the well-known impact of crystalline order.89,90 Among all of the reports of organic salts used in thin films PVs there has been little evidence for any crystalline nature of thin films. Fig. 8a demonstrates two of the organic salts that we have explored, for example, have been shown to be amorphous glasses. However, we and others have reported several crystal structures from slow-solution growth, as shown in Fig. 8b–e, so that it is possible that tuning the crystalline content in films could be used to further enhance electronic properties and efficiencies.17,60,68,73,91
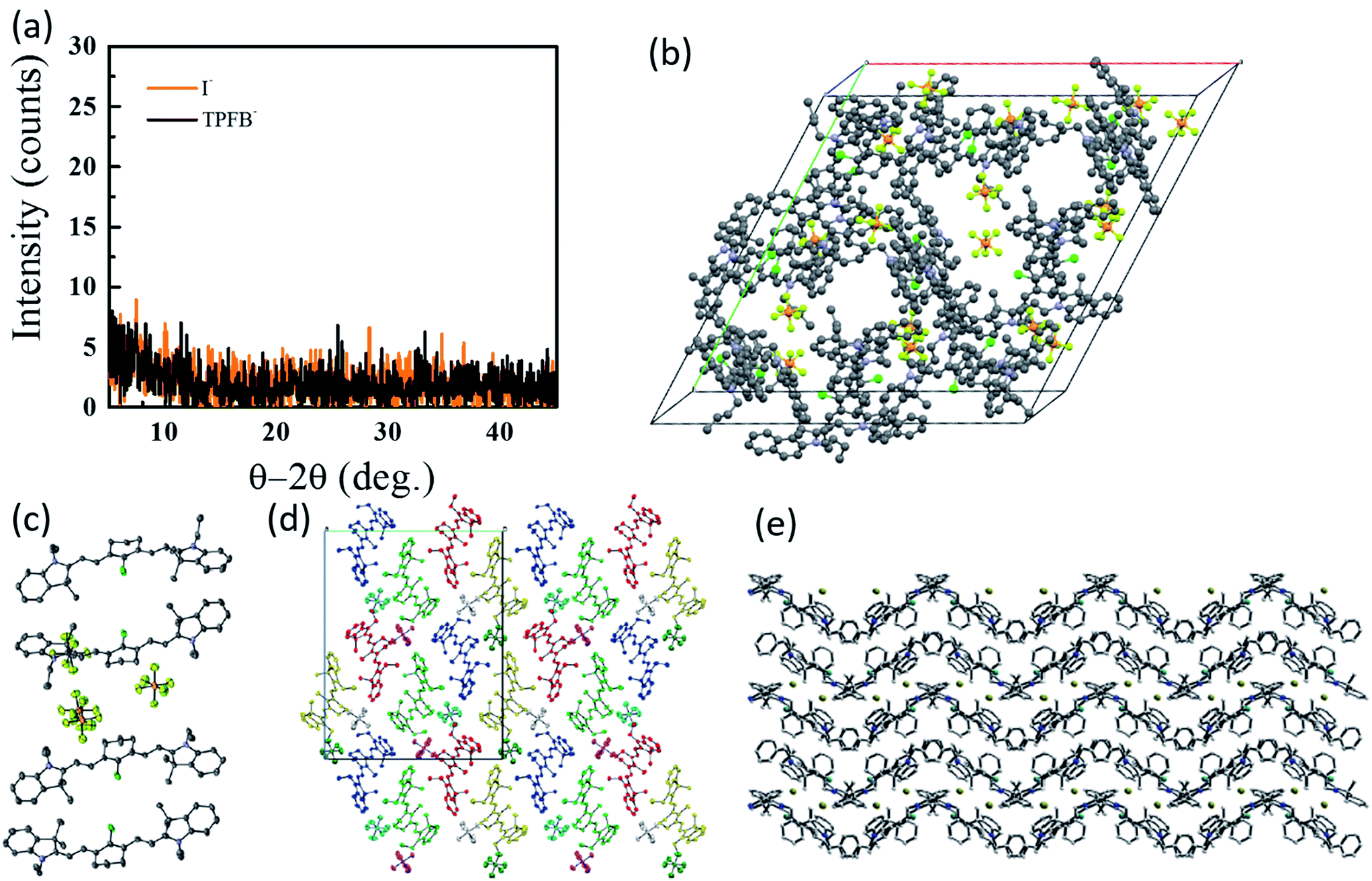 | ||
| Fig. 8 (a) Representative XRD data for 3-I and 3-TPFB demonstrating amorphous thin-film structures.68 Solved crystal structures for (b) 3-PF6,68 (c) the asymmetrical structure of 4-PF6,60 (d) 4-PF6 where colors represent equivalent symmetry,60 and (e) 8-Br.73 | ||
Organic salt transparent photovoltaics
Perhaps the most promising and exciting application of organic salts is in transparent photovoltaics (TPVs), which have emerged recently as a new class of photovoltaic devices.92,93 Due to the outstanding capabilities of organic salts to absorb selectively in the NIR spectrum, both in absorption breadth and EQE, TPVs with organic salts have demonstrated average visible transmission (AVT) as high as 70% and device efficiencies greater than 2% as shown in Fig. 9. This is approaching the highest efficiencies and transparencies reported for non-salt TPVs of 4–5%. Nonetheless, the salts have demonstrated the deepest photoresponse of any organic semiconductors and therefore could also play a role in transparent multijunction cells.94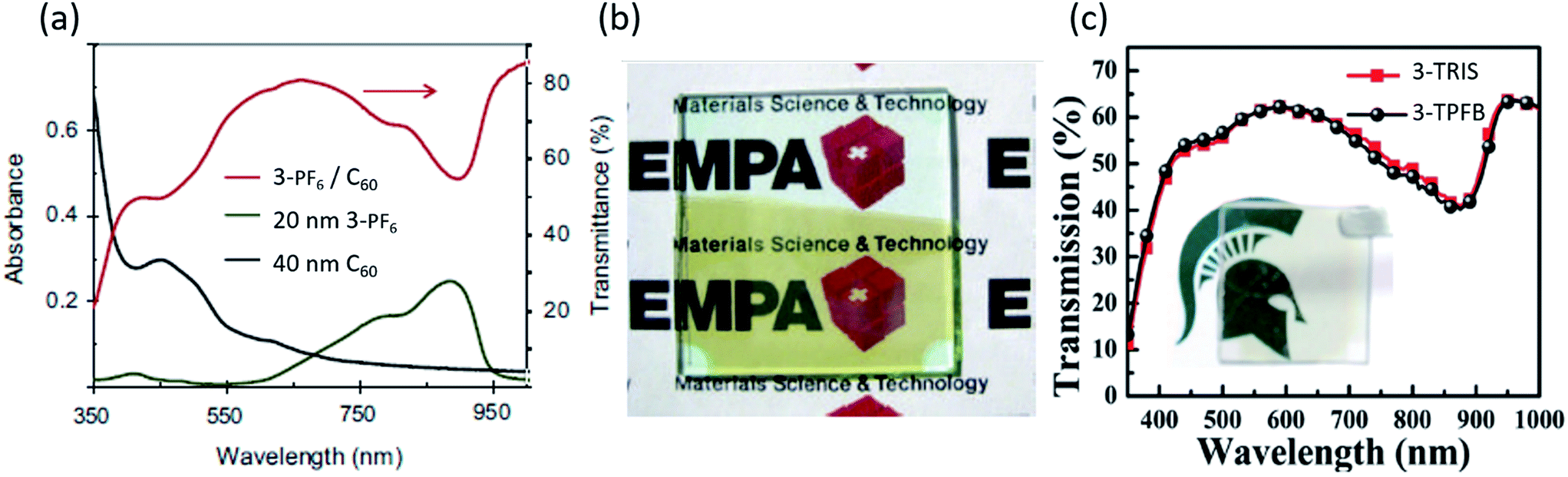 | ||
| Fig. 9 (a) Absorption and transmission spectra for transparent photovoltaic devices utilizing organic salt 3-PF6.95 (b) Picture of a transparent device represented in (a).95 (c) Transmission spectra for 3-TPFB and 3-TRIS based devices with a picture shown in the inset.68 | ||
