
CO2-looping in biomass pyrolysis or gasification
Yafei
Shen
 *a,
Dachao
Ma
b and
Xinlei
Ge
a
*a,
Dachao
Ma
b and
Xinlei
Ge
a
aJiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, Jiangsu Engineering Technology Research Center of Environmental Cleaning Materials, School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Nanjing 210044, China. E-mail: shen1225@nuist.edu.cn
bSchool of Environment, Guangxi University, Nanning 530004, China
First published on 29th June 2017
Abstract
Thermochemical technologies for converting biomass into energy or chemicals mainly consist of combustion, pyrolysis, and gasification. This review summarizes the recent advances in biomass pyrolysis and gasification by using CO2 as a reaction medium. The CO2-looping pyrolysis or gasification of biomass is compared with conventional processes. In the integrated valorization of biomass by pyrolysis or gasification, CO2 can play a vital role in each stage, mainly including biomass pyrolysis, biomass/biochar gasification, biochar activation, and tar cracking/reforming. CO2 as a reaction medium can significantly improve the thermal efficiency of biomass pyrolysis. Pyrolysis in CO2 results in deep decomposition of biomass compared to pyrolysis in N2. Also, CO2 has an affinity to react with hydrogenated and oxygenated groups, leading to biochar with a higher specific surface area (SBET). Thus, exploiting CO2 as a reaction medium in biomass pyrolysis provides an attractive option for enhanced generation of syngas and tuned adsorption capability of biochar. In addition, the CO2 pyrolysis of biomass can enhance the thermal cracking of harmful organic compounds, thus suppressing of the formation of benzene derivatives (e.g., volatile organic compounds) and polycyclic aromatic hydrocarbons. In general, CO2 gasification of biomass serves a dual purpose of reducing pollution and generating syngas. In addition, introducing CO2 with steam as a gasifying agent can enhance CO production. The internal mineral species (e.g., K, Ca) in biomass can also improve the reactivity of char gasification and tar reforming in the entire gasification processes.
1. Introduction
Global environmental issues such as climate change triggered by anthropogenic greenhouse gases have prompted unprecedented researches associated with carbon capture and storage and renewable energies to alleviate various global environmental problems.1–4 Harnessing biofuels has been paid much attention due to its intrinsic carbon neutrality, along with political support such as renewable fuel standard that expedited the rapid employment of biofuels. In parallel, the concept of biorefining has been developed since our demand for carbon to sustain the current industrial infrastructure still continues to grow.2,4 Despite significant technical advances, biomass as an energy feedstock has been considered as quite challenging due to its heterogeneous matrices and highly functionalized structures.1,5 In this case, concrete technical methodologies for producing fuels from biomass have not been established. In general, biomass can be converted to biofuels via various pathways such as biochemical (e.g., cellulosic-ethanol production) or thermochemical processes (e.g., gasification, pyrolysis). However, pyrolysis or gasification has been recognized as one of the promising techniques for efficiently transforming biomass into various fuels or chemicals.6,71.1 Biomass pyrolysis
Biofuels derived from biomass pyrolysis have technical advantages over those from conventional biochemical conversion since the entire biomass can be used as the feedstock (rather than simple sugars) in only a few seconds (rather than hours or days).8 Commonly, biomass pyrolysis is induced by the heat transported from the surrounding gas to the fuel particle, causing thermal decomposition of it into a huge number of products. As illustrated in Fig. 1, during transient heating, temperature increases locally, leading to the evaporation of moisture (i.e. drying stage) and then to the progressive release of pyrolytic volatiles (i.e. primary pyrolysis stage). The primary volatiles (denoted by “1”) are produced from the thermal scission of chemical bonds in the individual biomass components, including cellulose, hemicellulose, lignin and extractives, and comprise permanent gas species (e.g., CO2, CO, CH4) and condensates at ambient conditions (several organics and water). Although each of the biomass components decomposes at faster rates in different temperature ranges, the overall primary pyrolysis stage is complete at relatively low temperatures (<500 °C), yielding a carbon-rich non-volatile solid (i.e. char). The char contains a significant part of the mineral matter of the parent fuel. Nevertheless, if the fuel is converted at higher temperatures, some of the primary volatiles released inside the particle can further participate in a variety of secondary reactions to form product “2”. | ||
| Fig. 1 Thermal conversion of solid biomass particle under inert atmosphere: drying, primary pyrolysis and secondary pyrolysis. The arrows indicate the main routes for the formation of products.9 | ||
Serial and parallel reactions either heterogeneously or homogeneously (e.g., cracking, reforming, dehydration, condensation, polymerization, oxidation and gasification) can occur.10,11 The distinction between intraparticle/primary pyrolysis and extraparticle/secondary pyrolysis is not perfect as the secondary reactions of volatiles can occur both in the pores of the particles and/or in the bulk gas. Thus, the primary and secondary reactions can occur simultaneously in different parts of a fuel particle. The char derived from the primary pyrolysis stage can be active during the secondary reactions, namely by catalytic conversion of organic vapors into light gases (cracking reactions) and secondary char (polymerization reactions). In addition, the char can be converted into gases by gasification reactions with H2O and CO2. Nevertheless, the rates of char gasification with H2O and CO2 are orders of magnitude lower than those of primary pyrolysis, so the conversion of char is limited during the release of volatiles. In comparison, the secondary conversion of the primary volatiles is a rapid process, mainly depending on the operational conditions, and it can exert a major influence on the ultimate composition and yields of the volatiles. In addition, primary fragmentation and shrinkage of the particles can occur in parallel with the above physicochemical processes. The composition of pyrolytic volatiles mainly includes H2O, CO2, CO, H2, CH4, other non-condensable hydrocarbons and condensable organic compounds (i.e. tar).
Pyrolysis can be considered as a sustainable biomass valorization method for energy production and carbon capture and sequestration.12 Pyrolysis defined as the thermal decomposition at an intermediate temperature (500–900 °C) under limited O2 can convert biomass into bio-oil, gases and biochar.13 The yields and properties of the pyrolytic products are influenced by many parameters including residence time, temperature, and heating rate. Based on the heating rate and reaction time, the pyrolysis process could be classified into three types, slow, fast and flash pyrolysis (as shown in Table 1).13–15 Slow pyrolysis can provide great environmental benefits since it produces more biochar, possibly applied for soil reclamation and carbon sequestration. However, fast pyrolysis has a better economic return because of the valuable biofuel production (e.g., bio-oil, syngas).16 In addition, pyrolysis is the initial chemical step in all biomass gasification and combustion processes.17
| Slow pyrolysis | Fast pyrolysis | Flash pyrolysis | |
|---|---|---|---|
| Heating rate (°C min−1) | 5–7 | 300–800 | >1000 |
| Pyrolysis temp. (°C) | 500–1200 | 600–900 | 600–1200 |
| Feedstock size (mm) | 5–50 | <3 | <0.5 |
| Feedstock residence time | >1 h | 0.5–10 s | <2 s |
| Pyrolysis setup | Fixed bed | Fixed or fluidized bed | Fluidized or entrained flow bed |
| Main products | Char | Oil | Gas |
In general, the mechanism for pyrolysis of lignocellulosic biomass can be divided into the mechanisms of three individual components.18 The decomposition of cellulose, hemicelluloses, and lignin depends mainly on heating rate, temperature, reactor type, residence time, and particle size. In biomass pyrolysis, hemicelluloses decompose initially at 470–530 K, whereas cellulose breaks down at 510–620 K, and decomposition of lignin occurs at 550–770 K.14 The mechanism of cellulose pyrolysis can be characterized by depolymerization.19 Two basic reactions occur in the pyrolysis: one is the decomposition and charring of the cellulose in slow pyrolysis;20 the other is rapid volatilization with levoglucosan formation in fast pyrolysis.21 The cellulose is initially depolymerized into oligosaccharides, and then the glucosidic bonds of the oligosaccharides are broken to form D-glucopyranose, undergoing an intramolecular rearrangement to form levoglucosan (LG).22 The LG decomposes via three ways (i.e., C–O scission, C–C scission, and LG dehydration) to form the small-molecular-weight compounds.23–31 At 667–1327 K, the dehydration, with the formation of a H2O molecule composed of an OH group from C2 and an H atom from C3, is a preferred pathway for LG decomposition (Fig. 2), whereas the C–O bond scission mechanism agrees better with that of C–C bond scission. The pathway of C–O bond scission starts with the breakage of C6–O1 and C1–O5 bonds to form CO simultaneously (Fig. 2). The C–C bond scission pathway has the highest energy barrier among the three LG decomposition pathways (Fig. 2).32
 | ||
| Fig. 2 Levoglucosan (LG) decomposition mechanism in the pyrolysis of cellulose: LG dehydration, C–O bond scission, and C–C bond scission.32 | ||
The pyrolysis mechanism of hemicelluloses is very similar to that of cellulose, which starts with the depolymerization of polysaccharide chains to form oligosaccharides, following the cleavage of the xylan chain in the glycosidic linkage and rearrangement of the produced molecules to afford 1,4-anhydro-D-xylopyranose.33 After that, 1,4-anhydro-D-xylopyranose is further decomposed to form small-molecular-weight compounds, e.g., furfural and two- and three-carbon fragments.34–36 More details of hemicellulose pyrolysis have been reviewed in terms of experimental studies on various hemicellulose feedstocks and reactors, reaction mechanisms for pyrolysis of hemicellulose and its model compounds unraveled by computational efforts, and global kinetic modeling as well as mechanistic modeling of hemicellulose pyrolysis.37
The structure of lignin is more complex than those of cellulose and hemicellulose, leading to a complex decomposition mechanism.38–41 Lignin pyrolysis consists of drying, fast degradation, and slow degradation stages. Phenolic compounds, i.e., guaiacol type, phenol (P-) type, syringol (S-) type, and catechol (C-) type, are the main products of lignin pyrolysis.40 The dominant pathway of free radical reaction has been widely regarded as one of the most important mechanisms for lignin pyrolysis.42–45 As shown in Fig. 3, in the pyrolysis process, the free radicals are generated in the breaking of the β-O-4 linkage of the lignin molecules, which is the initial step for the reaction of free radical chain.43,46 The radicals can capture the protons from other species with weak C–H or O–H bonding (e.g., C6H5–OH) and form decomposed products (e.g., vanillin and 2-methoxy-4-methylphenol). Thereafter, the radicals can be passed to other species for chain propagation. Finally, the chain reactions can be terminated, once a stable compound is formed by the collision of two radicals.
 | ||
| Fig. 3 Lignin decomposition mechanism in pyrolysis process.42 | ||
Besides cellulose, hemicelluloses, and lignin, inorganic species can significantly influence the pyrolysis of biomass. For example, K and Cl can vaporize at a relatively low temperature, whereas Ca and Mg are ionically or covalently bound with organic molecules and vaporize at high temperatures.47 P, S, and N are covalently bound with complex organic compounds within the cells of plants48 that can be decomposed at low pyrolysis temperatures. Biomass pyrolysis can also be regarded as an autocatalytic process, since some inorganic constituents in biomass, especially alkali and alkaline earth metals (such as K, Ca, and Mg), have significant catalytic effects on biomass pyrolysis.49 K catalyzes the secondary cracking of the pyrolytic volatiles,13 producing more gas compounds (e.g., CO, H2, CH4, CO2), and further resulting in biochar cracking. As for lignocellulosic biomass, the autocatalysis of the inherent inorganic species alone is insufficient to lead to bio-oil and biochar with the desired properties. Further studies are required to develop efficient catalysts for the selective conversion of biomass to bio-oil with high heating values and biochar with the desired functional groups and porous structure. The use of efficient catalysts may allow the reduction of the pyrolysis temperature and residence time and improve the qualities of both bio-oil and biochar. More recently, Liu et al.50 critically reviewed the fates of the main chemical elements (C, H, O, N, P, Cl, S, and metals) in biomass during the pyrolysis process.
The integration of various technologies can work as leverage in promoting a “circular economy”, aiming at improving both resource use and operation efficiency. Moreover, using an integrated process can overcome defects in each individual process. Thus, the integrated process of anaerobic digestion with pyrolysis can contribute to a new highly effective conversion.51 The integration of anaerobic digestion with pyrolysis can allow the bio-conversion of the aqueous phase of pyrolysis liquor derived from the pyrolysis of solid digestate.52 In addition, biochar has the ability to catalyze anaerobic digestion by means of mitigating mild ammonia inhibition, supporting archaeal growth and the methanization of the biochar labile carbon.53 Therefore, the integrated process of anaerobic digestion with pyrolysis will open up innovative pathways for efficient valorization of lignocellulosic biomass.54 In general, lignocellulosic biomass through anaerobic digestion can be converted into biogas (mainly CH4 and CO2). After pyrolysis, biomass can be degraded into syngas (mainly H2 and CO2), bio-oil and biochar. Through such integration, resource use efficiency can be significantly improved. On the one hand, after anaerobic digestion, more energy in the feedstocks is transferred into pyrolysis volatiles and biogas.55 Biochar derived from anaerobic digestate pyrolysis has lower heating value than biochar from pyrolysis of raw feedstocks. Besides, the biochar can fix heavy metals and nutrient elements such as P, K and Ca in digestate, preventing them from being released into aqueous phase.56 In the anaerobic digestion process, some recalcitrant components of lignocellulose biomass (e.g., lignin) will prevent anaerobic microorganisms from degrading substrate. Thus, the anaerobic digestate still contains considerable energy.57 In this case, pyrolysis of anaerobic digestate not only reduces anaerobic digestate cost management, but further uses anaerobic digestate to recover higher bioenergy and produce more products including biochar, bio-oil and syngas.58 Through the life cycle assessment of lignocellulosic biomass pyrolysis integrated with anaerobic digestion, it was found that greenhouse gas emissions can be significantly reduced in the integrated process without worsening the abiotic resources depletion.59 In summary, the integration of anaerobic digestion with pyrolysis can offer further potentially synergistic combinations including use of anaerobic digestate for pyrolysis,60 biomethanation of syngas61 or use of pyrochars in anaerobic digestion.62 From an ecosystem perspective, functions carried out by two soil amendments (digestate or pyrochar) are different. While digestate is rich in degradable organic matter that will be slowly decomposed by soil microbes, pyrochar has very poor, easily degradable organic matter and its chemical form is particularly stable. Digestate gradually releases plant nutrients under microbial mineralization and its degradable organic matter is the primary source to start up the ecological “debris chain” (Fig. 4). Conversely, pyrochar can play an important role in carbon capturing and its long-term sequestration into soil, thus contributing to climate change mitigation.63
 | ||
| Fig. 4 Integration of anaerobic digestion with pyrolysis for bio-waste valorization and the final closing loop with the return of the organic and mineral by-products to agricultural soil (as soil amendment and/or fertilizer).63 | ||
The chemical mechanism of the integration of pyrolysis with anaerobic digestion for valorization of lignocellulosic biomass is illustrated in Fig. 5.64 The complexity of the metabolic pathways leading to biogas from biopolymers (namely cellulose, lignin, and hemicellulose) can be classified into four sequential steps: degradation of biopolymers into smaller molecules like monomers via hydrolysis, which are fermented principally into volatile fatty acids via acidogenesis, further digested into CH3COOH, CO2 and H2 (acetogenesis) that are converted into the final products CH4 and CO2 (methanogenesis). Upon pyrolysis, biopolymers are converted into gas (syngas), liquid (bio-oil) and solid (biochar). Herein, two main configurations can be identified in the integrated process of pyrolysis with anaerobic digestion. On the one hand, pyrolysis products are upgraded into biogas via anaerobic digestion, and pyrolysis is applied “upstream” to break down hemicellulose and lignin into smaller compounds accessible to biomethanation; on the other hand, anaerobic digestion products are upgraded via pyrolysis. Specifically, “downstream” pyrolysis can convert residual solid digestate from biogas plants into fuels and materials.
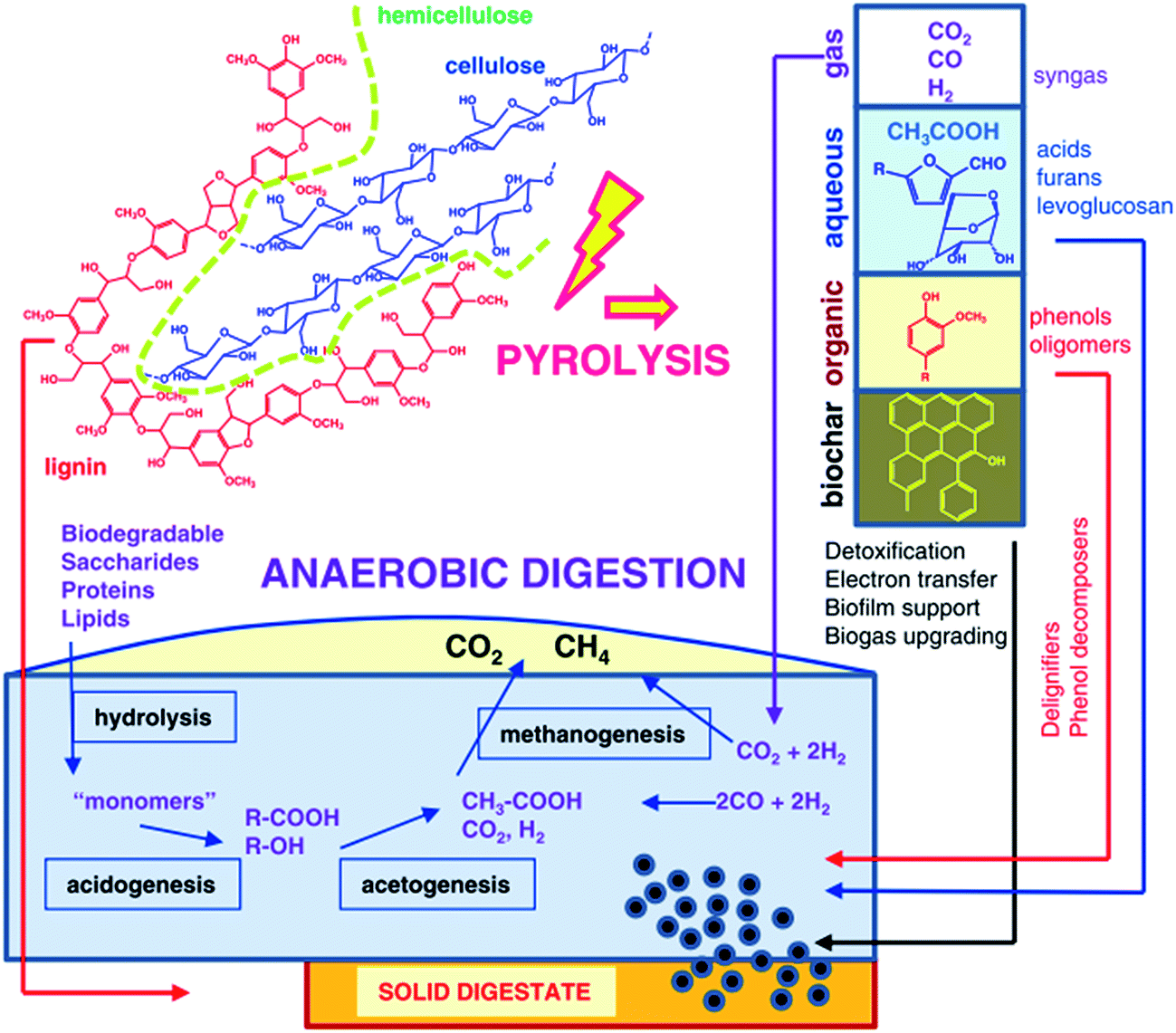 | ||
| Fig. 5 Schematic of chemical conversion in the integration of pyrolysis and anaerobic digestion.64 | ||
1.2 Biomass gasification
Biomass gasification is an intricate process involving drying the feedstock followed by pyrolysis, partial combustion of intermediates, and finally gasification of the resulting products. It is performed in the presence of a gasifying medium (e.g., air, O2, H2O or CO2), inside a gasifier. Biomass gasification reduces the C/H mass ratio resulting in increased calorific content of the product on account of enhanced H2 fraction.65 The gasifying medium plays a vital role in converting char and heavy hydrocarbons to low-molecular-weight gases, e.g., CO and H2. The quality and properties of the product mainly depend on the feedstock types and size, gasifying agent, temperature and pressure inside the reactor, design of gasifier and the presence of catalyst and sorbent, etc.66 There are many useful products from biomass gasification including syngas, heat, power, biofuels, fertilizer and biochar. Syngas can be further processed by the Fischer–Tropsch process into methanol, dimethyl ether and other chemical feedstocks.Over the past decade, biomass gasification has been developed to obtain value-added products such as syngas, H2, CH4 and chemical feedstocks.67 These gases can also be produced from biomass via biochemical routes. Thermochemical pathways have an edge over the other routes, as biochemical processes have issues with treating biomass rich in lignocellulose.68,69 In addition, they operate relatively slowly in batch mode and produce a dilute product stream, with large amounts of water recirculating. However, the thermochemical route can accommodate a more diverse range of biomass70 with a higher efficiency and a lower cost.71 One of the main limitations is the small range of products.70,71 At present, cutting-edge, innovative and economical techniques with high efficiencies are still urgently required for biomass gasification.72 Silkarwar et al.73 provided an assessment of the fundamentals such as feedstock types, the impacts of operating parameters, tar formation/cracking, and modelling methods for biomass gasification. Herein, various mechanisms for biomass gasification are comparatively discussed. The gasification process is an alternative thermochemical method that can convert biomass into gaseous products (e.g., syngas) at relatively high temperatures. The gasification routes are shown in Fig. 6A. The related reactions including heterogeneous and homogeneous ones in biomass gasification are depicted in Fig. 6B. Generally, processes in biomass gasification include drying and devolatilization, volatile and char combustion/gasification, and tar reforming with CO2 and steam.74
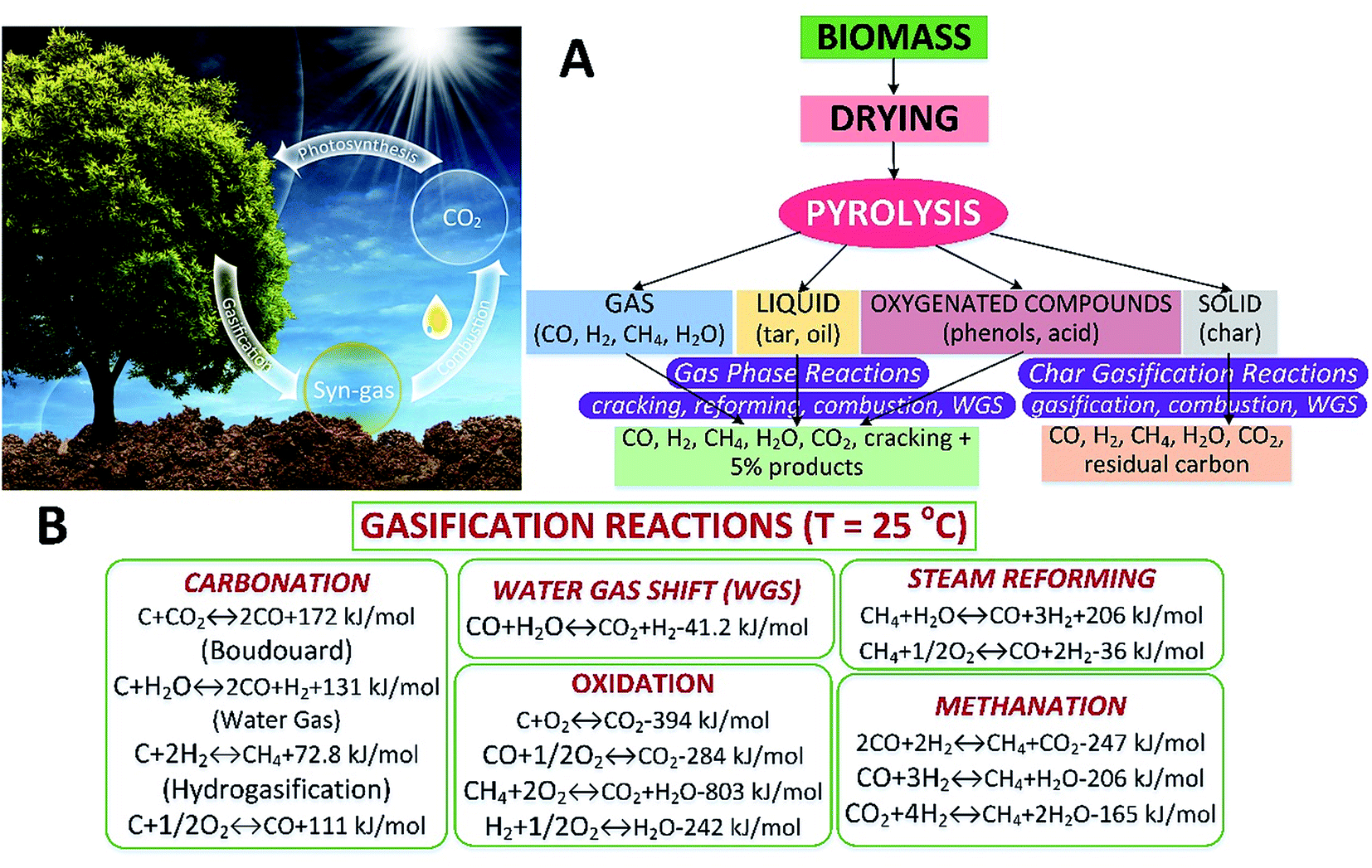 | ||
| Fig. 6 Biomass gasification routes (A) and reactions (B).73 | ||
Gasification is the process of partial combustion of biomass to produce the valuable syngas (e.g., CO, H2, CH4, and CO2) along with tar (i.e. condensable organic compounds) and other contaminants (e.g., NH3, H2S, and HCN).74,75 Gasification consists of three major steps: drying, devolatilization (pyrolysis), and gasification. Extensive research has been done to understand the effect of steam, air, oxygen, and their combinations on the process.76–80 However, the role of CO2 as an oxidizing agent has been explored in only a limited number of studies. Biomass gasification with CO2 as a gasifying agent is a promising way to relieve energy shortages and minimize CO2 emission. In Section 3, CO2 gasification of biomass will be further discussed.
1.3 Biomass tar
Condensable organic compounds referred to as “tar” are inevitably generated along with the producer gas in biomass pyrolysis or gasification. Tar is regarded as one of the main bottlenecks, which limits the commercialization of biomass pyrolysis and gasification technology. Tar can condense and polymerize to form more complex structures (e.g., coke). Tar represents a complex mixture of more than 100 compounds, including benzene, phenols, N-heterocyclic compounds, and polycyclic aromatic hydrocarbons (PAHs),81,82 which can be condensed with small particle impurities to form complex structures in downstream equipment, resulting in the mechanical breakdown of the entire pyrolysis/gasification system, or deactivation of catalysts.83 Furthermore, various aromatic and N-heterocyclic compounds in tars are toxic and act as environmental hazards. Tar can be divided into primary, secondary and tertiary tars.84,85 The tar yield of biomass gasification varies from 0.5 to 100 g m−3, mainly depending upon the reactors, operating conditions, and feedstock types.86 Tars are formed in the decomposition of cellulose, hemicelluloses, and lignin. The main components of primary tar include oxygenated hydrocarbons, and with increasing temperature, the oxygenated hydrocarbons are initially converted to light alkanes, olefins, and aromatics, and then transformed to higher hydrocarbons and larger PAHs with a further increase in temperature (Fig. 7).50 | ||
| Fig. 7 Formation of tar compounds at different reaction temperatures.50 | ||
Many approaches can be used for tar removal in the biomass pyrolysis/gasification process. Generally, these methods can be divided into two types: (1) physical methods using absorption (e.g., wet scrubbers) and adsorption (e.g., carbon materials) processes; (2) thermochemical cracking/reforming with catalysts.74,84,87 Among all these methods, thermochemical methods have received the most interest since tar can be transformed into useful gas products. Biochar has a high potential to be used as catalysts or supports for tar conversion.83,88 In general, catalytic conversion of tar is significantly influenced by tar components, thermal cracking/reforming conditions, and catalysts performance. Steam reforming of tar has been extensively considered, because more value-added H2 is generated through water gas and water gas shift reactions.84 As for the biomass gasification technology, steam or CO2 gasification agent can provide a necessary environment for tar conversion, and ensure that catalytic deactivation of biochar cannot be caused by the carbon deposition of tar during the reaction process. In the scope of this review, CO2 reforming of tar with catalysts will be further discussed in Section 5.
1.4 Biochar
The overall mechanism of biochar formation is characterized by the pyrolysis mechanisms of the biomass components of cellulose, hemicellulose, and lignin. As illustrated in Fig. 8, cellulose is initially depolymerized into oligosaccharides followed by the cleavage of the glucosidic bond to produce D-glucopyranose, which undergoes an intramolecular rearrangement to produce levoglucosan. Levoglucosan can form levoglucosenone via a dehydration process, which further proceeds through dehydration, decarboxylation, aromatization, and intramolecular condensation, to form solid biochar. Alternatively, levoglucosan undergoes a series of rearrangement and dehydration processes to form hydroxymethylfurfural, which further decomposes to produce more bio-oil and syngas. It can also undergo polymerization, aromatization and intramolecular condensation reactions to form biochar. The pyrolysis of hemicellulose is similar to that of cellulose. Compared to cellulose and the hemicelluloses, the structure of lignin is more complex, and this leads to a complex decomposition mechanism. As shown in Fig. 8, free radicals are generated by the breaking of the β-O-4 lignin linkage, suggesting the initiating step in the free radical chain reaction. The radicals can capture the protons of other species with weak C–H or O–H bonds (e.g., C6H5–OH) and form decomposed products such as vanillin and 2-methoxy-4-methylphenol. Later in the reaction timeline, the radicals are passed onto other species for further reaction, leading to chain propagation. Finally, the chain reactions are terminated when two radicals collide with each other to form stable compounds.89 The biochar can be fabricated into various functional materials.89 Particularly, the biochar can work as catalysts or supports for tar conversion in the biomass pyrolysis or gasification processes.90,91 | ||
| Fig. 8 Biochar formation mechanism: cellulose, hemicellulose and lignin pyrolysis.89 | ||
1.5 Purpose of this work
Thermochemical processes such as pyrolysis can effectively convert lignocellulosic biomass into value-added energy (e.g., syngas, bio-oil) and materials (e.g., biochar). CO2-assisted technologies for biomass processing can be designed to deliver optimal performance with maximal energy savings, so the biorefinery concept assumes the integrated valorization of biomass toward energy, materials, and chemicals.86 It can be found that CO2 acting as an agent participates in each stage (e.g., hydrolysis, pyrolysis, and gasification) through the entire thermochemical processes. High-pressure CO2 can act as an interesting solvent, which can be used in a great variety of reactions within the biorefinery concept starting from its use in direct biomass processing to its use as a catalyst for dehydration of biomass-derived carbohydrates along with its use in specific applications for biomass intermediary compounds.92 As for CO2 used in pyrolysis and gasification of lignocellulosic biomass, several works have been conducted, but there is still a lack of a comprehensive article on the fundamentals in this field. The purpose of this work is to evaluate the CO2 functions in the entire processes of biomass pyrolysis and gasification, mainly including biomass pyrolysis and gasification, biochar gasification, tar cracking/reforming and gas upgrading (as shown in Fig. 9). The research advances and challenges will be discussed. Furthermore, the environmental implications of CO2-assisted biomass pyrolysis and gasification will be highlighted. In addition, the future development trends and challenges will be presented. | ||
| Fig. 9 Schematic diagram of CO2-looping pyrolysis or gasification of biomass. | ||
2. CO2 pyrolysis of biomass
The thermochemical process of pyrolysis and gasification has been recognized as one of the promising techniques for biomass valorization, but one of the main limitations is that it involves very high temperature, and therefore is quite energy-intensive. To make it a more viable option in energy industry, technical advancements for enhancing the thermochemical efficiency are greatly required. Preferably, exploring the ways to use CO2 as a reaction medium and initial feedstock in the thermochemical process would be a desirable scenario that conforms to current carbon management strategies.93,94In several works, it has been demonstrated that pyrolysis of carbonaceous materials (e.g., biomass) under CO2 atmosphere has enhanced thermal efficiency compared to that under N2 atmosphere.95–106 Zhang et al.107 also studied fast pyrolysis of biomass in a fluidized bed gasifier at 550 °C under N2, CO2, CO, CH4 and H2 atmospheres. It was found that the liquid yield, composition and higher heating value mainly depend on the composition of the pyrolysis bath gas. Pyrolysis in CO2 can produce less char than that in others. The CO2 yield decreased compared to the yield obtained under N2. With regard to the liquid product distribution, the CO2 atmosphere led to the highest yield of acetic acid compared to the others. The acid products yield under N2 was 9% while it increased to 16% under CO2. In addition, the ketones yield increased slightly from 15% to 17% while the phenol yield decreased from 33% to 26%. Two possible mechanisms were proposed: the CO2 reacted either with the active volatiles or with the biochar. The former was more plausible in view of the pyrolysis temperature.
Biomass pyrolysis in the presence of CO2 can cause deep decomposition compared to pyrolysis in pure N2, possibly due to either char gasification by CO2 or the fact that CO2 hinders polymerization reaction and secondary char formation by reacting or cracking tar compounds that may lead to its formation. Moreover, CO2 has an affinity to react with hydrogenated and oxygenated groups, leading to a carbon-rich char. The possible mechanisms in biomass pyrolysis with the CO2 insertion are proposed in Fig. 10. In summary, introducing CO2 in the pyrolysis medium alongside N2 can enhance CO production as a result of homogeneous and heterogeneous reactions of CO2 with gases, tars and char.95
 | ||
| Fig. 10 Possible mechanisms of biomass pyrolysis with CO2 insertion.95 | ||
The pyrolysis of biomass can also be conducted after the initial biorefinery processes. As for the production of bioethanol, lignocellulosic biomass is generally subjected to various pre-treatment methods such as mechanical comminution, acid pre-treatment, ammonia fiber explosion, alkaline extraction, and ionic liquids pre-treatment.108–113 The pre-treated biomass can be hydrolyzed to sugars using cellulose enzymes at 45–50 °C under pH 5.0.114 After that, sugars are converted into bioethanol with 20–50% yield via fermentation with bacteria, yeast, or fungi.110,115 Furthermore, the solid residue after cellulosic ethanol production can be considered for energy recovery by thermal processing such as pyrolysis or gasification. Kim et al.102 studied the pyrolysis of wastes generated through saccharification of oak tree using CO2 as reaction medium. The production of bioethanol was evaluated via saccharification and fermentation of oak tree pretreated with H2SO4, NH3, or NaOH using a yeast (Pichia stipites). Then, the effects of CO2 on pyrolysis of biomass residues were studied. The effect of CO2 was most noticeable in syngas, as the ratio of CO and H2 exhibited a 20- to 30-fold increase at >550 °C. Particularly, the CO/H2 ratio of bio-waste pyrolysis in CO2 is nearly 1100% of that of pyrolysis in N2 at 720 °C. In addition, the use of CO2 as pyrolysis medium does not enhance syngas production but reduces tar formation by thermal decomposition of volatile organic compounds (VOCs) and reaction between VOCs and CO2.
Lee et al.106 also studied the susceptibility of CO2-assisted pyrolysis of various biomasses, including macroalgae (red seaweed), microalgae (C. vulgaris), major constituents of lignocellulosic biomass (cellulose, lignin, hemicellulose), and lignocellulosic biomass (spent coffee ground, corn stover, oak wood). From TGA, the thermal degradation rate of nine different samples in N2 and CO2 is nearly identical, which can be evidenced by the thermal decomposition rate (shown as the slope in Fig. 11), implying that the CO2 co-feed impact on physical aspects such as onset and end temperature of thermal decomposition of biomass is negligible. And the final mass conversion (residual mass at 800 °C) of biomass and coal is nearly the same, suggesting the rate of volatilization via thermolysis (bond dissociation) is not affected by CO2 even at different heating rates and for different types of biomass, since the different rate of volatilization induced by CO2 in pyrolysis of biomass may be reflected by the different thermal degradation rate. Besides, the expected Boudouard reaction is not initiated, which can be explained by the heterogeneous reaction (i.e. slow reaction kinetics between solid phase carbon and gaseous phase of CO2), since the Boudouard reaction is thermodynamically favored at temperatures higher than 720 °C.97,99 Therefore, it also suggests that the reaction kinetics of the Boudouard reaction is highly contingent on the structural biochar matrix.
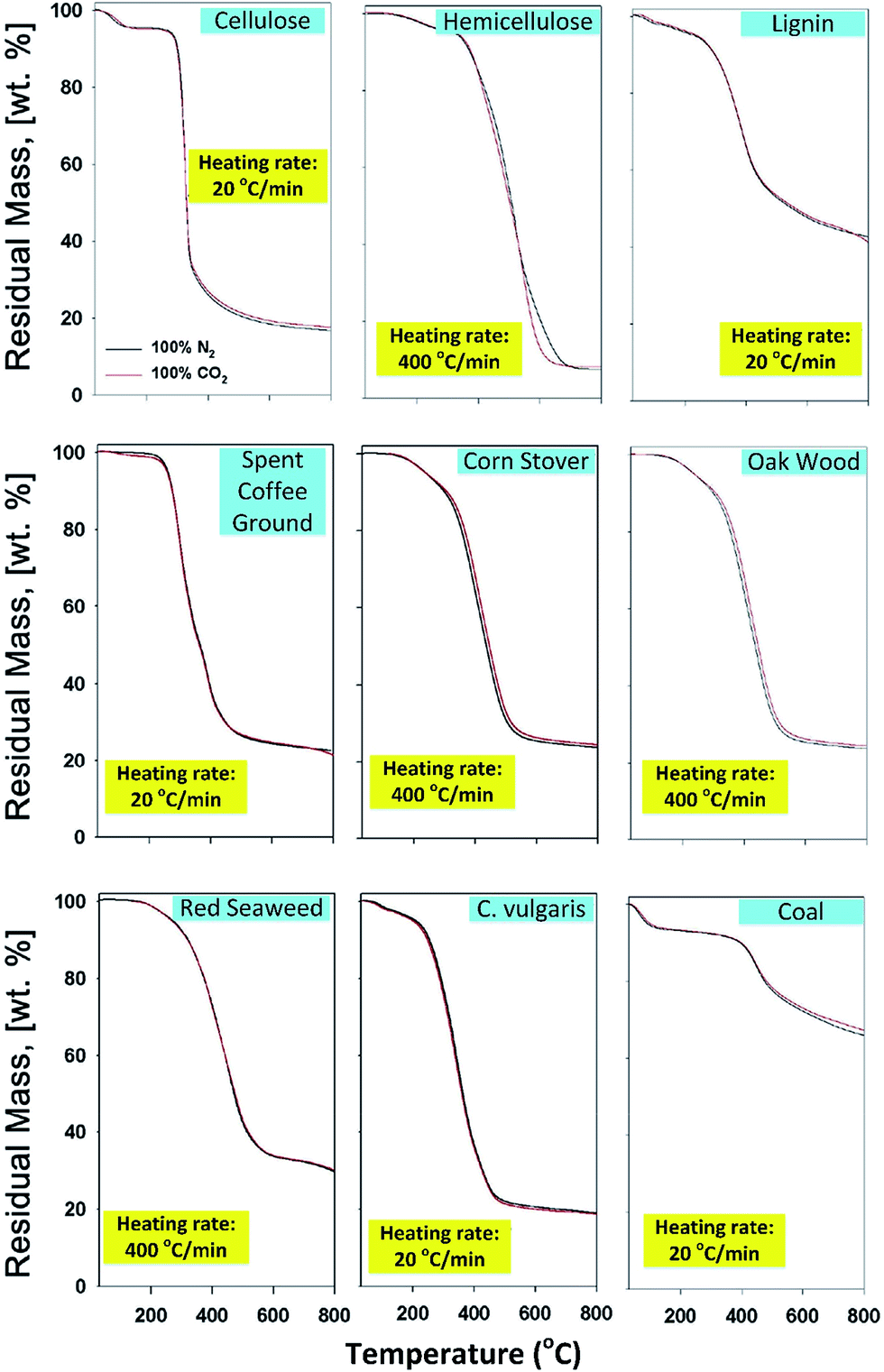 | ||
| Fig. 11 Representative thermograms of various biomasses subjected to slow pyrolysis (20 °C min−1) and fast pyrolysis (400 °C min−1) in N2 and CO2 environments.106 | ||
However, the morphology of biochar generated from pyrolysis of each biomass in N2 and CO2 was noticeably different (Fig. 12a and b). Biochar generated from pyrolysis of biomass in the CO2 atmosphere could develop more pores, thus slightly increasing the specific areas of the biochar.105,106 Furthermore, the differential scanning calorimetry (DSC) curves in N2 and CO2 were quite different in the temperature regime indicating volatilization. An exothermic reaction from 250 to 500 °C in CO2 pyrolysis of oak wood noticeably occurred, suggesting several chemical aspects induced by CO2. For example, carbonization and volatilization in CO2 pyrolysis of oak wood occurred independently since the thermal degradation rate in Fig. 11 and 12c in CO2 was nearly the same. In other words, morphology modification induced by CO2 did not influence the rate of volatilization via bond scission.106 In addition, the biochar derived from pyrolysis of biomass in CO2 environment showed a comparatively high surface area and more porous structure as adsorbent materials.101
 | ||
| Fig. 12 SEM images of biochar generated from pyrolysis of oak wood (a) in N2 and (b) in CO2, and (c) TGA and DSC curves of oak wood pyrolysis in N2 and CO2.106 | ||
The gas evolution of syngas follows the very typical pyrolytic patterns of biomass in CO2 atmosphere: the concentration of H2 is proportional to pyrolytic temperatures since the thermal cracking via dehydrogenation occurs predominantly at high pyrolytic temperatures.97 These typical behaviors provide a favorable condition for forming high content of aromatic chemical species in the oil.116 Similarly, dehydrogenation is also favorable for carbonization.116 In these respects, the concentration of CO is substantially lower than that of H2 (as shown in Fig. 13). However, the gas evolution of major gases is discrepant with the typical pyrolytic patterns.117 The concentration of CO is rapidly increased at above 500 °C. Indeed, this enhanced generation of CO is initiated at >450 °C, but its magnitude is not comparable to that which occurs at >500 °C. For example, the enhanced generation of CO in CO2 can reach up to 1200% at 600 °C for oak wood (Fig. 13a). As for syngas production from biomass pyrolysis at higher temperatures, CO2 can enhance the generation of syngas (e.g., CO, H2) via the thermal cracking of VOCs and the reaction between VOCs and CO2. The identified roles of CO2 lead to the enhanced generation of CO and the subsequent reduction of condensable tar.106 Besides, at lower temperatures (<450 °C), the gas yield can be slightly reduced while the biochar and bio-oil yields marginally improve in CO2.118 Nevertheless, the Boudouard reaction and VOCs or tar cracking occur with difficulty at much lower temperatures. Through the study of the CO2-assisted co-pyrolysis of coal and biomass,119,120 the influence of CO2 could effectively occur at above 550 °C, which leads to VOC and tar reduction, thus enhancing generation of syngas and modifying the ratio of CO/H2.
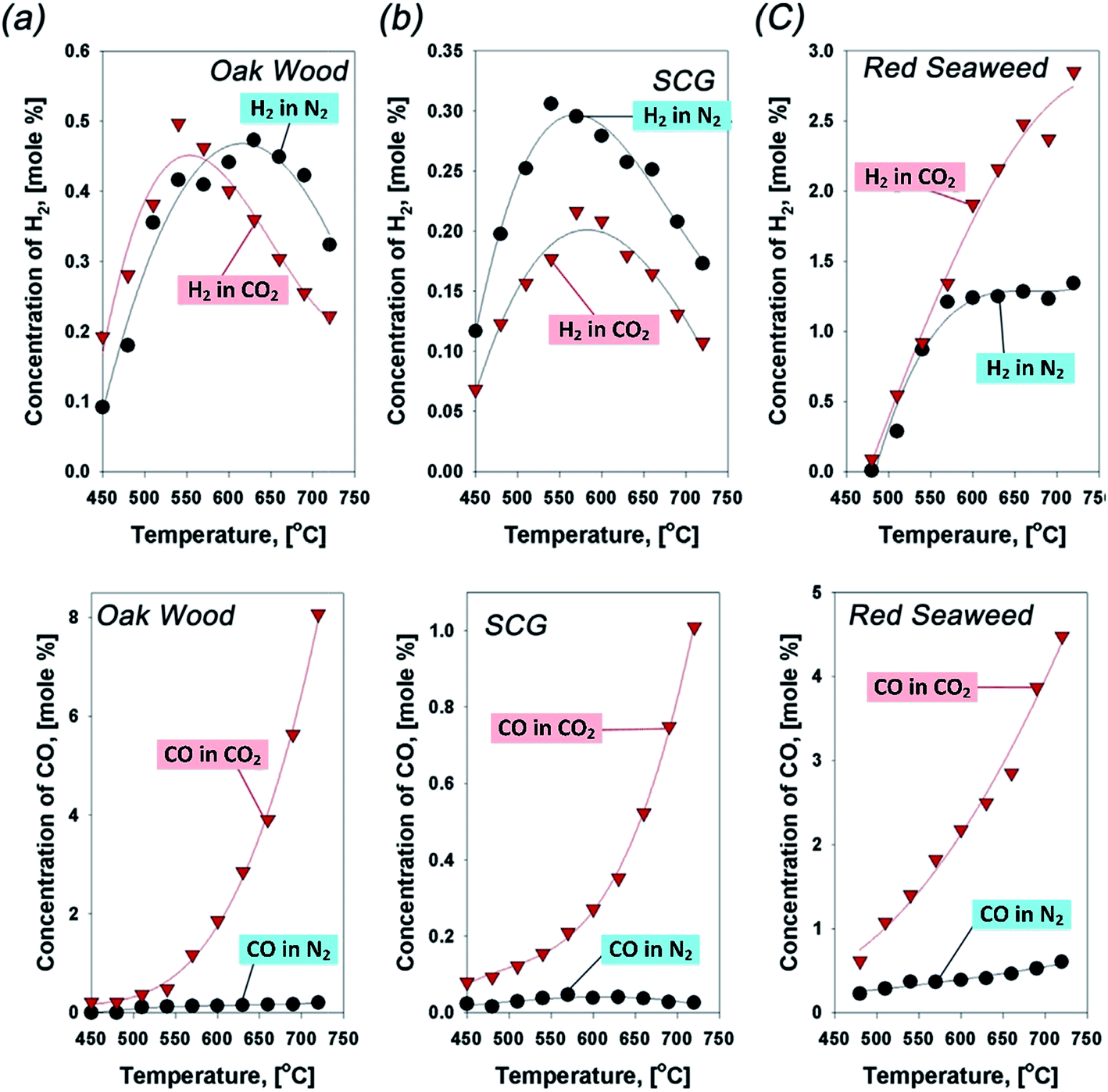 | ||
| Fig. 13 Concentration profiles of H2 and CO in N2 (black line) and CO2 (red line) evolved from pyrolysis of (a) oak wood, (b) spent coffee ground (SCG), and (c) red seaweed.106 | ||
As a major product, bio-oil produced from pyrolysis of biomass is a complex assemblage of chemicals, mainly containing organic acids, aldehydes, ketones, furans, sugar-based components, phenolic compounds, etc.121,122 Miscellaneous oxygenates, sugars, and furans are generated from cellulose and hemicellulose. Esters, acids, alcohols, ketones, and aldehydes are produced from the decomposition of the miscellaneous oxygenates, sugars, and furans. Guaiacols and syringols come from lignin.123 The compositions of bio-oils from the pyrolysis of oil palm fibers (OPF) and oil palm fiber pellets (OPFP) at 450 °C are shown in Fig. 14a–d. In general, the chromatograms in the figure are similar to each other, indicating that the carrier gas and biomass pattern (i.e. needle-shape fibers or pellets) have no remarkable effects on the components in bio-oils produced from pyrolysis of biomass at relatively low temperatures (i.e. 450 °C). However, chemical components in tar generated from pyrolysis of woody biomass at 650 °C in CO2 were less than those in N2 (Fig. 14e), which is attributed to the enhanced thermal cracking induced by CO2.124
 | ||
| Fig. 14 GC-MS chromatograms of bio-oils from the pyrolysis of (a) OPF in N2, (b) OPFP in N2, (c) OPF in CO2, and (d) OPFP in CO2 at 450 °C;103 (e) of bio-oil generated from pyrolysis of woody biomass in N2 and CO2 at 650 °C.124 | ||
The presence of CO2 could enhance VOCs/tar cracking or bio-oil upgrading in the pyrolysis of biomass, but the catalysts used for catalytic pyrolysis (in situ) or catalytic reforming (ex situ) are still required to meet the demands of practical use.125–127 Cho et al.128,129 studied the pyrolysis of metal (e.g., Fe3+, Co2+) impregnated biomass using CO2 as a reaction medium. Generally, metal species loaded in the biomass have catalytic effects in the pyrolysis process. For example, the yield of CO at above 620 °C was enhanced via the catalytic effect of iron oxide (e.g., Fe3O4) formed during pyrolysis of spent coffee ground (SCG) in CO2. Moreover, evolution of organic compounds in liquid products (i.e. tar) was mitigated in CO2. In particular, caffeine in SCG was almost decomposed due to the synergistic effect of Fe3O4 and CO2.128 Furthermore, value-added biochar composites (e.g., biochar-Co) could be synthesized via one-step pyrolysis for other applications including catalysis and adsorption.129,130 More details can be found in some reviews89,131 on catalytic pyrolysis for syngas and biochar functionalized materials production.
3. CO2 gasification of biomass
As discussed above, gasification consists of three major steps: drying, devolatilization (pyrolysis), and gasification. Extensive research has been conducted on the effect of steam, air, oxygen, and their combinations in the gasification process.132,133 However, CO2 as an oxidizing agent has been explored for biomass gasification in only a limited number of studies.134–139 From the viewpoint of CO2 consumption and CO yield enhancement, the utilization of CO2 is more preferable as a gasifying agent. Besides, CO2 is a major component of flue gases from many industries. It is a major cause of global warming and leads to various health issues. Significantly, CO2 exists in the gaseous products from biomass gasification. Thus, recycling CO2 for biomass gasification serves a dual purpose of reducing pollution and generating syngas. A major drawback is an external source of heat that is constantly required to maintain the gasification temperature, since the heat is not supplied by partial combustion of biomass in air or oxygen.140Generally, biomass pyro-gasification is operated with steam or air as gasifying medium but can also be performed using CO2. It was proved that introducing CO2 with steam as a gasifying medium leads to an enhanced CO production.141 In the gas phase, CO2 can potentially react with hydrocarbons, such as methane (CH4), via a dry reforming reaction (1):
| CO2 + CH4 ↔ 2H2 + 2CO, ΔH298 K = +246.9 kJ mol−1 | (1) |
CO2 can also react with H2 according to the reverse water gas shift reaction (2):
| CO2 + H2 ↔ H2O + CO, ΔH298 K = +41.2 kJ mol−1 | (2) |
Finally, in a biomass gasifier, CO2 can react with the carbon of the char formed by the pyrolysis step, via the heterogeneous Boudouard reaction (3):
| CO2 + C ↔ 2CO, ΔH298 K = −179.5 kJ mol−1 | (3) |
Renganathan et al.142 performed a thermodynamic analysis of biomass gasification by CO2 or mixtures of CO2 with steam or O2 and identified a universal optimal operating temperature of 850 °C for minimum energy input. Also, it was found that the use of CO2 in biomass gasification in a fluidized bed gasifier increased substantially the carbon and energy conversion efficiency and decreased the amount of tars in the produced gas. The highest cold gas efficiency was achieved when biomass was gasified with CO2.135 In the case of rice straw gasification, the effect of different gasification agents H2O, CO2, O2 and N2 on the thermal efficiency was studied and it was found that the introduction of CO2 has a positive effect on the thermal efficiency of a gasifier at temperatures of 850 °C and above.141,143 In addition, Parvez et al.144 simulated the air-, steam- and CO2-enhanced gasification of rice straw by using Aspen Plus™ and compared them in terms of their energy, exergy and environmental impacts. It was found that the addition of CO2 had less influence on syngas yield compared with gasification temperature. At lower CO2/biomass mass ratios (<0.25), CO2-enhanced gasification had a lower gasification system efficiency (GSE) than air gasification (<22.1%). However, at higher CO2/biomass ratios, CO2-enhanced gasification showed higher GSE than conventional gasification. The GSE of CO2-enhanced gasification increased to 58.8% as the CO2/biomass ratio was raised to 0.87. It was also found that chemical exergy was 2.05–4.85 times higher than physical exergy. The syngas exergy increased with CO2 addition, mainly due to the increase in physical exergy. The maximum exergy efficiency occurred at 800–900 °C. For CO2-enhanced gasification, exergy efficiency was more sensitive to temperature than to CO2/biomass ratio. In addition, CO2-enhanced gasification can result in significant environmental benefits compared with steam gasification.
The major effects of CO2 on biomass pyrolysis are the increase of syngas yield and modification of the biochar textural properties. However, the char reactivity to O2, H2O and CO2 was practically the same as for char prepared in N2.95 Furthermore, Guizani et al.139 studied the whole woody biomass gasification process in a CO2 atmosphere at 850 °C. It was found that although the CO2 is present inside the particle during the pyrolysis stage, it has no noticeable impact, neither on the reaction rate nor on the char yield due to the relatively low temperature. The CO2 char gasification is the rate limiting step of the pyro-gasification reaction as its duration is near to 95% of the entire biomass conversion time.139
As we know, gasification of biomass often needs higher temperatures (normally above 800 °C). In particular, an external source of heat is constantly required to maintain the gasification temperature for CO2 gasification. Therefore, Sun et al.145,146 reported the integrated CO2 gasification of sludge using waste heat from hot steel slags. Here, the steel slags played two important roles of catalyst (strong crystallization ability due to high basicity: mass ratio of CaO to SiO2) and heat source. Significantly, the steel slag could inhibit pollutants such as SO2, NOx from being released from the decomposition of sludge.145,146 Besides, an integrated concept was proposed for industrial applications, mainly composed of a rotary cup atomizer (RCA) system, where the molten slags were granulated into small particles at 1550–1000 °C, and a sludge gasification system, where CO2 gasification of sludge occurred using hot slags at 1000–500 °C (Fig. 15).145
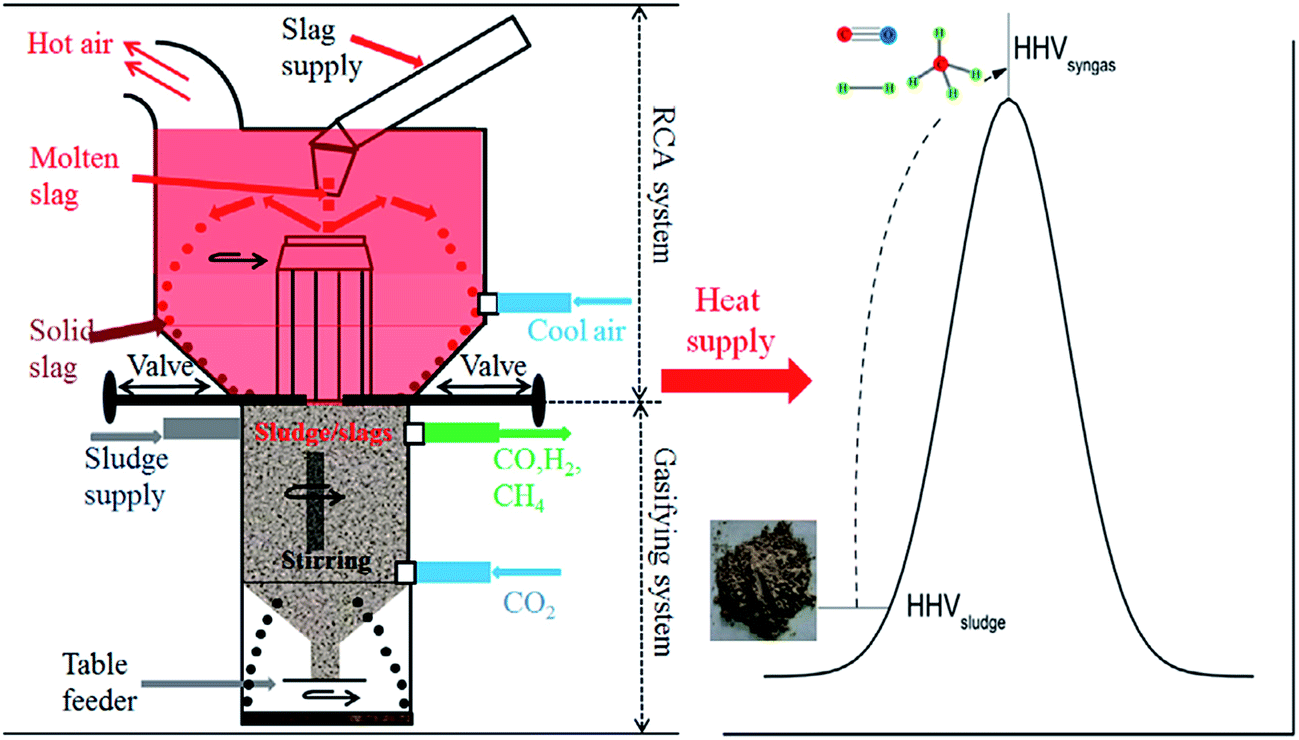 | ||
| Fig. 15 CO2 gasification of sludge using hot slags.145 | ||
To enhance the reactivity of biomass gasification in CO2, some researchers also studied the co-gasification of biomass and coal.147,148 It was found that synergy occurs when ashes are formed, followed by an almost complete gasification of biomass. Potassium (K) species in biomass ash play a catalytic role in promoting gasification reactivity of coal char, which is a direct consequence of synergy during co-gasification. The transfer of K from biomass to the surface of coal char occurs during co-pyrolysis/gasification. Biomass ash rich in silica can eliminate synergy in coal/biomass blends but not to the extent of inhibiting the reaction rate of the blended chars to make it slower than that of separated ones. In general, the best result in terms of synergy was the combination of low-ash coal and K-rich biomass.147 Kramb et al.149 also investigated the effects of Ca and K on CO2 gasification of birch wood in a fluidized bed. The CO2 gasification was performed after pyrolysis in N2. In this work, K in the biomass had a significant influence on the structure of the resulting char. However, K did not show a remarkable catalytic effect on the overall gasification reaction rate with CO2 due to the formation of a coke layer on the char surface. In contrast, Ca can increase the char conversion rate, which is likely the primary active catalyst in gasification of birch wood with CO2.
Entrained flow gasification, which plays an important role for large-scale utilization of carbonaceous materials, is typically performed at high temperatures (1200–1500 °C), normally higher than the ash fusion temperatures of the feedstocks.148 Although such high-temperature gasification is favorable for high carbon conversion and tar reduction, it may bring technological problems associated with ash melting such as sintering, agglomeration, slagging or corrosion during thermo-chemical reaction of biomass and coal.150,151 “Unburnt carbon”, called ungasified carbon, is formed in the char-slag/ash transition process as some unburnt carbon is encapsulated by the slag and excluded from slag discharge system, which is unfavorable for large-scale gasification. The process of solid particles transforming into slag is a crucial step for the stable operation of industrial gasifiers. Therefore, it is necessary to study in depth the particle fate and interactions between char matrix and minerals during the processes of char-slag/ash transition so as to further improve the gasification efficiency of biomass in gasifiers.
The char-slag/ash transition mechanisms were usually associated with biomass type, reaction temperature, etc.152 Moreover, the variations of morphological characters and reactivities of different biomasses at different temperatures have been studied by many researchers. Coda et al.153 analyzed the slagging behavior of woody ash in entrained-flow gasification conditions and found that a molten slag of woody ash was not prone to form at typical gasification temperatures of 1300–1500 °C, in spite of the presence of a relatively high content of low-melting alkaline species. It was concluded that the formation of high-temperature-melting species and the vaporization of low-melting alkali metal species during the char–slag transition process contributed to the poor slagging problem of woody ash in the reaction conditions close to those of industrial gasification. Metallic compounds in ash always play a vital role in the processes of char-slag/ash transition.154 For instance, alkali and alkaline earth metals (AAEM) in biomass ash, such as K, Na, Ca, had positive effects on biomass gasification.155–157 Ding et al.148 studied the characteristics of the morphological changes and interactions in char, slag and ash during CO2 gasification of rice straw and lignite. Rice straw particles exhibited shrinkage of particle form at the reaction temperature (1000 °C). And the variation of the existing state of K along with char–slag/ash transition can well explain the reactivity differences between raw char and demineralized char.
AAEM (e.g., K) in biomass has been considered as a natural and economical catalyst for thermochemical processes such as pyrolysis and gasification. Co-gasification of biomass and non-biomass feedstocks has been widely studied to investigate their interaction, including synergistic effects, inhibition effects, and no interaction. It was found that co-gasifying switchgrass (high K content) with coal char (high ash content) showed an inhibition effect during CO2 gasification.158 This was attributed to sequestration of the mobile alkali elements by the reaction with aluminosilicate minerals in coals to form inactive alkali aluminosilicates, such as KAlSi3O8 and KAlSiO4. K was not accessible to accelerate the gasification reaction. The catalytic activity was evident as excess alkali (K/Al > 1) present in the feed mixture to satisfy the stoichiometric requirements of these deactivation reactions.158 Therefore, fossil fuels with low ash contents have a high potential to be co-gasified with a K-rich biomass to benefit from the synergistic effect. Fernandes et al.159 quantified the synergistic/antagonistic behavior occurring in the co-gasification of non-biomass feedstocks (ash-free coal and fluid coke) with K-rich biomass (switchgrass). The acceleration of the gasification rate of the non-biomass feedstock followed a linear function of the switchgrass conversion. And the inhibition of the switchgrass conversion did not show a clear trend because it depends upon the non-biomass feedstock and temperature. The K in switchgrass acts as a catalyst significantly increasing the gasification rates for the fluid coke or ash-free coal in the mixture.159 The K catalyst can take oxygen from the reaction gas (step 1 in Fig. 16) and transfer it to the surface where oxygen reacts with carbon to form carbon monoxide (step 2). Step 3 is site regeneration, which requires a certain K mobility (intra- and interparticle), designated non-specifically as K∼. Note that K–C, K∼, and CO–K+ represent generalized sites (i.e., reduced, reduced, and oxidized, respectively) with the required K–C contact but an unknown stoichiometry. Once biomass-based carbon is converted, K can either move to the next C within the biomass or move to the non-biomass feedstock (step 3 or 3′ in Fig. 16, respectively).159
 | ||
| Fig. 16 Simplified mechanism of K-catalyzed CO2 gasification (oxygen transfer cycle), including intra- and interparticle K transfer.159 | ||
In summary, only unreacted AAEM can act as a catalyst for enhancing coal gasification. And coal minerals can increase the reactivity of coal. However, when they react with K in biomass, K is deactivated causing an inhibition effect. A schematic of switchgrass mineral (e.g., K2CO3) two-fold effects on coal gasification is displayed in Fig. 17.160 It was also demonstrated that K-rich biochar can act as a catalyst for tar reduction in biomass gasification.161 CO2 reforming of tar with biochar-based catalysts will be discussed in Section 5.
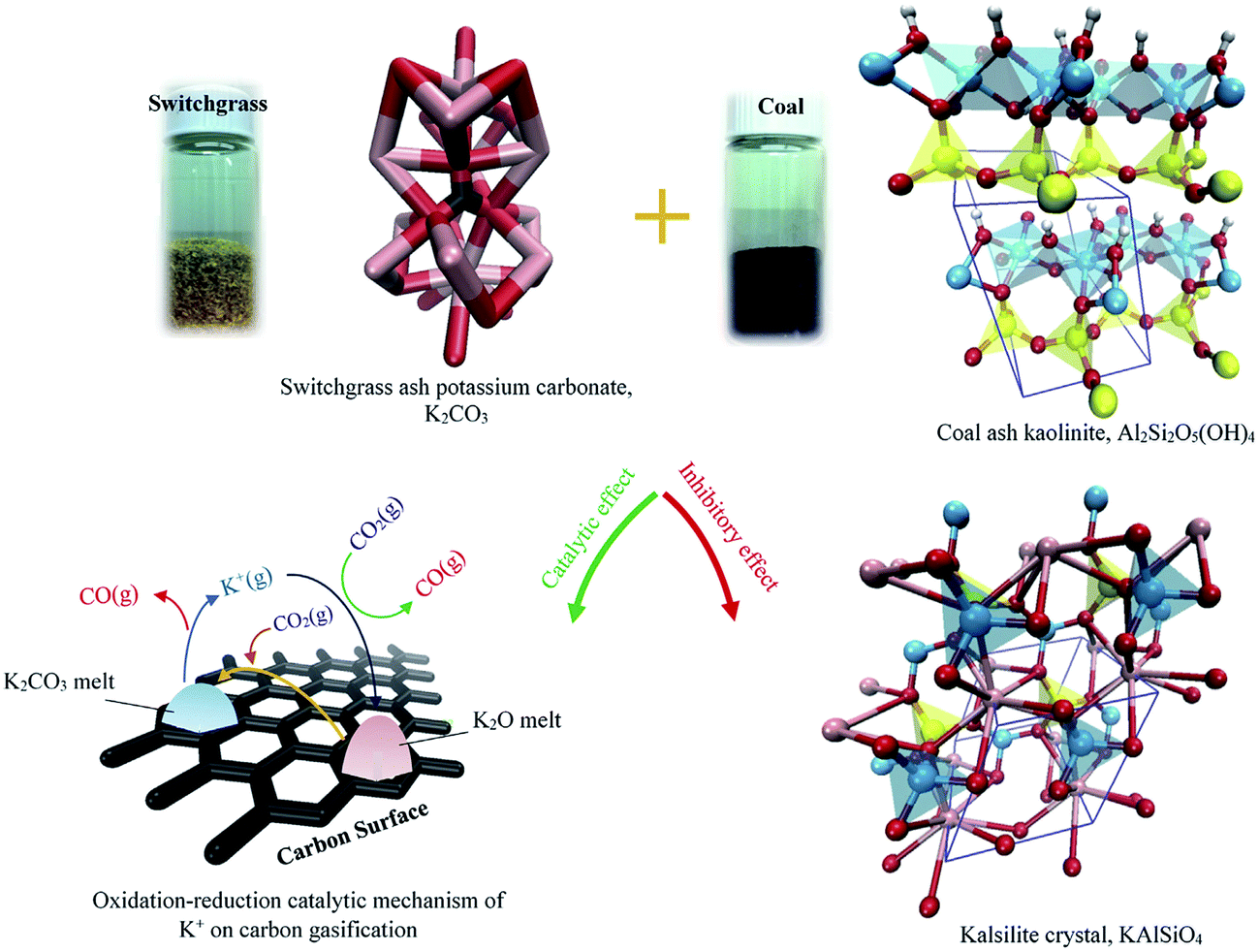 | ||
| Fig. 17 Schematic of two-fold effects of ash minerals (e.g., K2CO3) in switchgrass on coal gasification. Crystal elements: red: oxygen; white: hydrogen; pink: potassium; yellow: silicon; cyan: aluminum.160 | ||
Many studies, both experimental and theoretical, suggested that catalytic activity of alkali metal salts can be well explained by RedOx (reduction–oxidation) cycles of M2CO3, M(g), M2O, and MOH.162,163 Notably, black liquor can be gasified in an entrained-flow gasifier at relatively low temperatures (i.e. 1000–1100 °C) due to the catalytic activity of alkali metal salts.164–166 These studies also showed very low tar concentrations in the product gas (10–200 ppm of C6H6). Considering the ability to recover alkali metal salts as an aqueous solution at the outlet of the gasifier, alkali-catalyzed gasification of biomass can be an alternative to produce clean product gas from the gasifier (as shown in Fig. 18). Recent studies have also proved that woody biomass impregnated with solutions of alkali metal salts showed an increase of char reactivity.167,168 Furthermore, Umeki et al.169 demonstrated that tar and soot can be reduced simultaneously using the catalytic activity of alkali metal species. Raw and alkali-impregnated sawdust were gasified in a laminar drop-tube furnace at 900–1400 °C in an N2–CO2 mixture. At 900–1100 °C, char, soot and tar decreased with an increase of temperature for raw and alkali-impregnated sawdust. In addition, K can inhibit the growth of PAHs and promote the decomposition of tar (with 1–2 aromatic rings). The catalytic activity of K for tar decomposition is present in both solid and gas phases. Since alkali salts can be recovered from the product gas as an aqueous solution, alkali-catalyzed gasification of woody biomass can be a promising process to produce clean syngas from the entrained-flow gasification process at a relatively low temperature.
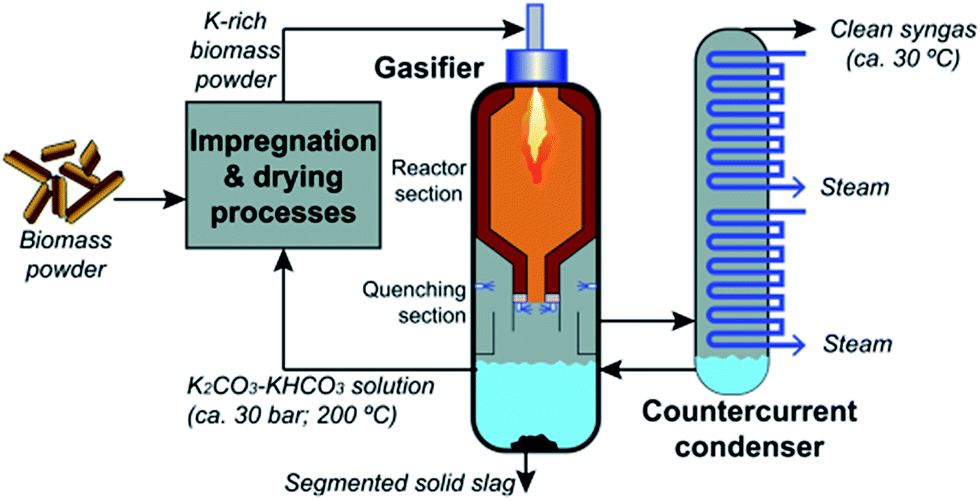 | ||
| Fig. 18 Alkali-catalyzed entrained-flow gasification of biomass along with catalyst recycling.169 | ||
As discussed above, K2CO3 as a carbonate salt shows a considerable catalytic activity for gasification of biomass or char. It was also reported that the stability of the carbonate salt can be maintained up to high temperatures of about 1073 K under CO2 atmosphere.170 Moreover, CO2 could reverse the salt oxides to liquid salt as in eqn (4). Thus, CO2 is expected to be a suitable gasifying agent for gasification with the molten salt. However, the CO2 gasification of char (eqn (5)) often requires slightly more energy input than steam gasification of char (eqn (6)). Therefore, additional catalytic enhancement by adding a heterogeneous catalyst to the molten salt would not increase syngas production rates and yields but would enable the process to operate at a lower temperature with smaller heat losses and less heat recovery requirement, thereby making the whole process more economical for large-scale construction. Regarding the heterogeneous metal catalysts for biomass gasification, noble metals such as Pt, Pd, and Rh show high reactivity but incur prohibitively high costs. Meanwhile, reasonably priced nickel catalysts are known to exhibit excellent catalytic enhancement of H2 production in many processes, such as biomass gasification,171 tar cracking/reforming,172 and CH4 reforming.173 In fact, nickel is not only selective for H2 production but also active for the enhancement of carbon conversion.174,175 Ratchahat et al.176 investigated the catalytic enhancement of syngas production rate and yield using a combined ternary eutectic carbonate salt–Ni/Al2O3 (MS–Ni) medium for CO2 gasification of cellulose, biomass wastes (chopstick, newspaper, sawdust), and sawdust char in a lab-scale reactor. The Ni/Al2O3 catalyst in MS–Ni medium can be active for H2 production and enhance CO production.
| M2O + CO2 ↔ M2CO3 (M = Li+, Na+, and K+) | (4) |
| C + CO2 ↔ 2CO, ΔH0 = +172.5 kJ mol−1 (@293 K) | (5) |
| C + H2O ↔ CO + H2, ΔH0 = +130 kJ mol−1 (@293 K) | (6) |
In MS–Ni, char could react with CO2 to CO via the Boudouard reaction. Char was also decomposed by the C–C cleavage reaction on the surface of the nickel catalyst. Reforming reactions of CH4 and hydrocarbons were enhanced by the nickel catalysts. Meanwhile, volatile gases could be dissociated in the molten salt (as illustrated in Fig. 19).176
 | ||
| Fig. 19 Mechanism of CO2 gasification of biomass in molten salt–Ni/Al2O3 (MS–Ni).176 | ||
Biomass gasification is a generic term encompassing several reactions occurring during biomass conversion, including drying, pyrolysis and residual char gasification steps. Char gasification is the rate limiting step in biomass gasification reactors. As for the entire gasification process of biomass, biochar gasification generally follows with the biomass pyrolysis process. For instance, char gasification occurs in the reduction zone in a downdraft gasifier (as illustrated in Fig. 20).177 Biochar is a porous, carbonaceous, non-organized material, which contains mainly C and, in lower proportions, O, H, N and minerals such as K, Ca, Na, Si and Mg. In the gasifiers, the char gasification reaction occurs with O2, H2O, CO2, and H2via the reactions of combustion, steam gasification, Boudouard reaction, and methanation, respectively. The gasification reaction is a heterogeneous reaction involving reactant gas diffusion inside the char, reaction on the char active sites and diffusion of the syngas out of the particle. The reaction can be also catalyzed in the presence of minerals such as the above mentioned K.178
 | ||
| Fig. 20 Downdraft “Imbert-style” gasification.177 | ||
The char porosity, its structural features, the nature of the surface functional groups as well as the presence of catalytic minerals affect its reactivity toward the reactant gases. These different char properties affecting the reactivity are classified in three categories: (1) the char textural properties of the porosity and pore size distribution; (2) the char structural properties of the carbonaceous structure and graphitization (ordering); (3) the char chemical properties of the surface functional groups as well as the catalytic mineral species. Based on reported works, the char reactivity is highly conditioned by its textural, structural and chemical properties. These characteristics are also highly coupled which makes the task of understanding the gasification reaction mechanisms even more difficult. Thus, several studies focus on the modeling of the char gasification reaction to determine the reaction kinetic parameters.179,180 Char gasification models are often semi-empirical ones, as they include a term accounting for the changes in the different char properties during the gasification,181 which reflect the ambiguity of this issue. To understand the phenomenology of char gasification, Guizani et al.182 further studied the char surface chemistry, structural and textural properties as well as mineral species behavior during gasification in CO2, H2O and CO2/H2O mixture. Fig. 21 shows the evolution of the char reactivity with the concentrations of K, Ca and Mg in different gasification atmospheres. Near-linear correlations were found between the char reactivity and the molar concentrations of these species in different atmospheres. The alkali metals (e.g., K) are more effective than Ca.183 As for Si-rich biomasses (e.g., rice husks, bagasse), much lower reactivities were observed. It was assumed that the formation of alkali silicates at low temperatures curtailed the catalytic action of K. In this work, quite interesting potential synergy was proved between CO2 and H2O during gasification as the presence of CO2 induced the departure of Si from the char, which is an inhibitor in steam gasification.182 Noteworthy, H2O and CO2 gasification reactions have different pathways.182,184 The principal characteristics and differences between CO2 and H2O gasification reactions are summarized in Table 2. In mixed atmosphere gasification, these two molecules cannot react independently because there are likely several competition and synergy interactions that lead to an apparent additive law of reactivity. The separate active site reaction mechanism is rather a sum of a more complicated synergy and inhibition interactions. It would be only a fortuitous correct mathematical representation of the char reactivity in mixed CO2–H2O. In addition, Chang et al.185 proved that intermittent addition of steam could increase the gasification reactivity of palm kernel shell biochar with CO2, which was influenced by the formation of char pore structure and size.
 | ||
| Fig. 21 Relationship between the char reactivity and molar concentration of K, Ca and Mg in the char (mol%) during the gasification with H2O, CO2 and their mixtures.182 | ||
| H2O gasification | CO2 gasification | |
|---|---|---|
| SFG | (1) Less ethers, anhydrides and phenols since the early gasification stages; (2) differences in peak positions compared to CO2 gasification | (1) More CO emitting FG; (2) much less hydrogenated char |
| Structure | Preferential removal of amorphous carbon forms | Lower selectivity towards the carbon forms |
| Texture | (1) Higher TSA at equivalent X and less damaged surface; (2) development of mesoporosity besides microporosity; (3) small micropore size distribution; (4) preferential development of 1 nm micropores | (1) Lower TSA and more damaged char surface; (2) higher proportion of micropores; (3) large micropore size distribution |
| Minerals | (1) Better retention of AAEM species; (2) higher retention of Si and Al | (1) Lower retention of AAEM species; (2) lower retention of Si and Al |
In respect of char gasification in CO2, many experimental and modelling works have been performed.186–192 Since the gasification reactivity of char is mostly determined by its carbonaceous structure, four kinetic models can be applied to describe the CO2 gasification behavior of chars: the volumetric model (VM), the grain model (GM), the random pore model (RPM) and the modified random pore model (MRPM). The RPM and MRPM can well describe the reactivity of different chars. However, when the peak gasification rate appears in high conversion range, the MRPM can perform better.188 Seo et al.193 studied the gasification of biochars with CO2 at different temperatures. It was found that the experimental data agreed well with the RPM. Fermoso et al.194 studied the effect of gasification temperature, pressure and CO2 concentration during gasification of biochars. It was found that among VM, GM and RPM, only the RPM accurately predicted the conversion of different char which can be well fitted by the Langmuir–Hinshelwood model. In general, RPM could well describe the gasification curve of char under CO2 atmosphere, but when peak reaction rate appears at high conversion range over 0.393, the discrepancy between experiment data and model calculation is significant. Therefore, based on RPM, many further investigations were carried out. Zhang et al.195 evaluated a semi-empirical kinetic model to reconcile with gasification reactivity profiles of biochars. It was found that the fitting parameters introduced in the MRPM well predicted the amount of active K in biochar. Zhang et al.196 developed a MRPM, which could be reduced to a traditional VM, GM, hybrid model and RPM by varying the model parameters. Alvarez et al.192 studied the CO2 gasification kinetics of rice husk char. Rice husk char gasification has a complex kinetic behavior, with the reactivity being strongly dependent upon the temperature and char conversion. The experimental results are fitted to four different kinetic models (namely homogeneous, nth order, RPM and MRPM). The RPM provides the best fit to the experimental evolution. In addition, the catalyst/biomass integration concept is demonstrated to promote the reduction of char residence time and energy consumption via the catalysis of the rate limiting step of the entire biomass gasification process.197 Both the in situ generation of char/Ni0 catalyst and the CO2 gasification of the char/Ni0 can occur at temperatures as low as 500 °C.197,198
4. CO2 activation of biochar
Biochar, a fine-grained, highly porous charcoal substance, is typically a byproduct from pyrolysis and gasification of biomass. Biochar can be used as an energy source due to its excellent fuel properties such as high energy density and good grindability.199,200 Biochar has also been used as an effective adsorbent for application in environmental remediation.201,202 In addition, there has been great interest in biochars as soil amendments to improve soil fertility and to increase soil carbon sequestration. In its application as a soil amendment and adsorbent, the effectiveness of biochar has been limited by its low surface area. Many researchers have attempted to produce activated carbons from biomass. The production of activated carbons has generally used two activation methods: physical and chemical activation. Physical activation uses oxidizing gases such as air, CO2, and steam,203 but chemical activation uses chemicals such as alkali hydroxides (NaOH/KOH),204,205 inorganic acids (H3PO4, HCl and H2SO4)206 or ZnCl2.207 In general, the carbons produced are mainly microporous with high specific surface areas (up to 3000 m2 g−1). However, the difficulties associated with the recovery of the activating agent as well as the requirement for a stringent wash of the adsorbents produced to avoid the corrosion of the equipment and interferences in the adsorption process would increase the overall cost of the process and hinder its implementation at large scales.208 Alvarez et al.203,209 studied the physical activation of rice husk pyrolysis char for the production of high surface area activated carbons. Thermal activation is known to be more environmentally friendly and less costly than chemical methods. This carbonaceous precursor is subjected to partial gasification by using steam and CO2 as activating agents.The mechanism of pore development in activated carbon could be summarized as two main antagonistic contributions to specific surface area (SBET), namely the formation and disappearance of micropores (as shown in Fig. 22).210 On the one hand, appropriate conditions (e.g., mass ratio) are favorable for the development of pore structure.211 Under this condition, the formation rate of micropores is greater than the disappearance rate, increasing the SBET of activated carbon. When the formation and deformation rates are equal, pore development can reach a state of dynamic equilibrium with maximum SBET. On the other hand, extreme conditions can accelerate the widening of micropores.210 Many micropores are destroyed through collapsing or merging into mesopores and macropores, leading to a reduction of SBET.212 In addition, it is demonstrated that the gasifying agent (e.g., CO2, steam) has a profound influence on the activated carbon porosity development. First, steam is more reactive and can produce activated carbon with greater N2 adsorption capacity. Second, steam can produce more mesopores with the increase of activation time. While steam generates micro-, meso-, and macropores from the early stages of the process, CO2 can produce highly microporous carbon, with broadening of the microporosity only for long activation times.213
 | ||
| Fig. 22 Possible mechanism of char activation.212 | ||
The effects of steam and CO2 activation on the nitrogen adsorption isotherms and pore size distribution (PSD) are shown in Fig. 23. CO2 can favor the development of new fine porosity followed by pore enlargement, while steam causes widening of micropores from earlier activation stages.214,215 The wide PSD of the activated carbons obtained by using both steam and CO2 is also related to the presence of a well-developed porous structure in the precursor. Therefore, steam can generate new very small pores in the initial stages of activation, which subsequently form larger pores and finally porous structure destruction. However, CO2 creates new micro- and mesopores for longer treatments.203,209
 | ||
| Fig. 23 Adsorption–desorption isotherms and pore size distribution of activated carbons obtained at different burn off levels with (A) H2O209 and (B) CO2.203 | ||
5. CO2 reforming of tar
Biomass tar removal has become one of the main challenges for biomass gasification.216,217 Among several methods for tar elimination, catalytic reforming is considered to be the most promising for large-scale applications, because of the high reaction rate, high reliability and increased quantity of usable gases.87 Efficient catalytic conversion of tar by inert carbon-based catalysts is an attractive method for commercializing this technique.83 In particular, pyrolysis biochar is widely used as a low-cost catalyst in the cracking/reforming of tar in the process of biomass gasification.83 During catalytic reforming of tar over biochar, even after the loss of catalytic activity through coking, the char samples can still be directly combusted, so as to recover the chemical energy of the catalyst, thus avoiding any reprocessing as a result of deactivation. Additionally, as for biomass gasification, steam or CO2 gasification agent provides a necessary reaction environment for the tar conversion, and ensures that catalytic deactivation of biochar cannot be caused by the carbon deposition of tar. The biochar catalytic activity in tar reforming is significantly affected by the AAEM species in the biochar.218–220 Steam reforming of tar has been widely used, since it produces syngas with high yield of H2 due to the water–gas-shift reaction.221 Nevertheless, few works on CO2 reforming of tar have been reported.222–225 Feng et al.226,227 studied the catalytic effects of AAEM (e.g., K, Ca) in biochars on tar reforming. Moreover, the effects of H2O and CO2 on homogeneous conversion and heterogeneous reforming of biomass tar over biochar were studied. Fig. 24 shows that conversion of tar compounds with K-loaded char is significant in H2O or CO2.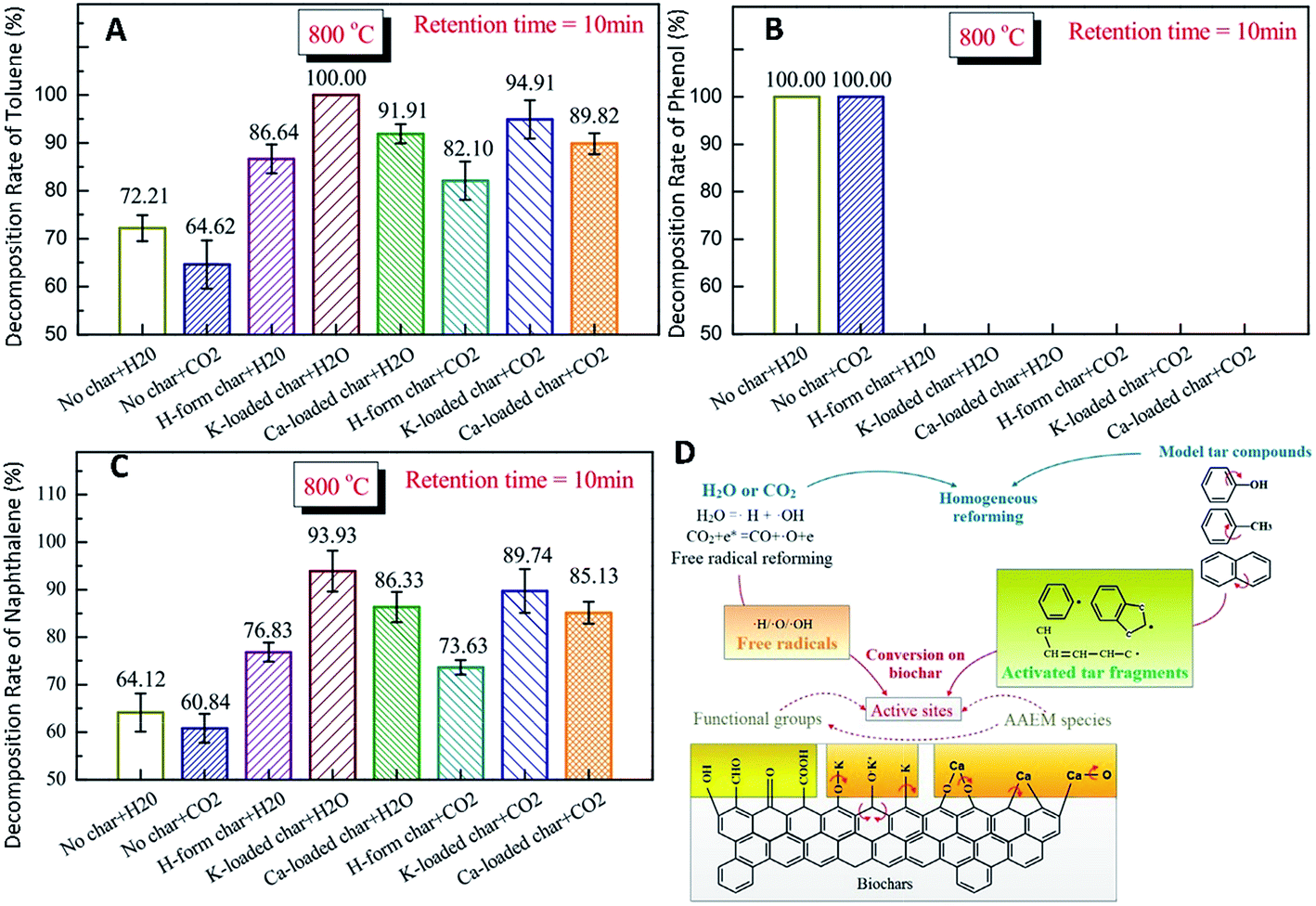 | ||
| Fig. 24 Decomposition rates of tar compounds (A) toluene, (B) phenol, and (C) naphthalene, and (D) mechanism of tar catalytic reforming with H-form/K-loaded/Ca-loaded chars in H2O/CO2 at 800 °C.226 | ||
As illustrated in Fig. 24D, the active oxygen atoms (O˙) for oxidative decomposition of hydrocarbons and intermediate products are mainly generated by the reaction CO2 + e* = CO + O˙ + e. Active OH free radicals (˙OH) can be formed by replacing the hydrogen atoms in hydrocarbons with oxygen atoms. In a steam atmosphere, O and OH free radicals can be formed by ionization of H2O (H2O = ˙H + ˙OH). Fracture of OH can form new H and O free radicals. However, the process is not strong. The H/O/OH atoms in the gas phase exist in radical form. These free radicals play a vital role in transforming AAEM species from metallic to radical forms. The CM–Ca and CM–K bonds may not be stable at high temperatures in H2O or CO2 and can be broken to generate active sites together with the release of O-containing species (e.g., COx) or aliphatic materials (e.g., CH4). Some of the AAEM species may leave the biochar particles (reactions (7) and (8)). New CM–Ca and CM–K bonds can be formed through recombination (reactions (11) and (12)). The CM–K and CM–Ca bonds can be continuously broken and reformed. Repeated CM–AAEM bond forming and bond breaking increases the concentration of active sites in the biochar particles, providing more chances for combination and cracking of tar.
| (CM–Ca–CM) = (–CM) + (–Ca–CM) | (7) |
| (–Ca–CM) = (–CM) + –Ca | (8) |
| (CM–K) = (–CM) + K | (9) |
| (–CM) = (–CM′) + gas | (10) |
| (–CM′) + (–Ca–CM) = (CM′–Ca–CM) | (11) |
| (–CM′) + K = (CM′–K) | (12) |
During volatilization, AAEM species leave the gas phase as K+ and/or Ca2+ radicals. The AAEM radicals can combine with activated tar fragments in the gas phase formed via dealkylation and depolymerization of tar. Due to the low stability of the species formed by combination, the H/O/OH free radicals can easily exchange with the AAEM species from the activated tar fragments to form light tar components. The presence of K and Ca, which lead to repeated bond forming and bond breaking of species from AAEM and tar fragments, causes tar conversion. After combination with free radicals, a series of bond-breaking and ring-opening reactions result in gradual tar decomposition and the formation of light hydrocarbons and small-molecule gases.228 The cracking and transformation of tar are promoted by reactions (7)–(12). Based on the free-radical theory,229 catalytic tar cracking occurs on the catalyst surface, and the formation of activated tar fragments in space could promote chemical reactions between tar fragments and H/O/OH free radicals. The reactants combined on the char active sites (AAEMs and some defects in the carbon structure) can easily participate in reforming reactions.230 For a biochar surface, the activated tar fragments can be combined on the active sites of the char matrix and take part in further reforming reactions. In sum, the pathways for H2O or CO2 reforming of tar by K and Ca in biochar can include direct homogeneous reforming and gasification on the biochar surface.
Furthermore, Feng et al.227 used H-form biomass (acid-pretreated biomass) with little AAEM to provide real tar components. For tar homogeneous reforming, temperatures of 700–900 °C are needed for tar reforming in the presence of H2O/CO2 (Fig. 25A). This process is considered to be a two-stage process. The first stage involves the decomposition and transformation of the active heteroatom-containing groups in tar, with the decomposition of dealkylated side chains, hydrocarbon molecular cyclization, aromatization reactions, etc. The products include short-chain aliphatic hydrocarbons, oxygen-containing small-molecule gases, and single-ring aromatic hydrocarbons. The second stage involves the reforming of tar, the dehydrogenation of cyclization products, the addition of acetylene and the growth, recombination and isomerization of aromatics. The two processes constitute the basis of the tar homogeneous reaction. The presence of reforming agents (i.e. H2O, CO2) can promote or inhibit the pyrolysis conversion of tar. The addition of H2O or CO2 can promote the generation of active free radicals such as ˙O, ˙OH, and ˙H. These free radicals can react with the active free tar fragments generated from the first stage of thermal decomposition, which suggests the importance of the H2O and CO2 reforming agents in the homogeneous conversion of biomass tar (as illustrated in Fig. 25B).
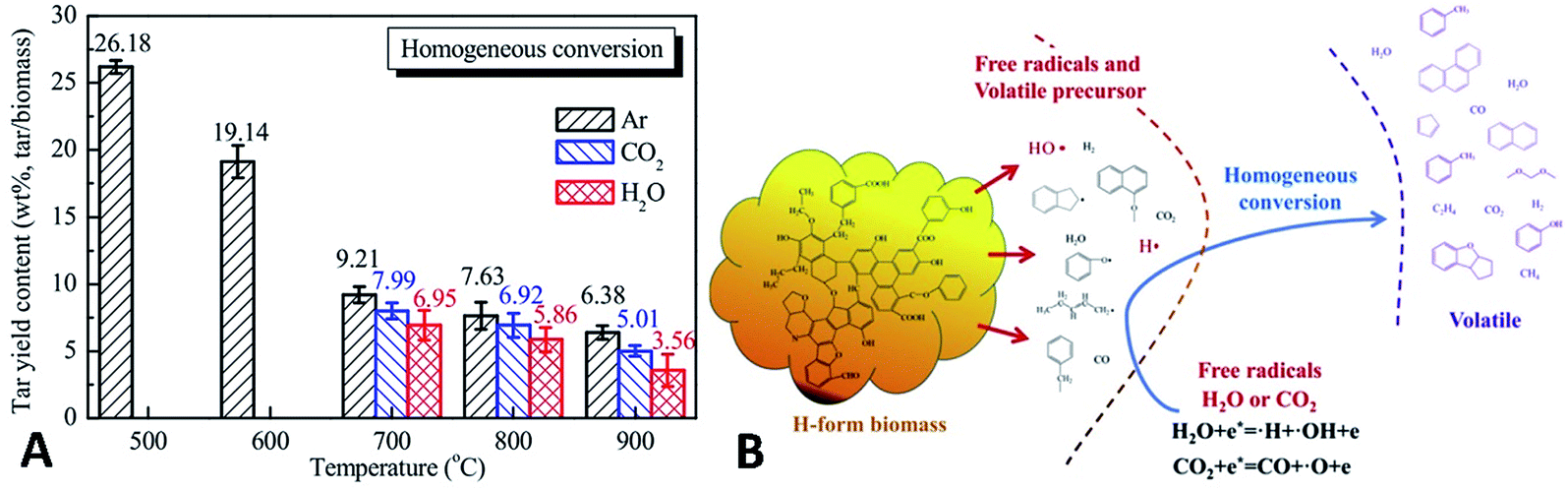 | ||
| Fig. 25 (A) Yields and (B) homogeneous conversion mechanism of biomass tar in Ar, H2O and CO2 at 500–900 °C.227 | ||
For heterogeneous reforming, the tar yield was significantly reduced with the biochar catalyst, being lower than that of homogeneous reforming. Moreover, CO2 reforming showed a similar tar reduction to H2O reforming during heterogeneous reforming over the biochar catalyst (Fig. 26A and B). The heterogeneous reforming mechanism of tar over biochar in the presence of H2O and CO2 at 800 °C is illustrated in Fig. 26C. H2O and CO2 dissociate in space to form many H/O/OH radicals, which play an important role in tar reforming. Tar, through the biochar layer, is adsorbed onto the acid–base active sites (oxygen-containing functional groups and AAEM catalysts). The attraction effect of the biochar matrix involves an electron pair shift in the tar molecules, which promotes the tar molecules to break at high temperatures. The tar adsorbed on the catalyst surface will catalytically crack to form free radicals. The chemical reactions between these free radicals permit new products. H2O and CO2 as the reforming agents in the biochar matrix result in the fragmentation of the smaller aromatic rings. The empty active sites, formed by bond cleavage, are gradually occupied by H/O/OH radicals, forming active groups (e.g., O-containing functional groups). In the presence of H2O/CO2, a significant amount of H/O/OH radicals in the vicinity can enter into the biochar structure. The AAEM catalysts migrate at different rates and transformation occurs from the biochar matrix onto the gas–solid interface or the gas phase. As the AAEM species are bonded with the C element on the char surface by the O element,226 H2O and CO2 react with these C elements resulting in AAEM–O bond cleavage followed by precipitation. The valence state of Ca results in a stronger bonding interaction with the biochar compared with K. Additionally, Ca migration and precipitation are more difficult than those of K. When tar adsorbs and then cleaves the AAEM–O bond and functional group bond on the biochar surface, an aromatic fragmented radical is formed as other free radicals are encountered. Also, the active AAEM species in the vicinity will continue to occupy active sites on the tar fragment groups, thus inhibiting their secondary polymerization. Furthermore, the H/O/OH radicals are exchanged to the AAEM, increasing the possibility for reforming of tar macromolecules. After the reaction, gas and light tars (CnHy/CO/H2) are formed, thus realizing the H2O or CO2 heterogeneous reforming of biomass tar over the biochar catalyst.227
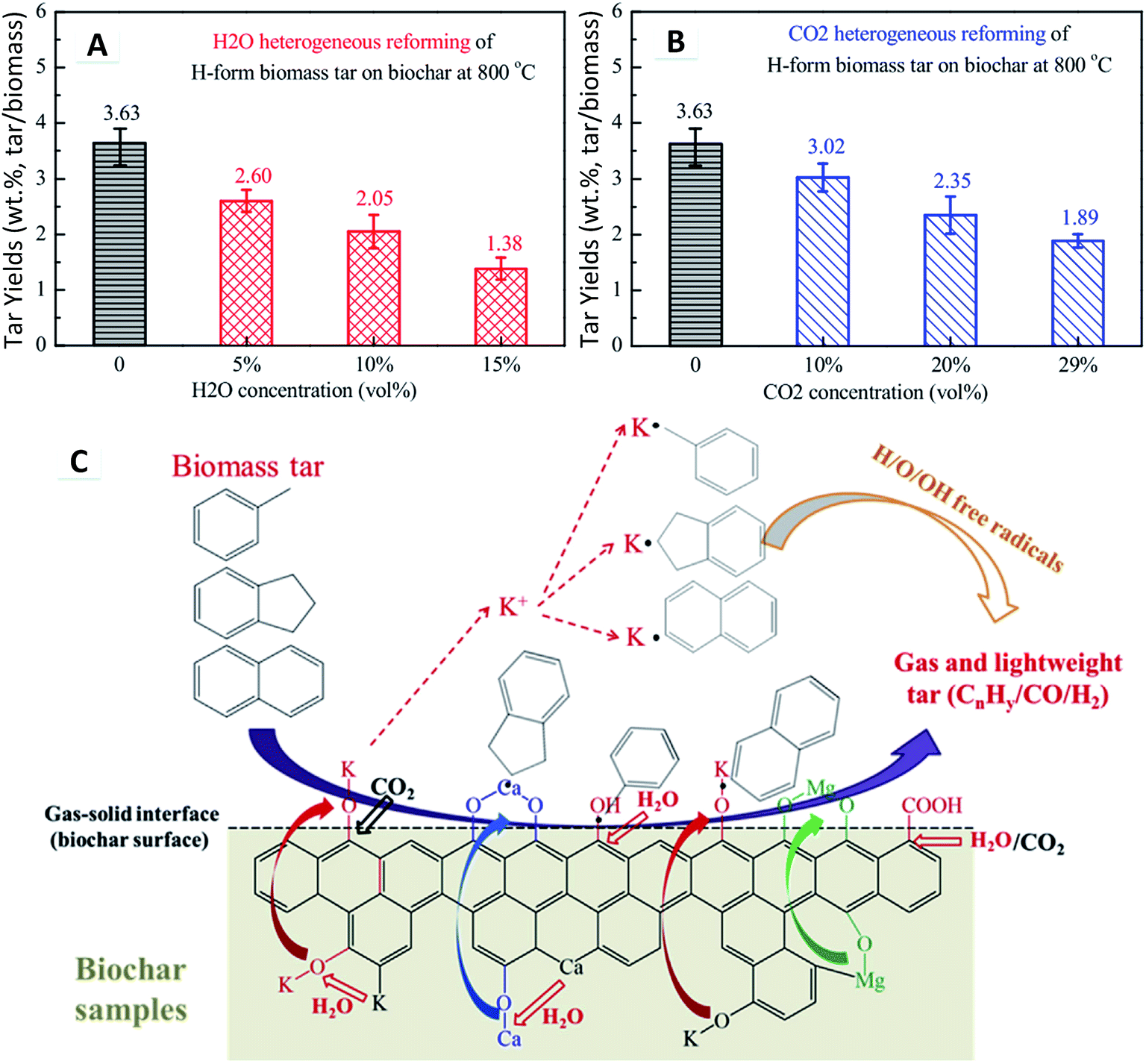 | ||
| Fig. 26 Tar yield during (A) H2O and (B) CO2 heterogeneous reforming on biochar. (C) Heterogeneous reforming mechanism of biomass tar over biochar in H2O and CO2 at 800 °C.227 | ||
6. Conclusions and prospective
Thermochemical processes such as pyrolysis or gasification can decompose carbon feedstocks in a controlled oxygen environment to transform them into gaseous (low molecular weight gases), liquid (high molecular weight compounds obtained after condensation), and solid (biochar) products. For example, pyrolysis is generally conducted in an inert environment (e.g., Ar, N2), and gasification is performed in a partial-oxidation environment (e.g., air, steam). In particular, steam gasification has attracted more attention for converting biomass into valuable syngas with high concentration of H2. This review summarized the recent progress in biomass pyrolysis and gasification using CO2 as a reaction medium. The CO2-looping pyrolysis and gasification of biomass were compared with conventional processes (e.g., N2 pyrolysis, steam gasification). In the integrated valorization of biomass by pyrolysis or gasification, CO2 can play a vital role in each stage mainly including biomass pyrolysis, biomass and biochar gasification, biochar activation, and tar cracking/reforming.CO2 as a reaction medium can significantly improve the thermal efficiency in the pyrolysis of biomass. Biomass pyrolysis in the presence of CO2 can result in deep decomposition compared to pyrolysis in pure N2, possibly due to either char gasification by CO2 or the fact that CO2 hinders polymerization reactions and secondary char formation by reacting or cracking tar compounds that may lead to its formation. Moreover, CO2 has an affinity to react with hydrogenated and oxygenated groups, leading to a carbon-rich char with a high specific surface area. Thus, exploiting CO2 as a reaction medium in biomass pyrolysis would provide an attractive option for the enhanced generation of syngas and tuned adsorption capability of biochar. In addition, CO2 biomass pyrolysis can enhance the thermal cracking of harmful organic compounds, thereby suppressing the formation of benzene derivatives (e.g., VOCs) and PAHs.231
Biomass gasification generally uses air, steam, oxygen, and their mixtures as gasifying agents to produce syngas at a desirable H2/CO ratio. Air is the cheapest gasifying agent as it is readily available at almost no cost but the syngas product has a very low heating value. The use of oxygen, in lieu of air, increases heating value of syngas product at the cost of oxygen purifying facility. Steam increases the yield of H2 production, but its use requires more heat input compared to air and oxygen gasification. CO2 as a candidate gasifying agent has the potential to further reduce CO2 emission. Also, the use of CO2 as a gasifying agent offers several other advantages, including (1) producing a more reactive char resulting in efficient gasification and reduction in the residual char, (2) being a less corrosive gasification medium compared with steam, and (3) offering more flexibility in syngas production suitable for various downstream applications. In general, CO2 gasification of biomass serves a dual purpose of reducing pollution and generating syngas. A major drawback is an external source of heat that is constantly required to maintain the gasification temperature, since heat is not supplied by partial combustion of biomass in air or oxygen. However, it has been proved that introducing CO2 with steam as a gasifying agent can lead to enhanced CO production. The inert mineral species (e.g., K, Ca) in biomass/biochar can also improve the reactivity of char gasification and tar reforming in the entire gasification processes. The technologies of pyrolysis or gasification of biomass using CO2 have attracted great attention, but they have still not been put into commercial applications. Future works should focus on the life cycle assessment of the integrated processes, including biomass pyrolysis for bio-oil and syngas production with activated carbon production (e.g., CO2 pyrolysis of biomass, CO2 activation of biochar) and biomass gasification for syngas production with tar reforming (e.g., CO2 gasification of biomass/biochar, CO2 reforming of tar). In addition, the CO2-looping technologies still face some challenges including higher cost of purified and compressed CO2. Actually, syngas including CO2 is required to be separated prior to its applications, so CO2 from syngas can be considered. Meanwhile, unpurified CO2 can be employed in real applications, so reducing the costs. The suitable mix ratio of CO2/N2 can be evaluated in different biomass pyrolysis or gasification cases. Finally, CO2 can be also captured from industrial flue gases, such as biomass gasification power plants.
Acknowledgements
This work is financially supported by the Startup Fund for Introducing Talent at NUIST (Grant 2243141501046), the National Science Foundation of China (Grant 91544220 and 21607079), and the National Science Foundation of the Jiangsu Higher Education Institutions (Grant 2151071609501).References
- D. W. Cho, S. H. Cho, H. Song and E. E. Kwon, Carbon dioxide assisted sustainability enhancement of pyrolysis of waste biomass: a case study with spent coffee ground, Bioresour. Technol., 2015, 189, 1–6 CrossRef CAS PubMed.
- D. P. Ho, H. H. Ngo and W. Guo, A mini review on renewable sources for biofuel, Bioresour. Technol., 2014, 169, 742–749 CrossRef CAS PubMed.
- E. E. Kwon, S. H. Cho and S. Kim, Synergetic sustainability enhancement via utilization of carbon dioxide as carbon neutral chemical feedstock in the thermochemical processing of biomass, Environ. Sci. Technol., 2015, 49, 5028–5034 CrossRef CAS PubMed.
- P. Srinivasan, A. K. Sarmah, R. Smernik, O. Das, M. Farid and W. Gao, A feasibility study of agricultural and sewage biomass as biochar, bioenergy and biocomposite feedstock: production, characterization and potential applications, Sci. Total Environ., 2015, 512–513, 495–505 CrossRef CAS PubMed.
- L. Xu, Q. Yao, J. Deng, Z. Han, Y. Zhang and Y. Fu, et al., Renewable N-heterocycles production by thermocatalytic conversion and ammonization of biomass over ZSM-5, ACS Sustainable Chem. Eng., 2015, 3, 2890–2899 CrossRef CAS.
- O. Das and A. K. Sarmah, Mechanism of waste biomass pyrolysis: effect of physical and chemical pre-treatments, Sci. Total Environ., 2015, 537, 23–34 CrossRef PubMed.
- C. Henkel, P. D. Muley, K. K. Abdollahi, C. Marculescu and D. Boldor, Pyrolysis of energy cane bagasse and invasive Chinese tallow tree (Triadica sebifera L.) biomass in an inductively heated reactor, Energy Convers. Manage., 2016, 109, 175–183 CrossRef CAS.
- M. S. Mettler, D. G. Vlachos and P. J. Dauenhauer, Top ten fundamental challenges of biomass pyrolysis for biofuels, Energy Environ. Sci., 2012, 5, 7797–7809 CAS.
- D. Neves, H. Thunman, A. Matos, L. Tarelho and A. Gómez-Barea, Characterization and prediction of biomass pyrolysis products, Prog. Energy Combust. Sci., 2011, 37, 611–630 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Pyrolysis of wood/biomass for bio-oil: a critical review, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- C. Di Blasi, Modeling chemical and physical processes of wood and biomass pyrolysis, Prog. Energy Combust. Sci., 2008, 34, 47–90 CrossRef CAS.
- J. W. Lee, B. Hawkins, D. M. Day and D. C. Reicosky, Sustainability: the capacity of smokeless biomass pyrolysis for energy production, global carbon capture and sequestration, Energy Environ. Sci., 2010, 3, 1695–1705 CAS.
- A. V. Bridgwater, Review of fast pyrolysis of biomass and product upgrading, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- B. V. Babu, Biomass pyrolysis: a state-of-the-art review, Biofuels, Bioprod. Biorefin., 2008, 2, 393–414 CrossRef CAS.
- T. Kan, V. Strezov and T. J. Evans, Lignocellulosic biomass pyrolysis. A review of product properties and effects of pyrolysis parameters, Renewable Sustainable Energy Rev., 2016, 57, 1126–1140 CrossRef CAS.
- P. Roy and G. Dias, Prospects for pyrolysis technologies in the bioenergy sector: a review, Renewable Sustainable Energy Rev., 2017, 77, 59–69 CrossRef CAS.
- S. Kersten and M. Garcia-Perez, Recent development in fast pyrolysis of lingo-cellulosic materials, Curr. Opin. Biotechnol., 2013, 24, 414–420 CrossRef CAS PubMed.
- A. Anca-Couce, Reaction mechanisms and multi-scale modelling of lignocellulosic biomass pyrolysis, Prog. Energy Combust. Sci., 2016, 53, 41–79 CrossRef.
- D. Shen, R. Xiao, S. Gu and K. Luo, The pyrolytic behavior of cellulose in lignocellulosic biomass: a review, RSC Adv., 2011, 1, 1641–1660 RSC.
- D. Shen, S. Gu and A. V. Bridgwater, The thermal performance of the polysaccharides extracted from hardwood: cellulose and hemicellulose, Carbohydr. Polym., 2010, 82, 39–45 CrossRef CAS.
- D. Shen and S. Gu, The mechanism for thermal decomposition of cellulose and its main products, Bioresour. Technol., 2009, 100, 6496–6504 CrossRef CAS PubMed.
- S. Li, J. Lyons-Hart, J. Banyasz and K. Shafer, Real-time evolved gas analysis by FTIR method: an experimental study of cellulose pyrolysis, Fuel, 2001, 80, 1809–1817 CrossRef CAS.
- P. R. Patwardhan, D. L. Dalluge, B. H. Shanks and R. C. Brown, Distinguishing primary and secondary reactions of cellulose pyrolysis, Bioresour. Technol., 2011, 102, 5265–5269 CrossRef CAS PubMed.
- X. Zhang, W. Yang and W. Blasiak, Thermal decomposition mechanism of levoglucosan during cellulose pyrolysis, J. Anal. Appl. Pyrolysis, 2012, 96, 110–119 CrossRef CAS.
- J. L. Banyasz, S. Li, J. Lyons-Hart and K. H. Shafer, Gas evolution and the mechanism of cellulose pyrolysis, Fuel, 2001, 80, 1757–1763 CrossRef CAS.
- F. Ronsse, X. Bai, W. Prins and R. C. Brown, Secondary reactions of levoglucosan and char in the fast pyrolysis of cellulose, Environ. Prog. Sustainable Energy, 2012, 31, 256–260 CrossRef CAS.
- M. S. Mettler, A. D. Paulsen, D. G. Vlachos and P. J. Dauenhauer, Pyrolytic conversion of cellulose to fuels: levoglucosan deoxygenation via elimination and cyclization within molten biomass, Energy Environ. Sci., 2012, 5, 7864–7868 CAS.
- Y.-C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, Kinetics and mechanism of cellulose pyrolysis, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- Z. Luo, S. Wang, Y. Liao and K. Cen, Mechanism study of cellulose rapid pyrolysis, Ind. Eng. Chem. Res., 2004, 43, 5605–5610 CrossRef CAS.
- J. B. Wooten, J. I. Seeman and M. R. Hajaligol, Observation and characterization of cellulose pyrolysis intermediates by 13C CPMAS NMR. A new mechanistic model, Energy Fuels, 2004, 18, 1–15 CrossRef CAS.
- R. Vinu and L. J. Broadbelt, A mechanistic model of fast pyrolysis of glucose-based carbohydrates to predict bio-oil composition, Energy Environ. Sci., 2012, 5, 9808–9826 CAS.
- X. Zhang, W. Yang and W. Blasiak, Kinetics study on thermal dissociation of levoglucosan during cellulose pyrolysis, Fuel, 2013, 109, 476–483 CrossRef CAS.
- D. Shen, S. Gu and A. V. Bridgwater, Study on the pyrolytic behaviour of xylan-based hemicellulose using TG-FTIR and Py-GC-FTIR, J. Anal. Appl. Pyrolysis, 2010, 87, 199–206 CrossRef CAS.
- B. Peters, Prediction of pyrolysis of pistachio shells based on its components hemicellulose, cellulose and lignin, Fuel Process. Technol., 2011, 92, 1993–1998 CrossRef CAS.
- Y. Peng and S. Wu, The structural and thermal characteristics of wheat straw hemicellulose, J. Anal. Appl. Pyrolysis, 2010, 88, 134–139 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, Product distribution from the fast pyrolysis of hemicellulose, ChemSusChem, 2011, 4, 636–643 CrossRef CAS PubMed.
- X. Zhou, W. Li, R. Mabon and L. J. Broadbelt, A critical review on hemicellulose pyrolysis, Energy Technol., 2017, 5, 52–79 CrossRef CAS.
- J.-Y. Kim, H. Hwang, J. Park, S. Oh and J. W. Choi, Predicting structural change of lignin macromolecules before and after heat treatment using the pyrolysis-GC/MS technique, J. Anal. Appl. Pyrolysis, 2014, 110, 305–312 CrossRef CAS.
- B. Li, W. Lv, Q. Zhang, T. Wang and L. Ma, Pyrolysis and catalytic pyrolysis of industrial lignins by TG-FTIR: kinetics and products, J. Anal. Appl. Pyrolysis, 2014, 108, 295–300 CrossRef CAS.
- Z. Ma, Q. Sun, J. Ye, Q. Yao and C. Zhao, Study on the thermal degradation behaviors and kinetics of alkali lignin for production of phenolic-rich bio-oil using TGA-FTIR and Py-GC/MS, J. Anal. Appl. Pyrolysis, 2016, 117, 116–124 CrossRef CAS.
- S. Wang, H. Lin, B. Ru, W. Sun, Y. Wang and Z. Luo, Comparison of the pyrolysis behavior of pyrolytic lignin and milled wood lignin by using TG-FTIR analysis, J. Anal. Appl. Pyrolysis, 2014, 108, 78–85 CrossRef CAS.
- M. Kosa, H. Ben, H. Theliander and A. J. Ragauskas, Pyrolysis oils from CO2 precipitated Kraft lignin, Green Chem., 2011, 13, 3196 RSC.
- S. Chu, A. V. Subrahmanyam and G. W. Huber, The pyrolysis chemistry of a [small beta]-O-4 type oligomeric lignin model compound, Green Chem., 2013, 15, 125–136 RSC.
- J. Cho, S. Chu, P. J. Dauenhauer and G. W. Huber, Kinetics and reaction chemistry for slow pyrolysis of enzymatic hydrolysis lignin and organosolv extracted lignin derived from maplewood, Green Chem., 2012, 14, 428–439 RSC.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, Lignin pyrolysis components and upgrading-technology review, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- H. Ben and A. J. Ragauskas, NMR characterization of pyrolysis oils from Kraft lignin, Energy Fuels, 2011, 25, 2322–2332 CrossRef CAS.
- J. Bourke, M. Manley-Harris, C. Fushimi, K. Dowaki, T. Nunoura and M. J. Antal, Do all carbonized charcoals have the same chemical structure 2. A model of the chemical structure of carbonized charcoal, Ind. Eng. Chem. Res., 2007, 46, 5954–5967 CrossRef CAS.
- H. Marschner and G. Rimmington, Mineral Nutrition of Higher Plants, Wiley Online Library, 1996 Search PubMed.
- S. Yaman, Pyrolysis of biomass to produce fuels and chemical feedstocks, Energy Convers. Manage., 2004, 45, 651–671 CrossRef CAS.
- W. Liu, W. Li, H. Jiang and H. Yu, Fates of chemical elements in biomass during its pyrolysis, Chem. Rev., 2017, 117, 6367–6398 CrossRef CAS PubMed.
- Q. Feng and Y. Lin, Integrated processes of anaerobic digestion and pyrolysis for higher bioenergy recovery from lignocellulosic biomass: a brief review, Renewable Sustainable Energy Rev., 2017, 77, 1272–1287 CrossRef CAS.
- T. Hübner and J. Mumme, Integration of pyrolysis and anaerobic digestion – use of aqueous liquor from digestate pyrolysis for biogas production, Bioresour. Technol., 2015, 183, 86–92 CrossRef PubMed.
- J. Mumme, F. Srocke, K. Heeg and M. Werner, Use of biochars in anaerobic digestion, Bioresour. Technol., 2014, 164, 189–197 CrossRef CAS PubMed.
- C. Torri and D. Fabbri, Biochar enables anaerobic digestion of aqueous phase from intermediate pyrolysis of biomass, Bioresour. Technol., 2014, 172, 335–341 CrossRef CAS PubMed.
- X. D. Song, D. Z. Chen, J. Zhang, X. H. Dai and Y. Y. Qi, Anaerobic digestion combined pyrolysis for paper mill sludge disposal and its influence on char characteristics, J. Mater. Cycles Waste Manage., 2017, 19, 332 CrossRef CAS.
- M. B. Folgueras, M. Alonso and R. M. Díaz, Influence of sewage sludge treatment on pyrolysis and combustion of dry sludge, Energy, 2013, 55, 426–435 CrossRef CAS.
- Y. Zheng, J. Zhao, F. Xu and F. Li, Pretreatment of lignocellulosic biomass for enhanced biogas production, Prog. Energy Combust. Sci., 2014, 42, 35–53 CrossRef.
- Y. Li, R. Zhang, Y. He, C. Zhang, X. Liu and C. Chen, et al., Anaerobic co-digestion of chicken manure and corn stover in batch and continuously stirred tank reactor (CSTR), Bioresour. Technol., 2014, 156, 342–347 CrossRef CAS PubMed.
- S. Righi, V. Bandini and D. Marazza, et al., Life cycle assessment of high ligno-cellulosic biomass pyrolysis coupled with anaerobic digestion, Bioresour. Technol., 2016, 212, 245–253 CrossRef CAS PubMed.
- M. Inyang, M. Gao, P. Pullammanappallil, W. Ding and A. R. Zimmerman, Biochar from anaerobically digested sugarcane bagasse, Bioresour. Technol., 2010, 101, 8868–8872 CrossRef CAS PubMed.
- S. R. Guiot, R. Cimpoia and C. Gael, Potential of wastewater-treating anaerobic granules for biomethanation of synthesis gas, Environ. Sci. Technol., 2011, 45, 2006–2012 CrossRef CAS PubMed.
- J. Mumme, F. Srocke, K. Heeg and M. Werner, Use of biochars in anaerobic digestion, Bioresour. Technol., 2014, 164, 189–197 CrossRef CAS PubMed.
- F. Monlau, C. Sambusiti, N. Antoniou, A. Barakat and A. Zabaniotou, A new concept for enhancing energy recovery from agricultural residues by coupling anaerobic digestion and pyrolysis process, Appl. Energy, 2015, 148, 32–38 CrossRef.
- D. Fabbri and C. Torri, Linking pyrolysis and anaerobic digestion (Py-AD) for the conversion of lignocellulosic biomass, Curr. Opin. Biotechnol., 2016, 38, 167–173 CrossRef CAS PubMed.
- C. Higman and M. v. d. Burgt, Gasification, Gulf Professional Publishing, Elsevier, USA, 2008 Search PubMed.
- P. Parthasarathy and K. S. Narayanan, Hydrogen production from steam gasification of biomass: influence of process parameters on hydrogen yield – a review, Renewable Energy, 2014, 66, 570–579 CrossRef CAS.
- P. Basu, Biomass Gasification and Pyrolysis, Elsevier, Oxford, 2010 Search PubMed.
- A. George, A. Brandt, K. Tran, S. M. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila and R. Parthasarathi, Design of low-cost ionic liquids for lignocellulosic biomass pretreatment, Green Chem., 2015, 17, 1728–1734 RSC.
- R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Characterization of milled wood lignin and ethanol organosolv lignin from miscanthus, Polym. Degrad. Stab., 2009, 94, 1632–1638 CrossRef CAS.
- H. L. Chum and R. P. Overend, Biomass and renewable fuels, Fuel Process. Technol., 2001, 71, 187–195 CrossRef CAS.
- K. J. Ptasinski, Thermodynamic efficiency of biomass gasification and biofuels conversion, Biofuels, Bioprod. Biorefin., 2008, 2, 239–253 CrossRef CAS.
- S. Heidenreich and P. U. Foscolo, New concepts in biomass gasification, Prog. Energy Combust. Sci., 2015, 46, 72–95 CrossRef.
- V. S. Silkarwar, M. Zhao, P. Clough, J. Yao, X. Zhang, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, An overview of advances in biomass gasification, Energy Environ. Sci., 2016, 9, 2939–2977 Search PubMed.
- Y. Shen, J. Wang, X. Ge and M. Chen, By-products recycling for syngas cleanup in biomass pyrolysis – an overview, Renewable Sustainable Energy Rev., 2016, 59, 1246–1268 CrossRef CAS.
- S. K. Sansaniwai, K. Pai, M. A. Rosen and S. K. Tyagi, Recent advances in the development of biomass gasification technology: a comprehensive review, Renewable Sustainable Energy Rev., 2017, 72, 363–384 CrossRef.
- Y.-D. Kim, C. W. Yang, B. J. Kim, K. S. Kim, J. W. Lee, J. H. Moon, W. Yang, T. U. Yu and U. D. Lee, Air-blown gasification of woody biomass in a bubbling fluidized bed gasifier, Appl. Energy, 2013, 112, 414–420 CrossRef CAS.
- A. A. Ahmad, N. A. Zawawi, F. H. Kasim, A. Inayat and A. Khasri, Assessing the gasification performance of biomass: a review on biomass gasification process conditions, optimization and economic evaluation, Renewable Sustainable Energy Rev., 2016, 53, 1333–1347 CrossRef CAS.
- J. Billaud, S. Valin, M. Peyrot and S. Salvador, Influence of H2O, CO2 and O2 addition on biomass gasification in entrained flow reactor conditions: experiments and modelling, Fuel, 2016, 166, 166–178 CrossRef CAS.
- D. Barisano, G. Canneto, F. Nanna, E. Alvino, G. Pinto, A. Villone, M. Camevale, V. Valerio, A. Battafarano and G. Braccio, Steam/oxygen biomass gasification at pilot scale in an internally circulating bubbling fluidized bed reactor, Fuel Process. Technol., 2016, 141, 74–81 CrossRef CAS.
- S. Sharma and P. N. Sheth, Air-steam biomass gasification: experiments, modeling and simulation, Energy Convers. Manage., 2016, 110, 307–318 CrossRef CAS.
- A. Dufour, P. Girods, E. Masson, S. Normand, Y. Rogaume and A. Zoulalian, Comparison of two methods of measuring wood pyrolysis tar, J. Chromatogr. A, 2007, 1164, 240–247 CrossRef CAS PubMed.
- L. Jia, Y. Le Brech, G. Mauviel, F. Qi, M. Bente-von Frowein, S. Ehlert, R. Zimmermann and A. Dufour, Online analysis of biomass pyrolysis tar by photoionization mass spectrometry, Energy Fuels, 2016, 30, 1555–1563 CrossRef CAS.
- Y. Shen, Chars as carbonaceous adsorbents/catalysts for tar elimination during biomass pyrolysis or gasification, Renewable Sustainable Energy Rev., 2015, 43, 281–295 CrossRef CAS.
- G. Guan, M. Kaewpanha, X. Hao and A. Abudula, Catalytic steam reforming of biomass tar: prospects and challenges, Renewable Sustainable Energy Rev., 2016, 58, 450–461 CrossRef CAS.
- C. Font Palma, Modelling of tar formation and evolution for biomass gasification: a review, Appl. Energy, 2013, 111, 129–141 CrossRef CAS.
- A. Gómez-Barea and B. Leckner, Gasification of Biomass and Waste, in Handbook of Combustion, Wiley-VCH, 2010 Search PubMed.
- Y. Shen and K. Yoshikawa, Recent progress in catalytic elimination of tar during biomass gasification or pyrolysis, Renewable Sustainable Energy Rev., 2013, 21, 371–392 CrossRef CAS.
- J. Lee, K.-H. Kim and E. E. Kwon, Biochar as a catalyst, Renewable Sustainable Energy Rev., 2017, 77, 70–79 CrossRef CAS.
- W. Liu, H. Jiang and H. Yu, Development of biochar-based functional materials: toward a sustainable platform carbon materials, Chem. Rev., 2015, 115, 12251–12285 CrossRef CAS PubMed.
- Y. Shen, P. Zhao, Q. Shao, D. Ma, F. Takahashi and K. Yoshikawa, In situ catalytic conversion of tar using rice husk char-supported catalysts for biomass pyrolysis/gasification, Appl. Catal., B, 2014, 152–153, 140–151 CrossRef CAS.
- Y. Shen, M. Chen, T. Sun and J. Jia, Catalytic reforming of pyrolysis tar over metallic nickel nanoparticles embedded in pyrochar, Fuel, 2015, 159, 570–579 CrossRef CAS.
- A. R. C. Morais, A. M. da C. Lopes and R. Bogel-Łukasik, Carbon dioxide in biomass processing: contributions to the green biorefinery concept, Chem. Rev., 2015, 115, 3–27 CrossRef CAS PubMed.
- A. C. D. Freitas and R. Guirardello, Use of CO2 as a co-reactant to promote syngas production in supercritical water gasification of sugarcane bagasse, J. CO2 Util., 2015, 9, 66–73 CrossRef CAS.
- Y. He, X. Ma and G. Zeng, Consequences of poly(vinyl chloride) presence on the thermochemical process of lignocellulosic biomass in CO2 by thermogravimetric analysis, Bioresour. Technol., 2015, 177, 346–354 CrossRef CAS PubMed.
- C. Guizani, F. J. Escudero Sanz and S. Salvador, Effects of CO2 on biomass fast pyrolysis: reaction rate, gas yields and char reactive properties, Fuel, 2014, 116, 310–320 CrossRef CAS.
- G. Pilon and J.-M. Lavoie, Pyrolysis of switchgrass (Panicum virgatum L.) at low temperatures within N2 and CO2 environments: product yield study, ACS Sustainable Chem. Eng., 2013, 1, 198–204 CAS.
- E. E. Kwon, S.-H. Cho and S. Kim, Synergetic sustainability enhancement via utilization of carbon dioxide as carbon neutral chemical feedstock in the thermochemical processing of biomass, Environ. Sci. Technol., 2015, 49, 5028–5034 CrossRef CAS PubMed.
- S.-H. Cho, K.-H. Kim, Y. J. Jeon and E. E. Kwon, Pyrolysis of microalgal biomass in carbon dioxide environment, Bioresour. Technol., 2015, 193, 185–191 CrossRef CAS PubMed.
- D.-W. Cho, S.-H. Cho, H. Song and E. E. Kwon, Carbon dioxide assisted sustainability enhancement of pyrolysis of waste biomass: a case study with spent coffee ground, Bioresour. Technol., 2015, 189, 1–6 CrossRef CAS PubMed.
- J. Kim, K.-H. Kim and E. E. Kwon, Enhanced thermal cracking of VOCs evolved from the thermal degradation of lignin using CO2, Energy, 2016, 100, 51–57 CrossRef CAS.
- D.-W. Cho, E. E. Kwon and H. Song, Use of carbon dioxide as reaction medium in the thermo-chemical process for the enhanced generation of syngas and tuning adsorption ability of biochar, Energy Convers. Manage., 2016, 117, 106–114 CrossRef CAS.
- J. Kim, J. Lee, K.-H. Kim, Y. S. Ok, Y. J. Jeon and E. E. Kwon, Pyrolysis of wastes generated through saccharification of oak tree by using CO2 as reaction medium, Appl. Therm. Eng., 2017, 110, 335–345 CrossRef CAS.
- W.-H. Chen and B.-J. Lin, Characteristics of products from the pyrolysis of oil palm fiber and its pellets in nitrogen and carbon dioxide atmospheres, Energy, 2016, 94, 569–578 CrossRef CAS.
- J. Lee, Y. F. Tsang, J.-I. Oh, S.-R. Lee and E. E. Kwon, Evaluating the susceptibility of pyrolysis of monosaccharide, disaccharide, and polysaccharide to CO2, Energy Convers. Manage., 2017, 138, 338–345 CrossRef CAS.
- J. Lee, X. Yang, S.-H. Cho, J.-K. Kim, S. S. Lee, D. C. W. Tsang, Y. S. Ok and E. E. Kwon, Pyrolysis process of agricultural waste using CO2 for waste management, energy recovery, and biochar fabrication, Appl. Energy, 2017, 185, 214–222 CrossRef CAS.
- J. Lee, J.-I. Oh, Y. S. Ok and E. E. Kwon, Study on susceptibility of CO2-assisted pyrolysis of various biomass to CO2, Energy, 2017 DOI:10.1016/j.energy.2017.01.155.
- H. Zhang, R. Xiao, D. Wang, G. He, S. Shao, J. Zhang and Z. Zhong, Biomass fast pyrolysis in a fluidized bed reactor under N2, CO2, CO, CH4 and H2 atmospheres, Bioresour. Technol., 2011, 102, 4258–4264 CrossRef CAS PubMed.
- A. Hendriks and G. Zeeman, Pretreatments to enhance the digestibility of lignocellulosic biomass, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS PubMed.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- E. Shirkavand, S. Baroutian, D. J. Gapes and B. R. Young, Combination of fungal and physicochemical processes for lignocellulosic biomass pretreatment-a review, Renewable Sustainable Energy Rev., 2016, 54, 217–234 CrossRef CAS.
- X. Meng, Q. Sun, M. Kosa, F. Huang, Y. Pu and A. J. Ragauskas, Physicochemical structural changes of poplar and switchgrass during biomass pretreatment and enzymatic hydrolysis, ACS Sustainable Chem. Eng., 2016, 4, 4563–4572 CrossRef CAS.
- Y. Zheng, J. Shi, M. Tu and Y.-S. Cheng, Principles and development of lignocellulosic biomass pretreatment for biofuels, Advances in Bioenergy, 2017, 2, 1–68 Search PubMed.
- F. Xu, J. Sun, N. V. S. N. Murthy Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Transforming biomass conversion with ionic liquids: process intensification and the development of a high-gravity, one-pot process for the production of cellulosic ethanol, Energy Environ. Sci., 2016, 9, 1042–1049 CAS.
- Y. Sun and J. Cheng, Hydrolysis of lignocellulosic materials for ethanol production: a review, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- R. Sindhu, P. Binod and A. Pandey, Biological pretreatment of lignocellulosic biomass – an overview, Bioresour. Technol., 2016, 199, 76–82 CrossRef CAS PubMed.
- E. E. Kwon, J.-I. Oh and K.-H. Kim, Polycyclic aromatic hydrocarbons (PAHs) and volatile organic compounds (VOCs) mitigation in the pyrolysis process of waste tires using CO2 as a reaction medium, J. Environ. Manage., 2015, 160, 306–311 CrossRef CAS PubMed.
- T. Kan, V. Strezov and T. J. Evans, Lignocellulosic biomass pyrolysis: a review of product properties and effects of pyrolysis parameters, Renewable Sustainable Energy Rev., 2016, 57, 1126–1140 CrossRef CAS.
- B. Biswas, R. Singh, J. Kumar, R. Singh, P. Gupta, B. B. Krishna and T. Bhaskar, Pyrolysis behavior of rice straw under carbon dioxide for production of bio-oil, Renewable Energy, 2017, 1–9 Search PubMed.
- S.-H. Cho, J. Lee, K.-H. Kim, Y. J. Jeon and E. E. Kwon, Carbon dioxide assisted co-pyrolysis of coal and lingo-cellulosic biomass, Energy Convers. Manage., 2016, 118, 243–252 CrossRef CAS.
- S. Zellagui, C. Schönnenbeck, N. Zouaoui-Mahzoul, G. Leyssens, O. Authier, E. Thunin, L. Porcheron and J.-F. Brilhac, Pyrolysis of coal and woody biomass under N2 and CO2 atmospheres using a drop tube furnace - experimental study and kinetic modeling, Fuel Process. Technol., 2016, 148, 99–109 CrossRef CAS.
- P. K. Kanaujia, Y. K. Sharma, M. O. Garg, D. Tripathi and R. Singh, Review of analytical strategies in the production and upgrading of bio-oils derived from lignocellulosic biomass, J. Anal. Appl. Pyrolysis, 2014, 105, 55–74 CrossRef CAS.
- J. S. Kim, Production, separation and applications of phenolic-rich bio-oil – a review, Bioresour. Technol., 2015, 178, 90–98 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- J. Lee, Y. F. Tsang, J.-I. Oh, S.-R. Lee and E. E. Kwon, Evaluating the susceptibility of pyrolysis of monosaccharide, disaccharide, and polysaccharide to CO2, Energy Convers. Manage., 2017, 138, 338–345 CrossRef CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Catalytic fast pyrolysis of lignocellulosic biomass, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- H. Li, Z. Fang, R. L. Smith Jr and S. Yang, Efficient valorization of biomass to biofuels with bifunctional solid catalytic materials, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- W.-J. Liu, K. Tian, H. Jiang, X.-S. Zhang, H.-S. Ding and H.-Q. Yu, Selectively improving the bio-oil quality by catalytic fast pyrolysis of heavy-metal-polluted biomass: take copper (Cu) as an example, Environ. Sci. Technol., 2012, 46, 7849–7856 CrossRef CAS PubMed.
- D.-W. Cho, J. Lee, K. Yoon, Y. S. Ok, E. E. Kwon and H. Song, Pyrolysis of FeCl3-pretreated spent coffee grounds using CO2 as a reaction medium, Energy Convers. Manage., 2016, 127, 437–442 CrossRef CAS.
- D.-W. Cho, G. Kwon, Y. S. Ok, E. E. Kwon and H. Song, Reduction of bromate by cobalt-impregnated biochar fabricated via pyrolysis of lignin using CO2 as reaction medium, ACS Appl. Mater. Interfaces, 2017, 9, 13142–13150 CAS.
- D.-W. Cho, G. Kwon, K. Yoon, Y. F. Tsang, Y. S. Ok, E. E. Kwon and H. Song, Simultaneous production of syngas and magnetic biochar via pyrolysis of paper mill sludge using CO2 as reaction medium, Energy Convers. Manage., 2017, 145, 1–9 CrossRef CAS.
- Y. Shen, Carbothermal synthesis of metal-functionalized nanostructures for energy and environmental applications, J. Mater. Chem. A, 2015, 3, 13114–13188 CAS.
- V. S. Sikarwar, M. Zhao, P. S. Fennell, N. Shah and E. J. Anthony, Progress in biofuel production from gasification, Prog. Energy Combust. Sci., 2017, 61, 189–248 CrossRef.
- H. de Lasa, E. Salaices, J. Mazumder and R. Lucky, Catalytic steam gasification of biomass: catalysts, thermodynamics and kinetics, Chem. Rev., 2011, 111, 5404–5433 CrossRef CAS PubMed.
- P. Chaiwatanodom, S. Vivapatarakij and S. Assabumrungrat, Thermodynamic analysis of biomass gasification with CO2 recycle for synthesis gas production, Appl. Energy, 2014, 114, 10–17 CrossRef CAS.
- M. Pohořely, M. Jeremiáš, K. Svoboda, P. Kameníková, S. Skoblia and Z. Beno, CO2 as moderator for biomass gasification, Fuel, 2014, 117, 198–205 CrossRef.
- L. Xiao, X. Zhu, X. Li, Z. Zhang, R. Ashida, K. Miura, G. Luo, W. Liu and H. Yao, Effect of pressurized torrefaction pretreatments on biomass CO2 gasification, Energy Fuels, 2015, 29, 7309–7316 CrossRef CAS.
- N. Sadhwani, S. Adhikari and M. R. Eden, Biomass gasification using carbon dioxide: effect of temperature, CO2/C ratio, and the study of reactions influencing the process, Ind. Eng. Chem. Res., 2016, 55, 2883–2891 CrossRef CAS.
- M. Fernandez-Lopez, D. Lopez-Gonzalez, M. Puig-Gamero, J. L. Valverde and L. Sanchez-Silva, CO2 gasification of dairy and swine manure: a life cycle assessment approach, Renewable Energy, 2016, 95, 552–560 CrossRef CAS.
- C. Guizani, O. Louisnard, F. J. Escudero Sanz and S. Salvador, Gasification of woody biomass under high heating rate conditions in pure CO2: experiments and modelling, Biomass Bioenergy, 2015, 83, 169–182 CrossRef CAS.
- J. D. Martínez, E. E. Silva Lora, R. V. Andrade and R. L. Jaén, Experimental study on biomass gasification in a double air stage downdraft reactor, Biomass Bioenergy, 2011, 35, 3465–3480 CrossRef.
- B. Prabowo, K. Umeki, M. Yan, M. R. Nakamura, M. J. Castaldi and K. Yoshikawa, CO2-steam mixture for direct and indirect gasification of rice straw in a downdraft gasifier: laboratory-scale experiments and performance prediction, Appl. Energy, 2014, 113, 670–679 CrossRef CAS.
- T. Renganathan, M. Yadav, S. Pushpavanam, R. Voolapalli and Y. Cho, CO2 utilization for gasification of carbonaceous feedstocks: a thermodynamic analysis, Chem. Eng. Sci., 2012, 83, 159–170 CrossRef CAS.
- B. Prabowo, M. Aziz, K. Umeki, H. Susanto, M. Yan and K. Yoshikawa, CO2-recycling biomass gasification system for highly efficient and carbon-negative power generation, Appl. Energy, 2015, 158, 97–106 CrossRef CAS.
- A. M. Parvez, I. M. Mujtaba and T. Wu, Energy, exergy and environmental analyses of conventional, steam and CO2-enhanced rice straw gasification, Energy, 2016, 94, 579–588 CrossRef CAS.
- Y. Sun, Integrated carbon dioxide/sludge gasification using waste heat from hot slags: syngas production and sulfur dioxide fixation, Bioresour. Technol., 2015, 181, 174–182 CrossRef CAS PubMed.
- Y. Sun, Q. Liu, H. Wang, Z. Zhang and X. Wang, Role of steel slags on biomass/carbon dioxide gasification integrated with recovery of high temperature heat, Bioresour. Technol., 2017, 223, 1–9 CrossRef CAS PubMed.
- Y. Zhang, Y. Zheng, M. Yang and Y. Song, Effect of fuel origin on synergy during co-gasification of biomass and coal in CO2, Bioresour. Technol., 2016, 200, 789–794 CrossRef CAS PubMed.
- L. Ding, Y. Gong, Y. Wang, F. Wang and G. Yu, Characterization of the morphological changes and interactions in char, slag and ash during CO2 gasification of rice straw and lignite, Appl. Energy, 2017, 195, 713–724 CrossRef CAS.
- J. Kramb, A. Gómez-Barea, N. DeMartini, H. Romar, T. R. K. C. Doddapaneni and J. Konttinen, The effects of calcium and potassium on CO2 gasification of birch wood in a fluidized bed, Fuel, 2017, 196, 398–407 CrossRef CAS.
- A. Monti, V. N. Di and G. Venturi, Mineral composition and ash content of six major energy crops, Biomass Bioenergy, 2008, 32, 216–223 CrossRef CAS.
- C. G. Vassileva and S. V. Vassilev, Behavior of inorganic matter during heating of Bulgarian coals 1: lignites, Fuel Process. Technol., 2005, 86, 1297–1333 CrossRef CAS.
- Y. Niu, H. Tian and S. Hui, Ash-related issues during biomass combustion: alkali-induced slagging, silicate melt-induced slagging (ash fusion), agglomeration, corrosion, ash utilization, and related countermeasures, Prog. Energy Combust. Sci., 2016, 52, 1–61 CrossRef.
- B. Coda, M. K. Cieplik, P. J. Wild and J. H. A. Kiel, Slagging behavior of wood ash under entrained-flow gasification conditions, Energy Fuels, 2007, 21, 3644–3652 CrossRef CAS.
- S. H. Li and K. J. Whitty, Physical phenomena of char-slag transition in pulverized coal gasification, Fuel Process. Technol., 2012, 95, 127–136 CrossRef CAS.
- C. Dupont, T. Nocquet, J. A. D. Costa and C. Verne-Tournon, Kinetic modelling of steam gasification of various woody biomass chars: influence of inorganic elements, Bioresour. Technol., 2011, 102, 9743–9748 CrossRef CAS PubMed.
- P. Lahijani, Z. A. Zainal and A. R. Mohamed, CO2 gasification reactivity of biomass char: catalytic influence of alkali, alkaline earth and transition metal salts, Bioresour. Technol., 2013, 144, 288–295 CrossRef CAS PubMed.
- L. A. Calderón, J. Garza and J. F. Espinal, Theoretical study of sodium effect on the gasification of carbonaceous materials with carbon dioxide, J. Phys. Chem. A, 2015, 119, 12756–12766 CrossRef PubMed.
- R. Habibi, J. Kopyscinski, M. S. Masnadi, J. Lam, J. R. Grace, C. A. Mims and J. M. Hill, Energy Fuels, 2013, 27, 494–500 CrossRef CAS.
- R. Fernades, J. M. Hill and J. Kopyscinski, Determination of the synergism/antagonism parameters during co-gasification of potassium-rich biomass with non-biomass feedstock, Energy Fuels, 2017, 31, 1842–1849 CrossRef.
- M. S. Masnadi, J. R. Grace, X. T. Bi, C. J. Lim and N. Ellis, From fossil fuels towards renewables: inhibitory and catalytic effects on carbon thermochemical conversion during co-gasification of biomass with fossil fuels, Appl. Energy, 2015, 140, 196–209 CrossRef CAS.
- R. S. Postma, S. R. A. Kersten and G. van Rossum, Potassium-salt-catalyzed tar reduction during pyrolysis oil gasification, Ind. Eng. Chem. Res., 2016, 55, 7226–7230 CrossRef CAS.
- D. W. McKee, Mechanisms of the alkali metal catalysed gasification of carbon, Fuel, 1983, 62, 170–175 CrossRef CAS.
- J. A. Moulijn, M. B. Cerfontain and F. Kapteijn, Mechanism of the potassium catalysed gasification of carbon in CO2, Fuel, 1984, 63, 1043–1047 CrossRef CAS.
- I. Landälv, R. Gebart, B. Marke, F. Granberg, E. Furusjӧ, P. Lӧwnertz, O. G. W. Ohrman, E. L. Sӧrensen and P. Salomonsson, Two years experience of the BioDME project – a complete wood to wheel concept, Environ. Prog. Sustainable Energy, 2014, 33(3), 744–750 CrossRef.
- Y. Jafri, E. Furusjӧ, K. Kirtania and R. Gebart, Performance of a Pilot-Scale Entrained-Flow Black Liquor Gasifier, Energy Fuels, 2016, 30(4), 3175–3185 CrossRef CAS.
- O. Öhrman, C. Häggstrӧm, H. Wiinikka, J. Hedlund and R. Gebart, Analysis of trace components in synthesis gas generated by black liquor gasification, Fuel, 2012, 102, 173–179 CrossRef.
- M. Perander, N. DeMartini, A. Brink, J. Kramb, O. Karlstrӧm, J. Hemming, A. Moilanen, J. Konttinen and M. Hupa, Catalytic effect of Ca and K on CO2 gasification of spruce wood char, Fuel, 2015, 150, 464–472 CrossRef CAS.
- K. Kirtania, J. Axelsson, L. Matsakas, P. Christakopoulos, K. Umeki and E. Furusjӧ, Kinetic study of catalytic gasification of wood char impregnated with different alkali salts, Energy, 2017, 118, 1055–1065 CrossRef CAS.
- K. Umeki, G. Häggstrӧm, A. Bach-Oller, K. Kirtania and E. Furusjӧ, Reduction of tar and soot formation from entrained-flow gasification of woody biomass by alkali impregnation, Energy Fuels, 2017, 31, 5104–5110 CrossRef CAS.
- R. I. Olivares, C. Chen and S. Wright, The thermal stability of molten lithium–sodium–potassium carbonate and the influence of additives on the melting point, J. Sol. Energy Eng., 2012, 134, 041002 CrossRef.
- F. L. Chan and A. Tanksale, Review of recent developments in Ni-based catalysts for biomass gasification, Renewable Sustainable Energy Rev., 2014, 38, 428–438 CrossRef CAS.
- Y. Shen, M. Chen, T. Sun and J. Jia, Catalytic reforming of pyrolysis tar over metallic nickel nanoparticles embedded in pyrochar, Fuel, 2015, 159, 570–579 CrossRef CAS.
- E. Baktash, P. Littlewood, R. Schomäcker, A. Thomas and P. C. Stair, Alumina coated nickel nanoparticles as a highly active catalyst for dry reforming of methane, Appl. Catal., B, 2015, 179, 122–127 CrossRef CAS.
- S. Ratchahat, S. Kodama, W. Tanthapanichakoon and H. Sekiguchi, Combined molten salt-Ni/Al2O3 as synergistic medium for high-quality syngas production, Chem. Eng. J., 2015, 278, 224–233 CrossRef CAS.
- T. Kodama, Y. Isobe, Y. Kondoh, S. Yamaguchi and K. I. Shimizu, Ni/ceramic/molten-salt composite catalyst with high-temperature thermal storage for use in solar reforming processes, Energy, 2004, 29, 895–903 CrossRef CAS.
- S. Ratchahat, S. Kodama, W. Tanthapanichakoon and H. Sekiguchi, CO2 gasification of biomass wastes enhanced by Ni/Al2O3 catalyst in molten eutectic carbonate salt, Int. J. Hydrogen Energy, 2015, 40, 11809–11822 CrossRef CAS.
- A. N. Rollinson and M. K. Karmakar, On the reactivity of various biomass species with CO2 using a standardised methodology for fixed-bed gasification, Chem. Eng. Sci., 2015, 128, 82–91 CrossRef CAS.
- C. Dupont, S. Jacob, K. O. Marrakchy, C. Hognon, M. Grateau, F. Labalette and D. D. S. Perez, How inorganic elements of biomass influence char steam gasification kinetics, Energy, 2016, 109, 430–435 CrossRef CAS.
- D. Lopez-Gonzalez, M. Fernandez-Lopez, J. L. Valverde and L. Sanchez-Silva, Gasification of lignocellulosic biomass char obtained from pyrolysis: kinetic and evolved gas analyses, Energy, 2014, 71, 456–467 CrossRef CAS.
- G. Lopez, J. Alvarez, M. Amutio, A. Arregi, J. Bilbao and M. Olazar, Assessment of steam gasification kinetics of the char from lignocellulosic biomass in a conical spouted bed reactor, Energy, 2016, 107, 493–501 CrossRef CAS.
- C. D. Blasi, Combustion and gasification rates of lignocellulosic chars, Prog. Energy Combust. Sci., 2009, 35(2), 121–140 CrossRef.
- C. Guizani, M. Jeguirim, R. Gadiou, F. J. E. Sanz and S. Salvador, Biomass char gasification by H2O, CO2 and their mixture: evolution of chemical, textural and structural properties of the chars, Energy, 2016, 112, 133–145 CrossRef CAS.
- J. Kramb, N. DeMartini, M. Perander, A. Moilanen and J. Konttinen, Modelling of the catalytic effects of potassium and calcium on spruce wood gasification in CO2, Fuel Process. Technol., 2016, 148, 50–59 CrossRef CAS.
- C. Guizani, F. J. Escudero Sanz and S. Salvador, Influence of temperature and particle size on the single and mixed atmosphere gasification of biomass char with H2O and CO2, Fuel Process. Technol., 2015, 134, 175–188 CrossRef CAS.
- G. Chang, J. Xie, Y. Huang, H. Liu, X. Yin and C. Wu, Gasification reactivity and pore structure development: effect of intermittent addition of steam on increasing reactivity of PKS biochar with CO2, Energy Fuels, 2017, 31, 2887–2895 CrossRef CAS.
- L. Wang, J. Sandquist, G. Varhegyi and B. M. Güell, CO2 Gasification of Chars Prepared from Wood and Forest Residue: A Kinetic Study, Energy Fuels, 2013, 27, 6098–6107 CrossRef CAS.
- Z. Bouraoui, M. Jeguirim, C. Guizani, L. Limousy, C. Dupont and R. Gadiou, Thermogravimetric study on the influence of structural, textural and chemical properties of biomass chars on CO2 gasification reactivity, Energy, 2015, 88, 703–710 CrossRef CAS.
- G. Wang, J. Zhang, J. Shao, Z. Liu, H. Wang, X. Li, P. Zhang, W. Geng and G. Zhang, Experimental and modeling studies on CO2 gasification of biomass chars, Energy, 2016, 114, 143–154 CrossRef CAS.
- D. Vamvuka and S. Sfakiotakis, Gasification Reactivity and Mass Spectrometric Analysis of Gases of Energy Crop Chars under a CO2 Atmosphere, Energy Fuels, 2015, 29, 3215–3223 CrossRef CAS.
- K. Kirtania and S. Bhattacharya, CO2 gasification kinetics of algal and woody char procured under different pyrolysis conditions and heating rates, ACS Sustainable Chem. Eng., 2015, 3, 365–373 CrossRef CAS.
- C. Wu, V. L. Budarin, M. Wang, V. Sharifi, M. J. Gronnow, Y. Wu, J. Swithenbank, J. H. Clark and P. T. Williams, CO2 gasification of bio-char derived from conventional and microwave pyrolysis, Appl. Energy, 2015, 157, 533–539 CrossRef CAS.
- J. Alvarez, G. Lopez, M. Amutio, J. Bilbao and M. Olazar, Kinetic study of carbon dioxide gasification of rice husk fast pyrolysis char, Energy Fuels, 2015, 29, 3198–3207 CrossRef CAS.
- D. K. Seo, S. K. Lee, M. W. Kang, J. Hwang and T. Yu, Gasification reactivity of biomass chars with CO2, Biomass Bioenergy, 2010, 34, 1946–1953 CrossRef CAS.
- J. Fermoso, C. Stevanov, B. Moghtaderi, B. Arias, C. Pevida and M. G. Plaza, et al., High-pressure gasification reactivity of biomass chars produced at different temperatures, J. Anal. Appl. Pyrolysis, 2009, 85, 287–293 CrossRef CAS.
- Y. Zhang, S. Kajitani, M. Ashizawa and K. Miura, Proposal of a semi-empirical kinetic model to reconcile with gasification reactivity profiles of biomass chars, Fuel, 2008, 87, 457–481 Search PubMed.
- J. L. Zhang, G. W. Wang, J. G. Shao and H. B. Zuo, A modified random pore model for the kinetics of char gasification, BioResources, 2014, 9, 3497–3507 Search PubMed.
- Y. Richardson, S. T. Tanoh, A. Julbe and J. Blin, Improving the kinetics of the CO2 gasification of char through the catalyst/biomass integration concept, Fuel, 2015, 154, 217–221 CrossRef CAS.
- Y. Shen, M. Ding, X. Ge and M. Chen, Catalytic CO2 gasification of rice husk char for syngas and silica-based nickel nanoparticles production, Ind. Eng. Chem. Res., 2015, 54, 8919–8928 CrossRef CAS.
- H. Abdullah and H. Wu, Biochar as a fuel: 1. Properties and grindability of biochars produced from the pyrolysis of mallee wood under slow-heating conditions, Energy Fuels, 2009, 23, 4174–4181 CrossRef CAS.
- H. Abdullah, K. A. Mediaswanti and H. Wu, Biochar as a fuel: 2. Significant differences in fuel quality and ash properties of biochars from various biomass components of mallee trees, Energy Fuels, 2010, 24, 1972–1979 CrossRef CAS.
- X. Tan, X. Tan, Y. Liu, G. Zeng, X. Wang, X. Hu, Y. Gu and Z. Yang, Application of biochar for the removal of pollutants from aqueous solutions, Chemosphere, 2015, 125, 70–85 CrossRef CAS PubMed.
- M. B. Ahmed, J. L. Zhou, H. H. Ngo, W. Guo and M. Chen, Progress in the preparation and application of modified biochar for improved contaminant removal from water and wastewater, Bioresour. Technol., 2016, 214, 836–851 CrossRef CAS PubMed.
- J. Alvarez, G. Lopez, M. Amutio, J. Bilbao and M. Olazar, Physical Activation of Rice Husk Pyrolysis Char for the Production of High Surface Area Activated Carbons, Ind. Eng. Chem. Res., 2015, 54, 7241–7250 CrossRef CAS.
- O. Pezoti, A. L. Cazetta, K. C. Bedin, L. S. Souza, A. C. Martins, T. L. Silva, O. O. S. Junior, J. V. Visentainer and V. C. Almeida, NaOH-activated carbon of high surface area produced from guava seeds as a high-efficiency adsorbent for amoxicillin removal: kinetic, isotherm and thermodynamic studies, Chem. Eng. J., 2016, 288, 778–788 CrossRef CAS.
- G. Singh, I. Y. Kim, K. S. Lakhi, P. Srivastava, R. Naidu and A. Vinu, Single step synthesis of activated bio-carbons with a high surface area and their excellent CO2 adsorption capacity, Carbon, 2017, 116, 448–455 CrossRef CAS.
- Y. Shen, P. Zhao and Q. Shao, Porous silica and carbon derived materials from rice husk pyrolysis char, Microporous Mesoporous Mater., 2014, 188, 46–76 CrossRef CAS.
- H. Zhang, Y. Yan and L. Yang, Preparation of activated carbon from sawdust by zinc chloride activation, Adsorption, 2010, 16, 161–166 CrossRef CAS.
- Q. Lin, H. Cheng and G. Chen, Preparation and characterization of carbonaceous adsorbents from sewage sludge using a pilot-scale microwave heating equipment, J. Anal. Appl. Pyrolysis, 2012, 93, 113 CrossRef CAS.
- J. Alvarez, G. Lopez, M. Amutio, J. Bilbao and M. Olazar, Upgrading the rice husk char obtained by flash pyrolysis for the production of amorphous silica and high quality activated carbon, Bioresour. Technol., 2014, 170, 132–137 CrossRef CAS PubMed.
- V. Fierro, G. Muñiz, A. H. Basta, H. El-Saied and A. Celzard, Rice straw as precursor of activated carbons: activation with orthophosphoric acid, J. Hazard. Mater., 2010, 181, 27 CrossRef CAS PubMed.
- T. H. Liou and S. J. Wu, Characteristics of microporous/mesoporous carbons prepared from rice husk under base- and acid-treated conditions, J. Hazard. Mater., 2009, 171, 693 CrossRef CAS PubMed.
- X. Song, Y. Zhang and C. Chang, Novel method for preparing activated carbons with high specific surface area from rice husk, Ind. Eng. Chem. Res., 2012, 51, 15075–15081 CrossRef CAS.
- J. F. González, S. Román, C. M. González-García, J. M. V. Nabais and A. L. Ortiz, Porosity development in activated carbons prepared from walnut shells by carbon dioxide or steam activation, Ind. Eng. Chem. Res., 2009, 48(16), 7474–7481 CrossRef.
- F. Rodriguez-Reinoso, M. Molina-Sabio and M. T. Gonzalez, The use of steam and CO2 as activating agents in the preparation of activated carbons, Carbon, 1995, 33, 15 CrossRef CAS.
- Y. Zhu, J. Gao, Y. Li, F. Sun, J. Gao, S. Wu and Y. Qin, Preparation of activated carbons for SO2 adsorption by CO2 and steam activation, J. Taiwan Inst. Chem. Eng., 2012, 43, 112 CAS.
- V. Skoulou, E. Kantarelis, S. Arvelakis, W. Yang and A. Zabaniotou, Effect of biomass leaching on H2 production, ash and tar behavior during high temperature steam gasification (HTSG) process, Int. J. Hydrogen Energy, 2009, 34, 5666–5673 CrossRef CAS.
- A. Sarıoğlan, Tar removal on dolomite and steam reforming catalyst: benzene, toluene and xylene reforming, Int. J. Hydrogen Energy, 2012, 37, 8133–8142 CrossRef.
- T. Matsuhara, S. Hosokai, K. Norinaga, K. Matsuoka, C.-Z. Li and J.-I. Hayashi, In situ reforming of tar from the rapid pyrolysis of a brown coal over char, Energy Fuels, 2009, 24, 76–83 CrossRef.
- S. Hosokai, K. Norinaga, T. Kimura, M. Nakano, C.-Z. Li and J.-I. Hayashi, Reforming of volatiles from the biomass pyrolysis over charcoal in a sequence of coke deposition and steam gasification of coke, Energy Fuels, 2011, 25, 5387–5393 CrossRef CAS.
- T. Sueyasu, T. Oike, A. Mori, S. Kudo, K. Norinaga and J.-I. Hayashi, Simultaneous steam reforming of tar and steam gasification of char from the pyrolysis of potassium-loaded woody biomass, Energy Fuels, 2011, 26, 199–208 CrossRef.
- G. Guan, M. Kaewpanha, X. Hao and A. Abudula, Catalytic steam reforming of biomass tar: prospects and challenges, Renewable Sustainable Energy Rev., 2016, 58, 450–461 CrossRef CAS.
- T. Chen, H. Liu, P. Shi, D. Chen, L. Song, H. He and R. L. Frost, CO2 reforming of toluene as model compound of biomass tar on Ni/Palygorskite, Fuel, 2013, 107, 699–705 CrossRef CAS.
- X. Bao, M. Kong, W. Lu, J. Fei and X. Zheng, Performance of Co/MgO catalyst for CO2 reforming of toluene as a model compound of tar derived from biomass gasification, J. Energy Chem., 2014, 23, 795–800 CrossRef.
- L. Li, Z. Song, X. Zhao, C. Ma, X. Kong and F. Wang, Microwave-induced cracking and CO2 reforming of toluene on biomass derived char, Chem. Eng. J., 2016, 284, 1308–1316 CrossRef CAS.
- X. Xu, E. Jiang, M. Wang and Y. Xu, Dry and steam reforming of biomass pyrolysis gas for rich hydrogen gas, Biomass Bioenergy, 2015, 78, 6–16 CrossRef CAS.
- D. Feng, Y. Zhao, Y. Zhang, S. Sun, S. Meng, Y. Guo and Y. Huang, Effects of K and Ca on reforming of model tar compounds with pyrolysis biochars under H2O or CO2, Chem. Eng. J., 2016, 306, 422–432 CrossRef CAS.
- D. Feng, Y. Zhao, Y. Zhang and S. Sun, Effects of H2O and CO2 on the homogeneous conversion and heterogeneous reforming of biomass tar over biochar, Int. J. Hydrogen Energy, 2017, 42, 13070–13084 CrossRef CAS.
- L. Devi, K. J. Ptasinski and F. J. Janssen, Pretreated olivine as tar removal catalyst for biomass gasifiers: investigation using naphthalene as model biomass tar, Fuel Process. Technol., 2005, 86, 707–730 CrossRef CAS.
- J. Howard, Fundamentals of coal pyrolysis and hydropyrolysis, Chem. Coal Util., 1981, 2, 665–784 Search PubMed.
- F.-J. Wang, S. Zhang, Z.-D. Chen, C. Liu and Y.-G. Wang, Tar reforming using char as catalyst during pyrolysis and gasification of Shengli brown coal, J. Anal. Appl. Pyrolysis, 2014, 105, 269–275 CrossRef CAS.
- J. Lee, D. Choi, Y. F. Tsang, J.-I. Oh and E. E. Kwon, Employing CO2 as reaction medium for in situ suppression of the formation of benzene derivatives and polycyclic aromatic hydrocarbons during pyrolysis of simulated municipal solid waste, Environ. Pollut., 2017, 1–8 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |