DOI:
10.1039/C7SE00195A
(Communication)
Sustainable Energy Fuels, 2017,
1, 1516-1523
3-D vertically aligned few layer graphene – partially reduced graphene oxide/sulfur electrodes for high performance lithium–sulfur batteries†
Received
11th April 2017
, Accepted 7th July 2017
First published on 13th July 2017
Abstract
3-D vertically aligned few-layered graphene (FLGs) nanoflakes synthesised using microwave plasma enhanced chemical vapour deposition are melt-impregnated with partially reduced graphene oxide-sulfur (PrGO-S) nanocomposites for use in lithium–sulfur batteries. The aligned structure and the presence of interconnected micro voids/channels in the 3-D FLG/PrGO-S electrodes serves as template not only for the high sulfur loading (up to 80 wt%, areal loading of 1.2 mg cm−2) but also compensates for the volume changes occurring during charge–discharge cycles. The inter-connectivity of the electrode system further facilitates fast electronic and ionic transport pathways. Consequently, the binder-free 3-D FLG/PrGO-S electrodes display a high first-cycle capacity (1320 mA h g−1 at C/20), along with excellent rate capability of ∼830 mA h g−1 and 700 mA h g−1 at 2C and 5C rates, respectively. The residual functional groups of PrGO (–OH, –C–O–C– and –COOH) facilitate fast and reversible capture of Li+ ions while confining the polysulfide shuttles, thus, contributing to excellent cycling capability and retention capacity. The 3D electrodes demonstrate excellent capacity retention of ∼80% (1040 mA h g−1 at C/10) over 350 charge–discharge cycles. Comparatively, the 2-D planar PrGO-S electrodes displayed poor electronic conductivity and can only provide 560 mA h g−1 after 150 cycles, thereby further highlighting the vital role of the electrode morphology in improving the electrochemical performance of Li–S batteries.
1. Introduction
As the next-generation energy storage materials, lithium–sulfur (Li–S) batteries have become increasingly attractive owing to their high gravimetric density (2600 W h kg−1) and specific capacity (1671 mA h g−1), in addition, sulfur is a highly cost-effective and environmentally benign element.1 However, the overall performance of current Li–S batteries is impeded by inherently poor electronic and ionic conductivity of sulfur and the dissolution of higher-order polysulphides phases (Li2Sn (8 ≥ n ≥ 2)) during potential cycling which cause irreversible loss of active material. This is especially so for the conventional Li–S battery cathodes, consisting of sulfur, conductive carbon and binder, where it is difficult to suppress the dissolution of lithium polysulfide in liquid electrolyte and stabilize the active material in the cathode matrix. During the continuous charge–discharge cycles, these polysulfides diffuse into liquid electrolyte and shuttle through the separator to lithium anode, and finally precipitate as an insulating layer (Li2S2 and/or Li2S) over the electrodes.2–5 With increasing current density and charge–discharge cycles, this further increases the interfacial charge transfer resistance, lowers the overall columbic efficiency and degrades the rate and life cycle performance.6–10
To expedite the reversible electrochemical reaction and to achieve high rate performance, both the sulfur and polysulphides must maintain the ionic and electronic conduction within the electrode matrix.6,11 This requires: (i) confining the insulating sulfur and polysulfide shuttles in electronically conductive electrode matrix, (ii) facile ionic network around encapsulated sulfur formed by the liquid electrolyte in the pores of the composite electrode matrix and (iii) efficient charge transfer reaction between the liquid electrolyte and the active material. To this effect, various matrices including mesoporous carbons,11,12 microporous carbon spheres,13,14 nanotube/fibres,15,16 activated carbon;17 polar metal oxides such as TiO2, SiO2, Al2O3,18,19 have been investigated. More recently, quasi-2-D metal carbides (such as Ti2C) and the 2-D nanosheets of reduced Graphene Oxide (rGO),20–22 owing to their superior electrical conductivity, large surface area and surface tailorability, have emerged as promising materials for confining lithium polysulphides while improving the material utilisation, rate and cyclic stability.23 It is well established that the applications of sp2 carbon in the area of energy storage are highly dependent not only on their superior intrinsic physical properties, such as mechanical strength, electrical and thermal conductivity, but also on their tunable chemical properties.24–26 Recent reports have shown that the surface modified carbon fibres, graphene oxide sheets and polymer coatings facilitate better polysulfide anion adhesion on the surface, thereby enhancing the active material utilisation and life cycle capability.27,28 Studies have shown that besides physically confining elemental sulfur and lithium polysulfides in the meso/microporous frameworks, the performance of liquid electrolyte based batteries is also governed by electrode morphology. Furthermore, higher performance requires facile ionic (through liquid electrolyte) and electronic transport (through conductive additive).29–33 Pioneering work by Nazar et al. has established the close relationship between the electrode microstructure and the subsequent electrochemical performance, which further necessitates the demand for microstructured electrodes with enhanced ionic and electronic tortuosity combined with high sulfur loading.6,11,19,34,35 Thus, it is still an on-going challenge to prepare high performance electrodes with high specific capacity at high rates due to shorter diffusion lengths and reduced interfacial contact resistance.
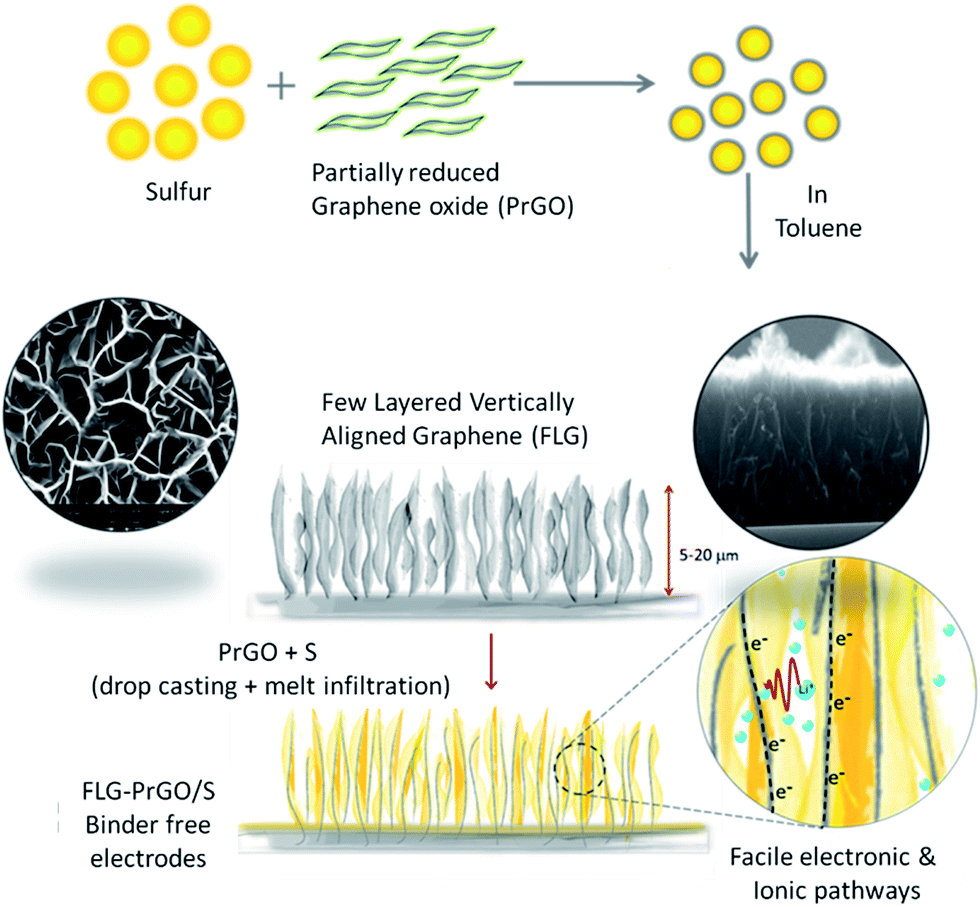
Herein, we report novel binder-free 3-D vertically aligned electrodes of few layered graphene (FLG) nanoflakes with interconnected micro voids/channel, filled with partially reduced graphene oxide-sulfur (PrGO-S) nanocomposites for high performance Li–S batteries (schematic shown in Fig. 1). The melt-infiltrated PrGO-S nanocomposites (see ESI†) within a vertically aligned FLG network, provide several key advantages over the conventional 2-D planar electrode morphology, including: (i) facilitation of improved electrical conductivity and high sulfur loading (∼80 wt% wrt PrGO/FLG as confirmed by TGA (ESI, Fig. S1†)) in the interconnected micro-porous FLG network; (ii) ease of electrolyte accessibility owing to its highly interconnected nature and (iii) uniform distribution of voids/channels confining the polysulfide shuttle within the cathode matrix and improving the active material utilisation to facilitate facile ionic and electronic pathways.16,36,37 This simple and facile method to fabricate the 3-D FLG/PrGO-S electrodes offers great flexibility in controlling and tuning of electrode thickness and free volume based on the variation of growth parameters of the FLGs (see ESI, Fig. S2†). Furthermore, the presence of epoxide (C–O–C), hydroxyl (–OH) and carboxyl (–COOH) groups on the PrGO surface is expected to further enhance the sulfur and lithium polysulfide immobilization during the discharge process. Drop casting of PrGO/S nanocomposite on the FLG is expected to have additional advantages; firstly, the large surface area of PrGO will act as a barrier layer between the electrode and electrolyte; with the functional groups such as epoxide, hydroxyl and carboxyl on the PrGO physically sandwiching the sulfur (and its polysulfides) within the micro void/channelled FLG structure. Secondly, the epoxide groups on the PrGO surface further enhances the immobilization of sulfur and polysulfides during the discharge process.38
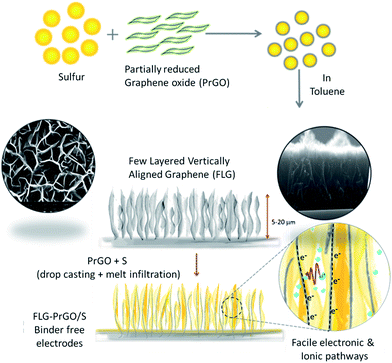 |
| | Fig. 1 Schematic illustration of the preparation of PrGO/S composite and fabrication vertically aligned FLG/PrGO-S electrode via drop casting and melt infiltration method. Top and cross sectional scanning electron micrograph of FLG electrode shows the interconnected network of micro voids/channels and the high aspect ratio of graphene sheets. Furthermore, the proposed facile (electronic and ionic) charge transport within interconnected porous structures is illustrated. | |
2. Experimental
2.1 Synthesis of partially reduced graphene oxide
Graphene oxide was synthesised using a modified Hummers' method.39,40 For the preparation of partially reduced graphene oxide, the as prepared GO was reduced using L-ascorbic acid (L-AA) as reported in the literature.41,42 Typically, 0.1 mg mL−1 aqueous solution of GO was prepared and L-AA was added such that the GO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L-AA ratio was 1:10. Upon complete dissolution of L-AA in the GO solution, the mixture was further heated for 60 minutes at 95 °C with continuous stirring. As the reaction took place, the brown solution was no longer homogeneous and black precipitates started to appear. Upon the completion of reaction, the solution was cooled down and the precipitate was separated and washed repeatedly with water using filtration and centrifugation. The material thus obtained (PrGO) was dried overnight in a vacuum oven (50 °C) for further use.
:L-AA ratio was 1:10. Upon complete dissolution of L-AA in the GO solution, the mixture was further heated for 60 minutes at 95 °C with continuous stirring. As the reaction took place, the brown solution was no longer homogeneous and black precipitates started to appear. Upon the completion of reaction, the solution was cooled down and the precipitate was separated and washed repeatedly with water using filtration and centrifugation. The material thus obtained (PrGO) was dried overnight in a vacuum oven (50 °C) for further use.
2.2 Synthesis of vertically aligned few layer graphene nanoflakes
The growth of few layered graphene (FLG) nanoflakes was carried out in a 1.5 kW, 2.45 GHz SEKI microwave plasma enhanced chemical vapour deposition system.43,44 Stainless steel substrates (SS316L) cleaned using acetone and isopropanol alcohol were used as substrates, which were placed on top of a Si wafer to allow plasma irradiation to higher temperatures. Once the samples were loaded, the chamber was pumped down to a base pressure of ∼2 × 10−3 Torr after which nitrogen (N2) plasma pre-treatment of substrate was carried out at 700 W for certain duration of time. CH4 was then injected at the end of pre-treatment time while the microwave power was simultaneously increased to 800 W. During the growth time of 60 seconds, the substrate temperature exceeded 1250 °C as monitored by an optical pyrometer mounted on top of the chamber. Post deposition, the samples were allowed to cool down to room temperature under a N2 atmosphere. The vertical alignment of the resulting nanostructures is a unique feature of the microwave plasma CVD route. Unlike the thermal CVD route wherein the alignment is due to the crowding and van der Waals forces, the alignment in microwave CVD route is attributed to the plasma induced electric field effects and has been discussed in detail in our previous works.43,44 It should be noted that the microwave plasma deposition route can be used for catalyst free growth of graphene nanoflakes on any substrate which can sustain the high temperatures and plasma bombardment encountered during the growth process such as metallic foils, carbon cloth etc.
2.3 Preparation of vertically aligned FLG/PrGO-S electrodes
The fabrication of vertically aligned FLG/PrGO-S electrode involves the following two steps. Firstly, predetermined amounts of commercially available sulfur (93 wt%) (Sigma Aldrich) and synthesised PrGO powders (7 wt%) were mixed in toluene (0.1 gm of PrGO/S in 50 mL of toluene) and ball milled using a Fritsch planetary ball-mill under an argon atmosphere for 60 to 90 minutes to obtain a homogenous mixture. This was followed by melt infiltration at 130 °C under argon atmosphere with subsequent drop casting of PrGO/S on the vertically aligned FLGs to prepare the binder-free 3-D FLG/PrGO-S electrodes.
2.4 Microstructural characterisation
Structural studies on the 3-D FLG/PrGO-S electrodes were performed using Titan Cubed Cs corrected electron microscope with a resolution of 0.08 nm. Inbuilt high resolution EDX was used for mapping the distribution of carbon and sulphur. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Axis Ultra utilising an Al Kα radiation source (1486.6 eV). XPS spectra were obtained with a spot size of 200 μm. High resolution scans of individual elements and the survey spectra were measured at a pass energy of 50 eV and 200 eV, respectively. The scans were averaged over 10 sweeps. Thermo Nicolet Nexus 670 FT-IR spectrometer was used for FTIR studies wherein the spectra was measured for 128 scans (DRIFT mode) at a resolution of ±4 cm−1. The thermogravimetric analysis of the sulfur content in the PrGO/S electrodes was measured using TGA carried out on a Netzsch5 STA F3 system. The measurement was carried out from room temperature until 800 °C at 5 °C min−1 ramp rate in air.
2.5 Electrochemical measurements
The vertically aligned 3-D FLG/PrGO-S electrodes were tested within the voltage range of 1.8–2.6 V against lithium metal using Maccor Battery Tester. Prior to electrochemical testing, electrodes were dried in vacuum. Cells were assembled using a homemade vacuum flange type assembly in an argon-filled glove box using polypropylene separator. The electrolyte used was 1.0 M lithium bis-trifluoromethanesulfonylimide (LiTFSI) in 1,3-dioxolane and 1,2-dimethoxyethane (1:1 by volume) with 3 wt% LiNO3 additive.45 To completely wet the electrode, approximately 100 μl of electrolyte was used. To compare the effect of 3D electrode morphology, a standard PrGO/S composite (85 wt% sulfur) electrode was prepared by mixing PrGO/sulfur composite with 85 wt% sulphur and carbon black (5 wt%) with PVDF (10 wt%) binder in NMP and casted onto carbon coated copper foil.
3. Results and discussion
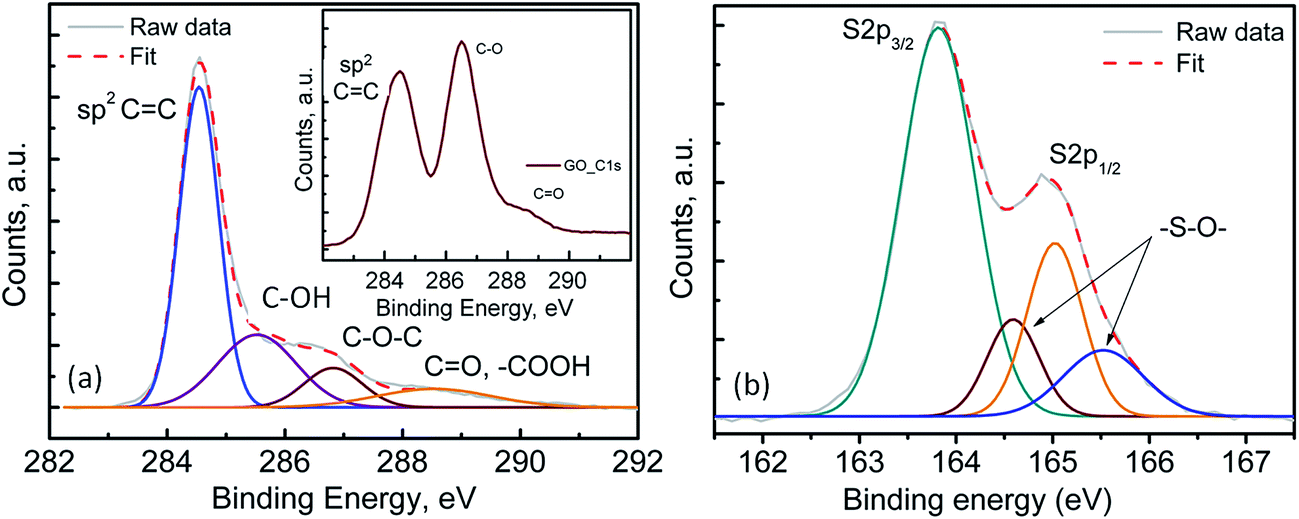
Fig. 2(a) shows the transmission electron microscopy (TEM) image of the pristine FLGs, wherein the samples show a predominantly knife-edge structure with a thick (15–20 nm) base constantly narrowing down as it goes along the axial growth direction till it reaches the top with 1–3 layered graphene.43,44 It should be noted that these highly interconnected FLGs synthesised (areal mass density (0.073 mg cm−2)) using a microwave plasma technique are largely composed of carbon only (98.8 at%) with a small amount of adsorbed oxygen (1.2 at%) (see Fig. S3†).43,44 For the PrGO/S composites, Fig. 2(b–d) shows the sub-micrometer sulfur particles wrapped by partially reduced graphene oxide (PrGO) sheets with the elemental mapping revealing a homogenous dispersion of sulfur on the PrGO sheets. Owing to the melt infiltration process utilised for the 3-D FLG/PrGO-S synthesis, a thin layer of sulfur nanoparticles as well as larger discrete particles were observed embedded in the matrix as evident from the elemental mapping and energy dispersive X-ray (EDX) analysis, which further confirms the high sulfur/carbon ratio (see Fig. S4, ESI†). For the 3-D FLG/PrGO-S electrodes, the mild L-ascorbic acid reduction (starting GO, C:O = 2.1) led to the removal of a significant amount of oxygen functional groups (PrGO, C:O = 4.6) leaving behind residual C–O–C, –COOH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (Fig. 3(a)).41,43–47 These residual groups have been previously suggested to immobilise sulfur and subsequently free lithium polysulfides during the charging–discharging process.24,38,48 Furthermore, peaks attributed to –S–O– bonding were observed in both the S 2p (164.6 and 165.5 eV) and O 1s spectrum (∼530.6 eV, Fig. S3, ESI†), thus confirming the electronic interactions occurring between sulfur and the PrGO matrix (Fig. 3(b)).49 The presence of the –S–O– bonding was further corroborated by Fourier Transform Infra-red Spectroscopy (FTIR) studies, which revealed peak at 1030 cm−1 (Fig. S5, ESI†). Similar results of interaction between GO and sulfur have been reported by Li et al.49 and Guo et al.50 wherein the –S–O bonding helps in anchoring sulfur and intermediate polysulfide products during the cycling process. Recent reports have utilised the epoxide and hydroxyl groups on the basal plane of GO in addition to the edge dominant carbonyl and carboxyl groups for the immobilisation of sulfur and its discharge products.51 However, the presence of these functional groups also leads to the poor electronic conductivity of GO.24,51 Thus, for most of the previous reports with GO/S composites, the achieved rate performances are generally below the rate of 2C (1C = 1675 mA g−1).24,52 On the contrary, reduced GO (RGO) or graphene with intrinsically higher electronic conductivity has also been employed to prepare RGO/S or G/S composites for improving rate performance.24 However, RGO or graphene does not have the abundant functional groups that can bind sulfur and its discharge products. Thus, to obtain the optimal balance of the presence of functional groups and the electronic conductivity of the graphene matrix, the partially reduced graphene oxide (PrGO) is ideal. The electrochemical performance of the 3-D FLG/PrGO-S electrode was evaluated using galvanostatic discharge–charge measurements at variable current densities from 80 mA g−1 (C/20) to 8000 mA g−1 (5C) within the voltage window of 1.8 to 2.6 V versus Li/Li+. As shown in Fig. 4, the first discharge of PrGO/FLG-S electrode at C/20 illustrates the typical two step charge discharge behaviour shown by Li–S batteries (corresponding to the conversion of elemental sulfur into long-chained (Li2Sn, 4 ≤ n ≤ 8), and short chained (Li2Sn, n < 4) polysulfides), and a significantly high discharge capacity of 1350 mA h g−1 at ∼80 wt% sulfur loading was achieved. This is higher than the value reported for electrochemically synthesised vertically aligned sulfur-graphene nanowalls (1261 mA h g−1 at C/8).49 Similarly, Wang et al. reported a first discharge capacity of 1611 mA h g−1 albeit at a much slower rate of 50 mA g−1 (C/33).53 Ni et al. reported sulfur-rGO nanocomposites with a high initial discharge capacity of ∼1473 mA h g−1 at 0.1C but after subsequent cycles at the same C-rate, the discharge capacity of their electrodes dropped to 1230 mA h g−1.54 For the first and third discharge cycles (Fig. 4), it can be observed that the voltage plateau and corresponding cathodic peak shifts (from 2.08 V to 2.1 V and from 2.35 V to 2.38 V) towards equilibrium. This shift in cathodic peak could be attributed to the lowering of internal resistance of the cell due to the conversion of ‘insulating’ elemental sulfur present in the electrode into the polysulfide species, followed by their subsequent dissolution into the liquid electrolyte (resulting into slightly shorter voltage plateau and lower capacity).54 Furthermore, an intermediate discharge plateau and corresponding cathodic peak at 2.15 V was observed, resulting in a three-step discharge profile, which is consistent with earlier reports.55,56 This intermediate peak has only been observed in first few charge–discharge cycles and disappears with subsequent cycles as shown in Fig. S7 (ESI†). Barchasz et al. have previously shown that this intermediate cathodic peak at ∼2.1 V (vs. Li/Li+) is due to the formation of S42− and S32− species resulting from the reduction of the S62− and S3˙− species formed at first discharge plateau.55 Interestingly, they have observed this intermediate peak only at high scan rates for electrodes with high carbon content, whereas the peak started to disappear as the carbon content in electrode decreased.55 Considering the PrGO/FLG electrodes in this study have only 20% carbon content, the presence of this intermediate peak (at 2.15 V) even at low currents (C/20) is attributed to the enhanced sulfur immobilisation within the PrGO/FLG matrix due to the presence of oxygen functional (carbonyl and carboxyl) groups, which actively form S–O bonds facilitating the chemical adsorption of sulfur. As discussed above (XPS, Fig. 3), Song et al. too have evidenced the formation of S–O bonds between the oxygen functional groups and sulfur due to the low enthalpy changes required for such bond formation, through X-ray absorption and density functional theory investigations.57 In our case, the PrGO shows a significant oxygen content (C:O ratio of 4.6), as compared to the pristine GO (C:O ratio of 2.1) which provides the conductive, high surface area and alongside the FLGs, further decreases the need for a large amount of carbon to immobilize the high S-content.
O groups (Fig. 3(a)).41,43–47 These residual groups have been previously suggested to immobilise sulfur and subsequently free lithium polysulfides during the charging–discharging process.24,38,48 Furthermore, peaks attributed to –S–O– bonding were observed in both the S 2p (164.6 and 165.5 eV) and O 1s spectrum (∼530.6 eV, Fig. S3, ESI†), thus confirming the electronic interactions occurring between sulfur and the PrGO matrix (Fig. 3(b)).49 The presence of the –S–O– bonding was further corroborated by Fourier Transform Infra-red Spectroscopy (FTIR) studies, which revealed peak at 1030 cm−1 (Fig. S5, ESI†). Similar results of interaction between GO and sulfur have been reported by Li et al.49 and Guo et al.50 wherein the –S–O bonding helps in anchoring sulfur and intermediate polysulfide products during the cycling process. Recent reports have utilised the epoxide and hydroxyl groups on the basal plane of GO in addition to the edge dominant carbonyl and carboxyl groups for the immobilisation of sulfur and its discharge products.51 However, the presence of these functional groups also leads to the poor electronic conductivity of GO.24,51 Thus, for most of the previous reports with GO/S composites, the achieved rate performances are generally below the rate of 2C (1C = 1675 mA g−1).24,52 On the contrary, reduced GO (RGO) or graphene with intrinsically higher electronic conductivity has also been employed to prepare RGO/S or G/S composites for improving rate performance.24 However, RGO or graphene does not have the abundant functional groups that can bind sulfur and its discharge products. Thus, to obtain the optimal balance of the presence of functional groups and the electronic conductivity of the graphene matrix, the partially reduced graphene oxide (PrGO) is ideal. The electrochemical performance of the 3-D FLG/PrGO-S electrode was evaluated using galvanostatic discharge–charge measurements at variable current densities from 80 mA g−1 (C/20) to 8000 mA g−1 (5C) within the voltage window of 1.8 to 2.6 V versus Li/Li+. As shown in Fig. 4, the first discharge of PrGO/FLG-S electrode at C/20 illustrates the typical two step charge discharge behaviour shown by Li–S batteries (corresponding to the conversion of elemental sulfur into long-chained (Li2Sn, 4 ≤ n ≤ 8), and short chained (Li2Sn, n < 4) polysulfides), and a significantly high discharge capacity of 1350 mA h g−1 at ∼80 wt% sulfur loading was achieved. This is higher than the value reported for electrochemically synthesised vertically aligned sulfur-graphene nanowalls (1261 mA h g−1 at C/8).49 Similarly, Wang et al. reported a first discharge capacity of 1611 mA h g−1 albeit at a much slower rate of 50 mA g−1 (C/33).53 Ni et al. reported sulfur-rGO nanocomposites with a high initial discharge capacity of ∼1473 mA h g−1 at 0.1C but after subsequent cycles at the same C-rate, the discharge capacity of their electrodes dropped to 1230 mA h g−1.54 For the first and third discharge cycles (Fig. 4), it can be observed that the voltage plateau and corresponding cathodic peak shifts (from 2.08 V to 2.1 V and from 2.35 V to 2.38 V) towards equilibrium. This shift in cathodic peak could be attributed to the lowering of internal resistance of the cell due to the conversion of ‘insulating’ elemental sulfur present in the electrode into the polysulfide species, followed by their subsequent dissolution into the liquid electrolyte (resulting into slightly shorter voltage plateau and lower capacity).54 Furthermore, an intermediate discharge plateau and corresponding cathodic peak at 2.15 V was observed, resulting in a three-step discharge profile, which is consistent with earlier reports.55,56 This intermediate peak has only been observed in first few charge–discharge cycles and disappears with subsequent cycles as shown in Fig. S7 (ESI†). Barchasz et al. have previously shown that this intermediate cathodic peak at ∼2.1 V (vs. Li/Li+) is due to the formation of S42− and S32− species resulting from the reduction of the S62− and S3˙− species formed at first discharge plateau.55 Interestingly, they have observed this intermediate peak only at high scan rates for electrodes with high carbon content, whereas the peak started to disappear as the carbon content in electrode decreased.55 Considering the PrGO/FLG electrodes in this study have only 20% carbon content, the presence of this intermediate peak (at 2.15 V) even at low currents (C/20) is attributed to the enhanced sulfur immobilisation within the PrGO/FLG matrix due to the presence of oxygen functional (carbonyl and carboxyl) groups, which actively form S–O bonds facilitating the chemical adsorption of sulfur. As discussed above (XPS, Fig. 3), Song et al. too have evidenced the formation of S–O bonds between the oxygen functional groups and sulfur due to the low enthalpy changes required for such bond formation, through X-ray absorption and density functional theory investigations.57 In our case, the PrGO shows a significant oxygen content (C:O ratio of 4.6), as compared to the pristine GO (C:O ratio of 2.1) which provides the conductive, high surface area and alongside the FLGs, further decreases the need for a large amount of carbon to immobilize the high S-content.
 |
| | Fig. 2 (a) TEM image of pristine FLG showing the variation of graphene layers with a knife-edge like structure, (b and c) low and high magnification TEM image of PrGO-S nanocomposite with the inset showing the diffraction pattern corresponding to crystalline nature of the composite, inset in (c): selected area diffraction pattern (d) cross-sectional SEM image of the 3-D FLG PrGO/S electrode, (e) dark-field STEM image of nanocrystalline sulfur wrapped inside the PrGO sheet with the corresponding elemental mapping demonstrating the uniform coating of sulfur for the PrGO/S nanocomposites. The TEM-EDX is provided in ESI Fig. S4.† | |
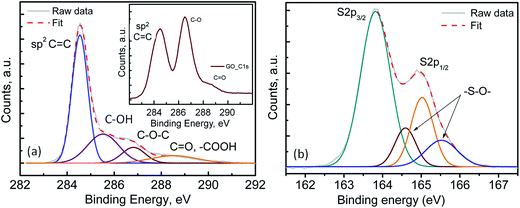 |
| | Fig. 3 X-ray photoelectron spectra of 3-D FLG PrGO/S electrode, (a) deconvolution of C 1s core level spectra of PrGO, inset shows the C 1s spectra of pristine GO, (b) deconvolution of the S 2p spectra showing the –S–O– bonding for the PrGO-S nanocomposites. | |
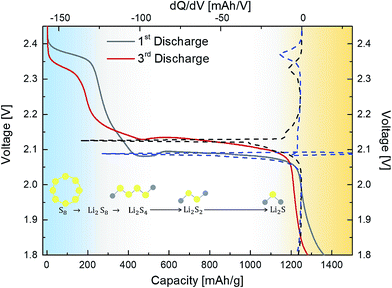 |
| | Fig. 4 Discharge voltage profile of the 3-D FLG PrGO/S electrode at C/20 within the voltage window of 2.6–1.8 V (vs. Li) combined with the corresponding differential capacity plot on the top axis. Typical voltage vs. capacity profiles illustrating typical conversion of elemental sulfur to polysulfide anion during various stage of discharge. (I) The conversion of solid sulfur to soluble polysulphides; (II) conversion of polysulphides to solid Li2S2; (III) conversion of solid Li2S2 to solid Li2S. | |
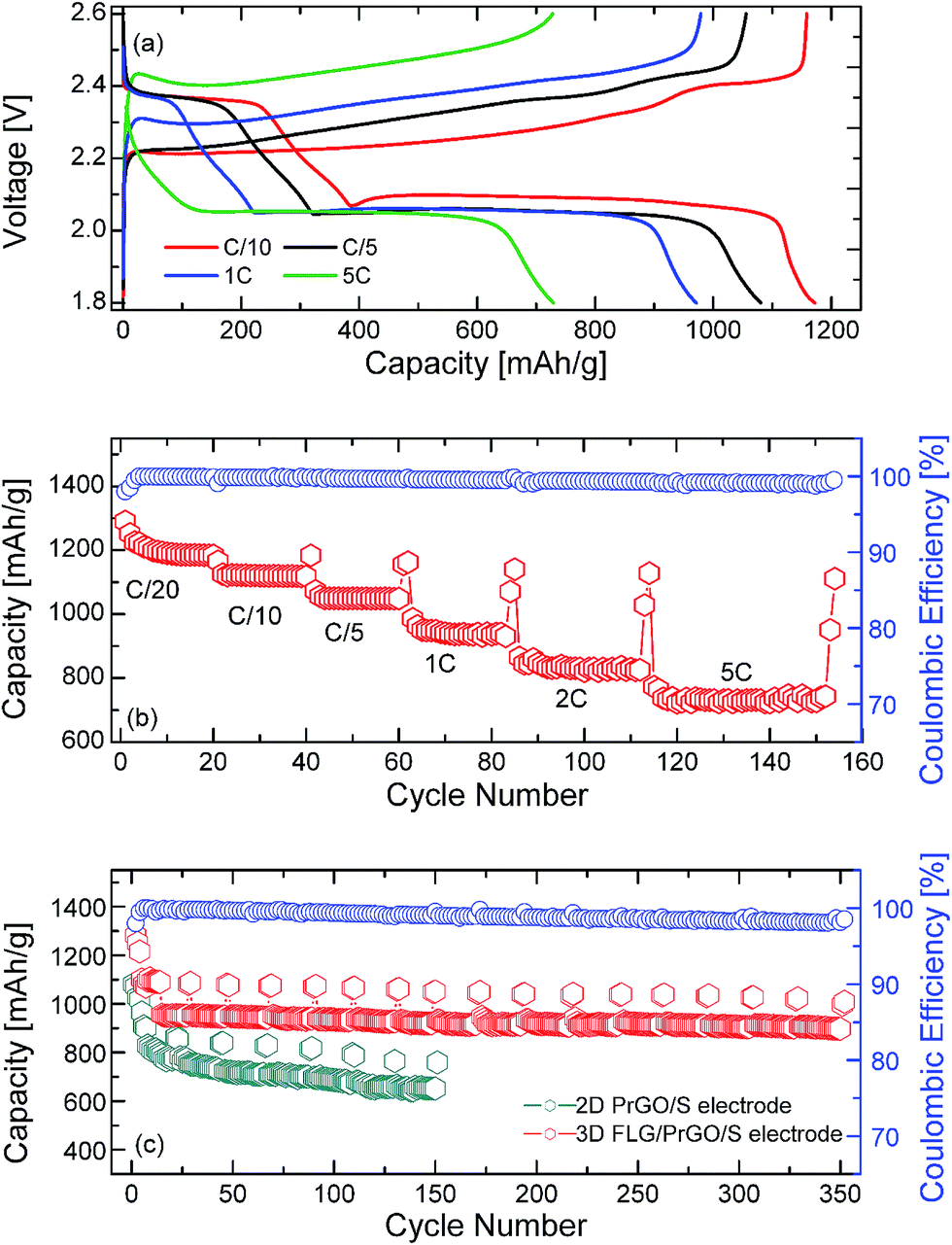
After a few charge–discharge cycles at C/20, the same 3-D PrGO/FLG-S electrode was further tested at different C rates (C/10 to 5C). As the current density was increased to C/10, the specific capacity dropped to ∼1160 mA g−1 (Fig. 5). However, even after 350 cycles, the 3-D FLG/PrGO-S electrode still retained an excellent discharge capacity of ∼1040 mA h g−1 and 880 mA h g−1 at C/10 and 1C, respectively. The values thus obtained at C/10 are higher than those observed for 3-D vertically aligned interconnected carbon nanosheets infiltrated with nearly 70 wt% sulfur,58 interleaved expanded graphite-embedded sulfur nanocomposite59 and are comparable to those obtained utilising a binder-free polymer encapsulated sulfur in an aligned CNT matrix.60 Interestingly, as the charge current density further increased to 2C and 5C (constant discharge at C/5), the 3-D FLG/PrGO-S electrode continued to deliver high specific capacity of ∼830 mA h g−1 and 700 mA h g−1, respectively (Fig. 5). However, in the case of high C rates, contrary to the slow C rate (C/20) charge–discharge profile where the second discharge plateau is almost three times that of the first plateau, it seems that the first plateau (2.35 V vs. Li/Li+) almost disappeares at the 5C rate, thereby making the second plateau appear to be larger than the first. This reduction of first plateau may be because of the intermediate species (such as unstable polysulfides and radicals (S3˙−)) not getting enough time to evolve and actively participate in the electrochemical reaction. Similar modified (dis)charge profiles at high C rate have been observed in previous reports as well.63,64 Nonetheless, various complex processes are involved during charge–discharge of Li–S battery and are affected by factors such as scan rate, dissolution of active material in electrolyte, sulfur to carbon ratio, additives etcetera. Specifically identifying a single factor for charge discharge behaviour at high C rate requires further extensive analysis. As compared to the other reported values of 400 mA h g−1@8C, and 315 mA h g−1@2C, 738 mA h g−1@3C, these are highly appreciable values.49,51,58 Such excellent rate capability at high currents in 3-D electrodes can be attributed to low charge-transfer resistance (both ionic and electronic resistance) and low lithium polysulfide dissolution.59,61,62 The discharge performance of the planar 2-D PrGO/S composites with equivalent sulfur loading (∼80 wt%) was carried out using similar charge–discharge sequences as that for the 3-D FLG/PrGO/S electrodes. The 2-D PrGO/S electrode showed an initial discharge capacity of 1080 mA h g−1 at C/10 rate, as compared to an initial value of 1320 mA h g−1 for the 3D FLG/PrGO-S electrode, as shown in Fig. 5. However, after subsequent cycles, the capacity continued to drop, suggesting dissolution of soluble Li-polysulfides in liquid electrolyte, causing irreversible loss of active material from the cathode.34 In fact, as the charge current density increased from C/10 to 1C, the capacity continued to drop, and just after 150 cycles, the 2-D planar geometry electrodes could deliver only ∼560 mA h g−1. This significant difference (of >200 mA h g−1) in the capacity of planar PrGO-S electrodes, and excellent cycling and rate performances demonstrated by the 3-D FLG/PrGO-S electrodes corroborates our earlier assumption that the 3-D microstructure with interconnected ionic and electronic transport pathways lowers the overall charge transport resistance and facilitates excellent electrical and ionic transport in entire electrode matrix.58–61 Additionally, a comparison of specific capacities from the literature with different types of carbon substrates can be found in Table 1. It can be clearly observed from the table the 3-D FLG/PrGO-S electrode offers some of the highest reported values for the Li–S electrodes. For both the 3-D and 2-D electrodes, the role of residual functional groups on the surface is of crucial importance. For pristine graphene oxide, lack of sufficient π-conjugation leads to poor electronic conductivity and as such it cannot be used for Li–S batteries.24,47,48 However, upon controlled reduction, the aromatic π–π bonding is partially restored, leading to improved conductivity. The residual oxygen functional groups on the PrGO not only provide the lithium polysulfide occupying sites but also the requisite hydrophilicity to facilitate the adsorption of such polysulfides.24,38,48
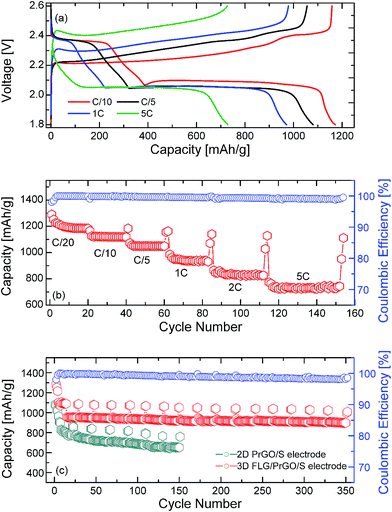 |
| | Fig. 5 (a) Charge and discharge profile of the 3-D FLG PrGO/S electrode at different rates from C/10 to 5C, (b) rate performance and corresponding coulombic efficiency of the FLG/PrGO-S electrode at different charging discharging rates, (from 1C to 5C rate, electrodes were discharged at C/10), (c) life cycle profile of 2-D PrGO/S and 3-D FLG/PrGO-S electrodes at C/10 and 1C rate with corresponding coulombic efficiency calculations for the 3-D FLG/PrGO-S electrodes. | |
Table 1
Comparison of specific capacities reported in the literature for various carbon substrates
| Electrode morphology |
Free standing carbon paper |
3D carbon composite |
Free standing graphene sponge |
Free standing honeycomb–graphene |
Vert. aligned carbon nano sheets |
Vert. aligned graphene nano walls |
Disordered carbon nanotube |
Layer-by-layer stacking |
Sandwiched Li2S electrodes |
3D gyroidal carbon |
Carbon onion |
3D FLG –PrGO – sulfur |
| Sulfur content |
72 wt%, 3.6 mg cm−2 |
72 wt%, 2.3 mg cm−2 |
80 wt%, 11.2 mg cm−2 |
61 wt%, 0.8 mg cm−2 |
68.3 wt%, 1.0 mg cm−2 |
66 wt%, 1.3 mg cm−2 |
40 wt% |
42.8 wt%, 1.9 mg cm−2 |
23 wt%, 1.0 mg cm−2 |
45 wt%, 0.8 mg cm−2 |
53 wt%, 2 mg cm−2 |
80 wt%, 1.2 mg cm−2 |
| Capacity/mA h g−1 (C-rate) (cycle) |
400 (2C); 950 (C/5) (200 cycle) |
917 (1C); (200 cycle) |
580 (C/10) 480 (C/10) (300 cycle) |
730 (C/5); (200 cycle) |
844 (C/2); (300 cycle) |
400 (4C); (240 cycle) |
∼600 (C/8); (100 cycle) |
785 (2C); 900 (C/5) (200 cycle) |
752 (C/2); 620 (C/2) (100 cycle) |
830 (C/10); (100 cycle) |
670 (C/10); (100 cycle) |
725 (5C); 880 (1C); 1040 (C/10) (350 cycle) |
| Potential window |
2.8–1.6 V |
3–1.5 V |
3–1.5 V |
3.0–1.5 V |
2.8–1.7 V |
3.0–1.0 V |
3.0–1.5 V |
2.8–1.5 V |
3–1.6 V |
2.8–1.5 V |
2.6–1.8 V |
2.6–1.8 V |
| LiNO3 |
0.2 M |
Yes (unreported value) |
No |
0.1 M |
0.1 M |
No |
No |
0.1 M |
0.1 M |
0.1 5 M |
0.25 M |
0.25 M |
| Reference |
Li Z.63 |
Li G.64 |
Lu S.65 |
Ni W.54 |
Rehman S.58 |
Li B.49 |
Guo J.66 |
Qie L.67 |
Fu Y.68 |
Werner J. G.69 |
Choudhury S.70 |
This work |
Furthermore, the post-mortem SEM (Fig. S6, ESI†) revealed that the 3-D FLG/PrGO-S electrode maintained excellent structural integrity even after 350 cycles at 1C, suggesting that the substantial micro-porosity within the FLG structure compensates the volume changes of sulfur during polysulfide formation. Moreover, as seen in the cross-sectional FIB milled image, Fig. S6(b),† the FLG structural morphology post-mortem, displays a marked difference for infiltrated samples as compared to pristine FLGs. The residual functional groups on the PrGO surface especially C–O and –COOH can act as the reaction centres with Li+ ions by rapidly and reversibly capturing them through surface absorption and surface redox reaction. Furthermore, with the on-going optimisation of height and porosity of the underlying FLG electrodes being currently undertaken, it is expected that the electrochemical performance of these 3D FLG/PrGO/S electrodes can be enhanced even further.
4. Conclusions
In conclusion, hybrid 3-D electrode structures consisting of FLGs melt-impregnated with PrGO-sulfur nanocomposites were successfully prepared with a high sulphur loading of ∼80 wt%. The 1–3 layered FLGs with a predominantly knife-edge structure were synthesised using a microwave plasma technique and largely composed of carbon (98.8 at%) with a small amount of adsorbed oxygen (1.2 at%). The melt infiltration process enabled deposition of a thin layer of sulfur nanoparticles as well as larger discrete particles embedded in the PrGO matrix as evident from the elemental mapping and EDX, which also confirmed the high sulfur/carbon ratio. Through the use of these multifunctional nanocomposites, a judicious balance of surface oxygen functional groups and 3-D architecture was achieved. This unique combination was able to facilitate electronic, ionic transport and ease of electrolyte accessibility along with confinement of the polysulfide shuttles within the matrix leading to excellent cycling capabilities. The resultant electrode not only displayed a high discharge capacity of 1350 mA h g−1 but also exhibited excellent capacity of ∼80% even after 350 cycles with ∼1040 mA h g−1 and 880 mA h g−1 at C/10 and 1C, respectively. On the other hand, comparative studies with 2-D planar PrGO-S electrodes displayed a relatively poor capacity retention of 560 mA h g−1 after 150 cycles, suggesting limited charge transport within 2D electrode matrix and dissolution of lithium polysulfide in to liquid electrolyte. Thus, the current study highlights the importance of the electrode microstructure and the presence of surface functional groups to improve active material utilisation and charge discharge performance at high currents (C-rates). Furthermore, these initial results suggest the sincere need to build fundamental understanding to establish electrode structure and electrochemical performance relationship and is being further explored by the group.
References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- R. D. Rauh, F. S. Shuker, J. M. Marston and S. B. Brummer, J. Radioanal. Nucl. Chem., 1977, 39, 1761–1766 CrossRef CAS.
- H. Yamin and E. Peled, J. Power Sources, 1983, 9(3), 281–287 CrossRef CAS.
- R. D. Rauh, K. M. Abraham, G. F. Pearson, J. K. Surprenant and S. B. Brummer, J. Electrochem. Soc., 1979, 126(4), 523–527 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
-
J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, New York, USA, 1999 Search PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- S. E. Cheon, K. S. Ko, J. H. Cho, S. W. Kim, E. Y. Chin and H. T. Kim, J. Electrochem. Soc., 2003, 150, A800–A805 CrossRef CAS.
- X. L. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- K. Xi, S. Cao, X. Peng, C. Ducati, R. V. Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192–2194 RSC.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 9592–9595 CrossRef CAS PubMed.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- S. Dörfler, M. Hagen, H. Althues, J. Tübke, S. Kaskel and M. J. Hoffmann, Chem. Commun., 2012, 48, 4097–4099 RSC.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Adv. Mater., 2011, 23, 5641–5644 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- C. J. Hart, M. Cuisinier, X. Liang, D. Kundu, A. Garsuch and L. F. Nazar, Chem. Commun., 2015, 51, 2308–2311 RSC.
- G. He, C. J. Hart, X. Liang, A. Garsuch and L. F. Nazar, ACS Appl. Mater. Interfaces, 2014, 6, 10917–10923 CAS.
- K. Xi, P. R. Kidambi, R. Chen, C. Gao, X. Peng, C. Ducati, S. Hofmann and R. V. Kumar, Nanoscale, 2014, 6, 5746–5753 RSC.
- G. Zhou, L. C. Yin, D. W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H. M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS PubMed.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- S. Yuan, Z. Guo, L. Wang, S. Hu, Y. Wang and Y. Xia, Adv. Sci., 2015, 2(8), 1500071 CrossRef PubMed.
- C. Tang, Q. Zhang, M. Q. Zhao, J. Q. Huang, X. B. Cheng, G. L. Tian, H. J. Peng and F. Wei, Adv. Mater., 2014, 26(35), 6100–6105 CrossRef CAS PubMed.
- M. Q. Zhao, X. F. Liu, Q. Zhang, G. L. Tian, J. Q. Huang, W. Zhu and F. Wei, ACS Nano, 2012, 6(12), 10759–10769 CrossRef CAS PubMed.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. D. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. She and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- C. Fongy, A. C. Gaillot, S. Jouanneau, D. Guyomard and B. Lestriez, J. Electrochem. Soc., 2010, 157, A885–A891 CrossRef CAS.
- M. Gaberscek and J. Jamnik, Solid State Ionics, 2006, 177, 2647–2651 CrossRef CAS.
- D. P. Singh, F. M. Mulder, A. M. Abdelkader and M. Wagemaker, Adv. Energy Mater., 2013, 3, 572–578 CrossRef CAS.
- P. A. Johns, M. R. Roberts, Y. Wakizaka, J. H. Sanders and J. R. Owen, Electrochem. Commun., 2009, 11, 2089–2092 CrossRef CAS.
- G. He, X. L. Ji and L. F. Nazar, Energy Environ. Sci., 2011, 4, 2878–2883 CAS.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- X. Xiao, P. Liu, J. S. Wang, M. W. Verbrugge and M. P. Balogh, Electrochem. Commun., 2011, 13(2), 209–212 CrossRef CAS.
- N. Li, H. Song, H. Cui and C. Wang, Nano Energy, 2014, 3, 102–112 CrossRef CAS.
- C. Zu, L. Li, J. Guo, S. Wang, D. Fan and A. Manthiram, Phys. Chem. Lett., 2016, 7, 1392–1399 CrossRef CAS PubMed.
- S. Sharma, A. Ganguly, P. Papakonstantinou, X. Miao, M. Li, J. L. Hutchison, M. Delichatsios and S. Ukleja, J. Phys. Chem. C, 2010, 114(45), 19459–19466 CAS.
- R. E. Priestley, A. Mansfield, J. Bye, K. Deplanche, A. B. Jorge, D. Brett, L. E. Macaskie and S. Sharma, RSC Adv., 2015, 5(102), 84093–84103 RSC.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. J. Fernandez-Merino, L. Guardia, J.
I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez Alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426–6432 CAS.
- N. Soin, S. S. Roy, S. Roy, K. S. Hazra, D. S. Misra, T. H. Lim, C. J. Hetherington and J. A. McLaughlin, J. Phys. Chem. C, 2011, 115(13), 5366–5372 CAS.
- N. Soin, S. S. Roy, C. O'Kane, J. A. McLaughlin, T. H. Lim and C. J. Hetherington, CrystEngComm, 2011, 13(1), 312–318 RSC.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- S. Sharma, M. N. Groves, J. Fennell, N. Soin, S. L. Horswell and C. Malardier-Jugroot, Chem. Mater., 2014, 26(21), 6142–6151 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CAS.
- J. W. Kim, J. D. Ocon, D. W. Park and J. Lee, J. Energy Chem., 2012, 22(2), 336–340 CrossRef.
- B. Li, S. Li, J. Liu, B. Wang and S. Yang, Nano Lett., 2015, 15, 3073–3079 CrossRef CAS PubMed.
- L. Zhang, L. Ji, P. A. Glans, Y. Zhang, J. Zhu and J. Guo, J. Phys. Chem. Phys., 2012, 14, 13670–13675 RSC.
- L. W. Ji, M. M. Rao, H. M. Zheng, L. Zhang, Y. C. Li, W. H. Duan, J. H. Guo, E. J. Cairns and Y. G. Zhang, J. Am. Chem. Soc., 2011, 133, 18522 CrossRef CAS PubMed.
- J. Rong, M. Ge, X. Fang and C. Zhou, Nano Lett., 2013, 14(2), 473–479 CrossRef PubMed.
- J. Z. Wang, L. Lu, M. Choucair, J. A. Stride, X. Xu and H. K. Liu, J. Power Sources, 2011, 196(16), 7030–7034 CrossRef CAS.
- W. Ni, J. Cheng, X. Li, Q. Guan, G. Qu, Z. Wang and B. Wang, RSC Adv., 2016, 6(11), 9320–9327 RSC.
- C. Barchasz, F. Molton, C. Duboc, J. C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84(9), 3973–3980 CrossRef CAS PubMed.
- M. Cuisinier, P. E. Cabelguen, S. Evers, G. He, M. Kolbeck, A. Garsuch, T. Bolin, M. Balasubramaniam and L. F. Nazar, J. Phys. Chem. Lett., 2013, 4, 3227–3232 CrossRef CAS.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- S. Rehman, X. Gu, K. Khan, N. Mahmood, W. Yang, X. Huang, S. Guo and Y. Hou, Adv. Energy Mater., 2016, 6(12), 1502518 CrossRef.
- Y. X. Wang, L. Huang, L. C. Sun, S. Y. Xie, G. L. Xu, S. R. Chen, Y. F. Xu, J. T. Li, S. L. Chou, S. X. Dou and S. G. Sun, J. Mater. Chem., 2012, 22(11), 4744–4750 RSC.
- W. Kong, L. Sun, Y. Wu, K. Jiang, Q. Li, J. Wang and S. Fan, Carbon, 2016, 96, 1053–1059 CrossRef CAS.
- R. Singhal, S.-H. Chung, A. Manthiram and V. Kalra, J. Mater. Chem. A, 2015, 3, 4530–4538 CAS.
- R. Chen, T. Zhao, J. Lu, F. Wu, L. Li, J. Chen, G. Tan, Y. Ye and K. Amine, Nano Lett., 2013, 13(10), 4642–4649 CrossRef CAS PubMed.
- Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. D. Lou, Nat. Commun., 2015, 6, 8850 CrossRef CAS PubMed.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- S. Lu, Y. Chen, X. Wu, Z. Wang and Y. Li, Sci. Rep., 2014, 4, 4629 CrossRef PubMed.
- J. Guo, Y. Xu and C. Wang, Nano Lett., 2011, 11(10), 4288–4294 CrossRef CAS PubMed.
- L. Qie and A. Manthiram, Adv. Mater., 2015, 27(10), 1694–1700 CrossRef CAS PubMed.
- Y. Fu, Y.-S. Su and A. Manthiram, Adv. Energy Mater., 2014, 4(1), 1300655 CrossRef.
- J. G. Werner, S. S. Johnson, V. Vijay and U. Wiesner, Chem. Mater., 2015, 27(9), 3349–3357 CrossRef CAS.
- S. Choudhury, M. Zeiger, P. Massuti-Ballester, S. Fleischmann, P. Formanek, L. Borchardt and V. Presser, Sustainable Energy Fuels, 2017, 1(1), 84–94 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00195a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *cd,
S.
Sharma
e,
S.
Basak
f,
S.
Sachdeva
*cd,
S.
Sharma
e,
S.
Basak
f,
S.
Sachdeva