Zhang et al. reported heptamethine (3)+/PF6− organic salt PVs with AVT for 450–670 nm of up to 67%, and observed that AVT was a strong function of cathode thickness and composition (Fig. 9a and b).95 A 1.5% efficient device with a non-transparent 100 nm Ag cathode was tested for cathodes of different thicknesses and Ag/Alq3 compositions. AVT increased from 42.8% (8 nm) to 47.9% (12 nm), and decreased to 39.2% (16 nm) and 31.0% (20 nm) for Ag cathodes. A 12/60 nm Ag/Alq3 cathode demonstrated the best AVT = 67.2% with PCE = 0.7%, Voc = 0.39 V, Jsc = 3.8 mA cm−2, and FF = 0.496. Fig. 9a and b show the absorption and transmission curves with an image of the transparent device. Simulations performed by Zhang calculated the optical field strength (E2) in the cathode layer to understand how Alq3 influenced the AVT, which depends greatly on E2 exiting the cathode and is similar to TPV work.93 Simulated 60 nm Alq3 had 81% transmittance of 568 nm light, compared to 79.8% experimentally. AVT was calculated from optical modeling data and demonstrated the highest transmittance for 12 nm Ag/60 nm Alq3 at 69.6%, in good agreement with experimental work. Minimal changes in AVT and PCE were observed for varying active layer thicknesses.
Veron et al. fabricated semitransparent OPVs with a heptamethine (4)+/TRIS− salt (1.4 eV bandgap).60 Using a Ag/Alq3 cathode, devices were demonstrated with maximum AVT = 66% from 450 to 670 nm, PCE = 2.2%, Jsc = 6.4 mA cm−2, Voc = 0.63 V, and FF = 0.54, a significant improvement from the 0.7% PCE for the heptamethine+/PF6− salt.
Suddard et al. subsequently created heptamethine (3)+/TPFB− organic salt (1.34 eV band gap) TPVs with a 2 nm Ag interfacial metal layer, 100 nm MoO3 and a 100 nm ITO cathode, demonstrating average device PCE = 0.8%, Jsc = 2.5 mA cm−2, Voc = 0.69 V, FF = 0.53 and AVT = 60.4%, and which had a voltage nearing the theoretical excitonic limit for OPVs. Fig. 9c demonstrates the transmission curve with an image of the device.68
Development of high efficiency TPVs requires energy harvesting deep into the NIR. Young et al. demonstrated organic salts with selective NIR absorption edges out to 1600 nm and NIR peak EQEs as high as 2.1%, shown in Fig. 4c.69 Absorption and photocurrent generation this deep had not been seen previously in any organic semiconductor and was thus a significant advancement in organic salt TPVs.96
Stability of organic salt photovoltaic devices
The longevity and stability of organic salt PVs are important factors in determining the potential and commercial viability of such devices. Wicht et al. studied the stability of bilayer organic salt and fullerene solar cells with both normal and inverted structures.97 Regular devices stored in the dark under N2 demonstrated a 10–20% drop off in Jsc and FF over 100 days, and a similar drop off in Voc if an Al cathode was used instead of Ag, which is less reactive because of the self-passivating nature of Al. Devices with PEDOT:PSS showed more degradation than those with MoO3 because PEDOT:PSS lowered the pH of the adjacent ITO layer, causing corrosion and allowing indium atoms to diffuse into the device. Ambient air testing demonstrated higher stability for inverted structures due to the increased protection provided for fullerene against O2, which can be reactive with fullerene but must first diffuse through the organic salt in the inverted structure. Organic salt thin films demonstrated a constant absorption spectra upon exposure to ambient conditions, showing photostability. Stress testing under heat and light demonstrated 30 to 60% degradation of Voc, FF, and Jsc in 17 days, while PCE stabilized at 15% of fresh devices. The T80 after a 5 day burn-in was 72 hours, compared to 10 hours for fresh devices.In addition to developing organic salt TPVs, Zhang et al. examined the stability of their heptamethine (3)+/PF6− based devices.95 A device with a 50 nm Ag cathode demonstrated the best protection against degradation for Voc, Jsc, and FF in the dark under N2. Devices with a 50 nm Ag cathode layer under 1 sun and N2 showed a 20% decrease in FF and Jsc, a slight drop off in Voc, and an overall 40% degradation in PCE in 100 hours, with a T80 of 30 hours.
Wicht et al. demonstrated that illumination can cause rapid degradation for particular cation/anion organic salt PVs, and mechanisms for this photodegradation were evaluated by Zhang et al.97,98 Trimethine (12)+/PF6− films showed no changes in absorption, confirming the photostability of organic salts noted by Wicht et al. MoO3 and C60 degradations were isolated via layer by layer 24 hour stress testing in devices and MoO3/C60 bilayer selective excitation. MoO3 photodegradation to Mo5+ accounted for the Voc decline and half of the Jsc decrease within 24 hours. Upon degradation to Mo5+ the work function changes from 5.6 eV to 5.3 eV, altering the energetics and hole extraction efficiency at the salt/MoO3 interface.
It is expected that different cation/anion pairings could play an important role in variations in the degradation effects due to the changing interface energetics. Indeed, recent work in our lab has shown promising lifetimes for organic salt PVs based on continuous AM1.5G illumination with maximum extrapolated lifetimes of years with little change in quantum efficiency (that will be published elsewhere). Nonetheless, the stability of organic salts as active layers remains an important criterion to enable organic salt PVs to become commercially viable.
Conclusion
Organic salts are an emerging class of active layers for organic photovoltaics and have demonstrated outstanding tunability in the structural, optical, and electronic properties. We have reviewed the historical development of such devices and discussed the exciting features organic salts bring to organic photovoltaics, including transparent devices, long lifetimes, ultra-deep NIR harvesting, and facile adjustment of energy levels. Most of the previous research in this field has focused on planar devices. Understanding thermodynamically-driven phase separations in organic salt donor–acceptor blends will be an important area of future investigation to enable the fabrication of high efficiency bulk heterojunction architectures. Future research should also focus on understanding the mechanisms of energy transport, the limitations of the binding energies, what role the ionic character has in exciton dissociation and migration, and how deep organic salts can absorb into the NIR. The development of solution processable organic salts with highly tunable properties can ultimately enable opportunities to approach the excitonic efficiency limit for OPVs while opening up new prospects for low cost transparent solar cells, photodetectors, and multijunction cells.Acknowledgements
Financial support for this work was provided by the National Science Foundation (CBET-1254662; and CBET-1511098), and by the American Physical Society (Stanford R. Ovshinsky Sustainable Energy Fellowship Award).Notes and references
- R. Hany, B. Fan, F. Castro, J. Heier, W. Kylberg and F. Nuesch, Prog. Photovoltaics Res. Appl., 2011, 19, 851 CrossRef CAS.
- X. Chen, J. Guo, X. Peng, M. Guo, Y. Xu, L. Shi, C. Liang, L. Wang, Y. Gao, S. Sun and S. Cai, J. Photochem. Photobiol., A, 2005, 171, 231 CrossRef CAS.
- B. Feng, D. Wan, L. Yan, V. Kadam, J. You and G. Gao, RSC Adv., 2016, 6, 66407 RSC.
- T. Geiger, H. Benmansour, B. Fan, R. Hany and F. Nuesch, Macromol. Rapid Commun., 2008, 29, 651 CrossRef CAS.
- B. Jedrzejewska and B. Michalak, Color. Technol., 2011, 127, 288 CAS.
- J. Lenhard, B. Hein and A. Muenter, J. Phys. Chem., 1993, 97, 8269 CrossRef CAS.
- M. Bedics, K. Mulhern, D. Watson and M. Detty, J. Org. Chem., 2013, 78, 8885 CrossRef CAS PubMed.
- W. Zhan, W. Wu, J. Hua, Y. Jing, F. Meng and H. Tian, Tetrahedron Lett., 2007, 48, 2461 CrossRef CAS.
- J. Lim, O. Przhonska, S. Khodja, S. Yang, T. Ross, D. Hagan, E. Van Stryland, M. Bondar and Y. Slominsky, Chem. Phys., 1999, 245, 79 CrossRef CAS.
- K. Funabiki, K. Yagi, M. Ueta, M. Nakajima, M. Horiuchi, Y. Kubota and M. Mastui, Chem.–Eur. J., 2016, 22, 12282 CrossRef CAS PubMed.
- A. Tatikolov, A. Ishchenko, S. Ghelli and G. Ponerini, Mater. Struct., 1998, 471, 145 CAS.
- W. Zhan, Y. Jin, X. Teng and H. Tian, Res. Chem. Intermed., 2008, 34, 229 CrossRef CAS.
- J. Bricks, A. Kachkovskii, Y. Slominskii, A. Gerasov and S. Popov, Dyes Pigm., 2015, 121, 238 CrossRef CAS.
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS.
- M. Henary and A. Levitz, Dyes Pigm., 2013, 99, 1107 CrossRef CAS.
- A. Mishra, R. Behera, P. Behera, B. Mishra and G. Behera, Chem. Rev., 2000, 100, 1973 CrossRef CAS PubMed.
- P. Bouit, E. Plazza, S. Rigaut, B. Guennic, C. Aronica, L. Toupet, C. Andraud and O. Maury, Org. Lett., 2008, 19, 4159 CrossRef PubMed.
- C. Li, C. Chueh, F. Ding, H. Yip, P. Liang, X. Li and A. Jen, Adv. Mater., 2013, 25, 4425 CrossRef CAS PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- S. Jenatsch, L. Wang, M. Bulloni, A. Veron, B. Ruhstaller, S. Altazin, F. Nuesch and R. Hany, ACS Appl. Mater. Interfaces, 2016, 8, 6554 CAS.
- C. Bertolino, G. Caputo, C. Barolo, G. Viscardi and S. Coluccia, J. Fluoresc., 2006, 16, 221 CrossRef PubMed.
- H. Jia, Y. Liv, S. Wang, D. Sun and L. Wang, RSC Adv., 2015, 5, 4681 RSC.
- A. Ellis, C. Mason, H. Lee, L. Strekowski, G. Patonay, H. Choi and J. Yang, Talanta, 2002, 56, 1099 CrossRef CAS PubMed.
- J. Flanagan, S. Khan, S. Menchen, S. Soper and R. Hammer, Bioconjugate Chem., 1997, 8, 751 CrossRef CAS PubMed.
- W. Zhan, H. Barnhill, K. Sivakumar, H. Tian and Q. Wang, Tetrahedron Lett., 2005, 46, 1691 CrossRef CAS.
- F. Hu, M. Cao, Z. Xu, J. Huang, D. Wu, W. Yang, S. Liu and J. Yin, RSC Adv., 2015, 5, 5982 RSC.
- H. Mustroph, M. Stollenwerk and V. Bressau, Angew. Chem., Int. Ed., 2006, 45, 2016 CrossRef CAS PubMed.
- R. Mujumdar, L. Ernst, S. Mujumdar, C. Lewis and A. Waggoner, Bioconjugate Chem., 1993, 4, 105 CrossRef CAS PubMed.
- N. Siraj, F. Hasan, S. Das, L. Kiruri, K. Gall, G. Baker and I. Warner, J. Phys. Chem., 2014, 118, 2312 CAS.
- L. Strekowski, M. Lipowska and G. Patonay, J. Org. Chem., 1992, 57, 4580 CrossRef.
- T. Osedach, A. Lacchetti, R. Lunt, T. Andrew, P. Brown, G. Akselrod and V. Bulovic, Appl. Phys. Lett., 2012, 101, 113303 CrossRef.
- Y. Zhao, G. Meek, B. Levine and R. Lunt, Adv. Opt. Mater., 2014, 2, 606 CrossRef CAS.
- B. Regmi, W. Galpothdeniya, N. Siraj, M. Webb, N. Speller and I. Warner, Sens. Actuators, B, 2015, 209, 172 CrossRef CAS.
- R. Lunt, T. Osedach, P. Brown, J. Rowehl and V. Bulovic, Adv. Mater., 2011, 23, 5712 CrossRef CAS PubMed.
- M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123 CrossRef CAS PubMed.
- J. Tisserant, R. Bronnimann, R. Hany, S. Jenatsch, F. Nuesch, R. Mezzenga, G. Bona and J. Heier, ACS Nano, 2014, 8, 10057 CrossRef CAS PubMed.
- J. Lee, X. Wang, H. Luo, G. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279 CrossRef CAS PubMed.
- A. Hains, Z. Liang, M. Woodhouse and B. Gregg, Chem. Rev., 2010, 110, 6689 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- A. Ghosh and T. Feng, US Pat. 4,127,738, 1978.
- G. Stepanova, L. Volkova, M. Gainullina and F. Yumakulova, Zh. Fiz. Khim., 1977, 51, 1771 CAS.
- D. Whorle and D. Meissner, Adv. Mater., 1991, 3, 129 CrossRef.
- A. Jana, J. Photochem. Photobiol., A, 2000, 132, 1 CrossRef CAS.
- F. Meng, K. Chen, H. Tian, L. Zuppiroli and F. Nuesch, Appl. Phys. Lett., 2003, 21, 3788 CrossRef.
- M. Kawasaki and S. Aoyama, Chem. Commun., 2004, 988 RSC.
- F. Nuesch, G. Tornare, L. Zuppiroli, F. Meng, K. Chen and H. Tian, Sol. Energy Mater. Sol. Cells, 2005, 87, 817 CrossRef.
- F. Nuesch, A. Faes, L. Zuppiroli, F. Meng, K. Chen and H. Tian, J. Mater. Sci., 2005, 40, 1353 CrossRef.
- F. Meng, J. Hua, K. Chen, H. Tian, L. Zuppiroli and F. Nuesch, J. Mater. Chem., 2005, 15, 979 RSC.
- F. Castro, A. Faes, T. Geiger, C. Graeff, M. Nagel, F. Nuesch and R. Hany, Synth. Met., 2006, 156, 973 CrossRef CAS.
- B. Fan, R. Hany, J. Moser and F. Nuesch, Org. Electron., 2008, 9, 85 CrossRef CAS.
- B. Fan, F. Castro, J. Heier, R. Hany and F. Nuesch, Org. Electron., 2010, 11, 583 CrossRef CAS.
- P. Bouit, D. Rauh, S. Neugebauer, J. Delgado, E. Piazza, S. Rigaut, O. Maury, C. Andraud, V. Dyakonov and N. Martin, Org. Lett., 2009, 11, 4806 CrossRef CAS PubMed.
- B. Fan, F. Castro, B. Chu, J. Heier, D. Opris, R. Hany and F. Nuesch, J. Mater. Chem., 2010, 20, 2952 RSC.
