
H2/O2 enzymatic fuel cells: from proof-of-concept to powerful devices
I.
Mazurenko
,
X.
Wang
,
A.
de Poulpiquet
and
E.
Lojou
 *
*
Aix Marseille Univ, CNRS, Bioénergétique et Ingenierie des Protéines, 31 Chemin Joseph Aiguier, 13009 Marseille, France. E-mail: lojou@imm.cnrs.fr
First published on 9th June 2017
Abstract
Intensive research during the last 15 years on mechanistic understanding of hydrogenases, the key enzyme for H2 transformation in many microorganisms, has authorized the concept of green energy production through H2/O2 enzymatic fuel cells (EFCs), in which enzymes are used as biodegradable and bioavailable biocatalysts. More recently, great effort has been put in the improvement of the interfacial electron transfer process between the enzymes and high surface area conductive materials in order to shift from a proof-of-concept to a usable power device. Herein, we analyze the main issues that have been addressed during the last 5 years to make this breakthrough. After a brief introduction on the structure of hydrogenases and bilirubin oxidases, a widely used enzyme for O2 reduction, we compare their activity with that of platinum. We introduce the first H2/O2 EFCs and discuss their main limitations mainly related to the sensitivity of hydrogenases to O2 and oxidative potentials. We then review the discovery of new enzymes in the biodiversity and the advances in the control of the functional immobilization of these enzymes on electrodes that have permitted to overcome these limitations. We finally present all the reported H2/O2 EFCs, with a critical discussion on the perspectives of such devices.
 I. Mazurenko, X. Wang, A. de Poulpiquet and E. Lojou | Elisabeth Lojou is a senior researcher in the Bioenergetic and Protein Engineering Laboratory (Aix Marseille University, CNRS) in France. Her group develops research in the domain of fundamental bioelectrochemistry, with a special interest in the electron transfer between functionalized nanostructured electrodes and redox enzymes, such as hydrogenases and multicopper oxidases. Since 5 years, it is engaged in the design of H2/O2 enzymatic fuel cells. From left to right: Xie Wang is a biochemist working as a second year PhD student on the energy metabolic chain of an acidophilic bacterium; Ievgen Mazurenko is a post-doctoral researcher. He obtained his joint PhD degree from the University of Lorraine (Nancy, France) and the Taras Shevchenko National University of Kyiv (Ukraine) in 2013. His research interests involve electrochemistry, nanomaterials and immobilized biomolecules with a view to designing various biodevices for energy and substance conversion; Elisabeth Lojou; Anne de Poulpiquet obtained her PhD degree at Aix-Marseille University (France) under the direction of Dr Elisabeth Lojou. After a postdoctoral at the Institute of Molecular Sciences in Bordeaux (France), she joined the Bioenergetic and Protein Engineering Laboratory as an assistant professor. Her research interests focus on coupling electrochemistry to optical techniques to study the electro-enzymatic reactivity. |
Introduction
Two years ago, within COP21, 195 countries adopted a first universal deal aiming at maintaining the global average temperature increase below 2 °C (source: https://unfccc.int/resource/docs/2015/cop21/eng/10a01.pdf). This ambition requires a transition from non-renewable carbon-based energy sources to clean and low-carbon energy sources. The emergence of renewable energy sources such as solar and wind ones also induces the need of sustainable energy carriers that would allow to deliver and store this energy notably during intermittent production. Hydrogen meets this requirement, presenting at the same time high energy density and harmless water generation upon reaction with abundant oxygen. Hydrogen can be produced from clean energy sources such as biomass, and is transportable either in a liquid or gas state, or combined with nanomaterials. H2 high energy conversion efficiency in fuel cells (above 60%) is one last applicative interest of this fuel. With all these advantages, is the competition with Li batteries the unique reason for slow development of H2-based fuel cells, especially for transportation? One drawback of proton exchange membrane fuel cells (PEMFCs) is that they rely on the requirement for catalysts, mainly based on noble metals such as platinum, to accelerate the reactions of H2 oxidation and O2 reduction. Pt use encounters availability, economic and political issues. During the last few decades, active research thus focused towards the decrease or even the replacement of Pt in fuel cells. Can we imagine enzymes doing the job? In microorganisms, enzymes are biocatalysts that very efficiently convert various substrates, e.g. to sustain microorganism energy. The identification, then purification, and finally immobilization on electrodes of some of these enzymes led to the concept of enzymatic fuel cells (EFCs), which would operate as PEMFCs, but with biocatalysts harboring only earth-abundant metals and offering a larger panel of available fuels and oxidants1 (Fig. 1). Actually, EFCs based on substances largely available in physiological fluids, such as glucose and O2, associated with the specific enzymes, i.e. glucose oxidase and multicopper proteins such as bilirubin oxidase (BOD), were the most developed systems since their proof of concept in 1964.2–4 Powering of medical devices was targeted with implanted EFCs, or more recently with systems disposed on lenses or on patch applications.5–9 Interest in the development of H2/O2 EFCs is much more recent, and motivated by the expected high energy output and applications in the powering of portable electronics.10,11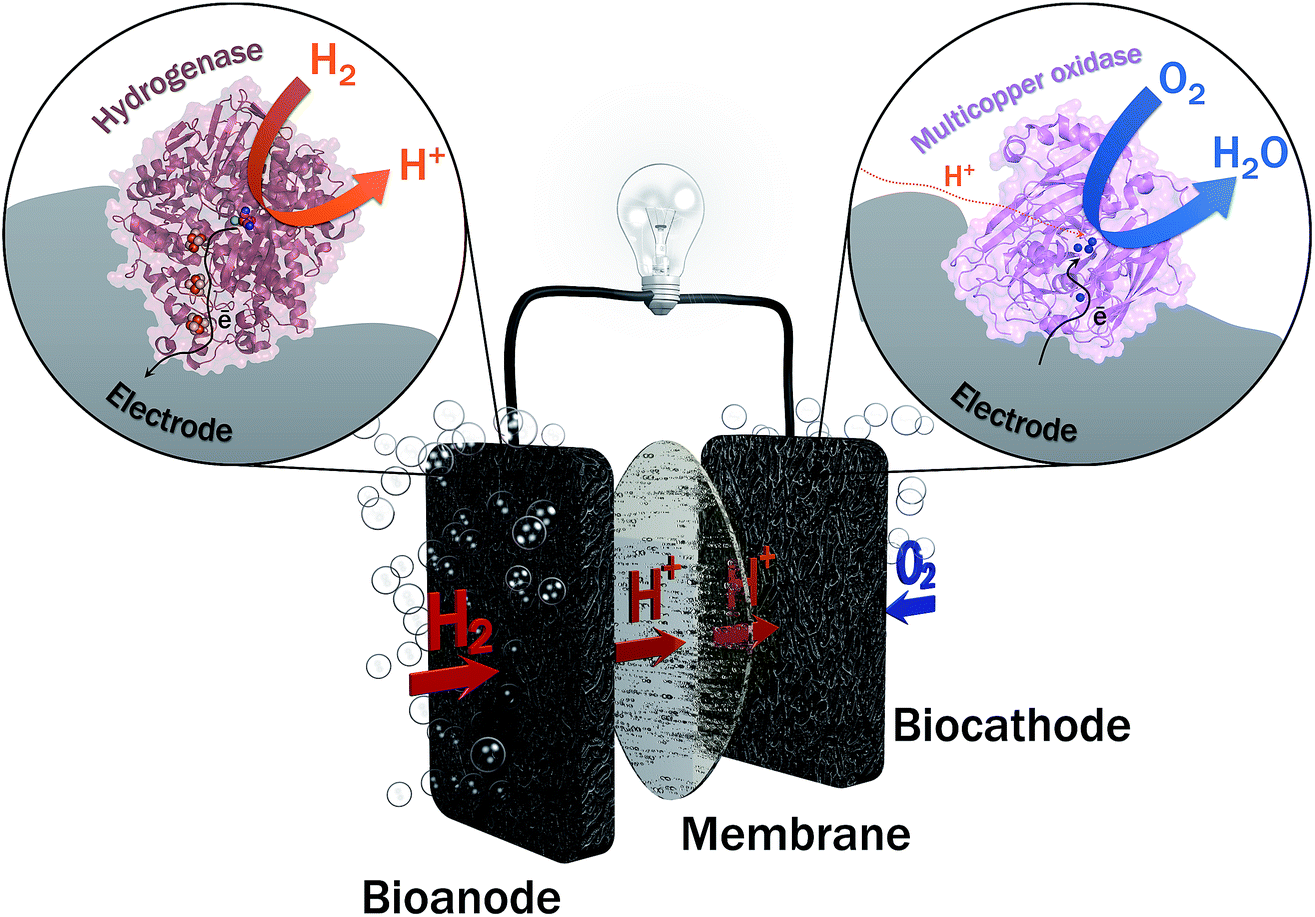 | ||
| Fig. 1 General scheme of the H2/O2 EFC. At the anode hydrogenase is the enzyme for H2 oxidation. Multicopper oxidase such as BOD reduces O2 into water at the cathode. A proton exchange membrane eventually separates the two compartments. | ||
In H2/O2 EFCs, hydrogenase, first recognized in 1931, is the key enzyme for H2 oxidation at the anode.12 However, H2/O2 EFCs were developed late compared with glucose/O2 EFCs, mainly because of the extreme oxygen sensitivity of most hydrogenases. Extensive research during the last decade towards the understanding of O2 sensitivity of hydrogenases and the discovery of new hydrogenases presenting outstanding properties including O2 tolerance and CO resistance, nowadays permit the envisioning of the development of H2/O2 EFCs.
This literature survey provides an excellent opportunity to emphasize the most recent improvements and breakthroughs in the knowledge of the involved enzymes in H2/O2 EFCs, in rational use of (nano)materials as the enzyme host matrix, in understanding of the interactions between these materials and enzymes required for fast electron transfer, and then in the resulting high power output of the related devices. It also allows to identify the remaining bottlenecks and future fundamental research directions required in this domain to fulfill the application requirements.
The proof-of-concept
The first H2/O2 enzyme-based fuel cell was not an entirely enzymatic one. Nevertheless, it was an initial proof that electricity could be generated by microorganisms presenting H2 metabolism, opening the way for a true H2/O2 EFC. Actually, Tsujimura et al.13 reported in 2001, i.e. 70 years after the discovery of hydrogenase activity in bacteria, a fuel cell based on sulfate-reducing cells at the anode and BOD at the cathode, both deposited on 0.22 cm3 carbon felts. Methyl viologen (MeV) and 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) redox mediators were used to mediate H2 oxidation and O2 reduction respectively. The fuel cell operated at 1.0 V with a current of 0.9 mA, exhibiting the highest open circuit voltage (OCV) ever reported for EFCs of 1.17 V. In this hybrid bacterial/enzymatic H2/O2 fuel cell, the anode activity towards H2 oxidation was suggested to be linked to the hydrogenase content of the periplasmic space of Desulfovibrio vulgaris Hildenborough (DvH). In our group, we further demonstrated that DvH was able to communicate with a pyrolytic graphite electrode (PG) in the presence of MeV in solution, exhibiting H2 oxidation or proton reduction activity depending on pH.14 The activity was measured with whole cells and periplasmic, membrane or cytoplasmic fractions, where different hydrogenases were identified. The highest contribution of the periplasmic hydrogenase in the overall catalytic current, and more specifically in the H2 uptake direction was proposed.15 Actually, H2 metabolism is widespread in many biotopes and in very different microorganisms. It is supported by the presence of many various hydrogenases. The occurrence of microorganisms gaining energy from H2 in air early suggested that some hydrogenases could even operate at high O2 concentrations.16 Hydrogenases differ in the metal content of the active site, which can be composed of Fe only, di-Fe or Ni and Fe atoms.17,18 Most often both [FeFe]- and [NiFe]-hydrogenases are identified in a given cell. They are located in different spaces and present a bias toward H2 uptake or evolution. Some of them have been recognized to be bidirectional and able to perform electron bifurcation to harness energy.19 In Desulfovibrio fructosovorans (Df), another sulfate-reducing bacterium, 6 different hydrogenases were identified in the genome! This indicates a complex but very efficient machinery that may be exploited in EFCs.Turning from bacterial cells to enzymes
Among the three classes of hydrogenases, i.e. [FeFe], [NiFe] and [Fe] hydrogenases, the [FeFe] hydrogenase presents the highest turnover rate for H2 evolution/uptake. Nonetheless, the [NiFe]-hydrogenase is the most wide-spread. It is present in all Proteobacteria as well as in Firmicutes, Cyanobacteria, Aquificae.19 It is also the most robust hydrogenase especially towards O2-tolerance, and its role in the microorganism is H2 uptake.17,18,20–22 Therefore, this class of hydrogenases is the most adapted for use in EFCs.[NiFe]-hydrogenases share a similar overall structure.18 The enzyme is composed of two subunits (Fig. 2). In addition, the membrane-bound [NiFe]-hydrogenases have a trans-membrane helix that serves as an anchor to the membrane. The large subunit harbors the [NiFe] active site, in which the Fe ion is coordinated by 1 CO and 2 CN− ligands, while two cysteine residues bridge the Ni and Fe atoms. A conserved pendant arginine would also act as a catalytic base for H2 activation.23 The small subunit contains three FeS clusters at a distance less than 13 Å and is able to transfer electrons from the active site to the physiological partner. Gas diffusion channels allowing H2 to reach the active site are identified. Nowadays, in-depth biochemical and spectroscopic characterization of [NiFe]-hydrogenases from different organisms allows to propose a global mechanism for H2 oxidation18,21 (Fig. 2). In brief, H2 reaches the Ni(II)Fe(II) active site (known as Ni-SI state) where it undergoes heterolytic cleavage resulting in a Ni(II)Fe(II) state with H− bridging the two atoms. Successive release of 2 protons and 2 electrons leads back to the initial Ni(II)Fe(II) state. However, some debates subsist concerning inactive states of the enzyme.24
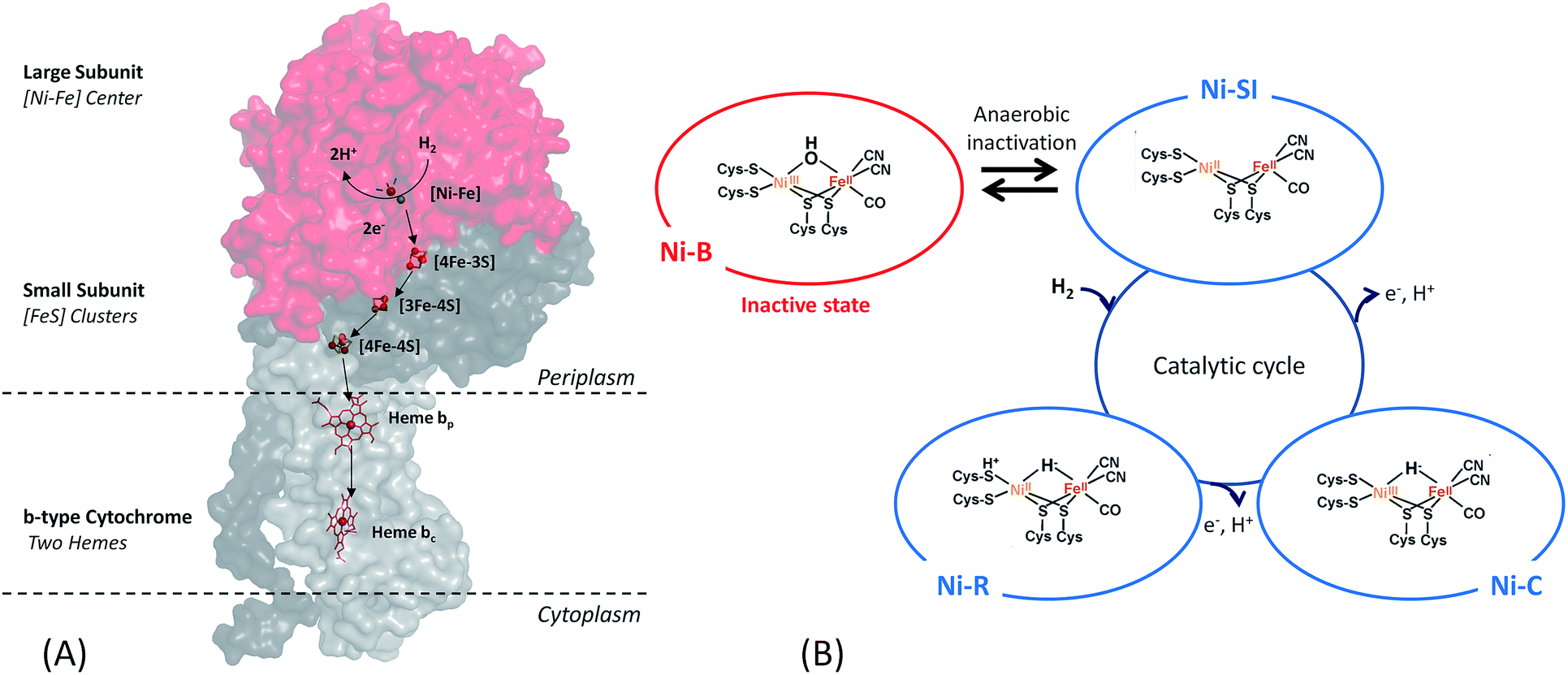 | ||
| Fig. 2 O2-tolerant [NiFe]-hydrogenases are key enzymes for H2 oxidation. (A) Structure of membrane-bound [NiFe]-hydrogenase showing the large subunit and the small subunit harboring the [NiFe] active site and the 3 FeS clusters, respectively, as well as the trans-membrane helix. Electron pathway from the [NiFe] active site through the FeS clusters and the diheme cytochrome b partner is also drawn. (B) States of the [NiFe]-hydrogenase involved in the catalytic cycle for H2 oxidation and Ni-B state formed at high potential in the absence of oxygen. | ||
Concerning O2 reduction, multicopper oxidases (MCOs) have been reported for many years as efficient bioelectrocatalysts reducing O2 into water.25–28 Laccase and BOD belong to this class of proteins, and they are found in many fungi and bacteria. They are both commonly used in sugar/O2 EFCs.4 However, BOD tends to surpass laccase because it is able to operate at neutral pH and is less sensitive to chloride at this pH.29 BOD contains four copper atoms, which are classified according to their geometric and spectroscopic properties: one T1 Cu, one T2 Cu coupled to a binuclear T3 Cu (the TNC center) (Fig. 3). As for hydrogenases, the global catalytic cycle is determined, although some states remain unclear, especially those formed in the presence of halides30–32 (Fig. 3). Briefly, O2 binds at the TNC center of the fully reduced BOD, and is reduced into water following two successive electronic steps involving a peroxy state and a state where a hydroxyl bridges the T3 copper atoms.27,28,33 The electrons are received at the T1 Cu and transferred to the TNC through a conserved (His-Cys-His) sequence.
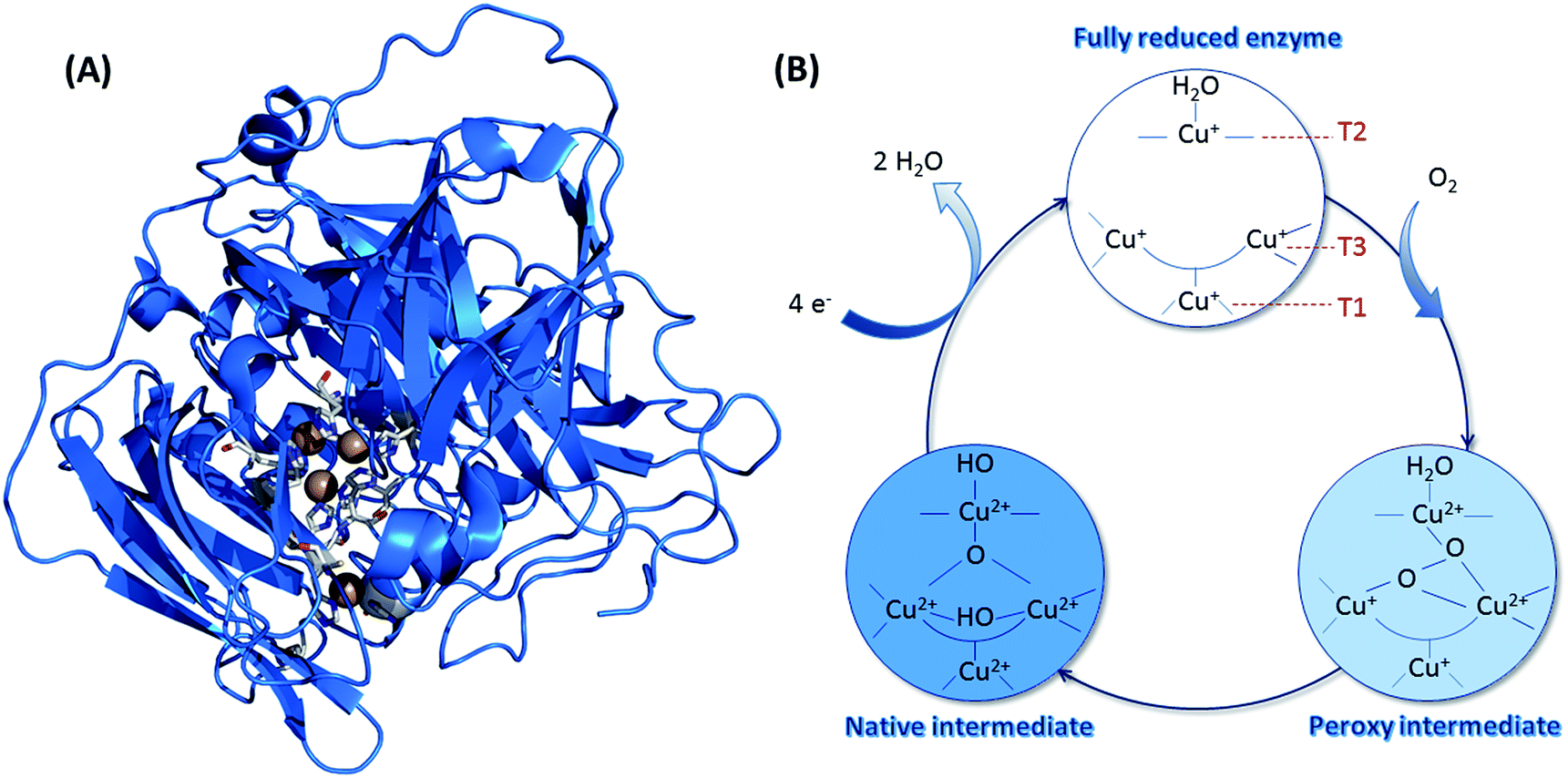 | ||
| Fig. 3 (A) Crystal structure of BOD from Myrothecium verrucaria (PDB ID: 2XLL). Copper atoms are represented by brown spheres, and their ligands appear as sticks with colors corresponding to the elements. Color code: white: carbon; blue: oxygen; red: nitrogen and yellow: sulfur. (B) Catalytic cycle involved in the mechanism of O2 reduction by BODs, with the different redox states of the three Cu atoms. | ||
Replacing platinum with enzymes?
The first report that envisioned hydrogenase as a biocatalyst in replacement of Pt is dated 1977.34 This was encouraged by the demonstration of hydrogenase ability to exchange electrons with an electrochemical interface to oxidize H2, at least with the aid of a redox mediator. This type of electron transfer process is named Mediated Electron Transfer (MET). At that time, the possibility of direct connection (Direct Electron Transfer (DET)) between the active site of the enzyme and the electrode was an open question34 (Fig. 4). DET between proteins less than 20 kDa and various electrodes was known since the 1970's.35 The issue of required orientation to expose the metal center close to the electrode was also clearly identified. However, the burying of the active site into the protein moiety of larger enzyme molecules (more than 50 kDa and 5 nm diameter if considered as globular) was long believed to preclude any DET. Until now, MET is still widely used to connect enzymes via small redox molecules either diffusing or immobilized mostly within polymers at the electrode.36,37 Many enzymes are still not directly addressable by electrochemistry. However, some of them, including hydrogenases and bilirubin oxidases, have been proved to be able to directly exchange electrons with electrodes such as pyrolytic graphite electrodes (PG).38,39 Before deeper explanations, which will be exposed later in this review, it was suggested that some enzyme molecules were able to adopt a favorable orientation for DET, i.e. placing the surface FeS (distal cluster) or Cu T1 at a tunneling distance from the electrode, for hydrogenase and BOD respectively. In the DET process, enzymatic rates are then measured directly as a current. | ||
| Fig. 4 Electrocatalysis using enzymes proceeds either via a direct enzyme connection at the electrode (DET, left), or through a redox mediator (MET) either diffusing (middle) or immobilized for example in hydrogels (right). | ||
The successful direct electric communication of enzymes on electrodes allowed comparison with Pt, a prerequisite towards further H2/O2 EFC developments. Accordingly, O2 reduction by the Pt electrode was compared to BOD immobilized on an electrode. An accurate comparison is complicated by the most often unknown quantity of enzyme participating to the current. However, based on an electroactive enzyme monolayer, the electrochemical data suggested that the enzyme should be several orders of magnitude more efficient than Pt.40–42 Net improvement in the overpotential required for O2 reduction was also expected by using BOD which has been maturated for bilirubin oxidation; hence presenting a high potential T1 Cu.43 Actually, the overpotential of O2 reduction was shown to be much lower for the enzyme compared to that of Pt, especially at neutral pH or moderate acidic pH.40,42 For BOD from the fungus Myrothecium verrucaria (Mv), an overpotential of 80 mV was measured at pH 5 against 210 mV for Pt under the same pH conditions. It should be also underlined that BOD outperformed Pt in complex media, for example, in air-saturated human blood.42 These results confirm the previous conclusion drawn from comparative O2 reduction efficiency between Pt and BOD wired in redox hydrogels.5,44 In this latter case, the enzyme loading is also far above the monolayer, allowing the envisioning of current densities in the same range as Pt-loaded carbon nanoparticles.
[NiFe]-hydrogenase from Allochromatium vinosum (Av) adsorbed on PG at 45 °C was compared to Pt under the same conditions.45 Under 1 atm H2, a similar catalytic current for H2 oxidation was recorded between the hydrogenase and Pt at an overpotential of 200 mV. The absence of non-catalytic signal for the hydrogenase precluded any quantification of the enzyme coverage. However, the similarity of current between the enzyme and Pt suggested a higher turnover rate for the enzyme to compensate for the low density of catalytic sites. At lower overpotentials, the enzyme was progressively less efficient as a result of interfacial and intramolecular electron transfer limitations, except when low H2 concentrations were used. It was also demonstrated that the enzyme surpassed Pt in the presence of CO. While CO immediately poisoned Pt, the hydrogenase recovered its activity after CO removal. The comparable efficiency of hydrogenase over Pt was confirmed later by Karyakin et al., even when the Pt-based electrode was operating under sulphuric acid condition.46 Woolerton et al. finally compared the efficiency of the membrane-bound hydrogenase from the aerobic bacterium Ralstonia eutropha (Re MbH) and that of Pt for oxidation of low levels of H2.47 Interestingly it was shown that the extremely high affinity of hydrogenase towards H2 (KM = 6 μM) resulted in the enzyme outperforming Pt for sub-atmospheric H2 levels. H2 oxidation was even recorded in mixtures of H2 and O2, under conditions where most hydrogenases were expected to be inactivated. An inactivation process of hydrogenase at high potentials, not occurring on Pt, limited however the usable potential range. Electrochemical data underlined that both the enzyme-based and Pt-based electrodes oxidized H2 and reduced O2, albeit the enzyme was much more efficient in H2 oxidation, thus again outperforming Pt in H2/O2 gas mixtures. At the same time, progressive inhibition of Re MbH by O2 was demonstrated. A fast recovery of the activity was however noticed after O2 removal, suggesting an O2-tolerance of this enzyme extracted from an aerobic bacterium. Last but not least, the Re MbH-based bioanode was demonstrated to be insensitive to CO.
The first enzymatic H2/O2 EFCs
Based on the efficiency of hydrogenases and MCOs, and on the ability of Re MbH to operate in air, the first H2/O2 EFC with no membrane separator was designed by Vincent et al. in 2005.48 The EFC operated at 30 °C, pH 5.6 in a beaker containing buffer flushed with H2 close to the bioanode, and air close to the biocathode. Re MbH and a laccase were deposited on graphite strips of 0.7 cm2. The OCV was 970 mV, and a maximum power of 5 μW was reached. Comparison with an EFC running with the hydrogenase from Av showed a maximum power of 0.2 μW.48 In addition to provide the proof that hydrogenase could be coupled to MCO in a H2/O2 EFC to produce electricity, this paper also underlined the requirement of an O2-tolerant hydrogenase, such as the membrane-bound [NiFe]-hydrogenase from Re. Afterwards, another H2/O2 EFC was demonstrated to be able to operate in 3% H2 in air, thanks to the peculiar properties of the membrane-bound hydrogenase from R. metallidurans.49 An OCV of 950 mV and a maximum power density of 5.2 μW cm−2 were recorded. Three cells in series were shown to power a wristwatch for 24 h. Even if a low power density was obtained due to very low enzyme coverage on graphite, these results clearly highlighted the great potential of such devices based on high H2 affinity, O2-tolerant MbH hydrogenases and high redox potential MCO.Limitations of the first H2/O2 EFCs
The initial biodevices also put forward the main limitations that had to be overcome in view of large scale development of H2/O2 EFCs. The first prerequisite is the use of efficient but stable enzymes. The anodic side which relies on sensitive hydrogenases is especially critical. The identification of the O2-tolerant Re MbH opened the route for new outstanding enzymes. This is the case of the membrane-bound [NiFe]-hydrogenase from Escherichia coli (EcHyd1) which was proved to be very active for H2 oxidation in air.50 O2-tolerance is not sufficient however. Wait et al. pushed forward the understanding of H2/O2 EFCs by comparing their performance in three different configurations: (i) fuel cell with a Nafion membrane separator fed with 100% H2 and 100% O2 in the anodic and cathodic compartments, respectively; (ii) a membrane-less fuel cell fed with 96% H2/4% O2 mixture; (iii) a membrane-less fuel cell fed with 4% H2/96% air mixture51 (Fig. 5). The bioelectrodes were based on EcHyd1 deposited on PG and Mv BOD immobilized at a carboxylic-modified PG. Important features can be extracted from these studies. In the first configuration, the OCV was 990 mV and the maximum power density was 63 μW cm−2 at 511 mV. In the second configuration, as expected, the cathode was limiting the EFC. O2 reduction at the anode occurred, and inactivation of EcHyd1 was visible at potentials higher than −200 mV vs. Ag/AgCl. Full power was restored after the polarization experiment. Conversely, in H2-poor gas mixtures (configuration iii) where the anode limited the EFC, complete anode inactivation occurred above −50 mV vs. Ag/AgCl, full power was not recovered after the polarization experiment and the anode potential was not restored. Typical characteristics of EFCs in the different configurations were demonstrated to be linked to the formation of the inactive state of the hydrogenase under O2, the so-called Ni-B state (Fig. 2). Many fundamental studies have been dedicated to the kinetics of formation of this inactive state, in which a hydroxyl bridges the Ni and Fe atoms. It is formed in all the hydrogenases either O2-sensitive or O2-tolerant, the difference being a faster reactivation rate under reducing conditions for O2-tolerant hydrogenases.50,52 One important conclusion of the EFC study was that Ni-B formation restricts the usage of H2/O2 EFCs to high-load devices for which the potential of the anode does not exceed critical values. Hydrogenase inactivation induces however a wider use restriction. Indeed, the Ni-B inactive state is also formed at high potential in the absence of O2. This state can be fully reactivated under reducing potentials. However above 200 mV vs. Ag/AgCl inactivation appears to be irreversible. As a consequence, in case of EFCs limited by the anode, even in the presence of a separator membrane, the very high potential reached by the anode may yield irreversible inactivation of the hydrogenase.53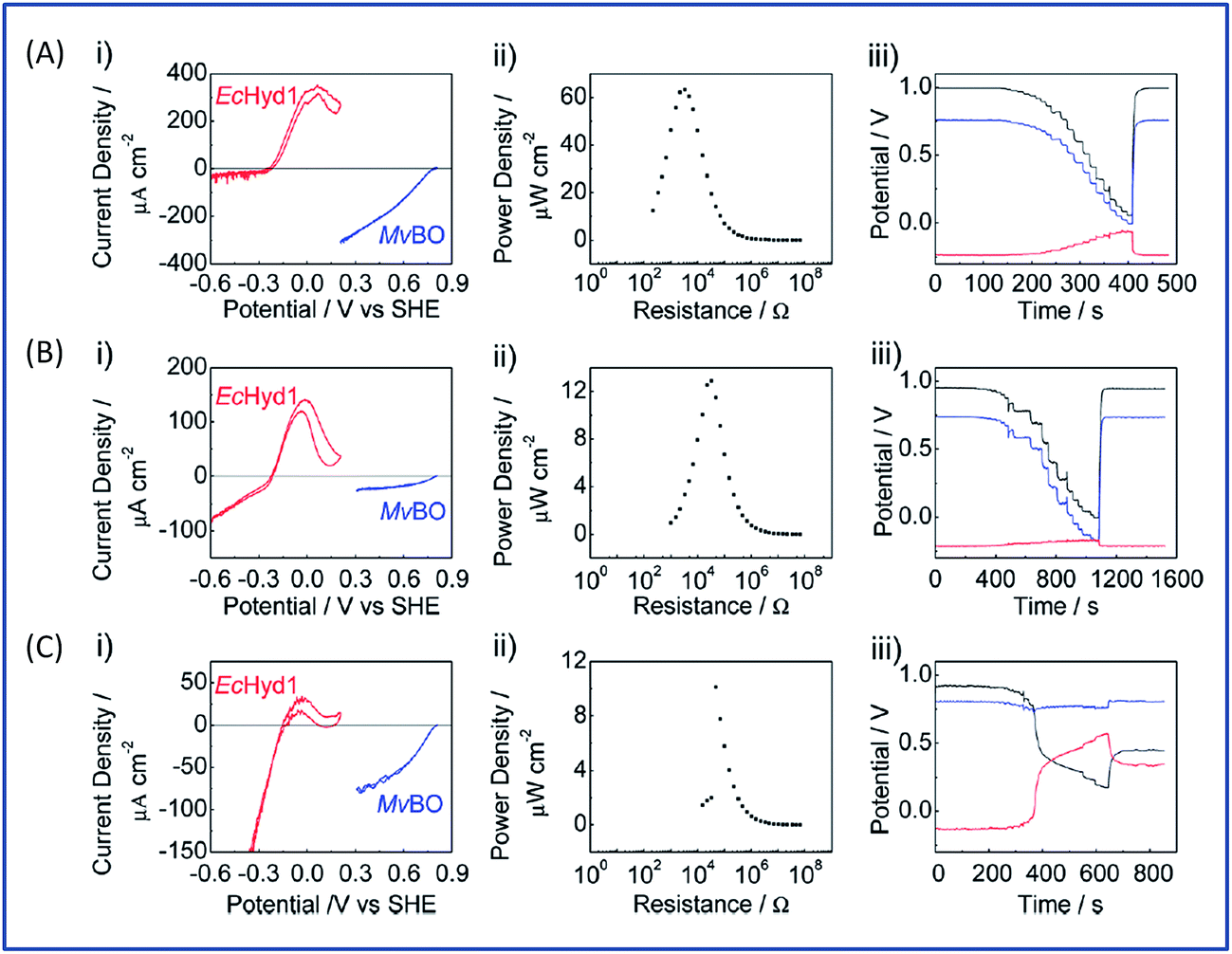 | ||
| Fig. 5 H2/O2 EFC operating in three different configurations and highlighting the effect of hydrogenase inactivation on the EFC performances. (A) Membrane EFC, 100% H2 and 100% O2 in the anodic and cathodic compartments; (B) membrane-less EFC, 96% H2 and 4% O2 in the anodic and cathodic compartments; (C) membrane-less EFC, 4% H2 and 96% air in the anodic and cathodic compartments. (i), (ii) and (iii) depict respectively in red catalytic H2 oxidation by Echyd1 and in blue catalytic O2 reduction by Mv BOD, power performances, and voltages of cell (black) and individual electrodes (anode in red and cathode in blue). Reprinted with permission from Wait et al., J. Phys. Chem. C, 2010, 114, 12003–12009. Copyright 2017 American Chemical Society. | ||
In addition to the main limitation associated with hydrogenase inactivation under O2 or under oxidizing conditions, the first H2/O2 EFCs also raised other critical issues. Tsujimura et al. concluded that mass transfer controlled the EFC performances, which could be only overcome by using porous materials as host matrices.13 Further, although enzymes were suggested to be much more active than Pt to compensate the lower surface coverage,45 the power reached in the EFCs clearly indicated the need to increase the enzyme coverage per geometric area.
Search in the biodiversity for new enzymes with outstanding properties
This review already underlined the benefit of enlarging the panel of enzymes in order to obtain new electrocatalytic properties. This is the case of the O2-tolerant [NiFe]-hydrogenases such as Re MbH and EcHyd1 which were proved to support high concentrations of O2 and to be insensitive to CO. These two properties allowed envisioning of their use in biotechnological devices, which had been quite unexpected for the “standard” sensitive hydrogenases originally purified and studied by electrochemistry. Besides Re MbH and EcHyd1, three more [NiFe]-hydrogenases have been identified presenting the same phenotypes of O2-tolerance and CO-insensitivity. They are present in bacteria able to sustain energy from an electron transfer chain coupling H2 oxidation to O2 reduction. The first one is extracted from the facultative anaerobe Salmonella enterica, and is homologous to EcHyd1.54 The second one is purified from the hyperthermophilic bacterium Aquifex aeolicus (Aa MbH).55 In addition to O2- and CO-tolerance, Aa MbH was demonstrated to be able to oxidize H2 on a temperature range from 25 °C up to 85 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56,57 (Fig. 6). The last one is purified from the aerobic marine bacterium Hydrogenovibrio marinus (Hm MbH), and was also shown to be thermostable.58 As a general rule, O2-tolerant hydrogenases are membrane-bound enzymes, a membrane cytochrome b being their physiological partner (Fig. 2). They present a net bias toward H2 oxidation against proton reduction, but oxidize H2 at potentials 100–150 mV higher than “standard” O2-sensitive hydrogenases, a drawback in view of application in EFCs. Many studies are still dedicated to the understanding of the origin of O2- and CO-tolerance in view of engineering O2-tolerant hydrogenases or in order to determine the molecular keys to synthesize biomimetic complexes. The crystal structures of four O2-tolerant hydrogenases definitely helped in the definition of some molecular determinants explaining the O2-tolerance.59–62 They are detailed in recent reviews.10,18,22,63,64 Briefly, the main exceptional feature of O2-tolerant hydrogenases compared to sensitive ones is the presence of an unusual [4Fe-3S] cluster close to the [NiFe] active site, coordinated by six cysteines instead of four, providing an electron-rich environment. This peculiar FeS cluster allows complete reduction of O2 into water, which can be evacuated through specific hydrophilic channels. However, this is not sufficient to totally account for O2-tolerance. Based on various mutations, specific amino acid residues in the canopy very close to the active site and at the end of the gas channels are also suggested to play a key role in the protection.23,65,66 The tetrameric structure of O2-tolerant hydrogenases might also act as a rescue mechanism, enabling electron transfer between inactivated and active monomers.67
56,57 (Fig. 6). The last one is purified from the aerobic marine bacterium Hydrogenovibrio marinus (Hm MbH), and was also shown to be thermostable.58 As a general rule, O2-tolerant hydrogenases are membrane-bound enzymes, a membrane cytochrome b being their physiological partner (Fig. 2). They present a net bias toward H2 oxidation against proton reduction, but oxidize H2 at potentials 100–150 mV higher than “standard” O2-sensitive hydrogenases, a drawback in view of application in EFCs. Many studies are still dedicated to the understanding of the origin of O2- and CO-tolerance in view of engineering O2-tolerant hydrogenases or in order to determine the molecular keys to synthesize biomimetic complexes. The crystal structures of four O2-tolerant hydrogenases definitely helped in the definition of some molecular determinants explaining the O2-tolerance.59–62 They are detailed in recent reviews.10,18,22,63,64 Briefly, the main exceptional feature of O2-tolerant hydrogenases compared to sensitive ones is the presence of an unusual [4Fe-3S] cluster close to the [NiFe] active site, coordinated by six cysteines instead of four, providing an electron-rich environment. This peculiar FeS cluster allows complete reduction of O2 into water, which can be evacuated through specific hydrophilic channels. However, this is not sufficient to totally account for O2-tolerance. Based on various mutations, specific amino acid residues in the canopy very close to the active site and at the end of the gas channels are also suggested to play a key role in the protection.23,65,66 The tetrameric structure of O2-tolerant hydrogenases might also act as a rescue mechanism, enabling electron transfer between inactivated and active monomers.67
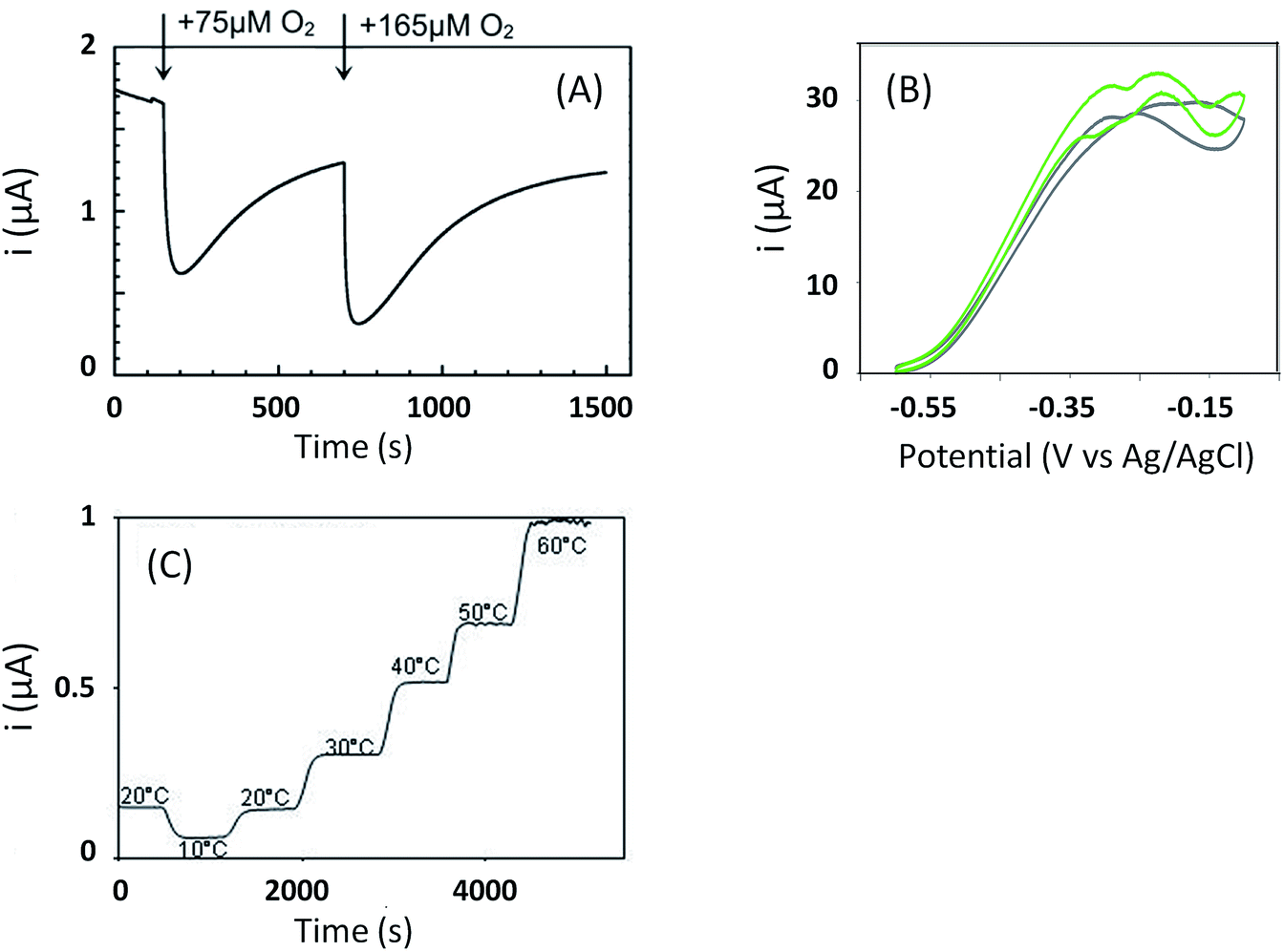 | ||
| Fig. 6 The membrane-bound [NiFe]-hydrogenase from Aquifex aeolicus presents outstanding properties. (A) O2-tolerance: H2 oxidation activity is recovered after O2 injection at Eapplied = 0 V vs. Ag/AgCl. (B) CO-resistance: H2 oxidation (grey curve) is unaffected by addition of CO (green curve). (C) T°-resistance: H2 catalytic oxidation current is stable on the 10–60 °C temperature range. (A) is reprinted with permission from Pandelia et al., J. Am. Chem. Soc., 2010, 132, 6991–7004. Copyright 2017 American Chemical Society. (C) is reprinted from Monsalve et al., ChemElectroChem, 2016, 3, 2179–2188. Copyright 2017 Willey. | ||
Biodiversity has been less explored for BODs, perhaps because the identification of BODs themselves already provided the key to overcome halide inhibition encountered by the laccases originally used for O2 reduction.29 Note however that the inhibition process of MCOs by halides is nowadays intensively revisited.30–32 Fungal Mv BOD is commercially available and is the most widely studied BOD, although some new BODs emerged such as those isolated from the bacterium Bacillus pumilus (Bp)68 and from the fungus Magnaporthe oryzae.69 Both of them were demonstrated to be thermostable, but the reason for such thermostability is not elucidated yet. However, the redox potential of T1 Cu, the entry point of electrons, was shown to be 100 mV lower than that of T1 Cu in Mv BOD,70 a feature that will result in lower OCV in EFCs.
How to address the issue of hydrogenase inactivation at high potentials?
As discussed before, anaerobic hydrogenase inactivation at high potentials restricts the devices that can be powered by H2/O2 EFCs, even if a membrane separator is maintained between the two cell compartments. This phenomenon is accelerated under O2 excess.50 Various directions have been explored to resolve this issue. The first one is a reconstitution of an in vivo reactivation mechanism at the electrode. We just discussed above that oligomeric forms of hydrogenase could provide the sink of electrons necessary to maintain the activity under oxidative conditions.67 The two dimers in the tetrameric forms would be able to share electrons through the distal FeS cluster. This is supported by a crystal structure of a dimer of EcHyd1 which underlines a distance between the two distal FeS clusters of each monomer compatible with electron transfer.71In vivo like strategy was used by Wait et al. under EFC conditions.51 Starting from the knowledge that an inactivated enzyme can be reactivated under reducing conditions, the idea consisted in the direct connection of a second hydrogenase-based anode to refill electrons to the deactivated anode. H2 oxidation at the second anode provided electrons required to reactivate the first inactivated bioanode. This resulted in the EFC recovery, and constitutes one solution to address anaerobic hydrogenase inactivation at high potentials. Another strategy was proposed by Ciaccafava et al. who demonstrated that O2-tolerant Aa MbH could be reactivated by light72 (Fig. 7). Actually, the magnitude of photocurrents was shown to be dependent on potential, and rate of photooxidation was shown to depend upon light intensity for potentials above Ni-B formation potential. The light-induced reactivation process of Aa MbH under turnover was thus hypothesized to promote the loss of the hydroxyl ligand bridging the Ni and Fe atoms. Unfortunately, attachment of Aa MbH to quantum rods did not allow the immobilized enzyme to assess the light induced reactivation because of the requirement of a redox mediator to oxidize H2.73 The last strategy explored to suppress anaerobic oxidative inactivation was reported by So et al.74,75 Both O2-tolerant Hm MbH and the O2-sensitive hydrogenase from Desulfovibrio vulgaris Miyazaki (Dv MF) were immobilized on gas diffusion electrodes made of Ketjen black (KB) deposited on carbon cloth. In this configuration, H2 is supplied from the gas phase, diminishing the limitation induced by low concentrations of hydrogen in buffer solutions. Electrochemical results were analyzed by a modeling approach that considered a competition between Ni-B formation from Ni-SI and catalytic reduction of Ni-SI by H2. While it remains unclear if H2 concentration in the gas diffusion electrode exceeds Henry's law saturation limit, it was proposed that it was sufficient to avoid the formation of the high potential inactive state for Hm MbH, and to limit its formation for Dv MF.74,75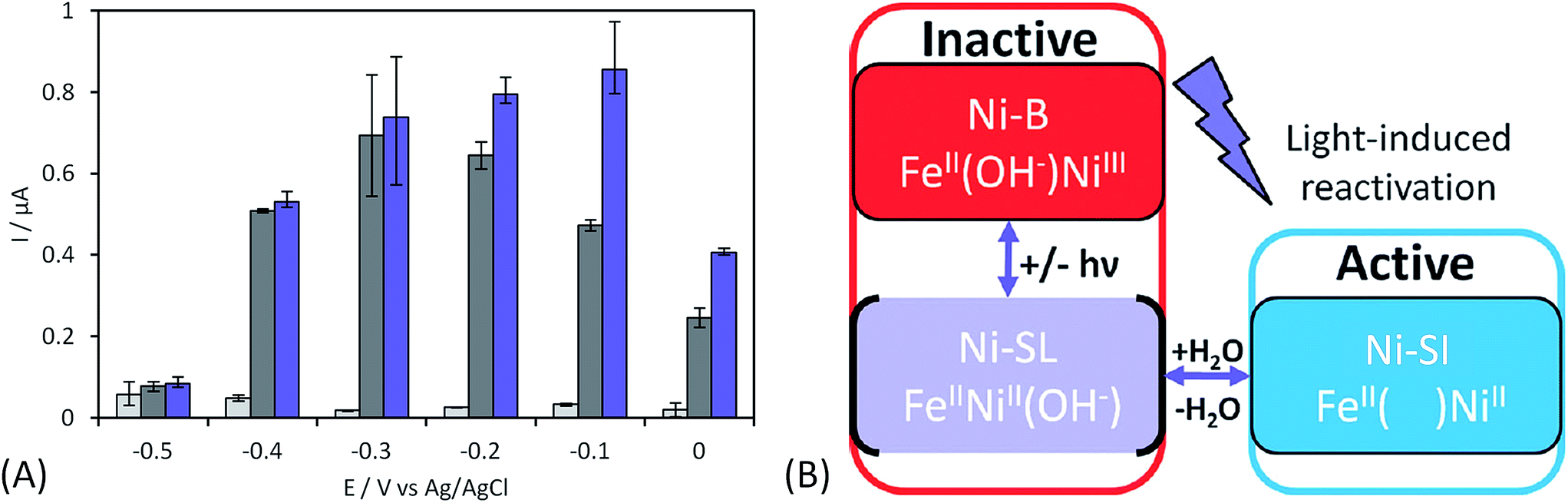 | ||
| Fig. 7 Light induced reactivation of Aa [NiFe]-hydrogenase and proposed mechanism. (A) Catalytic current obtained for H2 oxidation under illumination as a function of applied potential (purple columns) compared to catalytic current in the dark (grey columns) underlines that Ni-B can be reactivated by light. (B) Reactivation mechanism under irradiation is proposed to promote the loss of hydroxo ligand on the photo-induced Ni-SL intermediate from Ni-B to Ni-SI. Adapted from Ciaccafava et al., Phys. Chem. Chem. Phys., 2013, 39, 16463–16467. Copyright 2017 Royal Society of Chemistry. | ||
The strategies just discussed to overcome the issues of oxidative inactivation of hydrogenases are all based on DET between the enzyme and the electrode. DET is usually preferred to MET for fundamental studies because it allows to obtain kinetic data on the enzymatic catalysis. DET is also preferred nowadays to MET for biotechnological devices because of the lower overvoltage for the transformation of the substrate, and avoidance of redox mediators that are often toxic and not easily co-immobilized with enzyme on the electrode while retaining a high catalytic efficiency. MET is however the strategy used in vivo for electron transfer. Radu et al.76 immobilized membrane fractions of Re MbH including cytochrome b and quinone pool in tethered bilayer lipid membranes on gold electrodes. In such architecture, the physiological catalytic function of Re MbH is restored through a true electron transfer pathway from the [NiFe] active site to the electrode via the quinone pool. This elegant reconstitution demonstrated that hydrogenase did not inactivate at high potentials even under conditions of low H2 concentrations, a behavior completely different from the usual inactivation observed with purified Re MbH at the electrode. It was proposed that the higher oligomeric states inside the membrane prevent the Ni-B state formation by allowing one active oligomer to donate electrons, thus reactivating a neighboring inactive oligomer. However, in this configuration Re MbH is not placed in direct connection with the electrode, so it is difficult to determine the exact potential it is subjected to. In vitro, it was shown by Plumeré et al. that MET can be beneficial in the case of hydrogenase, because it prevents the formation of the Ni-B inactive state.77Dv MF O2-sensitive hydrogenase was embedded in redox hydrogels based on viologen entities. No deactivation at high potential was observed in the cyclic voltammograms. Spectroelectrochemical Fourier transform infrared experiments confirmed the absence of Ni-B formation as MET proceeds at high potentials. This protective process is most probably linked to the fact that the enzyme experiences the redox potential of viologen, but not the potential of the electrode. It should be noted that a similar result was observed by incorporation of Df O2-sensitive hydrogenase in a viologen-polypyrrole matrix some years before, although the protective role of the polymer matrix was not recognized at that time.78 The protective role of redox hydrogels may not be effective for O2-tolerant hydrogenases, however. Indeed, in this case higher redox potential mediators are required to fit the higher operational potentials of these hydrogenases. The potential of redox mediators may then be too high to prevent oxidative deactivation.
Use of nanomaterials to enhance the power
The enhancement of the currents delivered by the bioelectrodes is required to power real devices by EFCs. Actually, planar electrodes limit the amount of available enzymes, hence the catalytic currents. It is difficult to assess the maximum attainable current because in most cases no information on the true quantity of electroactive enzymes is available. Assuming an average turnover kcat between 100 and 1000 s−1, and a maximal amount of a monolayer of electrocatalytically active enzymes, current densities would be limited to around 1 mA cm−2. On the contrary, the use of nanomaterials presenting a high surface/volume ratio will a priori increase the loading of enzymes per geometric area unit. Although many works have been reported since more than 15 years in this domain, research towards 3D architectures for both enzymatic H2 oxidation and O2 reduction is still very active.3,4,79–82 The interest has moved from amplification of the catalytic current by an increase in the developed electroactive surfaces, to more fundamental queries on the relation between loading/orientation/conformation/activity of the enzyme inside the nanostructures. Both DET and MET are considered, but the issues that need to be addressed are different depending whether DET or MET process is used for the electrocatalysis. Certainly, this section will not be exhaustive because this is beyond the scope of this review, but it will give the recent main results and current open questions in the use of nanomaterials for hydrogenase and BOD bioelectrochemistry.A variety of nanomaterials
For a DET process, the aim is to increase the amount of enzymes able to communicate directly with the electrochemical interface. Thus nanomaterials behave as ideal platforms to increase the surface area, to ensure intimate electric contact with enzymes, and potentially decrease the tunneling distance between active sites or surface electronic relays and electrode. In the particular case of EFCs, the current densities are expected to be sufficiently high to achieve useful power. Accordingly, this imposes high specific conductivity of the electrode material to avoid ohmic losses. That is why two kinds of highly conductive nanomaterials are particularly often used in EFCs: carbon and gold nanomaterials (Fig. 8). | ||
| Fig. 8 A variety of nanomaterials can be used for enzyme immobilization. (A) Preparation of graphene and rGO (reproduced from Filip et al., Electrochim. Acta, 2014, 136, 340–354); (B) CNT forests (reproduced from Zelechowska et al., Sensors Actuators B, 2017, 240, 1308–1313); (C) AuNP layer-by-layer deposits (reproduced from Monsalve et al., Bioelectrochem., 2015, 106, 47–55); (D) nanoporous gold electrode obtained from dealloying process (reproduced from Siepenkoetter et al., Electroanal., 2016, 28, 2415–2423). Copyright 2017 Elsevier. | ||
Carbon-based nanomaterials are the most popular for bioelectrode design.83,84 The graphene sheet is the building block of most of the carbon nanostructures. It possesses a high conductivity and an extraordinary surface area estimated to be about 2630 m2 g−1.3 It can be wrapped to form fullerenes, rolled along the axis to form nanotubes and stacked to form graphite.85 Although it was considered as a very attractive material when discovered, the advantage of using graphene in bioelectrocatalysis has not been fully demonstrated yet.86 This may be due to the difficulty to obtain and handle perfect monolayer graphene sheets, and from the fact that most of the studies are performed with (electro)chemically reduced graphene oxide (rGO) which still contains some oxygen functions and has different electronic properties than graphene.84 It has never been used for hydrogenase immobilization, and very poor catalytic signals are most often observed after BOD immobilization on rGO sheets.87 Enhanced electroenzymatic currents could be rather attributed to the deposit of carbon nanotubes (CNTs) on graphene used as a platform.87,88 However, some recent studies tend to demonstrate the usefulness of graphene for enzymatic O2 reduction. Catalytic current density around 0.5 mA cm−2 was reported for O2 reduction by BOD integrated within rGO.89,90 Besides, enzyme surface coverage and the rate of interfacial electron transfer were shown to depend on the density of charges on the graphene surface. This point will be discussed further in the stability section.
CNTs appear to be the nanomaterials of choice for both BOD70,91 and hydrogenase immobilization,53,56,57,83,92–97 thanks to their high conductivity, chemical and thermal stability.97 CNTs can be either single-walled (SWCNTs) or multi-walled (MWCNTs). In both cases, enzymes can readily interact with them through electrostatic and van der Waals forces. The chirality and diameter of SWCNTs control their electrical conductivity, resulting in either semi-conducting or metallic materials. On the other hand, MWCNTs can only be metallic, which makes them a more attractive material for bioelectrocatalysis. Their nanometric size and curvature comparable with enzyme dimensions may also be a key point.79 CNTs can be arranged in a multitude of different geometries (isolated tubes, planar arrays, random networks, or forests) that are currently explored for H2 oxidation and O2 reduction.92,98,99 They can be compacted into disks,100 arranged to form flexible buckypaper films with μm thickness,101 deposited at electrode surfaces by layer-by-layer drop casting,53,94,96 or painted as an ink.102 Although catalytic currents are enhanced in all the cases, the respective role of increase in the electroactive surface area, in the heterogeneous electron transfer rate or in the loading of electroactive enzymes need to be elucidated.
Nano- and microfibers are other examples of carbon based nanomaterials being considered more recently, which provide a large surface area for potential attachment or entrapment of enzymes. Compared to CNTs, carbon nanofibers (CNFs) usually have smaller specific surface areas, but instead, they represent a bigger and more open macroporous structure thus allowing enhancement of substrate diffusion towards the immobilized enzymes. CNFs made by graphene cone stacks and coated on a PG electrode were used for Aa MbH immobilization. Such modification allowed a current density of 4.5 mA cm−2 for enzymatic H2 oxidation, which was one of the highest current density ever reported.103
Upon enzyme-loading increase, substrate depletion is likely to occur. This is especially critical when talking about highly active enzymes like hydrogenases and low solubility of gaseous substrates (e.g. H2 and O2) in water-based solutions. Apart from specific EFC design such as microfluidic substrate distribution, larger pores of the carbon support are thus desirable. In general, larger pores can be obtained through carbon felts (CFs), carbon cryogels, or carbon foams which present interesting properties of mechanical strength and stability.104–107 These macroscopic supports often have a defined geometric shape that can be easily modeled depending on the application. Furthermore, their mechanical stiffness facilitates manipulation. Large porosity in such materials, has a positive impact on the magnitude of catalytic current. However, it is difficult to relate this enhanced catalytic current to a larger amount of connected enzymes or to facilitated substrate diffusion. Different nanomaterials can be mixed to provide a hierarchical interconnected porous structure with the ability to reach a tunable pore size. Mesopores are desirable for enzyme anchorage while ensuring efficient substrate delivery in large macropores. It should be noted that micropores below 2 nm range are quasi-useless for EFC since they are too small for enzymes to penetrate and give rise to high capacitive current contribution which is usually undesirable except in some specific cases. In order to improve the favorable characteristics of porous materials, their macrostructure can thus be used as a primary framework towards 3D hierarchically structured materials by incorporation of a secondary structure of smaller dimensions like CNTs.108 For example, CNTs were deposited on the microfibers of carbon felts for H2 oxidation by Aa MbH.109 The developed electroactive surface evaluated by capacitance measurement was 5 times higher compared to that of the bare CF. In another work, a wet-spinning approach was used to fabricate electrodes in the form of microfibers, in which CNTs and enzymes for O2 reduction were directly imbricated.110,111 Pore size can otherwise be tuned and controlled using templated carbons such as MgO-based ones.112 Modeling has not been largely applied to such a system yet, but it will certainly help in the rationalization of porosity.
Gold nanoparticles (AuNPs) are another type of nanomaterials that have unique physicochemical characteristics and optical-electronic properties. They have been largely used for H2 oxidation and O2 reduction by enzymes. Their synthesis drives their properties, which can be tuned by changing their shapes, sizes and chemical environments. The most common preparations of AuNPs are either by physical “top-down” methods where the bulk is divided into smaller units, or by “bottom-up” chemical methods. The latter starts with a reduction of an Au-precursor creating metal clusters followed by their aggregation into particles and growth until the desired size.113 Despite the common acceptance that nanoparticles by themselves enhance electron transfer, a decisive study to clarify the influence of the size of nanoparticles in biocatalysis (current and reaction overpotential) was done by Pankratov et al.114 The authors modified bare Au electrodes with a sub-monolayer of AuNPs. It was found that AuNPs with a size at least four times bigger than the enzyme (20–80 nm) influenced neither the direct electron transfer rate nor the overpotential of O2 bioelectrocatalysis compared to bare Au electrodes. Similar results were obtained by immobilizing Aa MbH on AuNPs of two different sizes.106 In this latter study, multilayers of AuNPs were deposited on a gold electrode showing increasing catalytic currents for H2 oxidation up to 1.8 mA cm−2, i.e. 150 times more than the current recorded on the bare gold electrode. However, when reported to the real developed electroactive area, which can easily be deduced from simple cyclic voltammetry experiments in acidic solution, it was demonstrated that the enhancement in catalytic current was simply correlated to the surface area increase.
Other nanostructured materials are worth mentioning. Co-deposition of different nanomaterials is explored, such as AuNPs and CNTs deposited onto electrospun polyacrylonitrile microfibers.88 Nano- or mesoporous gold electrodes are promising materials. They were used to enhance DET with Mv BOD.115 As the diameter of the pores is expected to play a key role in the loading of electroactive enzymes, the pore size of the nanoporous gold electrode was varied between 4 and 78 nm thanks to Au/Ag dealloying processes at different temperatures.116 The catalytic current was shown to be the highest for a pore size of 8 nm, i.e. slightly higher than the BOD size. For higher pore dimension the current decreased. The authors concluded that BOD did not diffuse deeply inside the electrode, and that catalysis occurred mainly on the surface of the nanoporous electrode. The results can however suggest that 8 nm is the most suitable pore size for hosting a single enzyme molecule, thus favoring an excellent electrical connection.
Enzyme distribution in mesoporous and nanomaterials
There is a general assumption that enzymes are homogenously distributed throughout the entire nanomaterial volume with an optimal microenvironment to favor catalytic activity. Nonetheless, enzyme distribution should not only depend on the material properties, but also on the interactions the enzyme is subjected to during the immobilization process, and on the immobilization process itself. The correlation between enzyme distribution and electrochemical response is thus important to gather fundamental knowledge about substrate/enzyme diffusion restrictions that can compromise performance. This is also a prerequisite to optimize the loading of the electroactive enzyme. Indeed, even when enzymes are compacted with carbon particles into free-standing pellets, the intimate mixture does not preclude aggregates of inactive enzymes to be formed. This is especially true because large amounts of enzymes are generally used in this configuration.100 One relevant demonstration of non-homogeneous enzyme distribution in the porous matrix was given by the results obtained using fluorescence confocal microscopy to follow the distribution of malate dehydrogenase immobilized in macroporous chitosan. The images suggested that enzyme distribution was dependent on the interactions between the biomolecules and the charged polymer species that create a singular chemical microenvironment.117 Few studies however address such issues with hydrogenases and BODs. Complementary to electrochemistry measurements of O2 reduction using wet-spun carbon fibers directly fabricated with BOD and CNTs inside the fibers, Mateo–Mateo et al. corroborated the presence of Magnaporthe oryzae BOD within a fiber by confocal fluorescence microscopy experiments and transmission electron microscopy with EDX analysis of the cross section of CNT fibers. The images and EDX indicated the presence of BOD throughout the fiber, from its outer surface to its core110 (Fig. 9). | ||
| Fig. 9 (A) Scanning electron microscopy cross-sectional images of a spun CNT/enzyme fiber. Top and down are two images of the same fiber. (B) Superposition of (A) and of the images obtained using confocal fluorescence microscopy of the same cross sections with BOD labeled with a fluorescent dye. Adapted from Mateo–Mateo et al., ChemElectroChem., 2015, 2, 1908–1912. Copyright 2017 Willey. | ||
The embedding of enzymes and CNTs in the core fiber was shown to favor DET. However, these images show the total enzyme distribution and do not allow the deduction of the fraction of them participating actually in the biocatalysis. Armstrong et al. compared the electrocatalytic activity of EcHyd1 in a cylinder or in a half cylinder of compacted graphite particles. The twice less catalytic current in the half cylinder suggested a homogeneous distribution of enzymes inside the entire porous matrix.118 Nevertheless, the very few data on enzyme distribution in a porous matrix imposes development of tools to couple electrochemistry to surface imaging and other techniques to realize enzyme cartography of various bioelectrodes.
From enzyme orientation on planar electrodes…
As mentioned earlier, the large size of enzyme molecules and active sites buried within the protein shell often require the molecule to be adsorbed in specific orientations to perform electron transfer. It results in a low amount of electroactive enzyme molecules among adsorbed ones, and the appearance of a linear region on DET voltammograms due to the dispersion of enzyme orientations.119,120 From the practical point of view, the dispersion of orientations should be minimized and enzyme should be immobilized in a defined orientation bringing the surface electron relay as close as possible to the electrode surface in order to maximize the current densities. This can be done by rational electrode modification.In vivo, the orientation of an enzyme in a metabolic chain before electron transfer is most often driven by electrostatic interactions between interacting partners.121 Transitory complexes are formed which put the interacting redox sites at a minimal distance to optimize the electron transfer rate. This molecular recognition can serve as a model for chemical modification of electrochemical interfaces suitable for enzyme orientation. If the electron transfer chain is unknown, the examination of the secondary structure of the enzyme may help in the definition of the key amino acids for proper orientation. Hence, self-assembled-monolayers (SAM) on gold can be used to tune the chemical functions of the electrode in order to recognize the target residues. Let us consider the examples of Aa MbH and Mv BOD to illustrate the orientation issue on planar electrodes.
In the case of hydrogenase, the exit point of electrons consecutive to H2 oxidation is the distal FeS cluster. In our group, we examined the oriented immobilization of Aa MbH on SAM by electrochemistry and polarization modulated infrared reflection absorption spectroscopy (PMIRRAS).122 Briefly, H2 oxidation mainly proceeded through MET on hydrophobic SAMs, while both DET and MET were observed on hydrophilic SAMs (neutral, positive or negative). The two different electrochemical processes according to the SAM hydrophobicity were proved to be correlated to different orientations of the enzyme. Absence of DET on hydrophobic surfaces was explained by the presence of surfactant surrounding the trans-membrane helix. This helix is less than 15 Å from the FeS distal, thus defining a hydrophilic domain pushing away the exit point of electrons.123 Coexistence of DET and MET was attributed to a distribution of orientation: some enzymes are positioned at the electrode with the distal FeS cluster at a tunneling distance from the electrode surface, compatible with DET, and others are placed with the distal FeS cluster far from the electrode, thus requiring a redox mediator for electrocatalysis. This absence of enzyme orientation by electrostatic interactions is linked to the membrane position of the enzyme, which uses the hydrophobic trans-membrane helix for membrane anchorage with no intervention of electrostatic interactions. Molecular dynamics (MD) confirmed this assumption, underlining a low dipole moment whose direction fluctuates largely.124 MD analysis further underlined that orientations for DET should be preferred on positive interfaces (Fig. 10). Similar conclusions were drawn by Heidary et al. for Re MbH.125
 | ||
| Fig. 10 Orientation of dipole moment and large dipole moment fluctuation of Aa MbH obtained from MD (A) suggest a favored orientation for DET on positively charged interfaces, which is assessed by H2 oxidation by the enzyme immobilized on CNTs with different functionalities (B). (A) is adapted from Oteri et al., Phys. Chem. Chem. Phys., 2014, 16, 11318–11322. Copyright 2017 Royal Society of Chemistry. (B) is adapted from Monsalve et al., ChemElectroChem., 2016, 3, 2179–2188. Copyright 2017 Willey. | ||
A different scheme can be drawn for Mv BOD immobilization on SAMs. Mv BOD is a nice model, because its crystallographic structure has been resolved.126 The entry point of electrons is the T1 Cu. Mv BOD structure displays a dipole moment of 752 Debye pointing toward a highly positive patch formed by 4 arginine (pKa = 12.5) residues surrounding the T1 Cu center. Such an environment is expected to promote DET by electrostatic interactions on negatively charged interfaces. Accordingly, only the DET process was recorded for O2 reduction with Mv BOD immobilized on negatively charged SAMs, and only the MET process on positively charged SAMs, suggesting a specific orientation according to favorable electrostatic interactions.127,128
…to enzyme orientation in mesoporous structures and on nanomaterials
Turning back to one fundamental question: can nanostructuration help overcome the orientation limitation? If not, can the molecular determinants for enzyme orientation obtained on planar electrodes be extended to macroporous, meso-, and nanomaterials?The pore size effect on bioelectrocatalysis was recently modelled by Sugimoto et al.129 in the case of Mv BOD and Dv MF hydrogenase immobilized in mesoporous KB material. The authors concluded that an enzyme molecule encapsulated in pores of comparable size has a higher probability to be in electric connection whatever its orientation.129 On the other hand, for pore size much larger than enzyme dimensions, macroporous and planar electrodes gave similar electrochemical signals. The difference becomes more pronounced with bigger enzyme molecules. These findings have major consequences on the use of porous materials for bioelectrocatalysis, at least if one wants to take benefit from the porosity.
Quinson et al. compared various carbon-based materials (micron-sized graphite particles, CNTs, nanofibers and carbon blacks) showing different morphologies, graphitic structures and specific surface areas.83 Comparative H2 oxidation by immobilized EcHyd1 led to the main conclusion that there was no strong difference in the distribution of orientations of the enzyme as a function of the carbon type. In the case of the carbon blacks, this was explained by the size of the pores which was determined to be in the range 3.5–9 nm within all the carbon materials, i.e. in the same range than the enzyme, thus favoring the electrical contact with hydrogenase. In the particular case of CNTs, accurate comparison was not realized in this study. However, when enzymes are immobilized on CNTs either arranged within films or part of macro- or mesoporous structures, hierarchical porosity may induce heterogeneity of enzyme orientation. Hence, the demonstration of orientation distribution, and the tools to control efficient orientation is certainly the most important breakthrough in the domain for the last two years.
Monsalve et al.57 considered H2 oxidation by Aa MbH immobilized on different CNT surfaces. Not only can CNTs develop large surface areas, but their walls can be chemically modified by oxidative treatment, by π–π stacking of polyaromatic derivatives or by reduction of diazonium salts.84 Thus, they behave as versatile platforms for studying enzyme oriented immobilization. In the case of Aa MbH, three different CNT surfaces were studied, i.e. hydrophobic pristine CNTs, carboxylic- and amino-functionalized CNTs (renamed COOHCNT and NH2CNT respectively). The carboxylic functions were generated by oxidative treatment of CNTs, while amino ones were formed by π–π stacking of aminomethyl-pyrene, inducing negative and positive charges on the CNT walls at neutral pH, respectively. Films of the various CNTs were made by successive drop castings on a PG electrode. The surface area of the modified electrode was estimated from the capacitive current at a potential where faradic processes are minimal, and the catalytic currents were reported to the real electroactive surface area. In that way the catalytic current densities depend on the quantity of enzymes orientated favorably for DET on the CNTs. Only the hydrophilic CNT films, i.e. COOHCNTs or NH2CNTs, gave rise to enhanced catalytic current densities compared to the bare PG electrode. More interestingly, catalytic current density on the positively charged NH2CNTs was at least twice higher than that recorded on negatively charged COOHCNTs. These results provided experimental proof of a better orientation for DET of Aa MbH on positive interfaces, and underlined a full agreement with the model proposed on SAMs and confirmed by MD (Fig. 10).
Contrary to Aa MbH, many studies based on Mv BOD are available to assess the validity of molecular determinants determined on planar electrodes for porous ones. As the first answer, dos Santos et al. modified a PG electrode with different aromatic compounds, bearing amino or carboxyl groups,40 and studied enzymatic O2 reduction by coated Mv BOD. The roughness of the PG surface is sufficient to consider this electrode as non-planar albeit it cannot be considered as a true porous electrode. It was demonstrated that only modifiers containing carboxylates showed enhanced catalytic currents compared to bare PG electrode. This result is in agreement with data on SAMs. The use of aromatic compounds can furthermore play a role by mimicking the natural substrates of BOD.130 Unfortunately, in case of amino derivatives, the MET process was not evaluated, precluding the evaluation of the percentage of well oriented enzymes. Electrodeposited reduced graphene oxide was used as a platform for Mv BOD immobilization.89 High porosity was shown with macropores in the range of 10–20 μm within rGO aggregates. rGO was functionalized by diazonium salt reduction to provide naphtoic acid functionalities on the surface. DET for O2 reduction by Mv BOD was observed, although the addition of ABTS as a redox mediator gave rise to MET currents. This result would suggest a distribution of orientation of the enzyme on the surface. However, the immobilization was made in the presence of a coupling agent which can favor immobilization of enzymes in various orientations.
More insights into Mv BOD orientation in the porous matrix was gained by the use of CNT films. Bilirubin, the natural substrate of Mv BOD, as well as its functional analogues, was used to modify CNTs. With the DET/(DET + MET) ratio more than 90%, enhanced catalytic currents were attributed to the proper orientation of BOD.131,132 The strategy was extended by Lalaoui et al. by use of porphyrin-functionalized CNTs.91 The idea was to promote proper BOD orientation by use of bilirubin precursors. A non-catalytic pH-dependent signal attributed to T1 Cu was observed allowing us to propose the involvement of histidine residue in the electron transfer process. A kinetic model was used to fit the catalytic cyclic voltammograms.120 It was demonstrated that protoporphyrin IX, the closest to the bilirubin structure, enabled proper BOD orientation. At the same time, protoporphyrin derivatives deprived of ionisable carboxylic moieties resulted in much worse orientation arguing again for the importance of negative charge. Given the catalytic current and the coverage of electroactive BOD, a high value of kcat = 2400 s−1 was calculated. However, there is still some doubt whether the non-catalytic signals are related to enzymes effectively participating in catalysis. The same group reported the effect of the nature of chemical functions carried by CNTs on the electrocatalysis by Mv BOD.133,134 While on the negatively charged CNTs, DET occurred at the T1 Cu centre, O2 reduction was shown to occur at much lower potentials on positively charged CNTs. The latter catalytic process was attributed to a catalytic pathway through a partially oxidized form of the TNC, previously identified in the presence of chloride.135
Mazurenko et al. studied in depth the influence of the chemistry of CNTs on the O2 catalytic reduction by Mv BOD using an integrative approach that considers both the enzyme and the electrode surface70 (Fig. 11). Hydrophobic pristine CNTs, COOHCNTs and NH2CNTs were compared. Careful determination of the chemical composition and charge carried by each type of CNTs was made by XPS, and zeta potential was measured as a function of pH. The adsorption of Mv BOD was realized at different pH values in the range of pH 3–8, and the catalytic process for O2 reduction was quantified by electrochemistry and modelling before and after ABTS addition. The study also included the evolution of charges and dipole moments of the enzyme as a function of pH. This strategy allowed to provide a rational understanding of BOD/CNTs interactions. On NH2CNTs, MET/DET ratio increased with decreasing pHs, in relation with progressive appearance of positive charges on CNTs. However, no catalytic wave attributed to an electron pathway through the TNC was observed as stated in the former work by Lalaoui et al. The origin of the discrepancy is not clear yet. It was confirmed that carboxylic functions favor DET and narrow BOD distribution orientation, except at very low pH < 4 where the carboxylic functions are protonated. Similar results were obtained for BOD activity in carbon felts post-functionalized by COOHCNTs.109,136 It is noteworthy that the density of carboxylic functions can be also modulated by the length of CNTs, hence directly influencing O2 catalytic reduction by Mv BOD, with the maximum current obtained for the shortest CNTs, i.e. those bearing more negative charges.137
 | ||
| Fig. 11 Electrostatic interactions between Mv and Bp BODs and CNTs bearing various charges control the DET or MET processes for O2 reduction. Adapted from Mazurenko et al. ACS Appl. Mater. Interfaces, 2016, 8, 23074–23085. Copyright 2017 American Chemical Society. | ||
In parallel with Mv BOD, a thermostable Bp BOD was studied by Mazurenko et al.70 The crystal structure of this BOD is not resolved yet, but a model was constructed based on its sequence. From this model, a high dipole moment associated with a negative charge in a 15 Å sphere around the T1 Cu was determined in the pH range of 4–12. This configuration is totally different from that on Mv BOD, suggesting also a very different catalytic behavior when immobilized on CNTs. As expected from this structural consideration, no DET was obtained on negatively charged CNTs, while a high MET current was measured. Conversely, the ratio MET/DET was close to zero in the pH range of 3–8 used for Bp BOD adsorption (Fig. 11). DET was however also obtained on surfaces presenting weak total charges, as in the case of pristine CNTs or CNFs.103 These last results were attributed to nonpolar residues in the vicinity of T1 Cu.
KB electrodes were chemically modified to favor Mv BOD orientation towards efficient DET for O2 reduction. As obtained with CNTs, bilirubin coating was shown to enhance the catalytic current.138,139 A kinetic analysis of the DET signal led to the conclusion that almost all electroactive BODs adsorb in a proper orientation with some contribution of molecules in other orientations. In another report by the same group, several aromatic amines bearing various end-functions were linked to KB.140 The highest catalytic current was obtained with carboxylate end-function compared to –COOCH3 or –NH3+, with poor response in the case of –NH3+, again in agreement with the statement made on planar electrodes that negative functions promote DET on Mv BOD while positive ones prevent DET.
In conclusion, it appears that unless nanomaterials in which each enzyme molecule enters a pore of similar diameter to enzyme diameter are designed, surface chemical modification is required for enzyme orientation yielding efficient catalysis in porous electrodes. This general rule has been verified with other enzymes useful for H2/O2 EFCs. The immobilization of Df hydrogenase on amino-modified CNTs is fully consistent with a structure of the enzyme showing high value of dipole moment and a strong acidic patch of glutamate residues around the distal FeS.94,141 The case of Re MbH is more astonishing. Re and Aa MbHs share more than 60% of homology. Nevertheless, catalytic activity with Re MbH sharply dropped down to zero as the quantity of negative charges on the CNT surface increased.57 This unexpected result might be explained by Re MbH denaturation induced by strong negative electrode charge in the absence of detergent in the enzyme solution, contrary to Aa MbH. Further conformational studies of enzymes immobilized on strongly charged interfaces are required in the near future.
Generally speaking, rational functionalization of mesoporous and nanomaterials with a large developed surface had drastic consequences on the enhancement of enzymatic current densities. In many cases, the current densities reported for the projected surface area of the electrodes are nowadays close to 10 mA cm−2.
Case of MET in mesoporous and nanomaterials
The problem is somehow different when speaking about MET in mesoporous materials. In this case, enzymes in any orientations can a priori communicate with the electrode via the diffusing redox mediators or through electron hopping between the immobilized redox entities. Any conductive materials that can increase the loading of enzymes, while maintaining its activity and allowing substrate diffusion, should thus be suitable. In the particular case of redox polymers such as widely used redox hydrogels, the polymer itself can be made thick enough to incorporate a large amount of enzymes, although with very thick layers the entire film thickness does not take part in catalysis. Nevertheless, to further increase the currents, there is an advantage to deposit the hydrogel on porous materials as it was highlighted recently for photosystem II immobilized in the redox hydrogels on hierarchical indium tin oxide.142 Some other relevant examples are reported in the literature. CNT polyelectrolytes with viologen substituents acting as redox mediators were synthesized and reacted with Thiocapsa roseopersicina hydrogenase.143 It was proposed that CNTs act as nanoscale backbones with an increased surface area, and as electric wires for both redox mediator and hydrogenase. Oxidized CNTs were mixed with an Os-based redox polymer for Mv BOD encapsulation.144 Enhanced O2 reduction was shown to come from electron transfer through the conductive CNT networks which may assist the hopping process between the osmium entities. One main issue should be to determine the suitable pore size for the porous volume to be accessible. This is well illustrated in a very recent study dedicated to the electroactivity of enzyme embedded in an osmium polymer film deposited on/in nanoporous gold.145 Looking at the enzymatic current as a function of pore size in the range of 10–50 nm, it clearly appears that the polymer/enzyme biocomposite was unable to fully enter the pore structure.Bioelectrode stability: many origins, various solutions
The existence of living organisms relies on the constant renewal of enzymes,146 so that, in vivo, enzymes are functioning only during limited duration.37 It is thus expected that the stability of enzymatic electrodes is a key parameter that restricts EFCs to short term applications. As stated at the beginning of this review, screening biodiversity is the first answer for the discovery of new enzymes able to operate and be stable in various environments, including extreme ones. Otherwise, the stabilization of enzymes by immobilization on solid supports such as electrodes is still an open question. Sensitive tools are required to address this issue, and to be able to discriminate between the different origins of instability of bioelectrodes. Any variation in catalytic current needs to be attributed to higher/lower enzyme amount or more/less active enzyme, which can be itself related to enzyme reorientation, or reconformation, or denaturation. Some recent studies report the use of electrochemistry coupled to quartz crystal microbalance (e-QCM),147,148 or surface plasmon resonance (e-SPR)127 to determine the factors explaining the decrease in catalytic currents with time. It was established that this decrease is not related to enzyme release from the electrode. Enhanced stability by covalent attachment of the enzyme was then attributed to rigidification of the enzyme structure. Ellipsometry,114 PMIRRAS127 and interferometry148 suggested conformational changes in the enzyme structure upon immobilization. The influence of the applied potential, the drastic role of the negative charges used to properly orient the enzyme, and the influence of full enzyme coverage on the catalytic activity were also underlined. These studies have been limited to gold planar electrodes, so far. By modification of the negative charge density of reduced graphene oxide, it was nevertheless shown that the highest Mv BOD coverage was reached with the highest charge density while the highest heterogeneous ET was obtained with the lower charge density.91 This result may also reflect the negative effect of the charges used for enzyme orientation on its conformational stability.Otherwise, one of the main sources of instability in H2/O2 EFCs is the inhibition of hydrogenases by O2. All the [NiFe]-hydrogenases are inactivated under O2, but various states are formed with different reactivation rates according to the hydrogenase type. While the O2-tolerant hydrogenases such as Aa MbH are able to oxidize H2 in the presence of O2, this is not the case for O2-sensitive hydrogenases which require application of very low redox potentials to be reactivated. O2 inhibition of hydrogenases thus restricts the panel of enzymes suitable as anode catalysts in H2/O2 EFCs.
Three main directions have been proposed to overcome aerobic inactivation of hydrogenases. The first one takes profit of the porous matrix which may be used as an electrochemical barrier to O2. Xu et al. compacted CNTs alone or mixed with graphite (G/CNT) to design 0.3 mm thick disk electrodes.149 These electrodes contain pores with diameters in the 10–70 nm range, for which effusion of H2 occurs 4 times faster than O2 in the gaseous phase. Thanks to this property, the porous matrix seems to act as an O2 filter, and hydrogenases deeply embedded in the compact film become protected. Full activity of O2-tolerant EcHyd1 in 78% H2/22% air mixtures was thus observed. More importantly, it was demonstrated that even the O2-sensitive [NiFe]-hydrogenase from E. coli (Hyd2) was able to oxidize H2 in the presence of air. The larger the volume of mesopores, the more efficient the protection was. Depending however on the functionalities carried by CNTs, O2 reduction can be enhanced. This is the case of CNTs modified by π–π stacking of amino-pyrene derivatives on which enhanced O2 reduction most probably leads to reactive oxygen species (ROS). ROS were shown to be more deleterious than O2 for Aa MbH, thus imposing their trapping in the outer layers of the porous matrix.57 A second way of protection of hydrogenases against O2 is to design the EFC itself to restrict O2 diffusion to the anode. This is typically what is done within air-breathing hydrogen EFCs, in which O2 is supplied from the gaseous phase.140,150
We have focused our review on DET processes because they are mainly used in EFCs to avoid the drawbacks linked to redox mediators already discussed above (higher overpotentials, limited kinetics, co-immobilization, toxicity, etc.). Nevertheless, MET has long been and is still nowadays being used in many laboratories, either to study and take benefit of the specific interactions between enzymes and their natural partners – the case of soluble O2-sensitive hydrogenases and polyheme cytochrome c3 is particularly relevant151-or to overcome the difficulties related to enzyme orientation. Redox hydrogels based on osmium redox entities were in that way very popular to embed BODs.152,153 Whether the enzyme is more stable in such polymers compared to direct connection has not been discussed as far as we know. Hydrogenases were also previously incorporated in polymer films such as thin viologen films78,154 or phenothiazine-based polymers.155 Ciaccafava et al. suggested that the phenothiazine entity should play several roles and act as a mediator but also as a ROS scavenger to help Aa MbH to sustain hydrogen oxidation in the presence of oxygen. Morozov et al. showed O2 tolerance of the O2-sensitive hydrogenase from Thiocapsa roseopersicina when embedded in the viologen-based polymer.154 Viologen-based protection of O2-sensitive hydrogenases was recently rationalized by Plumeré et al.77,156–158 It was demonstrated that hydrogen oxidation induced catalytic O2 reduction by the reduced viologen entities at the polymer/solution interface. As a consequence, the bioanode displayed a half-life as high as 46 days. However, it also pointed out that, due to diffusional limitations, only a small fraction of the immobilized enzyme actually participates in the catalysis. ROS production in the film may be also questionable. As an alternative, an integral hydrogenase membrane complex might be used as a biocatalyst for H2 oxidation. As an illustration, Radu et al. studied quinone mediated H2 oxidation by the full heterotrimeric membrane-bound hydrogenase from Re, reconstituted on gold electrodes modified by tethered bilayer lipid membranes.76,159 In the presence of exogenous quinones, the oligomeric state of the enzyme was shown to accelerate enzyme reactivation after O2 addition. However, H2 oxidation occurs at high potential, determined by that of the quinone pool, with a very low current.
The new generation of H2/O2 enzymatic fuel cells
Progressive advancements in nano- mesoporous matrixes suitable for enzyme incorporation, and the understanding of how enzyme orientation proceeds in such environments, have led to progressive net improvements of H2/O2 EFCs. Table 1 summarizes all the reported EFCs since the proof-of-concept. It clearly appears that this is still an emergent device. For comparison, in the same period several hundreds of publications reported glucose/O2 EFCs.| Date | Bioanode | Biocathode | Conditions | Substrate | Membrane | Power density, mW cm−2 | OCV, V | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 2001 | DvH cells on CF MET by MeV in solution | Mv BOD on CF MET by ABTS | 100% H2–100% O2 | Yes | 0.4 | 1.17 | 2 h | 13 | |
| 2005 | Re MbH on PG | Tv LAC on PG | H2/air | No | 0.007 | 0.97 | nd | 48 | |
| 2006 | Rm CH34 MbH on PG | Tv LAC on PG | 3% H2 in air | No | 0.005 | 0.950 | nd | 49 | |
| 2010 | E. coli Hyd1 on PG | Mv BOD on PG modified by 6-amino-2-naphtoïc acid | (A) 100% H2–100% O2 (B) 96% H2–4% O2 (C) 4% H2 in air | (A) Yes, (B) and (C) No | (A) 0.063, (B) 0.013, (C) 0.010 | (A) 0.99, (B) 0.95, (C) 0.93 | nd | 51 | |
| 2012 | E. coli Hyd1 covalently attached to MWCNT modified with butyric acid pyrene | Mv BOD covalently attached to MWCNT modified with butyric acid pyrene | H2/air 80/20 mixture | No | 0.119 | ≈1 | 40% loss of power after 24 h at 0.98 V | 95 | |
| 2012 | Aa MbH covalently attached to SWCNT-COOH | Mv BOD covalently attached to SWCNT-COOH | Anode: 60 °C. Cathode: 25 °C | 100% H2–100% O2 | Yes | 0.300 | 1.1 | 40% loss of power after 24 h at 0.65 V | 53 |
| 2013 | E. coli Hyd1 in compacted mesoporous carbon | Mv BOD in compacted mesoporous carbon | Cathode/anode surface area = 3 | 78% H2–22% air | No | 1.67 (reported to the anode area) | 1.068 | 10% loss after 24 h at 0.8 V. 50% loss after one week | 118 |
| 2014 | Aa MbH in CNF | Bp BOD in CNF | Temperature range from 30 to 80 °C | 100% H2–100% O2 | Yes | 1.5 at 60 °C | 1.06 | 40% after 24 h at 0.5 V and 60 °C | 160 |
| 2014 | Dv MF in MeV hydrogel | Mv BOD covalently linked to carbon cloth | Cathode/anode surface area = 72 | 95% H2–5% O2 | No | 0.178 | 0.947 | 77 | |
| 2014 | Citrobacter sp. S-77 in carbon black | Pt (gas diffusion electrode) | PEMFC MEA, 60 °C | Membrane electrode assembly | 180 | 0.95 | nd | 161 | |
| 2015 | Aa MbH covalent attachment on AuNP | Mv BOD on AuNP | Anode: 60 °C. Cathode: 25 °C | 100% H2–100% O2 | Yes | 0.25 | 1.08 | nd | 106 |
| 2015 | CrHydA1 [FeFe]-hase in MeV-based hydrogels | Mv BOD on carbon cloth | Cathode/anode surface area = 200 | 95% H2–5% O2 | No | 0.225 (reported to anode area) | 1.08 | nd | 157 |
| 2015 | E. coli Hyd1 in mixtures of MWCNT and compacted graphite | Mv BOD in mixtures of MWCNT and compacted graphite | Cathode/anode surface area = 5 | 78% H2–22% O2 | No | 8 mW for 2[4 × 4] multi-cell | 2.09 | No decrease in the intensity of a powered LED for 8 h | 149 |
| 2015 | Aa MbH in CF modified by SWCNT-COOH | Mv BOD in CF modified by SWCNT-COOH | Anode: 60 °C. Cathode: 25 °C | 100% H2–100% O2 | Yes | 410 μW | 1.12 | Data sent wirelessly every 25 s during 7 h | 109 |
| 2015 | Aa MbH on naphtoic-modified MWCNT | Bp BOD on pristine MWCNT | Air-breathing cathode | 100% H2 | No | 0.72 at 40 °C | 0.95 | nd | 150 |
| 2016 | E. coli hyd1 in MWCNT | Mv BOD in MWCNT | Ionic liquid electrolyte | 96% H2–4% O2 | No | 0.007 | 0.86 | Stability for 9 h | 162 |
| 2016 | Dv Miyazaki MbH in Ketjen black modified with phenylenediamine | Mv BOD in Ketjen black modified with aminobenzoic acid | Gas diffusion system | H2/air | No | 6.1 | 1.12 | nd | 140 |
| 2016 | Aa MbH on Pyrene-NH2 modified MWCNT | Mv BOD on MWCNT-COOH | 15% H2–85% O2 | No | 0.07 | 0.95 | nd | 57 | |
| 2016 | Dv Miyazaki MbH in Ketjen black | Mv BOD in Ketjen black modified bilirubin | Gas diffusion system | H2/air | No | 8.4 (calculated from the half cells) | 1.14 | nd | 163 |
A first step was jumped in 2012 in F. Armstrong's laboratory in UK, and in our group. This step was followed by improvements of the bioelectrodes in the two laboratories during the next 4 years, to finally reach a useful power level. Initially, O2-tolerant hydrogenases and Mv BOD were immobilized on functionalized CNTs allowing great enhancement of the direct catalytic currents compared to bare PG. Covalent attachment between the carboxylic functions on CNTs and the enzymes improved the stability of the bioelectrodes. The design of the EFCs was however notably different in the two laboratories.
The EFC from Armstrong's group used EcHyd1 as anodic enzyme.95 No membrane was used to separate the two compartments and the EFC was fed with a 80/20 H2/air mixture. The average power density was 119 μW cm−2. The stability of the EFC was studied at a potential of 0.98 V. 40% loss was observed after 24 h of continuous operation. Fuel and oxidant supply was shown to limit the process. A gas bubbler was also required to be close to the cathode to compensate for the low oxygen availability. One year later, these last issues were partially overcome by using compacted mesoporous carbon in which enzymes were simply incorporated, and by varying the respective dimensions of the anode and cathode.117 Based on the total loading of enzyme, apparent TOFs of 6 s−1 and 0.4 s−1 were calculated for EcHyd1 and Mv BOD respectively. These values are much lower than expected, suggesting that enzymes buried inside the carbon disk would be not operational. Reproportion of the cathode vs. anode sizes allowed the increase of the power density to 1.7 mW cm−2. 90% of the EFC performance was maintained after 24 h at 0.8 V, and 54% after 7 days of continuous operation. Performance loss was suggested to be linked to enzyme inactivation, mainly because H2 depletion during catalysis led to a high local O2/H2 ratio.
Up-scaling was made in 2015 by arranging multiple cells in parallel or in series.149 The OCV and maximum power of a module composed of two stacks of four cells in parallel were 2.09 V and 7.84 mW respectively. The test bed was able to power an electronic clock and five red LEDs during 8 h with no decrease in light intensity. Miniaturization is another challenge for H2/O2 EFCs. Wang et al. recently reported a miniaturized EFC based on EcHyd1 and Mv BOD immobilized on the carbon electrode and maintained wet within a microvolume of buffer surrounded by an immiscible ionic liquid.162 Although a low OCV was observed, the small power density recorded, i.e. 4 μW cm−2, was stable for 9 h. This configuration allows operation in typically 6 μl of electrolyte, and opens up new avenues towards miniaturization of H2/O2 EFCs.
In our group, we first maintained the Nafion membrane separator and worked with 100% H2 and 100% O2 in the anodic and cathodic compartments, respectively.53 Should this membrane be maintained in a future device, the type of separator as a function of the geometry of the cell, and in particular as a function of pH should be particularly examined. Aa MbH was used at the anode, allowing it to operate between 25 °C and 80 °C. The concentration of BOD was varied on the cathode in order to study the three operating cell limitations, i.e. balance between the anode and cathode, anode limitation or cathode limitation. A potential zone where the EFC could efficiently operate was defined controlled by Aa MbH inactivation. The maximum power density was 300 μW cm−2 at 0.65 V. The EFC exhibited more than 60% of its power initial value after 24 h of continuous operation at 0.65 V. Otherwise, as amine groups were demonstrated to favour Aa MbH orientation, COOHCNTs were replaced by CNTs modified by π–π stacking of an amino-pyrene derivative.57 Membrane-less EFCs were designed with different H2/air mixtures up to 15% H2/85% air. Although the CNT film was not optimized, the EFC was demonstrated to operate under such extreme conditions for the hydrogenase, delivering a maximum power of 70 μW cm−2. This is an interesting result, since, as related above, platinum is not very efficient in poor H2 gas mixtures. Other nanomaterials such as AuNPs were used for the same enzyme immobilization. A maximum power density of 250 μW cm−2 at 0.8 V was obtained,106 but the behavior of Mv BOD on AuNPs was shown to severely limit the device. To reach the net power required for a real device, Monsalve et al. up scaled the bioelectrodes by using cylinders of carbon felts. Post functionalization by CNTs bearing carboxylic functionalities were suitable for high loading of active Aa MbH and Mv BOD. The OCV and short circuit current were 1.12 V and 765 μA, respectively. A maximum power of 410 μW was obtained. Associated with suitable electronic circuits, and in particular to a supercapacitor able to store the energy, this EFC was used to power a wireless transmission system. The device powered by the H2/O2 EFC was measuring in real time five different parameters and was sending them wirelessly to a laptop every 25 s for 7 h.
The optimal working temperatures of these former EFCs based on Aa MbH at the anode were 60 °C and 25 °C in the anodic and cathodic compartments, respectively. The limitation induced by denaturation of Mv BOD at temperatures higher than 40 °C was overcome by the use of the thermostable Bp BOD at the cathode.160 In a new EFC, de Poulpiquet et al. immobilized the enzymes in carbon nanofibers. A power density of 1.5 mW cm−2 was reached at 60 °C. Thanks to the thermostability of both Aa MbH and Bp BOD, the EFC was proved to be able to operate from 30 to 80 °C, i.e. in extreme situation for classical enzymes, enlarging the operational conditions of EFCs.
Are H2/O2 EFCs limited to O2-tolerant hydrogenases on the anodic side? Three different configurations offer the opportunity to enlarge the panel of hydrogenases suitable for the EFC (Fig. 12). The first one takes advantage of the protective role of mesoporous films against O2 discussed before. Thus, Xu et al. designed a membrane-less EFC based on the O2-sensitive hydrogenase from E. coli embedded in compacted CNTs.149 Under 89% H2–11% air, the EFC delivered a maximum volumetric power density close to 100 μW cm−3 at 0.7 V. The OCV was 1.114 V, a value higher than that one obtained with EcHyd1, in agreement with the comparative catalytic offsets of both enzymes. However, the stability was much less. More than 50% of the activity was lost after 6 h of operation, compared to 54% remaining activity under the same conditions after 7 days with the E. coli O2-tolerant hydrogenase.
 | ||
| Fig. 12 Main H2/O2 EFC configurations. (A) EFC with a membrane separator and direct connection of enzymes; (B) dual gas-diffusion EFC with direct connection of enzymes; (C) membrane-less EFC in low air/H2 gas mixtures with direct connection of enzymes; (D) membrane-less EFC with direct connection of BOD at the cathode and hydrogenase connected through a redox hydrogel at the anode. In (C) and (D) cathodes are oversized to compensate low cathode substrate availability. | ||
The second configuration consists of air-breathing systems that have been proved to be efficient in fuel cell technology.164 As this design was shown to be beneficial to prevent hydrogenase from inactivation, hydrogen/air-breathing EFC was evaluated with both O2-tolerant and O2-sensitive hydrogenases. Lalaoui et al. used Aa MbH and Bp BOD as thermostable enzymes immobilized on CNTs with suitable surface chemistry as defined above. The membrane-less air-breathing EFC was operated at 45 °C, and a maximum power density of 0.72 mW cm−2 was obtained at 0.6 V. The demonstration of hydrogenase protection by the air-breathing cathode design was even more relevant when an O2-sensitive hydrogenase was used instead of O2-tolerant hydrogenases. So et al. immobilized the O2-sensitive Dv MF hydrogenase and Mv BOD in KB deposited on waterproof carbon cloth.163 In this case, the anode was also supplied by H2 as a gas. As discussed before, this configuration was proposed to protect the hydrogenase from anaerobic inactivation. The performances of the bioanode and biocathode were evaluated separately and served to calculate the cell voltage and cell power density. Values of 1.14 V and 8.4 mW cm−2 at 0.7 V were calculated, for OCV and power density respectively. The same group further functionalized KB with carboxylic- and amino-functions to fit suitable orientation of Mv BOD and Dv MH, respectively.140 Polytetrafluoroethylene was mixed to KB in order to get the required hydrophobicity for gas permeation. Very interestingly, the dual gas-diffusion membrane-less hydrogen/air-breathing EFC displayed a high power density of 6.1 mW cm−2 at 0.72 V under quiescent conditions. Nevertheless, anaerobic inactivation of hydrogenase was observed at high current densities due to the hydrogen depletion. While being actively developed and very promising in terms of performance, air-breathing systems are however rarely evaluated for long-term stability of enzymes that might occur in direct contact with gaseous phase in such configuration.
The third configuration is based on the use of low potential redox hydrogels for entrapment of hydrogenases. The above-mentioned EFCs were based on direct connection of enzymes. Here, H2 oxidation proceeds through a MET process. Plumeré et al. constructed a membrane-less H2/O2 EFC based on the O2-sensitive Dv MF hydrogenase and Mv BOD at the anode and cathode, respectively.77 To be able to prove the protective role of the redox hydrogels against hydrogenase inactivation under extreme conditions, 95% H2–5% O2 gas mixture composition was used, and the cathode was oversized. Mv BOD was classically immobilized on carbon modified by carboxylic-functionalized CNTs. Under these conditions, a maximum power density of 180 μW cm−2 was obtained, with an OCV of 947 mV. More interestingly, the catalytic current delivered by the bioanode remained unaffected even when the EFC approached short-circuit conditions where the anode potential reaches very high oxidative values. The approach based on viologen-based redox hydrogels was extended to [FeFe] hydrogenase from Chlamydomonas reinhardtii, a hydrogenase irreversibly inactivated by O2. The designed EFC delivered a maximum power density of 225 μW cm−2.157
Conclusion and outlook
During the last decade, discovery of new hydrogenases and BODs in the biodiversity, associated with deep understanding of their efficient connection to electrochemical interfaces has allowed us to envision electricity production through H2/O2 EFCs (Fig. 12). Although these EFCs were developed later compared to the glucose/O2 devices, they rapidly outperformed the sugar-based EFCs. This fast improvement is most certainly related to the deep knowledge of the structure of the hydrogenases and the way they behave on electrodes acquired before their use in biotechnological devices. Having a high OCV linked to the nature of oxidant and fuel and the fast kinetics of the enzymes, they nowadays reach power levels compatible with use in real applications. Before commercialization however, there is still a lot of research to be done, to increase short-term and long-term stability and improve electrical connection. These two limitations impose to couple electrochemistry to surface spectroscopy to get a full image of the active enzymes on an electrode surface. Modeling is also required to get a rationalization of the interfaces, to predict a suitable porous matrix for enhanced electrical connection of enzymes and fast substrate diffusion. Recent development in small paper fuel cell would certainly be relevant for EFC designs.165Interestingly, the research in multi-centre redox enzymes has stimulated intensive research in biomimetic inorganic chemistry and artificial enzymes.166–169 As an example, until recent years, biomimetic catalysts of [NiFe]-hydrogenases were only able to catalyze H2 oxidation in organic solvents or in acidic aqueous solution.170,171 Very recently,172 a nickel–diphosphine complex was immobilized on functionalized CNTs. High activity toward H2 oxidation over a pH range of 0.3–7 was obtained, with a TOF of 22 s−1 at pH 7. A fuel cell was mounted based on this inorganic catalyst at the anode and Mv BOD at the cathode. At 25 °C and pH 5, the fuel cell delivered 1.85 mW cm−2 at 0.6 V, which compares well with the hydrogenase-based EFCs. Talking about advantages of enzymes over inorganic catalysts, one must be reminded, however, that single enzyme molecule activity outperforms that of platinum or any other chemical catalyst, provided a better long-term stability and more efficient connection are reached. They are widespread in the environment, even more than reported yet, suggesting new properties to be discovered and exploited soon.19 The occurrence of O2-tolerant hydrogenases purified from very different microorganisms underlines the relevance of screening biodiversity to look for new enzymes with new properties suitable for H2/O2 EFCs, such as tolerance to high potentials, pressure, halides, ionic strength, large range of operational temperatures, low pHs, etc. It should also be highly interesting to search for BODs with higher affinity to O2, higher reduction potentials, or being more active at neutral pHs. Mass production of these enzymes should not be a limitation as soon as commercialization appears on the market. Whatever their future, H2/O2 EFC development is a nice example of applicative research which is derived from fundamental ones, and which induces new fundamental research studies in an interdisciplinary approach. Other devices in the energy domain nowadays already take advantage or will take advantage in the close future of the advances in hydrogenase electrochemistry. This is the case of water splitting,173 CO2 enzymatic reduction174 or ammonia-producing H2/N2 EFC.175
Acknowledgements
The authors thank ANR for financial support (CAROUCELL-ANR-13-BIOME-0003-02). This work was also supported by A*MIDEX Marseille (ANR-11-IDEX-0001-02).Notes and references
- M. Rasmussen, S. Abdellaoui and S. Minteer, Enzymatic biofuel cells: 30 years of critical advancements, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- A. Yahiro, S. Lee and D. Kimble, Bioelectrochemistry. I. Enzyme utilizing biofuel cell studies, Biochim. Biophys. Acta, 1964, 88, 375–383 CAS.
- A. Babadi, S. Bagheri and S. Hamid, Progress on implantable biofuel cell: nano-carbon functionalization for enzyme immobilization enhancement, Biosens. Bioelectron., 2016, 79, 850–860 CrossRef CAS PubMed.
- S. Cosnier, A. Gross, A. Le Goff and M. Holzinger, Recent advances on enzymatic glucose/oxygen and hydrogen/oxygen biofuel cells : achievement and limitations, J. Power Sources, 2016, 325, 252–263 CrossRef CAS.
- N. Mano, J. Fernandez, Y. Kim, W. Shin, A. Bard and A. Heller, Oxygen is electroreduced to water on a « wired » enzyme electrode at a lesser overpotential than on platinum, J. Am. Chem. Soc., 2003, 125, 15290–15291 CrossRef CAS PubMed.
- S. Cosnier, A. Le Goff and M. Holzinger, Towards glucose biofuel cells implanted in human body for powering artificial organs: review, Electrochem. Commun., 2014, 38, 19–23 CrossRef CAS.
- E. Katz and K. MacVittie, Implanted biofuel cells operating in vivo-methods, applications and perspectives, Energy Environ. Sci., 2013, 6, 2791–2803 CAS.
- M. Falk, V. Andoralov, M. Silow, M. Toscano and S. Shleev, Miniature biofuel cell as a potential power source for glucose-sensing contact lenses, Anal. Chem., 2013, 85, 6342–6348 CrossRef CAS PubMed.
- Y. Ogawa, K. Kato, T. Miyake, K. Nagamine, T. Ofuji, S. Yoshino and M. Nishizawa, Organic transdermal iontophoresis patch with built-in biofuel cell, Adv. Healthcare Mater., 2015, 4, 506–510 CrossRef CAS PubMed.
- A. de Poulpiquet, D. Ranava, K. Monsalve, M. T. Giudici-Orticoni and E. Lojou, Biohydrogen for a new generation of H2/O2 biofuel cells: a sustainable energy perspective, ChemElectroChem, 2014, 1, 1724–1750 CrossRef CAS.
- P. Chenevier, L. Mugherli, S. Darbe, L. Darchy, S. DiManno, P. Tran, F. Valentino, M. Iannello, A. Volbeda, C. Cavazza and V. Artero, Hydrogenase enzymes: application in biofuel cells and inspiration for the design of noble-metal free catalysts for H2 oxidation, C. R. Chim., 2013, 16, 491–505 CrossRef CAS.
- M. Stephenson and L. Stickland, Hydrogenase: a bacterial enzyme activating molecular hydrogen: the properties of the enzyme, Biochem. J., 1931, 25, 205–214 CrossRef CAS PubMed.
- S. Tsujimura, M. Fujita, H. Tatsumi, K. Kano and T. Ikeda, Bioelectrocatalysis-based dihydrogen/dioxygen fuel cell operating at physiological pH, Phys. Chem. Chem. Phys., 2001, 3, 1331–1335 RSC.
- E. Lojou, M. C. Durand, A. Dolla and P. Bianco, Hydrogenase activity control at Desulfovibrio vulgaris cell-coated carbon electrodes: biochemical and chemical factors influencing the mediated electrocatalysis, Electroanalysis, 2002, 14, 913–922 CrossRef CAS.
- B. Pohorelic, J. Voordouw, E. Lojou, A. Dolla, J. Harder and G. Voordouw, Effects of deletion of genes encoding Fe-only hydrogenase of Desulfovibrio vulgaris Hildenborough on hydrogen lactate metabolism, J. Bacteriol., 2002, 184, 679–686 CrossRef CAS PubMed.
- B. Schink and I. Probst, Competitive inhibition of the membrane-bound hydrogenase of Alcaligenes eutrophus by molecular oxygen, Biochem. Biophys. Res. Commun., 1980, 95, 1563–1569 CrossRef CAS PubMed.
- P. Vignais and B. Billoud, Occurrence, classification and biological function of hydrogenases: an overview, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- C. Greening, A. Biswas, C. Carere, C. Jackson, M. Taylor, M. Stott, G. Cook and S. Morales, Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilized energy source for microbial growth and survival, ISME J., 2016, 10, 761–777 CrossRef CAS PubMed.
- H. Shafaat, O. Rüdiger, H. Ogata and W. Lubitz, [NiFe] hydrogenases: a common active site for hydrogen metabolism under diverse conditions, Biochim. Biophys. Acta, 2013, 1827, 986–1002 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, Structure and function of [NiFe] hydrogenases, J. Biochem., 2016, 160, 251–258 CrossRef CAS PubMed.
- L. Flanagan and A. Parkin, Electrochemical insights into the mechanism of NiFe membrane-bound hydrogenases, Biochem. Soc. Trans., 2016, 44, 315–328 CrossRef CAS PubMed.
- R. Evans, E. Brooke, S. Wehlin, E. Nomerotskaia, F. Sargent, S. Carr, S. Phillips and F. Armstrong, Mechanism of hydrogen activation by [NiFe] hydrogenases, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS PubMed.
- C. Greco, V. Fourmond, C. Baffert, P. Wang, S. Dementin, P. Bertrand, M. Bruschi, J. Blumberger, L. de Gioia and C. Leger, Combining experimental and theoretical methods to learn about the reactivity of gas-processing metalloenzymes, Energy Environ. Sci., 2014, 7, 3543–3573 CAS.
- E. Solomon, U. Sundaram and T. Machonkin, Multicopperoxidases and oxygenases, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS PubMed.
- E. Solomon, A. Augustine and J. Yoon, O2 reduction to H2O by the multicopper oxidases, Dalton Trans., 2008, 3921–3932 RSC.
- E. Solomon, Dioxygen binding, activation, and reduction to H2O by Cu enzymes, Inorg. Chem., 2016, 55, 6364–6375 CrossRef CAS PubMed.
- H. Komori and Y. Higuchi, Structural insights into the O2 reduction mechanism of multicopper oxidase, J. Biochem., 2015, 158, 293–298 CrossRef CAS PubMed.
- N. Mano and L. Edembe, Bilirubin oxidases in bioelectrochemistry: features and recent findings, Biosens. Bioelectron., 2013, 50, 478–485 CrossRef CAS PubMed.
- F. Tasca, D. Farias, C. Castro, C. Acuna-Rougier and R. Antiochia, Bilirubin oxidase from Myrothecium verrucaria physically absorbed on graphite electrodes. Insights into the alternative resting form and the sources of activity loss, PLoS One, 2015, 10, e0132181 Search PubMed.
- M. Dagys, A. Laurynenas, D. Ratautas, J. Kulys, R. Vidziunaite, M. Talaikis, G. Niaura, L. Marcinkeviciene, R. Meskys and S. Shleev, Oxygen electroreduction catalysed by lacase wired to gold nanoparticle via trinuclear copper cluster, Energy Environ. Sci., 2017, 10, 498–502 CAS.
- A. de Poulpiquet, C. Kjaergaard, J. Rouhana, I. Mazurenko, P. Infossi, S. Gounel, R. Gadiou, M. T. Giudici-Orticoni, E. Solomon, N. Mano and E. Lojou, Mechanism of chloride inhibition of bilirubin oxidases and its dependence on potential and pH, ACS Catal., 2017, 7, 3916–3926 CrossRef CAS.
- P. Agbo, J. Heath and H. Gray, Modeling dioxygen reduction at multicopper oxidase cathodes, J. Am. Chem. Soc., 2014, 136, 13882–13887 CrossRef CAS PubMed.
- S. Varfolomeev, A. Yaropolov, I. Berezin, M. Tarasevich and V. Bogdanovskaya, Bioelectrocatalysis. Hydrogenase as catalyst of electrochemical hydrogen ionization, Bioelectrochem. Bioenerg., 1977, 4, 314–326 CrossRef CAS.
- F. Armstrong, H. Hill and N. Walton, Direct electrochemistry of redox proteins, Acc. Chem. Res., 1988, 21, 407–413 CrossRef CAS.
- A. Heller, Electron-conducting redox hydrogels: design, characteristics and synthesis, Curr. Opin. Chem. Biol., 2006, 10, 664–672 CrossRef CAS PubMed.
- J. Masa and W. Schuhmann, Electrocatalysis and bioelectrocatalysis – Distinction without a difference, Nano Energy, 2016, 29, 466–475 CrossRef CAS.
- K. Vincent, A. Parkin and F. Armstrong, Investigating and exploiting the electrocatalytic properties of hydrogenases, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- J. Cracknell, K. Vincent and F. Armstrong, Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- L. Dos Santos, V. Climent, C. Blanford and F. Armstrong, Mechanistic studies of the blue Cu enzyme, bilirubin oxidase, as a highly efficient electrocatalyst for the oxygen reduction reaction, Phys. Chem. Chem. Phys., 2010, 12, 13962–13974 RSC.
- M. Tominaga, A. Sasaki and M. Togami, Laccase bioelectrocatalyst at a steroid-type biosurfactant-modified carbon nanotube surface, Anal. Chem., 2015, 87, 5417–5421 CrossRef CAS PubMed.
- S. Shleev, V. Andoralov, D. Pankratov, M. Falk, O. Aleksejeva and Z. Blum, Oxygen electroreduction versus bioelectroreduction: direct electron transfer approach, Electroanalysis, 2016, 28, 2270–2287 CrossRef CAS.
- A. Christenson, S. Shleev, N. Mano, A. Heller and L. Gorton, Redox potentials of the blue active sites of bilirubin oxidases, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1634–1641 CrossRef CAS PubMed.
- N. Mano, V. Soukharev and A. Heller, A laccase-wiring redox hydrogel for efficient catalysis of O2 electroreduction, J. Phys. Chem. B, 2006, 110, 11180–11187 CrossRef CAS PubMed.
- A. Jones, E. Sillery, S. Albracht and F. Armstrong, Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum electrode, Chem. Commun., 2002, 866–867 RSC.
- A. Karyakin, S. Morozov, O. Voronin, N. Zorin, E. Karyakina, V. Fateyev and S. Cosnier, The limiting performance characteristics in bioelectrocatalysis of hydrogenase enzymes, Angew. Chem., Int. Ed., 2007, 46, 7244–7246 CrossRef CAS PubMed.
- T. Woolerton and K. Vincent, Oxidation of dilute H2 and H2/O2 mixtures by hydrogenases and Pt, Electrochim. Acta, 2009, 54, 5011–5017 CrossRef CAS.
- K. Vincent, J. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. Armstrong, Electrocatalytic hydrogen oxidation by an enzyme at high carbon monoxide or oxygen levels, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951–16954 CrossRef CAS PubMed.
- K. Vincent, J. Cracknell, J. Clark, M. Ludwig, O. Lenz, B. Friedrich and F. Armstrong, Electricity from low-level H2 in still air – an ultimate test for an oxygen tolerant hydrogenase, Chem. Commun., 2006, 5033–5035 RSC.
- M. Lukey, A. Parkin, M. Roessler, B. Murphy, J. Harmer, T. Palmer, F. Sargent and F. Armstrong, How Escherichia coli is equipped to oxidize hydrogen under different redox conditions, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS PubMed.
- A. Wait, A. Parkin, G. Morley, L. dos Santos and F. Armstrong, Characteristics of enzyme-based hydrogen fuel cells using an oxygen-tolerant hydrogenase as the anodic catalyst, J. Phys. Chem., 2010, 114, 12003–12009 CAS.
- M. Pandelia, V. Fourmond, P. Tron-Infossi, E. Lojou, P. Bertrand, C. Leger, M. T. Giudici-Orticoni and W. Lubitz, Membrane-bound hydrogenase I from the hyperthermophilic bacterium Aquifex aeolicus: enzyme activation, redox intermediates and oxygen tolerance, J. Am. Chem. Soc., 2010, 132, 6991–7004 CrossRef CAS PubMed.
- A. Ciaccafava, A. de Poulpiquet, V. Techer, M. T. Giudici-Orticoni, S. Tingry, C. Innocent and E. Lojou, An innovative powerful and mediatorless H2/O2 biofuel cell based on an outstanding bioanode, Electrochem. Commun., 2012, 23, 25–28 CrossRef CAS.
- A. Parkin, L. Bowman, M. Roessler, R. Davies, T. Palmer, F. Armstrong and F. Sargent, How Salmonella oxidises H2 under aerobic conditions, FEBS Lett., 2012, 586, 536–544 CrossRef CAS PubMed.
- M. Brugna-Guiral, P. Tron, W. Nitschke, K.-O. Stetter, B. Burlat, B. Guigliarelli, M. Bruschi and M. T. Giudici-Orticoni, [NiFe] hydrogenases from the hyperthermophilic bacterium Aquifex aeolicus: properties, function, and phylogenetics, Extremophiles, 2003, 7, 145–157 CAS.
- X. Luo, P. Tron-Infossi, M. Brugna, M. T. Giudici-Orticoni and E. Lojou, Immobilization of the hyperthermophilic hydrogenase from Aquifex aeolicus bacterium onto gold and carbon nanotube electrodes for efficient H2 oxidation, J. Biol. Inorg Chem., 2009, 14, 1275–1288 CrossRef CAS PubMed.
- K. Monsalve, I. Mazurenko, C. Gutierrez-Sanchez, M. Ilbert, P. Infossi, S. Frielingsdorf, M. T. Giudici-Orticoni, O. Lenz and E. Lojou, Impact of Carbon Nanotube Surface Chemistry on Hydrogen Oxidation by membrane-bound oxygen-tolerant hydrogenases, ChemElectroChem., 2016, 3, 2179–2188 CrossRef CAS.
- K.-S. Yoon, K. Fukuda, K. Fujisawa and H. Nishihara, Purification and characterization of a highly thermostable, oxygen-resistant, respiratory [NiFe]-hydrogenase from a marine, aerobic, hydrogen-oxidizing bacterium Hydrogenovibrio marinus, Int. J. Hydrogen Energy, 2011, 36, 7081–7088 CrossRef CAS.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. Spahn, The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- Y. Shomura, K. Yoon, H. Nishihara and Y. Higuchi, Structural basis for a [4Fe-3S] cluster in the oxygen-tolerant membrane-bound [NiFe]-hydrogenase, Nature, 2011, 479, 253–256 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J. Mouesca, A. Parkin, M. Roessler, F. Armstrong and J. Fontecilla-Camps, X-ray crystallographic and computational studies of the O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed.
- L. Bowman, L. Flanagan, P. Fyfe, A. Parkin, W. Hunter and F. Sargent, How the structure of the large subunit controls function in an oxygen-tolerant [NiFe]-hydrogenase, Biochem. J., 2014, 458, 449–458 CrossRef CAS PubMed.
- J. Fritsch, O. Lenz and B. Friedrich, Structure, function and biosynthesis of O2-tolerant hydrogenases, Nat. Rev. Microbiol., 2013, 11, 106–114 CrossRef CAS PubMed.
- F. Armstrong, R. Evans, S. Hexter, B. Murphy, M. Roessler and P. Wulff, Guiding principles of hydrogenase catalysis instigated and clarified by protein film electrochemistry, Acc. Chem. Res., 2016, 49, 884–892 CrossRef CAS PubMed.
- E. Brooke, R. Evans, S. Islam, G. Roberts, S. Wehlin, S. Carr, S. Phillips and F. Armstrong, Importance of the active site “canopy” residues in an O2-tolerant [NiFe]-hydrogenase, Biochemistry, 2017, 56, 132–142 CrossRef CAS PubMed.
- A. Hamdan, P. P. Liebgott, V. Fourmond, O. Gutierrez-Sanz, A. De Lacey, P. Infossi, M. Rousset, S. Dementin and C. Léger, Relation between anaerobic inactivation and oxygen tolerance in a large series of NiFe hydrogenase mutants, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19916–19921 CrossRef PubMed.
- P. Wulff, C. Thomas, F. Sargent and F. Armstrong, How the oxygen tolerance of a [NiFe]-hydrogenase depends on quaternary structure, J. Biol. Inorg Chem., 2016, 21, 121–134 CrossRef CAS PubMed.
- E. Suraniti, S. Tsujimura, F. Durand and N. Mano, Thermophilic biocathode with bilirubin oxidase from Bacillus pumilus, Electrochem. Commun., 2013, 26, 41–44 CrossRef CAS.
- F. Durand, S. Gounel, C. Kjaergaard, E. Solomon and N. Mano, Bilirubin oxidase from Magnaporthe oryzae: an attractive new enzyme for biotechnological applications, Appl. Microbiol. Biotechnol., 2012, 96, 1489–1498 CrossRef CAS PubMed.
- I. Mazurenko, K. Monsalve, J. Rouhana, P. Parent, C. Laffon, A. Le Goff, S. Szunerits, R. Boukherroub, M. T. Giudici-Orticoni, N. Mano and E. Lojou, How the Intricate Interactions between Carbon Nanotubes and Two Bilirubin Oxidases Control Direct and Mediated O2 Reduction, ACS Appl. Mater. Interfaces, 2016, 8, 23074–23085 CAS.
- A. Volbeda, C. Damault, A. Parkin, F. Sargent, F. Armstrong and J. Fontecilla-Camps, Crystal structure of the O2-tolerant membrane-bound hydrogenase 1 from Escherichia coli in complex with its cognate cytochrome b, Structure, 2013, 21, 184–190 CrossRef CAS PubMed.
- A. Ciaccafava, C. Hamon, P. Infossi, V. Marchi, M. T. Giudici-Orticoni and E. Lojou, Light-induced reactivation of O2-tolerant membrane-bound [Ni-Fe] hydrogenase from the hyperthermophilic bacterium Aquifex aeolicus under turnover conditions, Phys. Chem. Chem. Phys., 2013, 39, 16463–16467 RSC.
- C. Hamon, A. Ciaccafava, P. Infossi, R. Puppo, P. Even-Hernandez, E. Lojou and V. Marchi, Synthesis and Enzymatic activity of an O2 resistant hydrogenase/CdSe@CdS quantum rod bioconjugate, Chem. Commun., 2014, 50, 4989–4992 RSC.
- K. So, Y. Kitazumi, O. Shirai, K. Kurita, H. Nishihara, Y. Higuchi and K. Kano, Gas-diffusion and direct-electron-transfer-type bioanode for hydrogen oxidation with oxygen-tolerant [NiFe]-hydrogenase as an electrocatalyst, Chem. Lett., 2014, 43, 1575–1577 CrossRef CAS.
- K. So, Y. Kitazumi, O. Shirai, K. Kurita, H. Nishihara, Y. Higuchi and K. Kano, Kinetic analysis of inactivation and enzyme reaction of oxygen-tolerant [NiFe]-hydrogenase at direct electron-transfer anode, Bull. Chem. Soc. Jpn., 2014, 87, 1177–1185 CrossRef.
- V. Radu, S. Frielingsdorf, S. Evans, O. Lenz and L. Jeuken, Enhances oxygen-tolerance of the full heterotrimeric membrane-bound [NiFe]-hydrogenase of Ralstonia eutropha, J. Am. Chem. Soc., 2014, 136, 8512–8515 CrossRef CAS PubMed.
- N. Plumeré, O. Rüdiger, A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Lubitz and W. Schuhmann, A redox hydrogel protects hydrogenase from high-potential deactivation and oxygen damage, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- J. Baur, A. Le Goff, S. Dementin, M. Holzinger, M. Rousset and S. Cosnier, Three-dimensional carbon nanotube-polypyrrole-[NiFe] hydrogenase electrodes for the efficient electrocatalytic oxidation of H2, Int. J. Hydrogen Energy, 2011, 36, 12096–12101 CrossRef CAS.
- A. Walcarius, S. Minteer, J. Wang, Y. Lin and A. Merkoci, Nanomaterials for bio-functionalized electrodes: recent trends, J. Mater. Chem. B, 2013, 1, 4878–4908 RSC.
- A. de Poulpiquet, A. Ciaccafava and E. Lojou, New trends in enzyme immobilization at nanostructured interfaces for efficient electrocatalysis in biofuel cells, Electrochim. Acta, 2014, 126, 104–114 CrossRef CAS.
- S. Minteer, P. Atanassov, H. Luckarift and G. Johnson, New materials for biological fuel cells, Mater. Today, 2012, 15, 166–173 CrossRef CAS.
- R. Milton, T. Wang, K. Knoche and S. Minteer, Tailoring biointerfaces for electrocatalysis, Langmuir, 2016, 32, 2291–2301 CrossRef CAS PubMed.
- J. Quinson, R. Hidalgo, P. Ash, F. Dillon, N. Grobert and K. Vincent, Comparison of carbon materials as electrodes for enzyme electrocatalysis: hydrogenase as a case study, Faraday Discuss., 2014, 172, 473–496 RSC.
- P. Unwin, A. Güell and G. Zhang, Nanoscale electrochemistry of sp2 carbon materials: from graphite and graphene to carbon nanotubes, Acc. Chem. Res., 2016, 49, 2041–2048 CrossRef CAS PubMed.
- A. Geim and K. Novoselov, The rise of graphene, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- J. Filip and J. Tkac, Is graphene worth using in biofuel cells?, Electrochim. Acta, 2014, 136, 340–354 CrossRef CAS.
- A. Navaee, A. Salimi and F. Jafari, Electrochemical pretreatment of amino-carbon nanotubes on graphene support as a novel platform for bilirubin oxidase with improved bioelectrocataytic activity towards oxygen reduction, Chem.–Eur. J., 2015, 21, 4949–4953 CrossRef CAS PubMed.
- A. Campbell, M. Jose, S. Marx, S. Cornelius, R. Koepsel, M. Islam and A. Russel, Improved power density of an enzymatic biofuel cell with fibrous supports of high curvatures, RSC Adv., 2016, 6, 10150–10158 RSC.
- C. Di Bari, A. Goni-Urtiaga, M. Pita, S. Shleev, M. Toscano, R. Sainz and A. De Lacey, Fabrication of high surface area graphene electrodes with high performance towards enzymatic oxygen reduction, Electrochim. Acta, 2016, 191, 500–509 CrossRef CAS.
- J. Filip, A. Andicsova-Eckstein, A. Vikartovska and J. Tkac, Immobilization of bilirubin oxidase on graphene oxide flakes with different negative charge density for oxygen reduction. The effect of GO charge density on enzyme coverage, electron transfer rate and current density, Biosens. Bioelectron., 2017, 89, 384–389 CrossRef CAS PubMed.
- N. Lalaoui, A. Le Goff, M. Holzinger and S. Cosnier, Fully oriented bilirubin oxidase on porphyrin-functionalized carbon nanotube electrodes for electrocatalytic oxygen reduction, Chem.–Eur. J., 2015, 21, 16868–16873 CrossRef CAS PubMed.
- M. A. Alonso-Lomillo, O. Rudiger, A. Maroto-Valiente, M. Velez, I. Rodriguez-Ramos, F. Munoz, V. Fernandez and A. De Lacey, Hydrogenase-coated carbon nanotubes for efficient H2 oxidation, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- F. Hoeben, I. Heller, S. Albracht, C. Dekker, S. Lemay and H. Heering, Polymyxin-coated an and carbon nanotube electrodes for stable [NiFe]-hydrogenase film voltammetry, Langmuir, 2008, 24, 5925–5931 CrossRef CAS PubMed.
- E. Lojou, X. Luo, N. Candoni, M. Brugna, S. Dementin and M. T. Giudici-Orticoni, Hydrogenases as biocatalysts for biofuel cells: efficient H2 oxidation at electrodes modified with carbon nanotubes, J. Biol. Inorg Chem., 2008, 13, 1157–1167 CrossRef CAS PubMed.
- S. Krishnan and F. Armstrong, Order-of-magnitude enhancement of an enzymatic hydrogen-air fuel cell based on pyrenyl carbon nanostructures, Chem. Sci., 2012, 3, 1015–1023 RSC.
- A. de Poulpiquet, A. Ciaccafava, K. Szot, B. Pillain, P. Infossi, M. Guiral, M. Opallo, M. T. Giudici-Orticoni and E. Lojou, Exploring properties of a hyperthermophilic membrane-bound hydrogenase at carbon nanotube modified electrodes for a powerful H2/O2 biofuel cell, Electroanalysis, 2013, 25, 685–695 CrossRef CAS.
- A. de Poulpiquet, A. Ciaccafava, S. Benomar, M. T. Giudici-Orticoni and E. Lojou, in Synthesis and applications of carbon nanotubes and their composites, ed. S. Suzuki, Intech, 2013, pp. 433–466 Search PubMed.
- W. Zhang, S. Zhu, R. Luque, S. Han, L. Hu and G. Xu, Recent development of carbon electrode materials and their bioanalytical and environmental applications, Chem. Soc. Rev., 2016, 45, 715–752 RSC.
- K. Zelechowska, B. Trawinski, S. Draminska, D. Majdecka, R. Bilewicz and B. Kusz, Oxygen biosensor based on carbon nanotubes directly grown on graphitic substrate, Sens. Actuators, B, 2017, 240, 1308–1313 CrossRef CAS.
- B. Reuillard, A. Le Goff, C. Agnes, M. Holzinger, A. Zebda, C. Gondran, K. Elouarzaki and S. Cosnier, High power enzymatic biofuel cell based on naphthoquinone-mediated oxidation of glucose by glucose oxidase in a carbon nanotube 3D matrix, Phys. Chem. Chem. Phys., 2013, 15, 4892–4896 RSC.
- S. Babanova, K. Artyushkova, Y. Ulyanova, S. Singhal and P. Atanassov, Design of experiments and principal component analysis as approaches for enhancing performance of gas-diffusional air-breathing bilirubin oxidase cathode, J. Power Sources, 2014, 245, 389–397 CrossRef CAS.
- F. Giroud, R. Milton, B. Tan and S. Minteer, Simplifying Enzymatic Biofuel Cells: Immobilized Naphthoquinone as a Biocathodic Orientational Moiety and Bioanodic Electron Mediator, ACS Catal., 2015, 5, 1240–1244 CrossRef CAS.
- A. de Poulpiquet, H. Marques-Knopf, V. Wernert, R. Gadiou, M. T. Giudici-Orticoni and E. Lojou, Carbon Nanofiber Mesoporous Films: Efficient Platforms for Bio-Hydrogen Oxidation in Biofuel Cells, Phys. Chem. Chem. Phys., 2014, 16, 1366–1378 RSC.
- S. Tsujimura, E. Suraniti, F. Durand and N. Mano, Oxygen reduction reactions of the thermostable bilirubin oxidase from Bacillus pumilus on mesoporous carbon-cryogel electrodes, Electrochim. Acta, 2014, 117, 263–267 CrossRef CAS.
- S. Fujita, S. Yamanoi, K. Murata, H. Mita, T. Samukawa, T. Nakagawa, H. Sakai and Y. Tokita, A repeatedly refuelable mediated biofuel cell based on a hierarchical porous carbon electrode, Sci. Rep., 2014, 4, 4937 CrossRef CAS PubMed.
- K. Monsalve, M. Roger, C. Gutierrez-Sanchez, M. Ilbert, S. Nitsche, D. Byrne-Kodjabachian, V. Marchi and E. Lojou, Hydrogen bioelectrooxidation on gold nanoparticle-based electrodes modified by Aquifex aeolicus hydrogenase: application to hydrogen/oxygen enzymatic biofuel cells, Bioelectrochemistry, 2015, 106, 47–55 CrossRef CAS PubMed.
- V. Flexer, N. Brun, M. Destribats, R. Backov and N. Mano, A novel three-dimensional macrocellular carbonaceous biofuel cell, Phys. Chem. Chem. Phys., 2013, 15, 6437–6445 RSC.
- J. Vivekananthan, R. Rincon, V. Kuznetsov, S. Pöller and W. Schuhmann, Biofuel-cell cathodes based on bilirubin oxidase immobilized through organic linkers on 3D hierarchically structured carbon electrodes, ChemElectroChem., 2014, 1, 1901–1908 CrossRef CAS.
- K. Monsalve, I. Mazurenko, N. Lalaoui, A. Le Goff, M. Holzinger, P. Infossi, S. Nitsche, J. Y. Lojou, M. T. Giudici-Orticoni, S. Cosnier and E. Lojou, A H2/O2 enzymatic fuel cell as a sustainable power for a wireless device, Electrochem. Commun., 2015, 60, 216–220 CrossRef CAS.
- C. Mateo-Mateo, A.-S. Michardière, S. Gounel, I. Ly, J. Rouhana, P. Poulin and N. Mano, Wet-spun bioelectronic fibers of imbricated enzymes and carbon nanotubes for efficient microelectrodes, ChemElectroChem., 2015, 2, 1908–1912 CrossRef CAS.
- A. Engel, C. Both, A. Cherifi, S. Tingry, D. Cornu, A. Peigney and C. Laurent, Enhanced performance of electrospun carbon fibers modified with carbon nanotubes: promising electrodes for enzymatic biofuel cells, Nanotechnology, 2013, 24, 245402 CrossRef PubMed.
- S. Tsujimura and K. Murata, Electrochemical oxygen reduction catalyzed by bilirubin oxidase with the aid of 2,2′-azinobis(3-ethylbenzothiazolin-6-sulfonate) on a MgO-template carbon electrode, Electrochim. Acta, 2015, 180, 555–559 CrossRef CAS.
- J. Polte, Fundamental growth principles of colloidal metal nanoparticles – a new perspective, CrystEngComm, 2015, 17, 6809–6830 RSC.
- D. Pankratov, R. Sundberg, D. Suyatin, J. Sotres, A. Barrantes, T. Ruzgas, I. Maximov, L. Montelius and S. Shleev, The influence of nanoparticles on enzymatic bioelectrocatalysis, RSC Adv., 2014, 4, 38164–38168 RSC.
- U. Salaj-Kosla, S. Pöller, Y. Beyl, M. D. Scanlon, S. Beloshapkin, S. Shleev, W. Schuhmann and E. Magner, Direct electron transfer of bilirubin oxidase at an unmodified nanoporous gold biocathode, Electrochem. Commun., 2012, 16, 92–95 CrossRef CAS.
- T. Siepenkoetter, U. Salaj-Kosla, X. Xiao, S. Belochapkine and E. Magner, Nanoporous gols electrodes with tuneable pore sizes for bioelectrochemical applications, Electroanalysis, 2016, 28, 2415–2423 CrossRef CAS.
- G. Martin, S. Minteer and M. Cooney, Spatial distribution of malate dehydrogenase in chitosan scaffolds, ACS Appl. Mater. Interfaces, 2009, 1, 367–372 CAS.
- L. Xu and F. Armstrong, Optimizing the power of enzyme-based membrane-less hydrogen fuel cells for hydrogen-rich H2–air mixtures, Energy Environ. Sci., 2013, 6, 2166–2171 CAS.
- C. Leger, A. Jones, S. Albracht and F. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef CAS.
- S. Hexter, F. Gray, T. Happe, V. Climent and F. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS PubMed.
- M. Roger, A. de Poulpiquet, A. Ciaccafava, M. Ilbert, M. Guiral, M. T. Giudici-Orticoni and E. Lojou, Reconstitution of supramolecular organization involved in energy metabolisms at electrochemical interfaces for biosensing and bioenergy production, Anal. Bioanal. Chem., 2014, 406, 1011–1027 CrossRef CAS PubMed.
- A. Ciaccafava, P. Infossi, M. Ilbert, M. Guiral, S. Lecomte, M. T. Giudici-Orticoni and E. Lojou, Electrochemistry, AFM and PM-IRRAS spectroscopy of immobilized hydrogenase: role of a trans-membrane helix on enzyme orientation for efficient H2 oxidation, Angew. Chem., Int. Ed., 2012, 51, 953–956 CrossRef CAS PubMed.
- A. Ciaccafava, A. De Poulpiquet, P. Infossi, S. Robert, R. Gadiou, M. T. Giudici-Orticoni, S. Lecomte and E. Lojou, A friendly detergent for H2 oxidation by Aquifex aeolicus membrane-bound hydrogenase immobilized on graphite and SAM-modified gold electrodes, Electrochim. Acta, 2012, 82, 115–125 CrossRef CAS.
- F. Oteri, M. Baaden, E. Lojou and S. Sacquin-Mora, Multiscale simulations give insight into the hydrogen in- and out-pathways of [NiFe] hydrogenases from Aquifex aeolicus and Desulfovibrio fructosovorans, J. Phys. Chem. B, 2014, 118, 13800–13811 CrossRef CAS PubMed.
- N. Heidary, T. Utesch, M. Zerball, M. Horch, D. Millo, J. Fritsch, O. Lenz, R. von Klitzing, P. Hildebrandt, A. Fischer, M. A. Mroginski and I. Zebger, Orientation-Controlled Electrocatalytic Efficiency of an Adsorbed Oxygen-Tolerant Hydrogenase, PLoS One, 2015, 10, e0143101 Search PubMed.
- J. Cracknell, J. A. T. McNamara, E. Lowe and C. Blanford, Bilirubin oxidase from Myrothecium verrucaria: X-ray determination of the complete crystal structure and a rational surface modification for enhanced electrocatalytic O2 reduction, Dalton Trans., 2011, 40, 6668–6675 RSC.
- C. Gutierrez-Sanchez, A. Ciaccafava, P. Y. Blanchard, K. Monsalve, M. T. Giudici-Orticoni, S. Lecomte and E. Lojou, Efficiency of Enzymatic O2 Reduction by Myrothecium verrucaria Bilirubin Oxidase Probed by Surface Plasmon Resonance, PMIRRAS, and Electrochemistry, ACS Catal., 2016, 6, 5482–5492 CrossRef CAS.
- P. Ramirez, N. Mano, R. Andreu, T. Ruzgas, A. Heller, L. Gorton and S. Shleev, Direct electron transfer from graphite and functionalized gold electrodes to T1 and T2/T3 copper centers of bilirubin oxidase, Biochim. Biophys. Acta, 2008, 1777, 1364–1369 CrossRef CAS PubMed.
- Y. Sugimoto, Y. Kitazumi, O. Shirai and K. Kano, Effects of mesoporous structures on direct electron transfer-type bioelectrocatalysis: facts and simulation on a three-dimensional model of random orientation of enzymes, Electrochemistry, 2017, 85, 82–87 CrossRef CAS.
- Y. Matanovic, S. Babanova, M. Seow Chavez and P. Atanassov, Protein-support interactions for rationally designed bilirubin oxidase based cathode: a computational study, J. Phys. Chem. B, 2016, 120, 3634–3641 CrossRef PubMed.
- R. Lopez, S. Babanova, Y. Ulyanova, S. Singhal and P. Atanassov, Improved interfacial electron transfer in modified bilirubin oxidase biocathodes, ChemElectroChem, 2014, 1, 241–248 CrossRef.
- Y. Ulyanova, S. Babanova, E. Pinchon, I. Matanovic, S. Singhal and P. Atanassov, Effect of enzymatic orientation through the use of syringaldazine molecules on multiple multi-copper oxidase enzymes, Phys. Chem. Chem. Phys., 2014, 16, 13367–13375 RSC.
- N. Lalaoui, M. Holzinger, A. Le Goff and S. Cosnier, Diazonium functionalisation of carbon nanotubes for specific orientation of multicopper oxidases : controlling electron entry points and oxygen diffusion to the enzyme, Chem.–Eur. J., 2016, 22, 10494–10500 CrossRef CAS PubMed.
- F. Giroud, K. Sawada, M. Taya and S. Cosnier, 5,5′-Dthiobis(2-nitrobenzoic acid) pyrene derivative-carbon nanotube electrodes for NADH electrooxidation and oriented immobilization of multicopper oxidases for the development of glucose/O2 biofuel cells, Biosens. Bioelectron., 2017, 87, 957–963 CrossRef CAS PubMed.
- C. Kjaergaard, S. Jones, S. Gounel, N. Mano and E. Solomon, Two-electron reduction versus one-electron oxidation of the type 3 pair in the multicopper oxidases, J. Am. Chem. Soc., 2015, 137, 8783–8794 CrossRef CAS PubMed.
- T. Le, V. Flaud, M. Bechelany, M. Cretin and S. Tingry, Optimal direct electron transfer between MWCNTs@COOH/BOD/chitosan layer and porous carbon felt for dioxygen reduction, Electrochim. Acta, 2017, 230, 373–381 CrossRef CAS.
- K. So, M. Onizuka, T. Komukai, Y. Kitazumi, O. Shirai and K. Kano, Significance of the length of carbon nanotubes on the bioelectrocatalytic activity of bilirubin oxidase for dioxygen reduction, Electrochim. Acta, 2016, 192, 133–138 CrossRef CAS.
- K. So, S. Kawai, Y. Hamano, Y. Kitazumi, O. Shirai, M. Hibi, J. Ogawa and K. Kano, Improvement of a direct electron transfer-type fructose/dioxygen biofuel cell with a substrate-modified electrode, Phys. Chem. Chem. Phys., 2014, 16, 4823–4829 RSC.
- K. So, Y. Kitazumi, O. Shirai and K. Kano, Analysis of factors governing direct electron transfer-type bioelectrocatalysis of bilirubin oxidase at modified electrodes, J. Electroanal. Chem., 2016, 783, 316–323 CrossRef CAS.
- H.-Q. Xia, K. So, Y. Kitazumi, O. Shirai, K. Nishikawa, Y. Higuchi and K. Kano, Dual gas-diffusion membrane- and mediatorless dihydrogen/air-breathing biofuel cell operating at room temperature, J. Power Sources, 2016, 335, 105–112 CrossRef CAS.
- F. Oteri, A. Ciaccafava, A. de Poulpiquet, E. Lojou, M. Baaden and S. Sacquin-Mora, Fluctuations in the dipole moment of membrane-bound hydrogenase from Aquifex aeolicus account for its adaptability to charged electrodes, Phys. Chem. Chem. Phys., 2014, 16, 11318–11322 RSC.
- K. Sokol, D. Mersch, V. Hartmann, J. Zhang, M. Nowaczyk, M. Rogner, A. Ruff, W. Schuhmann, N. Plumere and E. Reisner, Rational wiring of photosystem II to hierarchical indium tin oxide electrodes using redox polymers, Energy Environ. Sci., 2016, 9, 3698–3709 CAS.
- J. Liu, W.-J. Wu, F. Fang, N. Zorin, M. Chen and D.-J. Qian, Immobilization of hydrogenase on carbon nanotube polyelectrolytes as heterogeneous catalysts for electrocatalytic interconversion of protons and hydrogen, J. Nanopart. Res., 2016, 18, 220 CrossRef.
- H. Shin and C. Kang, Enhanced performance of the wired-bilirubin oxidase oxygen cathode with incorporation of carboxylated single-walled carbon nanotubes, Electrochim. Acta, 2014, 115, 599–606 CrossRef CAS.
- T. Siepenkoetter, U. Salaj-Kosla, X. Xiao, P. O Conghaile, M. Pita, R. Ludwig and E. Magner, Immobilization of redox enzymes on nanoporous gold electrodes: applications in biofuel cells, ChemPlusChem, 2017, 82, 553–560 CrossRef CAS.
- P. Saboe, P. Owen, E. Conte, M. Farell, G. C. Bazan and M. Kumar, Biomimetic and Bioinspired Approaches for Wiring Enzymes to Electrode Interfaces, Energy Environ. Sci., 2017, 10, 14–42 CAS.
- K. Singh, T. McArdle, P. Sullivan and C. Blanford, Sources of activity loss in the fuel cell enzyme bilirubin oxidase, Energy Environ. Sci., 2013, 6, 2460–2464 CAS.
- T. McArdle, T. McNamara, F. Fei, K. Singh and C. Blanford, Optimizing the mass-specific activity of bilirubin oxidase adlayers through combined electrochemical quartz crystal microbalance and dual polarization interferometry analyses, ACS Appl. Mater. Interfaces, 2015, 7, 25270–25280 CAS.
- L. Xu and F. Armstrong, Pushing the limits for enzyme-based membrane-less hydrogen fuel cells - achieving useful power and stability, RSC Adv., 2015, 5, 3649–3656 RSC.
- N. Lalaoui, A. de Poulpiquet, R. Haddad, A. Le Goff, M. Holzinger, S. Gounel, M. Mermoux, P. Infossi, N. Mano, E. Lojou and S. Cosnier, A membrane-less air-breathing hydrogen biofuel cell based on direct wiring of thermostable enzymes on carbon nanotube electrodes, Chem. Commun., 2015, 51, 7447–7450 RSC.
- E. Lojou, M. T. Giudici-Orticoni and P. Bianco, Direct electrochemistry and enzymatic activity of bacterial polyhemic cytochromes c3 incorporated in clay films, J. Electroanal. Chem., 2005, 579, 199–213 CrossRef CAS.
- M. Cadet, X. Brilland, S. Gounel, F. Louerat and N. Mano, Design of a highly efficient O2 cathode based on bilirubin oxidase from Magnaporthe oryzae, ChemPhysChem, 2013, 14, 2097–2100 CrossRef CAS PubMed.
- D. MacAodha, P. O Conghaile, B. Egan, P. Kavanagh and D. Leech, Membrane-less glucose/oxygen enzymatic fuel cells using redox hydrogel films containing carbon nanotubes, ChemPhysChem, 2013, 14, 2302–2307 CrossRef CAS PubMed.
- S. Morozov, O. Voronin, E. Karyakina, N. Zorin, S. Cosnier and A. Karyakin, Electrochem. Commun., 2006, 8, 851–854 CrossRef CAS.
- A. Ciaccafava, P. Infossi, M. T. Giudici-Orticoni and E. Lojou, Stabilization role of a phenothiazine derivative on the electrocatalytic oxidation of hydrogen via Aquifex aeolicus hydrogenase at graphite membrane electrodes, Langmuir, 2010, 23, 18534–18541 CrossRef PubMed.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumere and C. Leger, Mechanism of protection of catalysts supported in redox hydrogel films, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
- A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, A Redox Hydrogel Protects the O2-Sensitive [FeFe]-Hydrogenase from Chlamydomonas reinhardtii from Oxidative Damage, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- A. Ruff, J. Szczesny, S. Zacarias, I. Pereira, N. Plumere and W. Schuhmann, Protection and reactivation of the [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough under oxidative conditions, ACS Energy Lett., 2017, 2, 964–968 CrossRef.
- V. Radu, S. Frielingsdorf, O. Lenz and L. Jeuken, Reactivation from the Ni-B state in [NiFe] hydrogenase of Ralstonia eutropha is controlled by reduction of the superoxidised proximal cluster, Chem. Commun., 2016, 52, 2632–2635 RSC.
- A. de Poulpiquet, A. Ciaccafava, R. Gadiou, S. Gounel, M. T. Giudici-Orticoni, N. Mano and E. Lojou, Design of a H2/O2 biofuel cell based on thermostable enzymes, Electrochem. Commun., 2014, 42, 72–74 CrossRef CAS.
- T. Matsumoto, S. Eguchi, H. Nakai, T. Hibino, K.-S. Yoon and S. Ogo, [NiFe] hydrogenase from Citrobacter sp. S-77 surpasses platinum as an electrode for H2 oxidation reaction, Angew. Chem., Int. Ed., 2014, 53, 8895–8898 CrossRef CAS PubMed.
- Y. Wang, T. Esterle and F. Armstrong, Electrocatalysis by H2–O2 membrane-free fuel cell enzymes in aqueous microenvironments confined by an ionic liquid, RSC Adv., 2016, 6, 44129–44134 RSC.
- K. So, Y. Kitazumi, O. Shirai, K. Nishikawa, Y. Higuchi and K. Kano, Direct electron transfer-type dual gas diffusion H2/O2 biofuel cells, J. Mater. Chem. A, 2016, 4, 8742–8749 CAS.
- C. Lau, E. Adkins, R. Ramasamy, H. Luckarift, G. Johnson and P. Atanassov, Design of Carbon Nanotube-Based Gas-Diffusion Cathode for O2 Reduction by Multicopper Oxidase, Adv. Energy Mater., 2012, 2, 162–168 CrossRef CAS.
- C. Narvaez Villarrubia, F. Soavi, C. Santoro, C. Arbizzani, A. Serov, S. Rojas-Carbonell, G. Gupta and P. Atanassov, Self-feeding paper based biofuel cell/self powered hybrid μ-supercapacitor integrated system, Biosens. Bioelectron., 2016, 86, 459–465 CrossRef CAS PubMed.
- T. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: from biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- G. Caserta, S. Roy, M. Atta, V. Artero and M. Fontecave, Artificial hydrogenases: biohybrid and supramolecular systems for catalytic hydrogen production and uptake, Curr. Opin. Chem. Biol., 2015, 25, 36–47 CrossRef CAS PubMed.
- N. Coutard, N. Kaeffer and V. Artero, Molecular engineered nanomaterials for catalytic hydrogen evolution and oxidation, Chem. Commun., 2016, 52, 13728–13748 RSC.
- P. Rodriguez-Macia, A. Dutta, W. Lubitz, W. Shaw and O. Rüdiger, Direct comparison of the performance of a bio-inspired synthetic nickel catalyst and a [NiFe]-hydrogenase, both covalently attached to electrodes, Angew. Chem., Int. Ed., 2015, 54, 12303–12307 CrossRef CAS PubMed.
- T. Huan, R. Jane, A. Benayad, L. Guetaz, P. Tran and V. Artero, Bio-inspired noble metal-free nanomaterials approaching platinum performances for H2 evolution and uptake, Energy Environ. Sci., 2016, 9, 940–947 CAS.
- P. Tran, A. Morozan, S. Archambault, J. Heidkamp, P. Chenevier, H. Dau, M. Fontecave, A. Martinent, B. Jousselme and V. Artero, A noble metal-free proton exchange membrane fuel cell based on bio-inspired molecular catalysts, Chem. Sci., 2015, 6, 2050–2053 RSC.
- S. Gentil, N. Lalaoui, A. Dutta, Y. Nedelec, S. Cosnier, W. Shaw, V. Artero and A. Le Goff, Carbon-nanotube-supported bio-inspired nickel catalyst and its integration in hybrid hydrogen/air fuel cell, Angew. Chem., Int. Ed., 2017, 56, 1845–1849 CrossRef CAS PubMed.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. Fontecilla-Camps, A. W. Rutherford and E. Reisner, Wiring of photosystem II to hydrogenase for photoelectrochemical water splitting, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- J. Hadj-Said, M. Pandelia, C. Leger, V. Fourmond and S. Dementin, The Carbon Monoxide Dehydrogenase from Desulfovibrio vulgaris, Biochim. Biophys. Acta, 2015, 1847, 1574–1583 CrossRef CAS PubMed.
- R. Milton, R. Cai, S. Abdellaoui, D. Leech, A. De Lacey, M. Pita and S. Minteer, Bioelectrochemical Haber-Bosch process : an ammonia-producing H2/N2 fuel cell, Angew. Chem., Int. Ed., 2017, 56, 2680–2683 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |