Protein-repellent and antimicrobial nanoparticle coatings from hyaluronic acid and a lysine-derived biocompatible surfactant†
Received
29th January 2017
, Accepted 3rd May 2017
First published on 4th May 2017
Abstract
Biofilm formation triggered by uncontrolled protein adsorption, on medical devices is the leading cause of catheter-associated urinary tract infections (CAUTI) during implantation. Herein, we report a water-based, green and one-step strategy to functionalize surfaces of silicone catheters, poly(dimethylsiloxane) (PDMS), with antifouling and antimicrobial substances to avoid uncontrolled protein adsorption and microbial attachment. A novel synergetic formulation consisting of an anionic glycosaminoglycan (hyaluronic acid, HA) and a lysine-derived biocompatible cationic surfactant (Nε-myristoyl-lysine methyl ester, MKM) was prepared, resulting in the formation of nanoparticles (NPs, ca. 100–250 nm). Besides their high stability and long-lasting hydrophilicity in ambient and aqueous environments for 60 days, the nanometric layers (48 ± 3 nm) of HA–MKM NPs on PDMS showed no adsorption of BSA and lysozyme and substantially lower adsorption of fibrinogen as revealed by a quartz crystal microbalance with dissipation (QCM-D). In vitro antimicrobial test with S. aureus, E. coli, P. aeruginosa, P. mirabilis, C. albicans microbes under dynamic conditions revealed that the microbial growth was hampered by 85% compared with unmodified PDMS. Given the multiple functionalities, charges and diverse physiochemical properties of polysaccharide-lysine-based surfactant mixtures, this approach can be easily extended to the development of novel coatings on other silicone-based materials, thereby broadening potential applicability of PDMS-based biomaterials/devices in microfluidics, diagnostic biosensors and others.
1 Introduction
Uncontrolled protein adsorption on surfaces of medically related biomaterials (prosthetic devices, stents, contact lenses and catheters) that come into contact with blood or tissue components is a commonly known problem in biomedical and clinical applications. In implants like catheters, the irreversible attachment of pathogenic microorganisms and subsequent biofilm formation triggered by unspecific protein adsorption, and the associated risk of infection are notoriously difficult to treat and lead often to serious complications or rejection of the implants.1,2 Among others, catheter associated urinary tract infections (CAUTI) have shown to be amongst the most prevalent ones having the highest occurrence of all infections in health institutions. Statistical data indicates that amongst newly submitted patients, 23% of all infections are urinary tract infections, 80% of which are CAUTI.3 The current material of choice for urinary catheter implants production is PDMS, due to its advantageous inherent material properties such as biocompatibility, chemical and thermal stability, optical transparency and elastomeric properties.4 Despite these attractive characteristics, polydimethylsiloxane (PDMS) itself is prone to strong unspecific protein adsorption and microbial attachment after prolonged usage due to its hydrophobic nature.5 Consequently, different approaches have been devised to improve surface properties (e.g., wettability), minimize unspecific protein adsorption and biofilm formation.5 Some approaches integrate antibiotics,6,7 ammonium salts, triclosan8,9 or silver ions10 in the medical device; however, the latter approaches often suffer from drug resistance,11 burst release and increase in biofilm formation.5,12 Other approaches employ hydrophilic polymers as antifouling materials such as peptides,10,13 PEGlyation coating with modified polymers, zwitterionic groups (e.g., phosphorylcholine, carboxybetaine)14,15 or polyvinyl alcohol.16 These approaches might be efficient to prevent the adhesion of microbes to the implant's surface for a certain period of time without killing the microbes, finally resulting in biofilm formation. Therefore, the material that offers both antimicrobial and antifouling properties in a sustained manner that enhances the shelf-life of medical devices is imperative.
In a previous work, the complex between the polysaccharide hyaluronic acid (HA) and the lysine-based cationic surfactant (Nε-myristoyllysine methyl ester, MKM) has shown to have good antimicrobial activity when coated on functional cellulosic materials.17 In the subsequent study, the different protonation behavior and the binding capacity of MKM with HA in an eco-friendly solvent water were explored in detail by potentiometric titration and using the theoretical models.18 These results motivated us to extend the obtained knowledge to tailor the surface properties of PDMS without involving any harsh chemical modification techniques. In particular to develop a novel and single-step coating comprising of biocompatible nanoparticles from HA–MKM mixture, compared to many other studies that involves complex process with several toxic reagents, and multiple coating and treatment steps.5 Its potential as an antifouling and antimicrobial coating on PDMS surfaces has so far not been exploited. Considering the structural and physicochemical diversity of HA and cationic surfactant, in addition to the non-destructive surface modification, this study also aims to understand the interaction of proteins and attachment of microbes at the interfaces of PDMS under dynamic condition, simulating the real conditions to be faced by the catheters, in detail. HA is a linear anionic glycosaminoglycan19 and one of the chief components of the extracellular matrix in various cells and tissues, and has been extensively used in studies of antifouling coatings for biomedical applications.20 For example, silicon-based elastomers coated with HA were shown to reduce fibrinogen (Fib) adsorption by up to 70% compared to unmodified ones, owing to its hydrophilic nature. The antifouling properties of HA on silicone surfaces have also been successfully demonstrated by Wong and co-authors21 as well as by Yue and co-authors20 who chemically coupled HA to a surface of activated silicone and studied the interaction with human plasma proteins (e.g., HSA, and Fib) by protein assay kit, showing a 2.4 fold and 4.7 fold decrease in adhesion respectively when compared to untreated silicone. Even though HA has shown to greatly decrease protein adsorption on PDMS, its bioactive action can be increased by imparting antimicrobial activity to prevent and fight infections during the application of an implant. MKM used in this work as an antimicrobial substance, is natural and biodegradable. It is a cationic amphiphile and can be orally administrated with minimum impact unlike synthetic MKM, due to its low toxicity and non-irritating properties.22 The latter is very important in considering its usage as antibacterial and antifungal agents in human infections.
In the present work, synergetic NPs were prepared by electrostatic interactions between HA and MKM. The binding of formed particles from an aqueous dispersion on thin films of PDMS at different conditions were studied using QCM-D. The wettability, thickness, mass, charges, morphology and coating coverage was observed by contact angle, profilometry, titration, atomic force microscopy (AFM) and confocal laser scanning microscopy (CLSM) measurements. The concentration dependent protein adsorption was studied by QCM-D, using three proteins, namely BSA, fibrinogen and lysozyme. The antimicrobial activity of the HA–MKM coated PDMS surfaces against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Candida albicans, and Proteus mirabilis was tested according to ASTM E-2149-10.
2 Experimental section
2.1 Materials
Sylgard 184 Silicone elastomer Kit for the preparation of PDMS thin films and casted PDMS sheets was purchased from Dow Corning, USA. HA (the monomer molecular weight: 401.3 g mol−1) was purchased from Lex, Slovenia. MKM (molecular weight, M.W.: 406.5 g mol−1) was synthesized and generously provided by the Institute for Advanced Chemistry in Catalonia, Spain (Fig. 1).23 Quartz crystal microbalance (QCM) crystals coated with a gold layer (QSX301) were purchased from LOT-Oriel, Germany and used as received. Fluorescein isothiocyanate isomer I (FITC), bovine serum albumin (BSA), fibrinogen (Fib) from bovine plasma, lysozyme (Lyz) from chicken egg white, phosphate buffered saline (PBS, pH 7.4) and toluene (99.9%) were purchased from Sigma-Aldrich, Austria. All water used in this study was from a MilliQ-water system (18.2 MΩ cm at 25 °C) from Millipore (Billerica, USA). A list of abbreviations used in this paper is shown in Table 1.
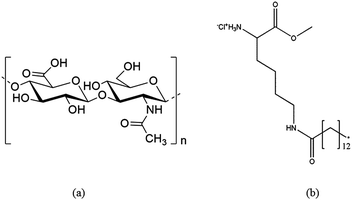 |
| | Fig. 1 The chemical structure of HA (a) and the MKM (b).23 | |
2.2 PDMS thin film and sheet preparation
The detailed procedure of the PDMS thin film preparation and characterization can be found elsewhere.24 PDMS sheets, for evaluating the antimicrobial activity of HA–MKM coatings, were prepared by a casting method. Briefly, 1 g of the Sylgard 184 component was added to 9 g of curing agent. The mixture was homogenized using a mechanical stirrer for 60 s and then casted into a plastic Petri dish (diameter: 145 mm). The Petri dish was placed in a vacuum drier at room temperature at a pressure of 100 mbar for 30 min to remove air bubbles from the mixture. Finally, the casted sheet was dried at 80 °C overnight. Circular samples of 2 cm in diameter were cut from the casted sheet with a circular cutting tool and used for further experiments.
2.3 Preparation of HA–MKM
The HA was dissolved in water at a concentration of 2.5 × 10−4 mol L−1 and shaken overnight. The MKM solution was prepared at a concentration of 5 × 10−4 mol L−1 in water. The NPs dispersion consisting of HA and MKM were prepared in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) by slowly adding the MKM solution to the HA solution. As a result, the final concentration of HA and MKM in the dispersion is 1.25 and 2.25 × 10−4 mol L−1. The solution was stirred constantly during the mixing. The pH of the dispersion prepared in this way is 6.8, which gives the complex a slightly positive charge.17 Throughout this work the particle dispersions or coatings are named as HA–MKM. The NPs dispersion was filtered using 0.45 μm PTFE syringe filter to remove large aggregates. Detailed information about the preparation of HA–MKM dispersion can be found elsewhere.18
:1 (v/v) by slowly adding the MKM solution to the HA solution. As a result, the final concentration of HA and MKM in the dispersion is 1.25 and 2.25 × 10−4 mol L−1. The solution was stirred constantly during the mixing. The pH of the dispersion prepared in this way is 6.8, which gives the complex a slightly positive charge.17 Throughout this work the particle dispersions or coatings are named as HA–MKM. The NPs dispersion was filtered using 0.45 μm PTFE syringe filter to remove large aggregates. Detailed information about the preparation of HA–MKM dispersion can be found elsewhere.18
2.4 Adsorption of HA–MKM on PDMS thin films
Two different conditions were chosen and investigated for the immobilization of HA–MKM-NPs on PDMS. Condition I: the PDMS coated sensors were mounted in the QCM-D flow cell and equilibrated with water until a constant frequency signal was established. The HA–MKM NPs dispersion was rinsed over the surface for 90 min followed by rinsing with water for 60 min. This was repeated twice to obtain three layers. The coating is designated in this work as HA–MKM (I). Condition II: the adsorption of HA–MKM NPs on PDMS was performed in a discontinuous process. After establishing a constant frequency signal with water for the PDMS coated sensors, the HA–MKM NPs were pumped through the QCM cell for 90 min followed by water for 60 min. After that, the crystals were taken out of the cell, blow dried with N2 gas, and dried in an oven at 40 °C for 30 min. This was repeated twice to obtain three layers. The coating is designated in this work as HA–MKM (II). The flow rate in the QCM-D cell was 0.1 mL min−1 and the temperature was 21 ± 0.1 °C for all conditions. Three independent PDMS coated films were used for each experiment.
2.5 Protein adsorption on uncoated and HA–MKM coated PDMS surfaces
The adsorption of BSA, Fib and Lyz dissolved in PBS (c = 0.1, 1 and 10 mg mL−1) on uncoated and HA–MKM coated PDMS surfaces was studied at physiological pH 7.4. The PDMS films (uncoated and HA–MKM coated) were assembled in the QCM flow cells and equilibrated with the PBS solution until a constant frequency signal was obtained. After this, the protein solutions were pumped through the flow cells for 60 min followed by rinsing with PBS buffer for 30 min at a flow rate of 0.1 mL min−1. All experiments were performed at 21 ± 0.1 °C. Each adsorption experiment has been performed in three parallels.
2.6 Antimicrobial activity
The antimicrobial activity of the uncoated and HA–MKM coated PDMS sheets was evaluated based on the colony count method against three Gram-negative bacteria (Escherichia coli (E. coli), Pseudomonas aeruginosa (P. aeruginosa), Proteus mirabilis (P. mirabilis)), one Gram positive bacterium (Staphylococcus sureus, S. aureus) and one fungus (Candida albicans, C. albicans) according to the ASTM E-2149-10 standard testing protocol, for determining the antimicrobial activity of immobilized antimicrobial agents under dynamic conditions. Briefly, the culture medium was prepared by mixing the nutrient agar, the nutrient broth, and the physiological solution. For the preparation of the nutrient agar, 8 g of nutrient broth, 5 g of NaCl and 15 g of agar were mixed with 1 L of water and autoclaved. For the preparation of the nutrient broth, the same procedure was carried out only without the addition of agar. The microorganism of interest was inoculated in 20 mL of the nutrient broth and shaken overnight at 37 °C and a speed of 180 rpm. The optical density of the broth was measured at 600 nm (OD600) and adjusted to 0.5 which equals 108 CFU mL−1. Prior to testing, the uncoated PDMS and HA–MKM NPs coated PDMS sheets were exposed to UV radiation for 20 min. The sterility was tested by pressing the irradiated silicone sample on the nutrient agar and incubating for 48 h at 37 °C. The PDMS sheets were transferred in sterile 150 mL Erlenmeyer flasks together with 50 mL of the nutrient broth and 50 μL of the microorganism broth. Immediately after the preparation (t0), 100 μL of the inoculum was transferred from the flask, diluted and spread over the nutrient agar plates. The inoculum in the flask was further incubated for 1 h at 37 °C and a shaking speed of 180 rpm. After 1 h (t1) the inoculum was prepared in the same way as described above for the t0 case. The microorganism colonies were counted after incubation of the plates for 2 days at 37 °C. The percentage of bacterial/fungal reduction rate (BRR/FRR) was calculated by the following eqn (1).| | 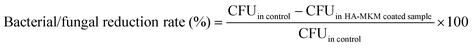 | (1) |
where control is the uncoated PDMS. All the experiments were performed in triplicates.
2.7 Analytical methods
The HA–MKM mean particle hydrodynamic diameters were determined by DLS using a Zetasizer Nano ZS device (Malvern Instruments Ltd, United Kingdom, wavelength: 633 nm, scattering angle: 173°). A QCM-D E4 from Q-Sense AB, Gothenburg, Sweden, was used for the adsorption studies of HA–MKM and proteins on PDMS thin films. The X-ray photoelectron spectroscopy (XPS) spectra of the uncoated and HA–MKM coated PDMS surfaces were recorded with a monochromatic K-Alpha spectrometer equipped with an Al X-ray source (1486.6 eV) operating with a base pressure in the range of 10−8 to 10−10 mbar. The layer thickness of the samples was determined by profilometry using a DEKTAK 150 Stylus Profiler from Veeco (Plainview, NY, USA). The surface morphology of the samples was characterized by atomic force microscopy (AFM) in tapping mode with an Agilent 5500 AFM multimode scanning probe microscope (Agilent, Santa Barbara, CA). A confocal laser scanning microscope (Leica TCS SP5 II laser scanning confocal microscope equipped with a LAS AF imaging software, Leica Microsystems, Germany) was used to observe the surface morphology of the stained samples. The potentiometric charge titration of the coated PDMS sheets was carried out with a two-burette instrument from Mettler Toledo T70, under inert atmosphere (nitrogen gas bubbling). A detailed description of each technique mentioned above can be found in ESI.†
Table 1 List of abbreviations
| Abbreviation |
Description |
| AFM |
Atomic force microscopy |
| BRR |
Bacterial reduction rate |
| BSA |
Bovine serum albumin |
| CAUTI |
Catheter associated urethral infection |
| CLSM |
Confocal laser scanning microscopy |
| DLS |
Dynamic light scattering |
| Fib |
Fibrinogen |
| FITC |
Fluorescin isothiocyanate isomer 1 |
| FRR |
Fungal reduction rate |
| HA |
Hyaluronic acid |
| MKM |
N
ε-Myristoyllysine methyl ester |
| HSA |
Human serum albumin |
| Lyz |
Lyzozyme |
| PBS |
Phosphate buffered saline |
| PDMS |
Polydymethyl siloxane |
| PEG |
Polyethylene glycol |
| PEI |
Polyethylenimine |
| QCM-D |
Quartz crystal microbalance with dissipation |
| SCA |
Static contact angle |
| XPS |
X-Ray photoelectron microscopy |
3 Results and discussion
3.1 Particle size, charge and stability of HA–MKM
The knowledge about the pH-dependent charge and structural behavior of HA–MKM has been elaborated in detail in our previous publications.18 As can be seen from the intensity dependent particle size curves in Fig. 2, the HA–MKM (see insert) exhibits particles of around 250 nm in size with a broad distribution from 100 to 400 nm. However, the number dependent curve reveals that the 90 nm particles represent more than 20% of all particles, while particles of 250 nm and above in size represent less than 5%. The potentiometric charge titration results (Fig. S1, ESI†) revealed that the HA–MKM are slightly positively charged at pH 6–7 (confirming that more MKM molecules are present on its surface), and have the net positive charge of 1.0 and a negative charge of 0.2 mmol g−1. In this scenario the surfactant molecules form micelles when bound to the HA backbone. This invites the HA polymer to wrap around the micelles forming insoluble nano-sized particles.18 The overall excess positive charge prevents further aggregation and allows the dispersions to be stable, when stored at room temperature, for 6 weeks. Since the stability of the NPs over long-term storage (shelf-life) is a highlighted issue for many biological and biomedical applications, we measured the particles size of the stored HA–MKM by DLS. The measurements showed that the particles size remain almost unchanged (98 ± 4 nm) even after prolonged storage (i.e., for 6 weeks), indicating that the dispersion is highly stable and no agglomeration of nanoparticles occurred, which in turn indicates the suitability of the NPs to be applied on PDMS surfaces at any given time without further modification.
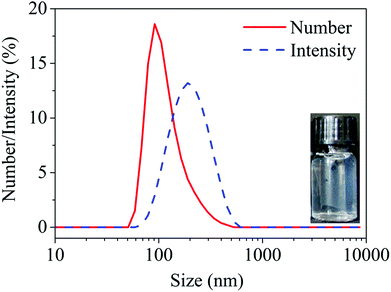 |
| | Fig. 2 Particle size of HA–MKM NPs by intensity and number of particles. | |
3.2 Coating of HA–MKM NPs on PDMS surface
3.2.1 QCM-D studies.
Our main target was to modify the PDMS surface with multifunctional coatings exhibiting mixed charged groups that would reduce or eliminate unspecific protein adsorption and concomitantly exhibit antimicrobial efficacy. For that purpose, we investigated the adsorption of HA–MKM, comprising anionic and cationic functional groups from HA and MKM, on PDMS thin films. The QCM-D frequency and dissipation response for the adsorption of HA–MKM on PDMS thin films (condition I–II) is shown in Fig. 3. When the HA–MKM is introduced into the QCM cells, an immediate steep decrease in the frequency and increase in the dissipation is observed. The frequency decrease (Δf3) and dissipation increase (ΔD3) started to reach a plateau already at t = 20 min for both conditions. Dissipation changes can be solely attributed to higher liquid densities and viscosities of polymers present in the NPs. After switching from the NPs dispersions to water, an increase in frequency and decrease in dissipation are observed, indicating the removal of loosely bound NPs from the surface. After rinsing for 90 min, the adsorbed mass is more or less the same in both cases (Δf3: −55 ± 2 Hz (condition 1), −60 ± 5 Hz (condition II)), as expected. The irreversible deposition of particles to the surface can be attributed to strong electrostatic interaction between the particles (positively charged) and the negatively charged PDMS surface (whose zeta potential, ζ, at pH 7, is −43 mV),24 in addition to other weaker forces such as hydrophobic and hydrogen-bonding interactions. Overall, for the continuous adsorption (condition I) there are no significant increases in the amount of deposition after the first step. For example, the frequency changes only slightly from −69 to −74 Hz and −77 Hz when the second and third dispersion was rinsed off the coating, an indication that no additional layers were created under these conditions. Interaction of dispersed species with already coated layers is weak and as a result the surface energy of these layers is low enough to prevent the adsorption of further NPs. It can be assumed that with the first adsorption step, the PDMS surface is already saturated by charged HA–MKM, preventing further deposition owing to steric electrostatic and presumably hydration repulsion of HA–MKM particles.25
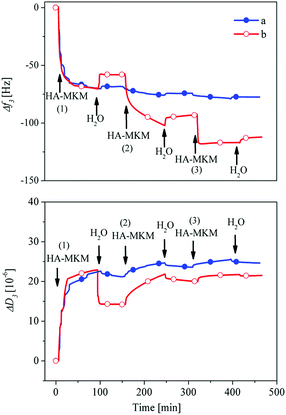 |
| | Fig. 3 QCM-D frequency and dissipation changes for adsorption of HA–MKM on PDMS films a: condition I, b: condition II. | |
Since the total adsorbed mass did not increase considerably under condition I, a three-step discontinuous adsorption process was adapted (condition II, Fig. 3b), where the coated layer from each adsorption step was dried at 40 °C for 30 min. In the latter case (condition II), interestingly the adsorption profile is notably different, especially after the first step. It has to be noted that the first layer was formed in the same way as in condition I. An enhanced deposition of HA–MKM is noticed during the second and third adsorption steps. As soon as HA–MKM is introduced a steep decrease in the frequency shift is observed. Upon rinsing with water major parts of the adsorbed nanoparticles remain on the surface, as determined by a minor increase in the frequency. This shows that the nanoparticles are strongly and irreversibly bound to the surface. The frequency is decreased from −62 to −103 Hz and −122 Hz after the second and third coating respectively, indicating that the drying allows the binding of additionally new nanoparticles to the surface by lowering the hydration repulsion and electrostatic interaction due to a reduced re-swelling in water. It is assumed that the water molecules surrounding the HA and MKM chains are removed upon drying, leading to a compaction of the coating as also observable by the lower f/D ratio after re-swelling compared to never swollen particle layers. The second approach allows successfully to build-up multiple layers of the same material (i.e., HA–MKM NPs) with variable functionalities, composition and charges.
The dissipation change is, generally, related to a specific conformation of the adsorbed polymer mass on the surface. As noticed in Fig. 3b, after the first step, the dissipation is not in direct correlation with the adsorbed amount. This indicates that the HA–MKM either binds less water when it is adsorbed onto the previous layer or a large amount of water is expelled from the film in the course of adsorption. A plausible explanation can be that the new nanoparticles penetrate into the previous layer and are adsorbed in the form of multilayers while expelling the water from the film. This can result in reorganization of the adsorbed particles/structure, changes in surface composition and charges; leading to an enhanced interaction and eventually to a densification of the layer, higher adsorbed mass and layer thickness (see Section 3.2.2). This in turn can lead to increased surface charges and hydrophilicity which are crucial to cause the unwanted adhesion of blood proteins and enzymes on the PDMS implant surface (see Section 3.3).
3.2.2 Confocal laser scanning and atomic force microscopy.
Fig. 4 (top) shows the florescence microscopy images (512 × 512 pixels) of uncoated (a) and coated PDMS thin films with three layers (b–d) of HA–MKM (II). Uncoated PDMS exhibits no detectable fluorescence under this condition since it cannot be labelled by FITC and unspecific binding is comparably low. A random distribution of particles with distinct space between them is observed with one layer of HA–MKM. With increasing number of coatings brighter and uniform fluorescence is observed. The increased fluorescence supports the findings of QCM-D, contact angle and thickness measurements where more material is bound to the surface after each coating step. In addition to CLSM, AFM was utilized to observe the morphology and the roughness of HA–MKM (II) coatings. The AFM phase images and cross-section profiles (obtained from height images) of uncoated and HA–MKM coated PDMS are given in Fig. 4 (middle and bottom). PDMS (a) shows a smooth surface with low roughness (Sq: 0.8 nm). After the first adsorption step (a), the inhomogeneous distribution of NPs as well as many large aggregates composed of HA–MKM and a higher surface roughness (Sq: 13.1 nm) are clearly visible. With the second and third step, in addition to the formation of small and larger cavities (pores), a more organized, densely packed dried NPs and a decreased surface roughness (Sq: 10.2 to 8.1 nm) are noticed. AFM findings are in good correlation with QCM-D, fluorescence thickness and contact angle data that showed also an increased adsorbed mass and uniform fluorescence signal with the increase in adsorption steps.
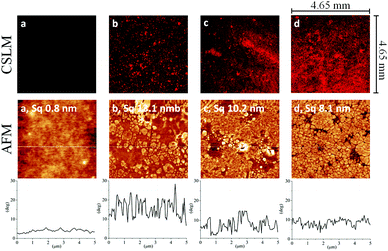 |
| | Fig. 4 CLSM (upper row) and AFM phase images (5 × 5 μm2 middle row) and their respective cross-section profiles (bottom row) of uncoated (a) and HA–MKM coated PDMS thin films, b: first, c: second, d: third layer. The Sq values were obtained from the height images. | |
3.2.3 XPS, profilometry, water contact angle and QCM-D.
The successful deposition of HA–MKM (II) on the PDMS and the surface properties of the coatings were further verified by XPS, profilometry, SCA(H2O) and QCM-D measurements. Table 2 shows the XPS results of HA–MKM coatings. For coated samples, nitrogen is detected, and the silicon content decreases with the number of HA–MKM layers compared to the uncoated one, indicating that the PDMS surface is covered with the nanoparticles. This is also supported by the increase in the oxygen/carbon ratio for coated samples. It should be mentioned that the detected nitrogen amount is originating from both the HA and the MKM present in the adsorbed nanoparticles. The nitrogen amount is increased as the coating step increased. On the other hand, silicon is still detected for the coated samples. These two observations suggest that the adsorbed nanoparticles are less evenly organized on the PDMS surface. This complement the CLSM and AFM results which showed the inhomogeneous distribution of nanoparticles and the presence of small and larger pores in all coated samples. The enrichment of surfaces with nitrogen under condition II is beneficial to increase the antimicrobial activity of the implant (see Section 3.4).
Table 2 XPS atomic surface concentration of oxygen, carbon, and nitrogen on PDMS and HA–MKM modified PDMS (condition II)
|
|
XPS surface concentration (atomic %) |
| C |
O |
Si |
N |
O/C ratio |
| PDMS |
50 ± 0.2 |
25 ± 0.1 |
25 ± 0.3 |
— |
0.5 |
| HA–MKM (layer 1) |
46.1 ± 0.5 |
30.7 ± 0.3 |
22.5 ± 0.2 |
0.7 ± 0.1 |
0.67 |
| HA–MKM (layer 2) |
44.2 ± 0.5 |
32.5 ± 0.5 |
20.1 ± 0.5 |
1.2 ± 0.2 |
0.74 |
| HA–MKM (layer 3) |
44.6 ± 0.8 |
34.8 ± 0.3 |
18.5 ± 0.7 |
2.1 ± 0.2 |
0.78 |
In accordance with the QCM-D dry mass, the film thickness (as-prepared, see Table 3) is also increased as a result of multilayer deposition from HA–MKM. Increase in the adsorbed mass is a direct indication that the surfaces are equipped with mixed charges from HA–MKM which also introduces hydrophilicity. The wettability of the surfaces with water increases (SCA(H2O): 100° to 50°) with an increasing amount of HA–MKM. This can be attributed to the charges and hydroxyl groups on the surface which causes reduced water contact angles as the number of coating increases. PDMS however still contributes to a certain hydrophobicity. On the whole, the above results support and are good in correlation with QCM-D data where an increase in adsorbed mass of HA–MKM is noticed after each adsorption step. To investigate the stability of each HA–MKM layer either in ambient or aqueous environments, the coated surfaces were exposed to air or water for 60 days. Table 3 shows that the film thickness and the SCA(H2O) of all three layers remains stable in air and water, thus displaying stable immobilization of the NPs and long-lasting hydrophilic characteristics. Permanent hydrophilization of PDMS and the presence of mixed charges are desired to prevent biofouling. Usually this is difficult to achieve since PDMS is well-known for its inherent hydrophobic recovery after surface hydrophilisation due to its surface rearrangement and low glass transition temperature.26 Through our approach, we could show that a robust hydrophilic HA–MKM coating on PDMS is possible without involving harsh chemical treatments or laborious multi-step surface modifications.
Table 3 Film thickness, and static water contact angles (SCA(H2O)) and QCM-D dry mass of HA–MKM NPs coated layers
|
|
Film thickness (nm) |
SCA(H2O) (°) |
QCM-D dry mass (μg cm−2) |
| As prepared |
In air (60 days) |
In water (60 days) |
As prepared |
In air (60 days) |
In water (60 days) |
| PDMS |
16 ± 1 |
15 ± 2 |
16 ± 3 |
110 ± 1 |
109 ± 1 |
107 ± 1 |
1.5 ± 0.1 |
| HA–MKM (layer 1) |
25 ± 2 |
24 ± 2 |
23 ± 3 |
100 ± 2 |
100 ± 1 |
101 ± 2 |
23.9 ± 3.2 |
| HA–MKM (layer 2) |
32 ± 4 |
32 ± 2 |
33 ± 3 |
75 ± 1 |
74 ± 2 |
74 ± 2 |
28.6 ± 1.3 |
| HA–MKM (layer 3) |
48 ± 3 |
47 ± 3 |
47 ± 2 |
50 ± 1 |
51 ± 2 |
50 ± 2 |
48.5 ± 1.6 |
3.3 Protein adsorption – antifouling properties
After functionalizing PDMS surfaces with stable coatings of HA–MKM under condition II, we investigated the adsorption of three model proteins (BSA, Fib and Lyz) on these surfaces using QCM-D. At pH 7.4, BSA (pI 4.7) and Fib (pI 5.2) are negatively charged and rich in deprotonated carboxylic acid groups; whereas Lyz (pI 9.4) is positively charged and also possesses a larger number of –OH and –NH2 groups. A large set of data on the low protein adsorption on differently treated PDMS surfaces is available in literature.5 However, the potential applicability of HA–MKM as antifouling coating exhibiting a combination of hydroxyl, carboxyl and amino groups and hydrophilic characteristics has not been demonstrated so far. Therefore, we became interested to test the unspecific protein adsorption onto theses surfaces. The time-dependent adsorption of BSA, Fib and Lyz (10 mg mL−1) at a physiological pH of 7.4 on uncoated and modified PDMS with three layers of HA–MKM is shown in Fig. 5. Protein adsorption is less on the HA–MKM coating. In all cases, the application of protein solutions on hydrophobic-PDMS causes a decrease in frequency and an increase in dissipation. The adsorption of BSA and Lyz is considerably lower than Fib, as reflected by a lower frequency (a) and dissipation change (b). For BSA and Lyz, the adsorption on PDMS reaches saturation significantly faster, already after 2 min of protein contact, and results in the lowest adsorbed mass. A probable explanation is that smaller proteins such as BSA (66 kDa) and Lyz (14.7 kDa) diffuse and reach the surface much faster followed by saturation, according to the Vroman effect.27 In contrast, for Fib, adsorption saturation is much slower and no plateau is reached after 50 min. These slower saturation and higher deposited mass can be, associated to: its higher molecular weight (250 kDa), and the structural rearrangements upon adsorption allowing for subsequent deposition of Fib on previously bound Fib layers.28 We propose here that since Fib is one of the key proteins involved in the blood coagulation cascade its nature favors deposition in more than a monolayer on surface.29 After rinsing with PBS buffer, the frequency and dissipation does not reach the baseline value anymore (Δf3: −16 Hz (BSA), −18 Hz (Lyz), −119 Hz (Fib)), indicating that all three proteins are irreversibly attached to the surface; due to various non-covalent interactions, which are a combination of van der Waals, hydrogen bonds, other dipol–dipol interactions and hydrophobic and electrostatic forces.30,31 It has to be noted that the methods used here cannot distinguish between the contributions of these forces to the overall negative Gibbs free energy of adsorption but non-polar interactions can be assumed to be the main component due to the hydrophobicity and low surface energy of the substrate.24 In contrast HA–MKM coated PDMS behaves very differently. Unlike BSA and Fib in the case of uncoated PDMS surfaces, Lyz interacts strongly with the HA–MKM coated surfaces upon introduction into the QCM cells; as reflected in the deep decrease and increase in the frequency and dissipation. Lyz as an enzyme that can cleave β-1–4-glycosidic bonds in the peptidoglycan of bacterial cell walls seems to have a much higher affinity to the molecularly similar hyaluronic acid in the HA–MKM coatings than the other proteins. Upon rinsing with buffer, the frequency and dissipation shifts are reversed and reach a baseline level for BSA and Lyz; indicating that the proteins are loosely bound to the surface, and rinsed off. Negative dissipation values after this rinsing indicate that the coating is less swollen and/or was partially removed from the surface and was substituted by a denser protein layer. These results show that with the hydrophilic HA–MKM coatings irreversible protein binding is very low and far better results are obtained than the results gained with the most chemical modifications used for antifouling coatings.32,33 For Fib, the adsorption is also almost reversible with approx. 90% protein removed from the surface (Δf3: 9 Hz; and ΔD3: −2 × 10−6). The resistance to unspecific protein adsorption can be attributed to electrostatic repulsion between the charged groups of HA–MKM and charged proteins, and the stability of the hydration layer on the surface. In addition, they also contain a large number of hydrophilic groups such as –OH, –CO–NH and –COOH groups, which impart hydrogen bond donor/acceptor.34–36 Both hydrophilic groups and mixed charges can bind a large amount of water molecules more strongly through electrostatically induced hydration and H-bonding interactions, and thus enhances the strength of hydration. HA–MKM coatings are therefore similar to zwitterionic materials which are known for their low unspecific binding.5
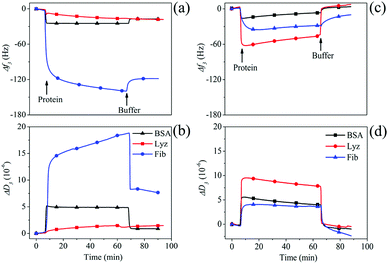 |
| | Fig. 5 QCM-D frequency and dissipation changes for the adsorption of proteins on uncoated (a and b) and HA–MKM coated (c and d) PDMS thin films. | |
To compare the antifouling performance of our coating with other surfaces which are known to undergo a strong unspecific protein adsorption, a positively charged polyethylenimine (PEI) coated layer was used to study the adsorption of blood plasma proteins. They were created on PDMS surface by the adsorption of PEI (0.5%, w/v, dissolved in PBS buffer) at pH 7.4 using QCM-D (see Fig. S2, ESI†). After creating a stable layer of PEI (Δf3 = −38 ± 2 Hz), the protein adsorption (10 mg mL−1) in PBS buffer (pH 7.4) was carried out as in the case of Fig. 5. Results show that all three proteins are adsorbed extensively and irreversibly onto PEI layer upon the introduction into the QCM cell. Rinsing with buffer solution showed no major desorption of the adsorbed protein layer as indicated by minor changes in the frequency and dissipation shift (see Fig. S3, ESI†). The protein adsorption is in the following order: Fib (−214 ± 4 Hz) > BSA (−143 ± 3 Hz) > Lyz (−32 ± 3 Hz). This trend is also seen for uncoated PDMS but more material is deposited on PEI layers compared to either uncoated or modified PDMS surfaces. The above values indicate that the PEI layer is highly prone to unspecific adsorption against all three proteins, as expected, in comparison with our coating where no or less protein adsorption is observed. We also correlated our results with the previously published data for neutral and hydrophilic surfaces of polyethylene glycol (PEG), a widely studied low-protein fouling material. While no BSA and Lyz adsorption are observed on HA–MKM coated surfaces, the PEG coated surfaces had a high BSA and Lyz (1 mg mL−1, in PBS buffer) adsorption of ca. 1.3 and ca. 2–5 μg cm−2, respectively, as revealed by QCM-D experiments.37 Similarly, the HA–MKM surfaces effectively prevent Fib adsorption down to a level of ca. 1.6 μg cm−2, which is almost similar to the value (ca. 1.3 μg cm−2) reported for PEG surfaces where the concentration of Fib used for the adsorption experiments is 10-fold lower than in our work.38 Overall, in comparison with positively charged PEI or neutral PEG surfaces, the HA–MKM NPs coated layers, exhibiting mixed charges, showed a better protein-repellent properties; demonstrating that it has huge potential as antifouling material.
Fig. 6 shows the comparison of wet QCM-D mass for the adsorption of three proteins at different concentrations (0.1, 1 and 10 mg mL−1) on both, uncoated and HA–MKM NPs coated surfaces. It can be seen that, the adsorbed BSA mass increases with increasing protein concentration on uncoated PDMS surfaces; which is also reflected for Lyz and Fib, but the adsorption with Fib leads to more adsorbed mass. No or very low BSA and Lyz are adsorbed on HA–MKM modified PDMS at all applied concentrations. For Fib, the unspecific adsorption is reduced by 90% compared to uncoated ones. On the whole, the HA–MKM exhibits excellent protein-repelling properties to both positively and negatively charged proteins, holding a vast potential for biocompatible applications that involve complex biological environments as for example the human body fluids.
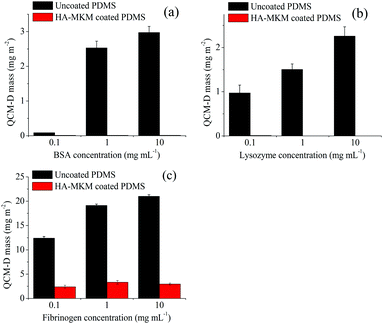 |
| | Fig. 6 Comparison of QCM-D wet mass (mg m−2) for the adsorption of BSA (a), Lyz (b) and Fib (c) at different concentrations. | |
3.4 Antimicrobial properties
Besides knowing that HA–MKM possesses antifouling characteristics, we also evaluated the antibacterial and antifungal properties against three Gram negative bacteria (E. coli, P. aeruginosa, P. mirabilis), one Gram positive bacterium (S. aureus) and one fungus (C. albicans) by using the colony count method. These properties were evaluated by using PDMS sheets that were coated with one and three layers of HA–MKM under condition II. Fig. 7 shows the reduction rates, of microbes calculated according to eqn (1), after exposure to uncoated and HA–MKM coated PDMS sheets at 37 °C for 48 h. Bare PDMS seems to prevent the growth of E. coli presumably because of inactivation though adsorption on the hydrophobic material.
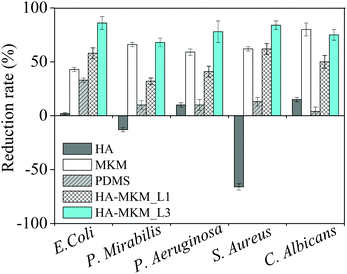 |
| | Fig. 7 Effect of uncoated and HA–MKM coated PDMS sheets on the reduction of five different microbes. MKM and HA were tested separately in solution for comparison. | |
The pure HA solution (0.05 mg mL−1) shows a promotion of growth for S. aureus (−66%) and P. mirabilis (−13%), and a reduction of growth for all other microorganisms E. coli: 2%, and P. aeruginosa: 10% and C. albicans (15%). Pure MKM solution (0.09 mg mL−1) results in a very substantial reduction of growth demonstrating the potential of this antimicrobial biocompatible surfactant. Three layers of HA–MKM on a PDMS sheet of 3.14 cm2 have a higher mass and (total) charge: 1758.5 ng and 3.45 × 10−2 mmol g−1 (calculated from charge titration) compared to first layer (526.2 ng and 1.03 × 10−2 mmol g−1). From charge titration of the coatings it is known that its composition is 16% HA and 84% MKM. Even though these amounts are lower than the mass of HA or MKM in the solution experiments a higher growth inhibition is observed for all bacteria. As expected, reduction rate is influenced by the number of HA–MKM layers deposited on the PDMS substrate. For example, three coatings of HA–MKM inhibited growth more substantially that one layer. The values of BRR of the three times coated PDMS substrate against S. aureus, P. aeruginosa and E. coli are 84, 78 and 86%, respectively, and the BRR against these three microbes reached 62, 40 and 58% when PDMS is coated with one layer of HA–MKM. In general, a much lower BRR is observed against P. mirabilis (30–45%) for both one and three layers of HA–MKM. The BRR for all microorganisms except for E. coli (33%) is below 15% for the untreated PDMS. HA–MKM is also active against the fungi, similar to bacteria, where a maximum reduction rate of 75 and 50% is obtained against C. albicans with three and one layer of HA–MKM compared with uncoated PDMS that shows low or negligible reduction rate. Further, it can be noticed that the fungal reduction rate of HA–MKM, coated three times on PDMS substrate, is almost comparable to pure MKM solution (80 vs. 75%) even if less was deposited, indicating that the activity of the deposited material is not lost. The reduction of the number of microbes clearly indicates the antimicrobial nature of the coating, which can be attributed to the presence of MKM. It has been shown that the cationic amphiphile like MKM molecules can interact with the cell membrane, and their activity against microbes is strongly depending on how well they can distort the phospholipid membrane whose structural integrity is important for biological viability.39,40 The mechanism of antimicrobial or antibacterial activity of cationic amphiphiles, in general, depends on two important factors. Firstly, the amount of cationic amphiphile that penetrates from the coating towards the bacterial membrane, which is, is presumably influenced by the electrostatic interaction between NH3+ of MKM and the phosphate groups in the phospholipid bilayer of the cell membrane. Secondly, the hydrophobic interactions of cationic amphiphiles adsorbed on the cell membrane and components of the membrane wall. Both interactions cause deformation of the cell membrane and subsequent disruption. Consequently, the microbial growth is prohibited and eventually causing cell death.41–43 It is worth noting that HA–MKM is active not only against Gram positive bacteria comprising one cytoplasmatic membrane but also against Gram negative bacteria that have of two bilayers such as the outer membrane and the inner cytoplasmatic membrane. In addition, activity against the multicellular fungus C. albicans is achieved. The synergetic effect of HA and MKM in the coatings is demonstrated by the fact that even though the amount of MKM in the coatings is lower than for pure MKM in solution and it is immobilized on the surface or released from it, it has a higher antimicrobial efficacy. It is therefore proposed that HA potentiates the MKM effect by providing a material that is camouflaging the MKM for the microorganisms tested. It still needs to be elucidated in further work how mechanisms could look like that cause such effect.
4. Conclusions
In this work, a new approach to modify PDMS with hydrophilic and biocompatible nanoparticles from hyaluronic acid and a lysine-derived surfactant was investigated. QCM-D experiments showed that the formed HA–MKM NPs are irreversibly bound on PDMS. The wettability of PDMS is increased by the coatings and an exceptional stability and long-lasting hydrophilicity for 60 days in the aqueous phase is observed. The coating minimizes the uncontrolled attachment of bovine serum albumin and fibrinogen and increases the interaction with Lyz due to the molecular structure of hyaluronic acid. The HA–MKM coated surfaces, however, showed compared to PDMS and PEI exceptionally low irreversible binding of all proteins after rinsing with buffer. The reduction of bacterial growth is the highest for PDMS coated with three layers of HA–MKM in particular against S. aureus, P. aeruginosa, E. coli and C. albicans. A synergetic effect is proposed that potentiated the efficacy of the incorporated surfactant by the concomitant action of hyaluronic acid. Owing to the excellent antifouling and antimicrobial properties, the coatings do have a very high potential to be applied on many types of medical devices in clinical applications.
Acknowledgements
L. Perez acknowledges Spanish Plan national I + D + I MAT2012-38047-C02-02. The authors are grateful to Mag. D. Reishofer and Assoc. Prof. Dr Thomas Grießer from the Technical University of Graz and University of Leoben for their help in regard of AFM, profilometry and XPS measurements. The authors are grateful for the financial support provided by the research program P2-0118 Textile chemistry and Slovenian Research Agency (project no. J2-7413). T. Mohan acknowledge the European Union WoodWisdom-NET+ project PShapes.
Notes and references
- A. Gristina, Science, 1987, 237, 1588–1595 CAS.
- D. R. Schmidt and W. J. Kao, Biomaterials, 2007, 28, 371–382 CrossRef CAS PubMed.
- J. Elvy and A. Colville, J. Infect. Prev., 2009, 10, 36–41 CrossRef.
- J. M. K. Ng, I. Gitlin, A. D. Stroock and G. M. Whitesides, Electrophoresis, 2002, 23, 3461–3473 CrossRef CAS PubMed.
- H. Zhang and M. Chiao, J. Med. Biol. Eng., 2015, 35, 143–155 CrossRef PubMed.
- K. Schumm and T. B. L. Lam, Cochrane Database Syst. Rev., 2008, CD004013, DOI:10.1002/14651858.CD004013.pub3.
- U. S. Ha and Y.-H. Cho, Int. J. Antimicrob. Agents, 2006, 28, 485–490 CrossRef CAS PubMed.
- Y. Liu, C. Leng, B. Chisholm, S. Stafslien, P. Majumdar and Z. Chen, Langmuir, 2013, 29, 2897–2905 CrossRef CAS PubMed.
- T. A. P. Gaonkar, L. B. S. Caraos and S. P. Modak, Infect. Control. Hosp. Epidemiol., 2007, 28, 596–598 CrossRef PubMed.
- J. Gilabert-Porres, S. Martí, L. Calatayud, V. Ramos, A. Rosell and S. Borrós, ACS Appl. Mater. Interfaces, 2016, 8, 64–73 CAS.
- F. N. Pirmoradi, J. K. Jackson, H. M. Burt and M. Chiao, Lab Chip, 2011, 11, 2744–2752 RSC.
- L. Santos, D. Rodrigues, M. Lira, M. E. C. D. R. Oliveira, R. Oliveira, E. Y.-P. Vilar and J. Azeredo, Cont. Lens Anterior. Eye, 2007, 30, 183–188 CrossRef PubMed.
- X. Yu, J. Xiao and F. Dang, Langmuir, 2015, 31, 5891–5898 CrossRef CAS PubMed.
- S.-B. Yeh, C.-S. Chen, W.-Y. Chen and C.-J. Huang, Langmuir, 2014, 30, 11386–11393 CrossRef CAS PubMed.
- A. Zhang, L. Cheng, S. Hong, C. Yang and Y. Lin, RSC Adv., 2015, 5, 88456–88463 RSC.
- D. Wu, Y. Luo, X. Zhou, Z. Dai and B. Lin, Electrophoresis, 2005, 26, 211–218 CrossRef CAS PubMed.
- M. Bračič, L. Pérez, R. I. Martinez-Pardo, K. Kogej, S. Hribernik, O. Šauperl and L. Fras Zemljič, Cellulose, 2014, 21, 2647–2663 CrossRef.
- M. Bračič, P. Hansson, L. Pérez, L. F. Zemljič and K. Kogej, Langmuir, 2015, 31, 12043–12053 CrossRef PubMed.
-
J.-M. Rabanel, N. Bertrand, S. Sant, S. Louati and P. Hildgen, in Polysaccharides for Drug Delivery and Pharmaceutical Applications, American Chemical Society, 2006, vol. 934, ch. 16, pp. 305–339 Search PubMed.
- Z. Yue, X. Liu, P. J. Molino and G. G. Wallace, Biomaterials, 2011, 32, 4714–4724 CrossRef CAS PubMed.
- I. Wong and C.-M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS PubMed.
- D. R. Nogueira, M. Mitjans, M. C. Morán, L. Pérez and M. P. Vinardell, Amino Acids, 2012, 43, 1203–1215 CrossRef CAS PubMed.
- L. Pérez, A. Pinazo, M. Teresa García, M. Lozano, A. Manresa, M. Angelet, M. Pilar Vinardell, M. Mitjans, R. Pons and M. Rosa Infante, Eur. J. Med. Chem., 2009, 44, 1884–1892 CrossRef PubMed.
- M. Bracic, T. Mohan, R. Kargl, T. Griesser, S. Hribernik, S. Kostler, K. Stana-Kleinschek and L. Fras-Zemljic, RSC Adv., 2014, 4, 11955–11961 RSC.
- H. Chen, J. R. Cox, H. Ow, R. Shi and A. Z. Panagiotopoulos, Sci. Rep., 2016, 6, 28553 CrossRef PubMed.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS PubMed.
- L. Vroman and A. L. Adams, Surf. Sci., 1969, 16, 438–446 CrossRef CAS.
- M. L. Clarke, J. Wang and Z. Chen, J. Phys. Chem. B, 2005, 109, 22027–22035 CrossRef CAS PubMed.
- A. Zbigniew, B.-S. Anna, Z. Paulina and W. Monika, Curr. Top. Med. Chem., 2014, 14, 702–729 CrossRef.
- K. Y. Chumbimuni-Torres, R. E. Coronado, A. M. Mfuh, C. Castro-Guerrero, M. F. Silva, G. R. Negrete, R. Bizios and C. D. Garcia, RSC Adv., 2011, 1, 706–714 RSC.
- W. Norde and J. Lyklema, J. Biomater. Sci., Polym. Ed., 1991, 2, 183–202 CrossRef CAS PubMed.
- L. Yang, L. Li, Q. Tu, L. Ren, Y. Zhang, X. Wang, Z. Zhang, W. Liu, L. Xin and J. Wang, Anal. Chem., 2010, 82, 6430–6439 CrossRef CAS PubMed.
- R. Marie, J. P. Beech, J. Vörös, J. O. Tegenfeldt and F. Höök, Langmuir, 2006, 22, 10103–10108 CrossRef CAS PubMed.
- X. Liu, R. Huang, R. Su, W. Qi, L. Wang and Z. He, ACS Appl. Mater. Interfaces, 2014, 6, 13034–13042 CAS.
- S. Yamanlar, S. Sant, T. Boudou, C. Picart and A. Khademhosseini, Biomaterials, 2011, 32, 5590–5599 CrossRef CAS PubMed.
- T. Mohan, R. Kargl, K. E. Tradt, M. R. Kulterer, M. Braćić, S. Hribernik, K. Stana-Kleinschek and V. Ribitsch, Carbohydr. Polym., 2015, 116, 149–158 CrossRef CAS PubMed.
- J. Jin, Y. Han, C. Zhang, J. Liu, W. Jiang, J. Yin and H. Liang, Colloids Surf., B, 2015, 136, 838–844 CrossRef CAS PubMed.
- J. Jin, W. Jiang, J. Yin, X. Ji and P. Stagnaro, Langmuir, 2013, 29, 6624–6633 CrossRef CAS PubMed.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- A. Colomer, A. Pinazo, M. A. Manresa, M. P. Vinardell, M. Mitjans, M. R. Infante and L. Pérez, J. Med. Chem., 2011, 54, 989–1002 CrossRef CAS PubMed.
- S. Tokura, K. Ueno, S. Miyazaki and N. Nishi, Macromol. Symp., 1997, 120, 1–9 CrossRef CAS.
- I. M. Helander, E. L. Nurmiaho-Lassila, R. Ahvenainen, J. Rhoades and S. Roller, Int. J. Food Microbiol., 2001, 71, 235–244 CrossRef CAS PubMed.
- S. Theapsak, A. Watthanaphanit and R. Rujiravanit, ACS Appl. Mater. Interfaces, 2012, 4, 2474–2482 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of DLS, QCM-D, XPS, profilometry, AFM, CLSM, potentiometric titration (for HA-MKM nanoparticles dispersion). See DOI: 10.1039/c7tb00311k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
K.
Kogej
b,
K.
Kogej