- E. Berner, T. Jager, T. Lanz, F. Nuesch, J. Tisserant, G. Wicht, H. Zhang and R. Hany, Appl. Phys. Lett., 2013, 102, 183903 CrossRef.
- O. Malinkiewicz, T. Grancha, A. Molina-Ontoria, A. Soriano, H. Brine and H. Bolink, Adv. Energy Mater., 2013, 3, 472 CrossRef CAS.
- C. Yap, M. Yahaya and M. Salleh, Sol. Energy, 2011, 85, 95 CrossRef CAS.
- N. Sabri, C. Yap, M. Yahaya and M. Salleh, J. Mater. Sci.: Mater. Electron., 2013, 24, 2183 CrossRef CAS.
- L. Wang, C. Hinderling, S. Jenatsch, F. Nuesch, D. Rentsch, R. Steim, H. Zhang and R. Hany, Polymer, 2014, 55, 3195 CrossRef CAS.
- A. Veron, H. Zhang, A. Linden, F. Nuesch, J. Heier, R. Hany and T. Geiger, Org. Lett., 2014, 16, 1044 CrossRef CAS PubMed.
- S. Jenatsch, T. Geiger, J. Heier, C. Kirsch, F. Nuesch, A. Paracchino, D. Rentsch, B. Ruhstaller, A. Veron and R. Hany, Sci. Technol. Adv. Mater., 2015, 16, 035003 CrossRef PubMed.
- J. Zhao, X. Yang, M. Cheng, S. Li, X. Wang and L. Sun, J. Mater. Chem., 2013, 1, 2441 RSC.
- F. Jradi, X. Kang, D. O'Neil, G. Pajares, Y. Getmanenko, P. Szymanski, T. Parker, M. El-Sayed and S. Marder, Chem. Mater., 2015, 27, 2480 CrossRef CAS.
- F. Jradi, D. O'Neil, X. Kang, J. Wong, P. Szymanski, T. Parker, H. Anderson, M. El-Sayed and S. Marder, Chem. Mater., 2015, 27, 6305 CrossRef CAS.
- D. Saccone, S. Galliano, N. Barbero, P. Quagliotto, G. Viscardi and C. Barolo, Eur. J. Org. Chem., 2016, 2244 CrossRef CAS.
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS.
- S. Singh and G. Sharma, Chem. Rec., 2014, 14, 419 CrossRef CAS PubMed.
- J. Suddard-Bangsund, C. Traverse, M. Young, T. Patrick, Y. Zhao and R. Lunt, Adv. Energy Mater., 2015, 1501659 Search PubMed.
- M. Young, J. Suddard-Bangsund, T. Patrick, N. Pajares, C. Traverse, M. Barr, S. Lunt and R. Lunt, Adv. Opt. Mater., 2016, 1, 1 Search PubMed.
- T. Engesser, M. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC.
- K. Landskron, Chem.–Eur. J., 2015, 21, 14258 CrossRef CAS PubMed.
- M. Demchuk, A. Ishchenko, V. Mikhailov and V. Avdeeva, Chem. Phys. Lett., 1988, 1, 99 CrossRef.
- P. Bouit, C. Aronica, L. Toupet, B. Guennic, C. Adraud and O. Maury, J. Am. Chem. Soc., 2010, 132, 4328 CrossRef CAS PubMed.
- G. Bulavko, N. Davidenko, N. Derevyanko and A. Ishchenko, High Energy Chem., 2015, 5, 331 CrossRef.
- H. Benmansour, A. Castro, M. Nigel, J. Heier, R. Hany and F. Nuesch, Chimia, 2007, 61, 787 CrossRef CAS.
- M. Lenes and H. Bolink, ACS Appl. Mater. Interfaces, 2010, 12, 3664 Search PubMed.
- S. Jenatsch, J. Groenewold, M. Dobeli, R. Hany, R. Crockett, J. Lubben, F. Nuesch and J. Heier, Adv. Mater. Interfaces, 2017, 1600891 CrossRef.
- D. Higgins, P. Reid and P. Barbara, J. Phys. Chem., 1996, 100, 1174 CrossRef CAS.
- R. Lunt, N. Giebink, A. Belak, J. Benziger and S. Forrest, J. Appl. Phys., 2009, 105, 053711 CrossRef.
- T. Mullenbach, K. McGarry, W. Luhman, C. Douglas and R. Holmes, Adv. Mater., 2013, 25, 3689 CrossRef CAS PubMed.
- S. Jenatsch, R. Hany, A. Veron, M. Neukom, S. Zufle, A. Borgschulte, B. Ruhstaller and F. Nuesch, J. Phys. Chem., 2014, 118, 17036 CAS.
- J. Risse, J. Heier, F. Nuesch and J. Moser, J. Mater. Chem. A, 2015, 3, 10935 Search PubMed.
- A. Devizis, J. Risse, R. Hany, F. Nuesch, S. Jenatsch, V. Gulbinas and J. Moser, J. Am. Chem. Soc., 2015, 137, 8192 CrossRef CAS PubMed.
- B. Singh, N. Marjanovic, G. Matt, S. Gunes, N. Sariciftci, A. Ramil, A. Andreev, H. Sitter, R. Schwodiauer and S. Bauer, Org. Electron., 2005, 6, 105 CrossRef.
- D. Anthopoulos, B. Singh, N. Marjanovic, N. Sariciftci, A. Ramil, H. Sitter, M. Colle and D. Leeuw, Appl. Phys. Lett., 2006, 89, 213504 CrossRef.
- K. Tennakone, P. Pitigala and A. Perera, RSC Adv., 2013, 3, 2770 RSC.
- P. Pitigala, M. Henary, E. Owens, A. Perera and K. Tennakone, J. Photochem. Photobiol., A, 2016, 325, 39 CrossRef.
- S. Berny, A. Topley, P. Tiwana, T. Cull, M. Carrasco-Orozco and N. Blouin, International Patent, WO2013167224, PCT/EP2013/001078, 2013.
- N. Karl, K. Kraft, J. Marktanner, M. Munch, F. Schatz, R. Stehle and H. Uhde, J. Vac. Sci. Technol., A, 1999, 17, 2318 CAS.
- N. Karl, Synth. Met., 2003, 133, 649 CrossRef.
- P. Bouit, C. Aronica, L. Guy, A. Martinez, C. Andraud and O. Maury, Org. Biomol. Chem., 2009, 7, 3086 CAS.
- R. Lunt and V. Bulovic, Appl. Phys. Lett., 2011, 98, 113305 CrossRef.
- R. Lunt, Appl. Phys. Lett., 2012, 101, 043902 CrossRef.
- C. Traverse, R. Pandey, M. Barr and R. Lunt, 2016, submitted.
- H. Zhang, G. Wicht, C. Gretener, M. Nigel, F. Nuesch, Y. Romanyuk, J. Tisserant and R. Hany, Sol. Energy Mater. Sol. Cells, 2013, 118, 157 CrossRef CAS.
- J. Zimmerman, V. Diev, K. Hanson, R. Lunt, E. Yu, M. Thompson and S. Forrest, Adv. Mater., 2010, 22, 2780 CrossRef CAS PubMed.
- G. Wicht, S. Bucheler, M. Dietrich, T. Jager, F. Nuesch, T. Offermans, J. Tisserant, L. Wang, H. Zhang and R. Hany, Sol. Energy Mater. Sol. Cells, 2013, 117, 585 CrossRef CAS.
- H. Zhang, A. Borgschulte, F. Castro, R. Crockett, A. Gerecke, O. Deniz, J. Heier, S. Jenatsch, F. Nuesch, C. Sanchez-Sanchez, A. Zoladek-Lemanczyk and R. Hany, Adv. Energy Mater., 2015, 5, 1400734 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |