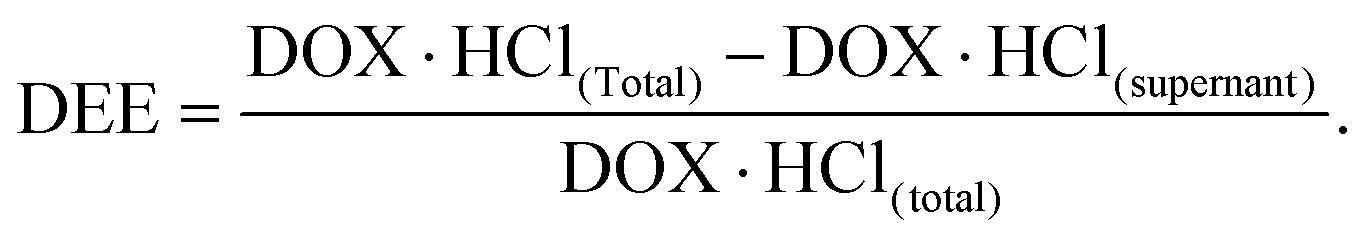DOI:
10.1039/C7BM00776K
(Paper)
Biomater. Sci., 2018,
6, 125-135
Screening of novel RGD peptides to modify nanoparticles for targeted cancer therapy
Received
24th August 2017
, Accepted 31st October 2017
First published on 3rd November 2017
Abstract
New targeted RGD peptides obtained by solid phase peptide synthesis (SPPS) were successfully screened by Molecular Operating Environment (MOE) and used for the building of the 6-O-carboxymethyl chitosan based carrier with an active target on the surface. CRGDYC-6-O-carboxymethyl chitosan based nanoparticles (NPs) loaded with doxorubicin hydrochloride (DOX) were successfully prepared by an ionic gelation method with the carrier synthesized before. Synthesis conditions and formulation parameters were optimized by determining the characteristics of nanoparticles including the particle size and drug encapsulation efficiency. 6-O-Carboxymethyl chitosan concentration, calcium chloride concentration and calcium chloride/6-O-carboxymethyl chitosan ratio all had effects on the particle size and drug encapsulation efficiency. Nanoparticles with an average diameter of 193.4 nm, an average drug loading efficiency of up to 69.5% and an average drug loading of up to 0.395% were prepared successfully with the optimal formulation. Flow cytometry and confocal microscopy analyses showed that the cellular uptake of DOX in human breast cancer cell lines (MCF-7) was higher in the CRGDYC-modified nanoparticles compared with the unmodified nanoparticles. In vivo imaging showed that the distribution of CRGDYC-modified nanoparticles in the tumor site was higher compared with the unmodified nanoparticles. These results suggest that CRGDYC-6-O-carboxymethyl chitosan may be a promising cancer targeting carrier which can enhance the intracellular uptake and cytotoxicity of the drug-loaded nanoparticles.
1. Introduction
Cancer has become the leading cause of death worldwide.1,2 As a major treatment of cancer, chemotherapy has undesirable side effects such as systemic toxicity3–5 and multidrug resistance.6,7 Nanoparticle based drug delivery,8–13 including liposomes,14 polymeric nanoparticles,15–19 micelles,20etc., is a hot-spot for cancer therapy, but its application is still limited due to the systemic toxicity and low delivery efficacy. In order to improve the selectivity of drug carrying vehicles, active targets (such as folate and RGD) have been used for the modification of nanoparticles.21–24
Solid phase peptide synthesis (SPPS)25,26 containing Boc-SPPS and Fmoc-SPPS is of the domination status in the peptide synthesis field. In this work, Fmoc-SPPS was selected for synthesis by virtue of its moderate reaction conditions and less side reaction. Molecular Operating Environment (MOE)27,28 is a drug design software with a comprehensive function. Comparison and certification of the docking manner was conducted due to the presence of the crystal structure data.
The tripeptide arginine-glycine-aspartate (Arg-Gly-Asp, RGD)29–31 specifically recognized by an αvβ3 integral protein with a high expression in a new vascular endothelial system29 is able to induce the apoptosis and death of the tumor neovessel. The c(RGDfv) with higher receptor affinity and binding specificity is more stable than linear RGD.32,33
Carboxymethyl chitosan, a water soluble derivative of chitosan, has great impact in the area of drug delivery and tissue engineering. Compared with chitosan, carboxymethyl chitosan has advantages on biocompatibility, biodegradability and antifungal activity, providing ample opportunities to the drug delivery.34–36
In this study, doxorubicin hydrochloride (DOX) was selected as a model drug. DOX is one of the most widely used anticancer drugs, however, it has serious side effects such as bone marrow inhibition and cardiotoxicity if applied directly.37–39 Hence, the enhancement of tumor targeting and the decrease of side effects of DOX by a preparation method is important.
The objective of the present study is to synthesize CRGDYC-6-O-carboxymethyl chitosan as a promising cancer targeting carrier for drug-loaded nanoparticles and conduct a systematic investigation of the nanoparticle. New targeted RGD peptides obtained by SPPS were successfully screened by MOE and used for the building of the 6-O-carboxymethyl chitosan based carrier with an active target on the surface. The ionic gelation method was used for the preparation of DOX-loaded CRGDYC-6-O-carboxymethyl chitosan based nanoparticles (NPs). The preparation method and formulation were optimized by determining the characteristics of nanoparticles including the particle size and drug encapsulation efficiency. A systematic investigation of the nanoparticle including physicochemical properties, morphology, in vitro release, in vitro cytotoxicity, cellular uptake study and in vivo imaging was also performed.
2. Materials and methods
2.1. Materials
Fmoc-Rink amide-MBHA resin, Fmoc-protected amino acid, thioanisole, anisole, ethylene dithioglycol (EDT), (1H-benzotriazol-1-yl)-N,N,N,N-tetramethyluronium hexafluorophosphate (HBTu), and 1-hydroxybenzotriazole (HOBt) were purchased from Zai Chuang Technology Co. (Shanghai, China). Acetonitrile (HPLC grade) was purchased from Burdick & Jackson (USA). Pancreatic enzyme was purchased from Sigma Chemical Co. (St Louis, MO, USA). RPMI1640 culture medium (Hyclone), fetal bovine serum (Hyclone), penicillin–streptomycin solution (Hyclone), and paraformaldehyde were purchased from LingFeng Chemical Reagent Co. (Shanghai, China). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), and 6-O-carboxymethyl chitosan were purchased from Hong Hai Biological Technology Co. (Qingdao, China). Doxorubicin hydrochloride (DOX) (batch number: HF111219) was purchased from Hua Feng Lian Bo Technology Co. (Beijing, China). N,N-Dimethylformamide (DMF) and trifluoroacetic acid (TFA), N-methyl pyrrolidone (NMP) (heavy steam reagent), N,N-diisopropylethylamine (DIPEA), phenol, iodine, ninhydrin, bromophenol blue, sodium chloride, and potassium phosphate monobasic were all analytical reagents.
2.2. Animals
Six Heps tumor bearing mice were obtained from the Comparative Medicine Centre of Yangzhou University (SCXK (Zhe) 2017-0016), and were randomly allocated in plastic healthy cages under a controlled environment (25 ± 2 °C, relative humidity of 70% ± 10%, and normal light–dark cycle) with free access to food and water. Rats were adapted to the experimental conditions for 1 week before the experiment. All procedures involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, NIH, USA). The animal experiment was approved by the Ethics Committee of China Pharmaceutical University.
2.3. Design and fitting of tumor targeting RGD
First of all, the integrin αvβ3 extracellular region and RGDfV cyclic peptide (cilengitide) compound crystal structure data were obtained from the protein data bank (PDB).40 And the cyclic peptide binding sites were found by MOE. Cilengitide was removed from 1L5G and docked freely into integrin αvβ3. Spatial location, structural form and key amino acid location of the conformation with lowest docking energy were compared with those of the original structure according to 30 kinds of different conformations and the corresponding rated power obtained by MOE. Cilengitide was removed from the original crystal–ligand complex again. CRGDWC, CRGDPC, CRGDFC, CRGDYC and CRGDHC designed before were docked freely into cilengitide. The spatial location, structural form and key amino acid location of the conformation with the lowest docking energy were observed. The docking energy was also investigated.
2.4. Synthesis and purification of CRGDYC and CRGDHC
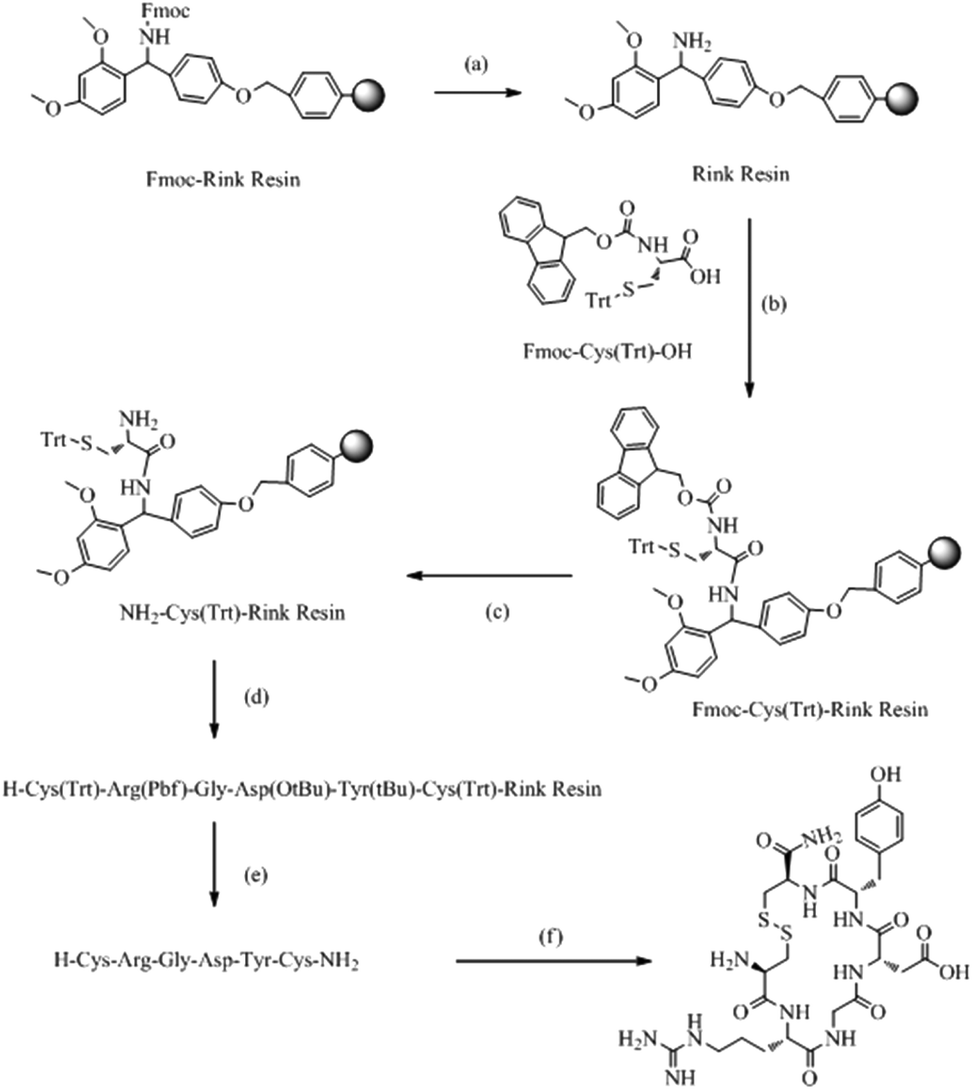
The synthesis was carried out as shown in Scheme 1. CRGDYC and CRGDHC were synthesized using a SPPS method.25,26 0.364 g of Fmoc-Rink amide-MBHA resin (0.55 mmol g−1) was swelled by adding 10 ml of DCM for 30 min. After removing DCM by filtration, the resin was rinsed cleanly with 10 ml of DMF, DCM, and DMF. The resin was deprotected by piperidine under microwave conditions. 0.586 g of Fmoc-Cys-OH (1.0 mmol), 0.383 g of HBTU (1.0 mmol), 0.133 g of HOBT (1.0 mmol) and 0.267 g of DIPEA (2.0 mmol) were dissolved in 10 ml of DMF. And the solution was added to the resin. After the reaction in a microwave reactor with a microwave power of 25 W and a temperature within 50 °C for 7 min, the mixture was cooled by using an air compressor. And then the reaction liquid was removed. The resin was washed three times with 7 ml of DMF and 7 ml of DCM respectively. 5 ml of 50% acetic anhydride/DCM (v/v) mixture solution and a catalytic amount of DMAP were added and stirred at room temperature for 1 h. The reaction liquid was removed by filtration. And the product was washed with DCM and dried under vacuum. The coupling degree was determined by the Fmoc method. Both the resin and resin–peptide compound with an Fmoc protection group were reacted with 25% piperidine/DMF solution in a microwave reactor with a microwave power of 15 W and a temperature within 50 °C for 7 min, and the mixture was cooled by using an air compressor. And then the reaction liquid was removed. Then 7 ml of 25% piperidine/DMF (v/v) solution was added and reacted for another 4 min with a microwave power of 25 W and a temperature within 75 °C. Then the mixture was cooled by using an air compressor. After removing the reaction liquid, the product was rinsed cleanly with DMF. Finally, both the resin and resin–peptide compound without an Fmoc protection group were obtained.
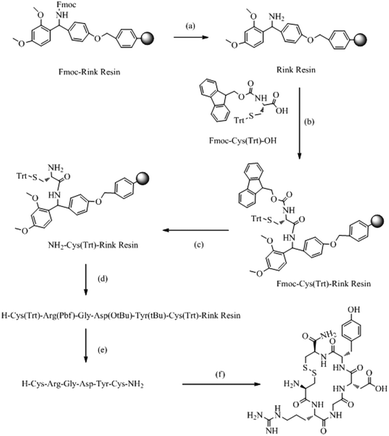 |
| | Scheme 1 Solid-phase synthesis of CRGDYC polypeptide. (a) 25% piperidine/DMF (v/v); (b) HBTU, HOBT, DIPEA; (c) 25% piperidine DMF (v/v); (d) repeated cycles of (a) and (b); (e) 90%TFA, (f) I2, NMP. | |
The peptide chain was extended using HBTU and HOBt as activators, and DIPEA as the activated alkali. After the reaction in a microwave reactor with a microwave power of 25 W and a temperature within 50 °C for 10 min, the reaction liquid was removed by filtration. The resin was washed three times with 10 ml of DCM and DMF. The coupling efficiency was qualitatively detected by the bromophenol blue method. The corresponding amino acid was attached to the resin by repeating the steps above. The coupling efficiency was measured. And if the chromogenic reaction showed negative, the reaction could enter the next circulation, whereas the coupled reaction would be repeated. 0.2 mmol of the N-terminal Fmoc-protected reduced resin obtained above was placed in a reactor with a microwave power of 10 W and a temperature within 45 °C for 10 min. Then 10 ml of anisole/NMP (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) solution containing 0.15 g of I2 was added. Then the mixture was cooled by using an air compressor. After removing the reaction liquid, the resin was rinsed cleanly with CCl4, DMF and DCM. The Fmoc protection group attached on the last N terminal residues was removed as described above. The resin was placed in the reactor, and then 10 ml of lysis solution reagent K (V(TFA):V(thioanisole):V(water):V(phenol):V(EDT) = 82.5:5:5:5:2.5) was added. The reactor was shaken at 0 °C for 30 min and then placed at room temperature for 2 h. The liquid was filtered and then washed three times with a small amount of TFA and DCM. The filtrates were merged. A white floccule was precipitated after adding a lot of ice ether to the filtrate. The product was formed by using a refrigerated centrifuge.
:50) solution containing 0.15 g of I2 was added. Then the mixture was cooled by using an air compressor. After removing the reaction liquid, the resin was rinsed cleanly with CCl4, DMF and DCM. The Fmoc protection group attached on the last N terminal residues was removed as described above. The resin was placed in the reactor, and then 10 ml of lysis solution reagent K (V(TFA):V(thioanisole):V(water):V(phenol):V(EDT) = 82.5:5:5:5:2.5) was added. The reactor was shaken at 0 °C for 30 min and then placed at room temperature for 2 h. The liquid was filtered and then washed three times with a small amount of TFA and DCM. The filtrates were merged. A white floccule was precipitated after adding a lot of ice ether to the filtrate. The product was formed by using a refrigerated centrifuge.
The targeted peptide cut from the resin was dissolved in a small amount of water to a concentration of 10 mg ml−1. After filtration with a 0.25 μm microporous membrane, the solution was purified by using a reversed phase prep-HPLC system (Shimadzu, Japan). The column was 340 mm × 28 mm, 5 μm. The mobile phase was a mixture of 0.1% TFA/water (v/v) and 0.1% TFA/acetonitrile (v/v) with a linear gradient changing volume ratio. The elution rate was 6.0 mL min−1 and the wavelength was set at 220 nm. The product was collected according to the target peak. After freezing and drying, the pure product was formed. Then the pure product was dissolved in a small amount of water to a concentration of 1 mg ml−1. After filtration with a 0.22 μm microporous membrane, the solution was analyzed by reversed phase HPLC (2010C, Shimadzu, Japan). The samples with a concentration of 0.05 mg ml−1 were chromatographed by the injection of a 20 μL sample into a C18 column (4.6 × 250 mm, 5 μm, Shimadzu, Japan). The mobile phase was a mixture of 0.1% TFA/water (v/v) and 0.1% TFA/acetonitrile (v/v) with a linear gradient changing volume ratio. The elution rate was 1.0 ml min−1 and the wavelength was set at 220 nm.
2.5. Screening of tumor cell targeted peptide
MCF-7 cells at a density of 1 × 105 cells per well were seeded in 24-well plates and incubated at 37 °C for 24 h. Then the culture medium was removed and serum-free incomplete culture medium was added. After preincubating for 20 min, both CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and 6-O-carboxymethyl chitosan based nanoparticles diluted with 1640 culture medium to a same concentration were added and the cells were incubated for another 1 h. Then the testing liquid was removed. After washing three times with serum-free culture medium, the cells were incubated with serum-free culture medium for 0 h, 1 h, and 2 h. Each time the upper testing liquid was removed and washed three times with serum-free culture medium. The cells were treated with 4% paraformaldehyde for 20 min and washed three times with PBS. Finally, the cells were observed by using an inverted fluorescence microscope. The fluorescence intensity of two groups of nanoparticles was compared.
MCF-7 cells at a density of 1 × 105 cells per well were seeded and incubated at 37 °C for 24 h. Then the medium was removed. Both 500 μl of CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and 6-O-carboxymethyl chitosan based nanoparticles diluted with serum-free DMEM culture medium were added. After incubating in a CO2 incubator at 37 °C for 1, 2, 4, and 6 h, the upper testing liquid was removed and washed three times with cold PBS. Trypsin was added for digestion and removed, and then the complete culture medium was added for the termination of digestion. The cells were blown and washed three times with PBS, and then determined by flow cytometry. The fluorescence intensity of two groups of nanoparticles was compared.
2.6. Preparation of CRGDYC modified 6-O-carboxymethyl chitosan
50 mg of 6-O-carboxymethyl chitosan was dissolved in 100 ml of water. Then a certain amount of EDC·HCl was added. After stirring for 5 min, 10 mg of CRGDYC polypeptide was added. The solution was stirred at a certain temperature for some time. Then 0.1 mol L−1 hydrochloric acid was added to adjust the pH for the precipitation of the product. The mixture was centrifuged by using a refrigerated centrifuge at 3000 rpm. The fluorescence absorption of the supernatant was measured. The precipitate was collected and dialysed for 48 h. The dialysate was replaced every 6 h. The product was obtained after the dialysate was freezed and dried. The amount of EDC·HCl, the stir time and the stir temperature after adding EDC·HCl were investigated. CRGDYC modified 6-O-carboxymethyl chitosan was detected qualitatively by infrared spectroscopy and nuclear magnetic resonance spectroscopy.
2.7. Preparation of CRGDYC-6-O-carboxymethyl chitosan based nanoparticles loaded with DOX·HCl
CRGDYC-6-O-carboxymethyl chitosan based nanoparticles loaded with DOX·HCl were prepared by an ionic gelation method.41,42 Briefly, 7.5 mg of lyophilized CRGDYC-6-O-carboxymethyl chitosan was dissolved in 5 mL of deionized water, and the pH of the solution was adjusted to 7.0. A certain amount of doxorubicin hydrochloride was added to the solution under magnetic stirring. Then a certain concentration of calcium chloride was added to the solution quickly by dropwise. After stirring at RT for 20 min, the CRGDYC-6-O-carboxymethyl chitosan based nanoparticle suspension loaded with DOX·HCl would be formed. The suspension was centrifuged at 16000 rpm for 30 min, and the precipitate was collected and lyophilized to obtain the nanoparticles. Blank nanoparticles were also prepared using the same protocol. Particle size distribution and drug encapsulation efficiency (DEE) were used to investigate the influence of the ratio of crosslinking agent calcium chloride and CRGDYC-6-O-carboxymethyl chitosan, the concentration of crosslinking agent calcium chloride, and the dosage of doxorubicin hydrochloride on nanoparticles.
2.8. Particle size distribution and zeta potential of the nanoparticles
Intensity averaged particle size and size distribution of the nanoparticles were measured by laser light scattering (Brookhaven Instruments, USA) at 25 °C. The average count rates of the samples were kept at 200–800 kcps to ensure the reliability of the results. The zeta potential of the nanoparticles was determined using a Zetaplus zeta potential analyzer (Brookhaven Instruments, USA) at 25 °C.
2.9. Morphology and drug encapsulation efficiency (DEE) of the nanoparticles
The morphology of CRGDYC-6-O-carboxymethyl chitosan based nanoparticles loaded with DOX·HCl was observed by using a transmission electron microscope (H-7650, HITACHI, Japan).
The speeding refrigerated centrifugation method was used to separate the unloaded DOX·HCl from the nanoparticles. The CRGDYC-6-O-carboxymethyl chitosan based nanoparticles loaded with DOX·HCl were centrifuged at 13000 rpm for 30 min. The supernatant was collected for determination. And the supernatant of blank nanoparticles was regarded as a reference solution.
DOX·HCl in the supernatant was determined by UV at 480 nm. The DEE value was calculated using the following equation:
2.10.
In vitro release
In vitro release studies were investigated by the centrifugation method. CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and 6-O-carboxymethyl chitosan based nanoparticles were placed in phosphate-buffered saline (PBS; pH 6.8) with shaking at 100 rpm for 72 h. At appropriate time intervals (2, 4, 6, 8, 12, 24, 36, 48 and 72 h), an appropriate amount of the release medium was withdrawn and replaced with an equal volume of fresh medium. The supernatant was collected for determination after ultracentrifugation. The accumulated release of DOX·HCl was quantitatively analyzed by UV at 480 nm.
2.11.
In vitro cytotoxicity of CRGDYC-6-O-carboxymethyl chitosan based nanoparticles
The in vitro cytotoxicity of CRGDYC-6-O-carboxymethyl chitosan based nanoparticles was determined by the MTT assay.43,44 Briefly, human breast cancer cell lines (MCF-7) were seeded at a density of 105 cells per well in 24-well plates and incubated at 37 °C for 24 h. Then the medium was removed and serum-free high glucose RPMI1640 medium, CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and 6-O-carboxymethyl chitosan based nanoparticles were added to the wells. After incubation at 37 °C for 48 h, 10 μl of MTT solution (5 mg ml−1 in PBS (pH 7.4)) was added to each well and the cells were incubated at 37 °C for another 4 h. Then the medium was removed, 150 μl of DMSO was added to the wells and the optical density (OD) of the resulting solutions was measured at 570 nm by using a microplate reader. The cells incubated only with serum-free medium were used as a blank control.
2.12 Cellular uptake study
The cellular uptake experiments were performed using confocal laser scanning microscopy (ZEISS LSM 700, Germany) and flow cytometry (BD FACS Calibur, USA).
2.12.1 Confocal laser scanning microscopy experiment.
MCF-7 cells at a density of 1 × 105 cells per well were seeded in a Petri dish for confocal laser scanning and incubated at 37 °C for 24 h. Then the culture medium was removed and serum-free incomplete culture medium was added. After preincubation for 20 min, both DOX·HCl-loaded CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and DOX·HCl-loaded 6-O-carboxymethyl chitosan based nanoparticles diluted with 1640 culture medium to a same concentration were added and the cells were incubated for another 1 h. Then the testing liquid was removed. After washing three times with serum-free culture medium, the cells were incubated with serum-free culture medium for 0 h, 1 h, and 2 h. Each time the upper testing liquid was removed and washed three times with serum-free culture medium. The cells were treated with 4% paraformaldehyde for 20 min and washed three times with PBS. Then, the slip was sealed by an anti-fluorescence quenching agent. Finally, the cells were imaged by confocal laser scanning microscopy.
2.12.2 Flow cytometry experiment.
MCF-7 cells at a density of 1 × 105 cells per well were seeded and incubated at 37 °C for 24 h. Then the medium was removed. Both 500 μl of DOX·HCl-loaded CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and DOX·HCl-loaded 6-O-carboxymethyl chitosan based nanoparticles diluted with serum-free DMEM culture medium were added. After incubation in a CO2 incubator at 37 °C for 1, 2, 4, and 6 h, the upper testing liquid was removed and washed three times with cold PBS. Trypsin was added for digestion and removed, and then the complete culture medium was added for the termination of digestion. The cells were blown and washed three times with PBS, and then determined by flow cytometry.
2.13.
In vivo imaging
In vivo imaging was performed on live mice using an in vivo fluorescence imaging system (KODAK, USA). Six Heps tumor bearing mice were randomly divided into two groups. NIRD-CRGDYC-6-O-carboxymethyl chitosan based nanoparticles and NIRD-6-O-carboxymethyl chitosan based nanoparticles (1 mg kg−1) were injected into the two groups respectively via the tail vein. Mice were anesthetized with urethane (mass concentration 20%) after 30 min, 45 min, 1 h, 2 h, 4 h, and 8 h, and then placed on the in vivo fluorescence imaging system. Fluorescence imaging was obtained by fluorescence shooting on mice with the same exposure intensity and time.
3. Results
3.1. Design of the tumor targeting RGD peptide and docking energy investigation
According to the docking fit by MOE, all the five peptides had a low docking energy indicating good combination with integrin αvβ3. All five have the potential to be a target for integrin αvβ3 with a high expression on the tumor cell surface and in new blood vessels. CRGDYC and CRGDHC had the lowest docking energy, so they had the potential to be a target of active targeting. The docking energy of the designed cyclic peptide is shown in Table 1.
Table 1 Docking energy of the designed cyclic peptide
| Cyclic peptide |
Docking energy |
| Cilengitide |
−4.3681 |
| CRGDFC |
−10.6487 |
| CRGDHC |
−12.1068 |
| CRGDPC |
−10.0833 |
| CRGDWC |
−11.1680 |
| CRGDYC |
−12.6320 |
3.2. Confirmation of the peptides after synthesis and purification
Peptides CRGDYC and CRGDHC with a purity higher than 95% were successfully synthesized as targets for active targeting.
3.3. Screening of tumor cell targeting peptides
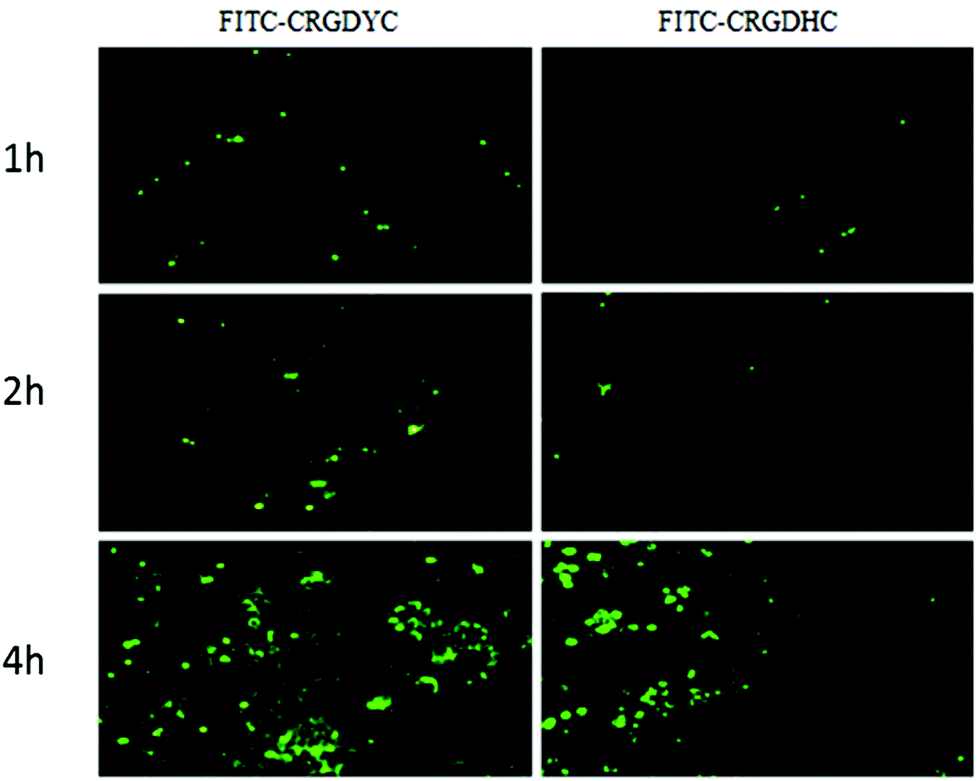
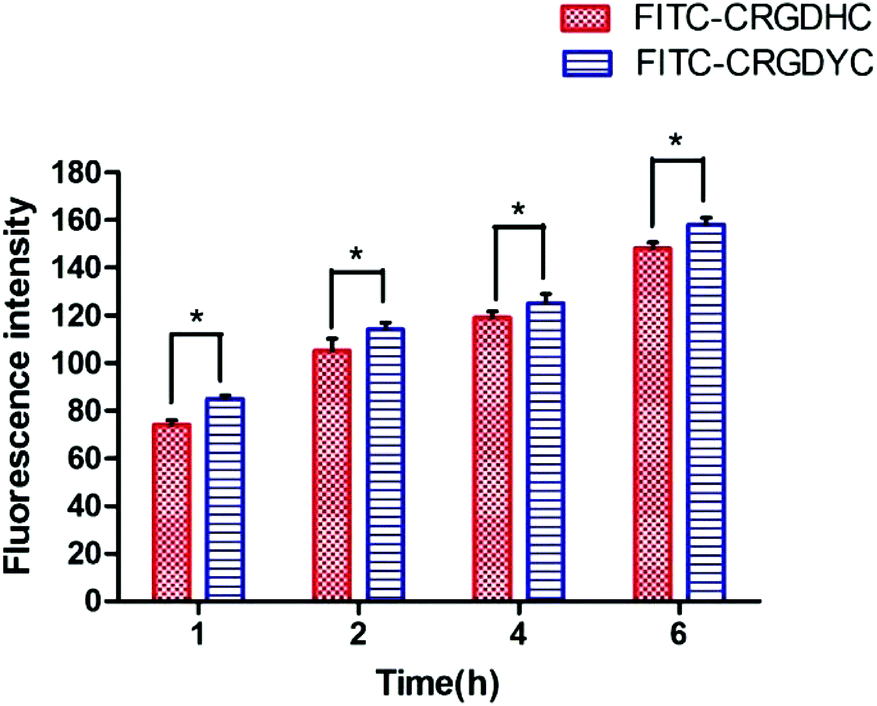
The result of the laser-scanning confocal experiment is shown in Fig. 1. After co-incubation of MCF-7 with FITC-CRGDYC and FITC-CRGDHC, the green fluorescence intensity of MCF-7 cells in the FITC-CRGDYC group was stronger than that in the FITC-CRGDHC group. And for the cells without DAPI staining, these images could show the significant difference between the cellular uptake of FITC-CRGDYC and FITC-CRGDHC. Besides, as shown in Fig. 2, the results of the flow cytometry experiment also showed that the cellular uptake of FITC-CRGDYC was higher than FITC-CRGDHC.
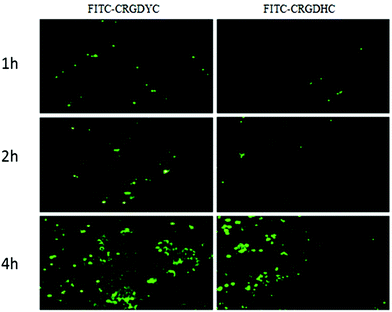 |
| | Fig. 1 Inverted fluorescence microscopy images detecting cellular uptake of FITC-CRGDYC and FITC-CRGDHC. | |
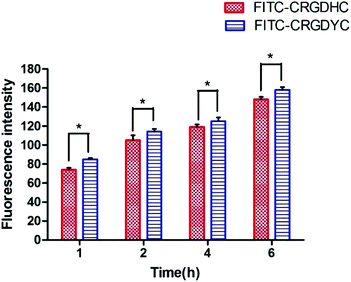 |
| | Fig. 2 Flow cytometry analysis of FITC-CRGDYC and FITC-CRGDHC (n = 3, *P < 0.05, **P < 0.01). | |
3.4. Methodology optimization of 6-O-carboxymethyl chitosan modified with CRGDYC
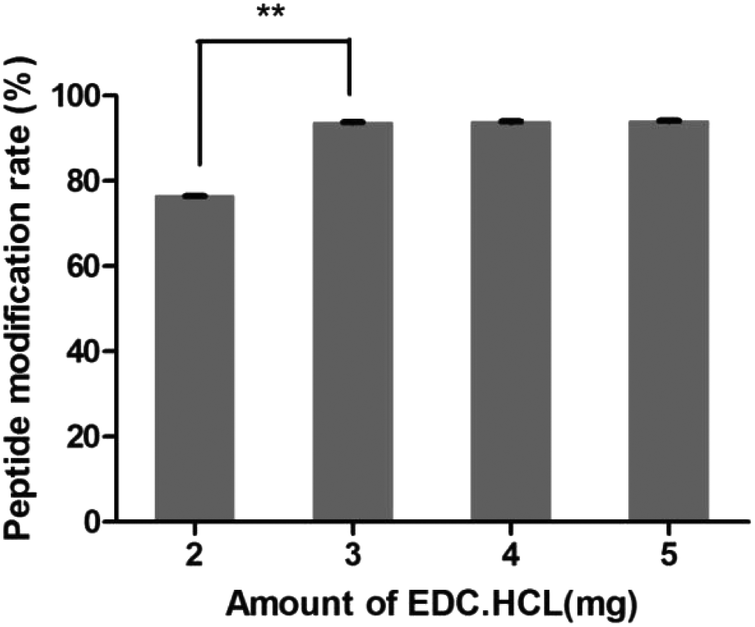
3.4.1 Amount of EDC·HCl.
As shown in Fig. 3, the peptide modification rate was relatively higher when 3 mg of EDC·HCl was added. So the amount of EDC·HCl was chosen as 3 mg.
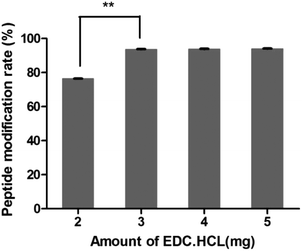 |
| | Fig. 3 Amount investigation of EDC·HCl (n = 3, **P < 0.01). | |
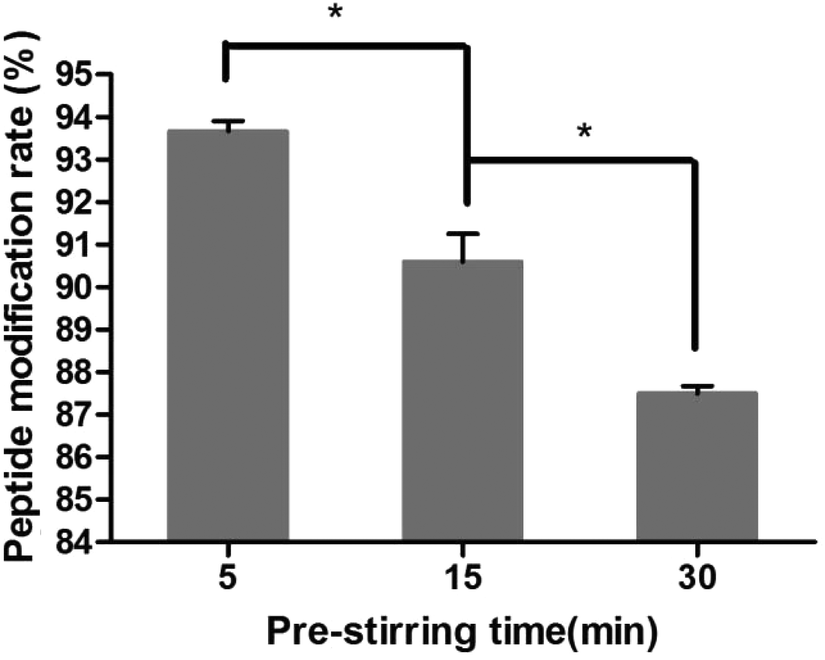
3.4.2. Investigation of pre-stir time after addition of EDC·HCl.
The effect of pre-stirring time after adding EDC·HCl is shown in Fig. 4. The peptide modification amount decreased from 93.7% to 87.6% with an increase in activation time, indicating that the amide bond between the amino and 6-carboxyl of the chitosan was formed. So the activation time after adding EDC·HCl was chosen as 5 min.
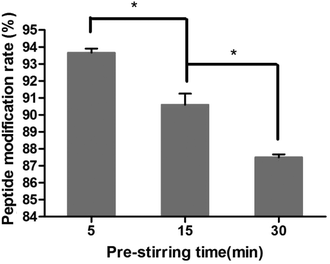 |
| | Fig. 4 Effect of pre-stirring time on the peptide modification amount (n = 3, *P < 0.05). | |
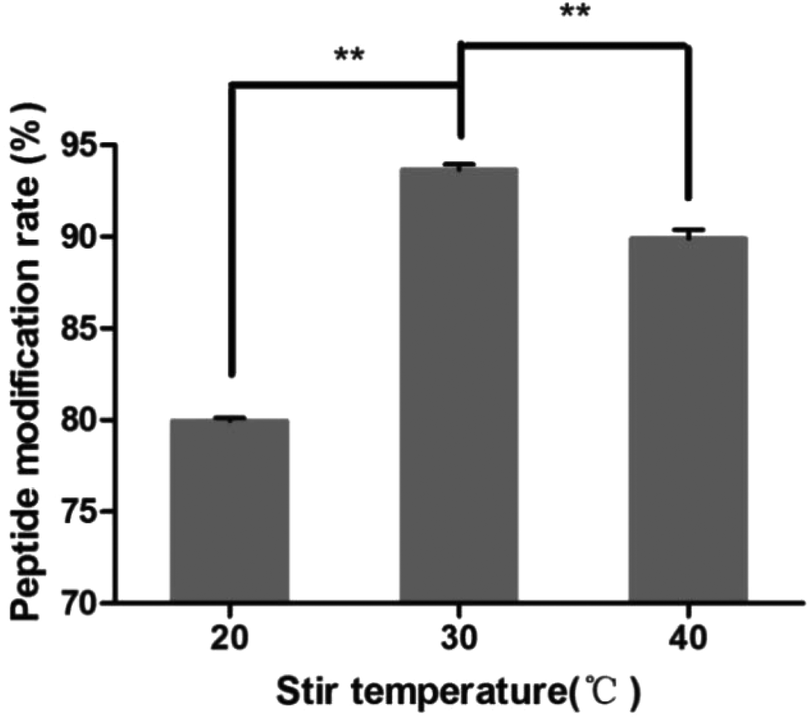
3.4.3 Investigation of stir temperature.
The results of different stir temperatures are shown in Fig. 5. The peptide modification rate was the highest at 30 °C. So the reaction temperature was chosen as 30 °C.
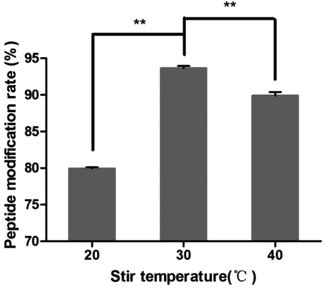 |
| | Fig. 5 Effect of stir temperature on the peptide modification amount (n = 3, **P < 0.01). | |
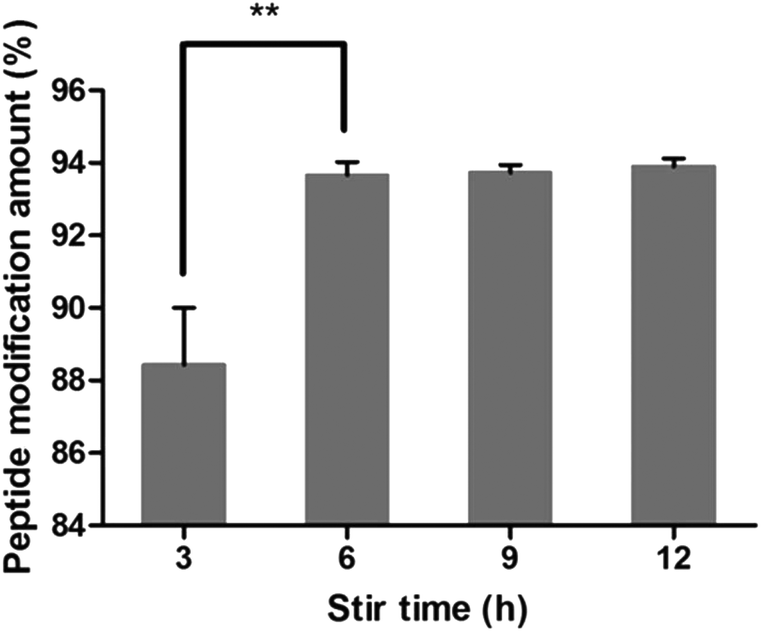
3.4.4. Investigation of stir time.
As shown in Fig. 6, the peptide modification amount increased in the first 6 hours and then maintained steady with the increase in stir time, indicating that the reaction was almost completed in 6 h. So the stir time was chosen as 6 h.
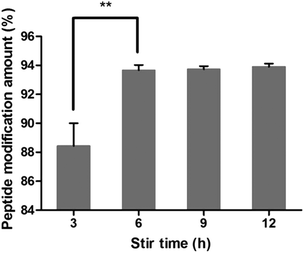 |
| | Fig. 6 Effect of stir time on peptide modification amount (n = 3, **P < 0.01). | |
3.5. Formulation optimization of CRGDYC-6-O-chitosan nanoparticles loaded with doxorubicin hydrochloride
3.5.2. Effect of the cross-linking agent concentration on nanoparticles.
The effect of different calcium chloride solution concentrations on physicochemical properties of nanoparticles is shown in Table 3. The CaCl2/CRGDYC-6-O-carboxymethyl chitosan ratio was 4:3.
Table 3 Effect on particle size and DEE of the CaCl2 concentration
| Concentration of CaCl2 solution (mg ml−1) |
Volume (ml) |
Particle size (nm) |
RSD (%) |
DEE (%) |
DLE (%) |
RSD (%) |
| 5 |
2 |
203.9 |
0.26 |
70.4 |
0.350 |
3.53 |
| 4 |
2.5 |
202.4 |
1.06 |
70.5 |
0.395 |
0.95 |
| 3 |
3.33 |
199.6 |
0.78 |
70.9 |
0.403 |
1.03 |
| 2 |
5 |
193.4 |
0.67 |
72.6 |
0.412 |
1.21 |
| 1 |
10 |
621.8 |
7.06 |
— |
— |
— |
As shown in Table 3, the particle size of nanoparticles decreased from 203.9 nm to 193.4 mm, and the DEE increased from 70.4% to 72.6% with the decrease in CaCl2 solution concentration from 5 mg ml−1 to 2 mg ml−1. Taken together, the optimal concentration of CaCl2 solution was chosen as 2 mg ml−1.
3.5.3. Effect of the doxorubicin hydrochloride dosage on nanoparticles.
The properties of nanoparticles with different drug dosages, including 20 μg ml−1, 30 μg ml−1, and 40 μg ml−1, are shown in Table 4. The CaCl2/CRGDYC-6-O-carboxymethyl chitosan ratio was 4:3 and the concentration of CaCl2 solution was 2 mg ml−1. As shown in Table 4, with the increase in drug dosage, the particle size of nanoparticles increased from 193.4 nm to 259.4 mm, DLE decreased slightly and EE decreased apparently, indicating that the doxorubicin hydrochloride encapsulation capacity of nanoparticles was limited and basically saturated at a concentration of 20 μg ml−1. The influence of three factors including DLE, DEE and particle size was taken into account together, and the optimal doxorubicin dosage was chosen as 20 μg ml−1.
Table 4 Effect of the doxorubicin concentration on particle size and DEE
| Concentration of doxorubicin (μg ml−1) |
Average particle size (nm) |
RSD (%) |
DEE (%) |
DLE (%) |
RSD (%) |
| 20 |
193.4 |
0.67 |
69.5 |
0.395 |
0.95 |
| 30 |
240.1 |
1.16 |
61.79 |
0.346 |
0.947 |
| 40 |
259.4 |
1.12 |
40.40 |
0.316 |
1.88 |
3.6. Morphology and structure of DOX·HCl-loaded CRGDYC-6-O-chitosan nanoparticles
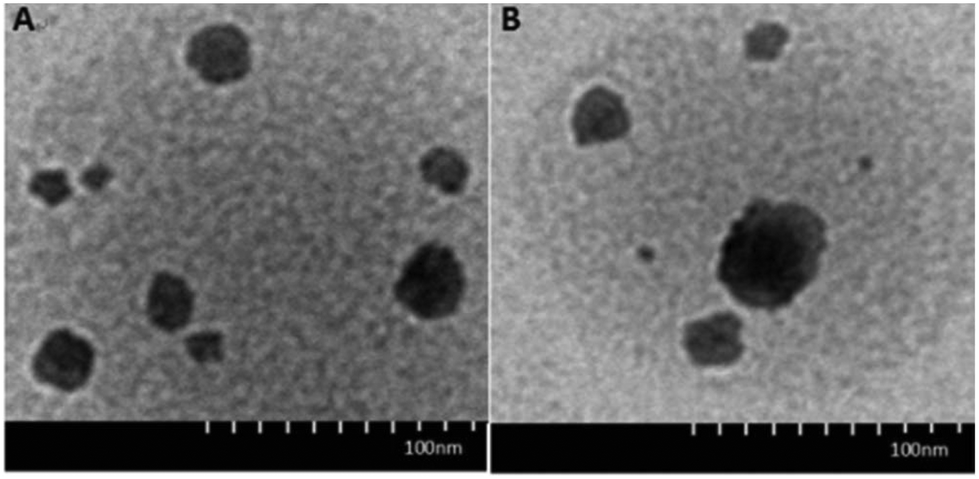
The morphology and structure of DOX·HCl-loaded CRGDYC-6-O-chitosan nanoparticles was investigated using a transmission electron microscope (H-7650, HITACHI, Japan). As shown in Fig. 7, the particles with a diameter of about 190 nm had a spherical structure.
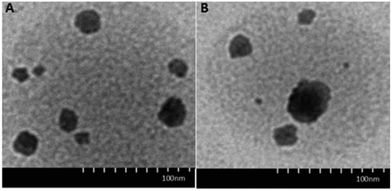 |
| | Fig. 7 TEM images of 6-O-carboxymethyl chitosan nanoparticles (A) and CRGDYC-6-O-carboxymethyl chitosan nanoparticles (B). | |
3.7.
In vitro release
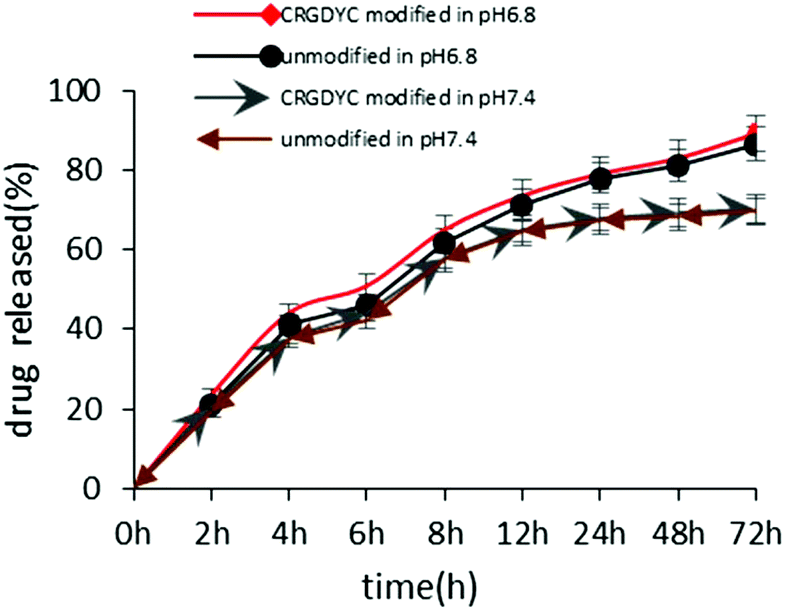
The in vitro release of DOX from 6-O-chitosan nanoparticles is shown in Fig. 8. The results showed that there was no significant difference in the release behavior of the two nanoparticles. The cumulative release after 72 h in the release medium of pH 6.8 was about 85%, and the two curves were very close, indicating that the modification of CRGDYC did not affect the release of doxorubicin hydrochloride. The release was relatively slow at pH 7.4 and reached about 70% after 72 h.
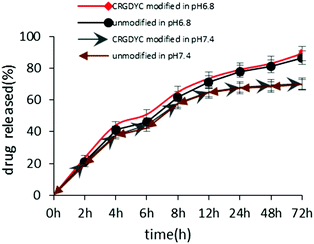 |
| | Fig. 8 Accumulated drug released profile of DOX from CRGDYC-6-O-chitosan nanoparticles and 6-O-chitosan nanoparticles in pH 6.8 and pH 7.4 media. | |
3.8. Cytotoxicity studies
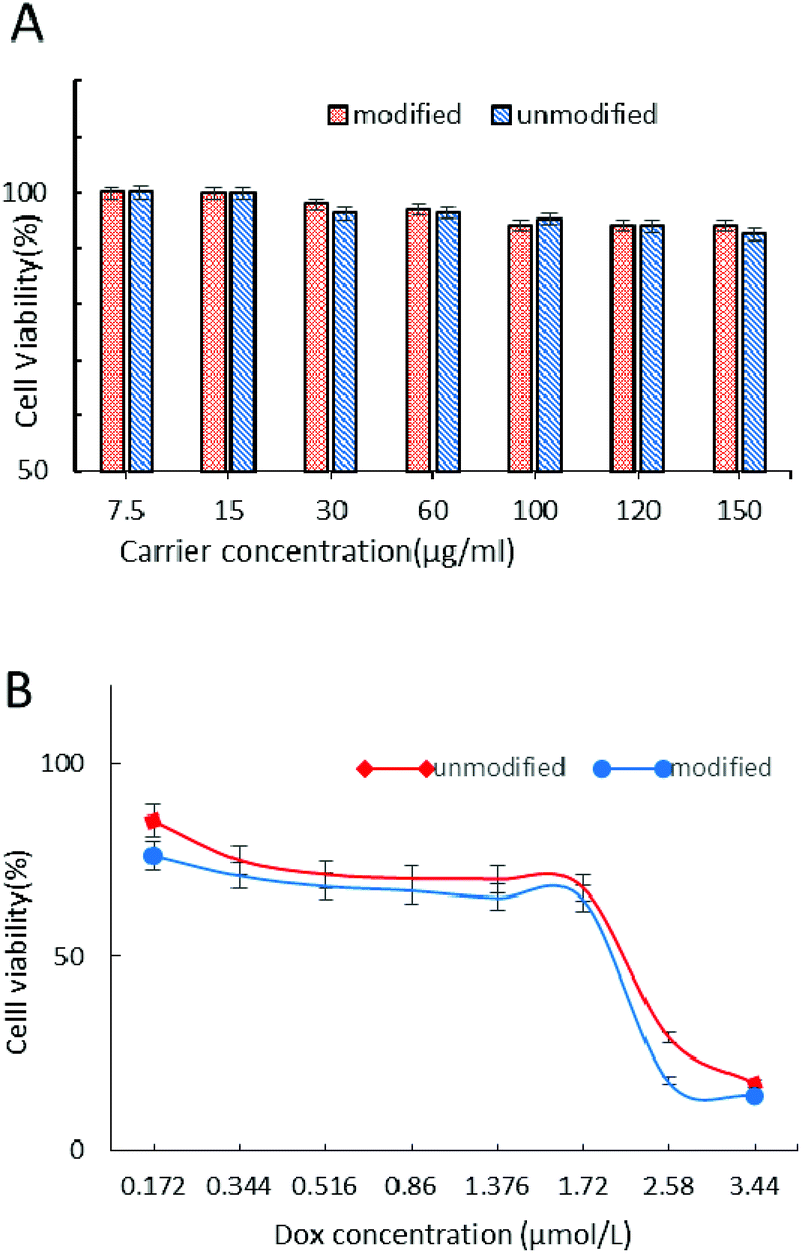
The cytotoxicity of carriers and DOX-loaded nanoparticle drug delivery systems was evaluated by the MTT test in MCF-7 cells after 24 h incubation (Fig. 9). From Fig. 9A we can see that the CRGDYC-6-O-chitosan nanoparticles and 6-O-chitosan nanoparticles had little effect on the activity of MCF-7, and even at the highest concentration, the cell survival rate was still higher than 85%, indicating that this drug carrier can be a low cytotoxicity and security drug delivery system.
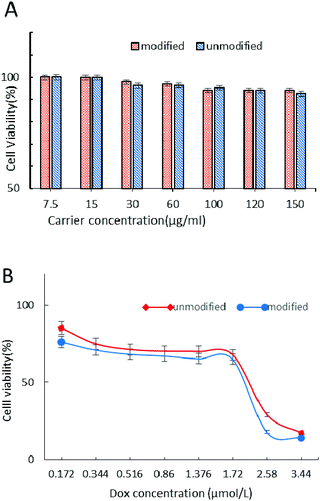 |
| | Fig. 9 Viability of MCF-7 cells treated with carriers (A) and DOX-loaded nanoparticles (B). Cell viability was determined by the MTT assay. Cells incubated only with serum-free medium were used as blank control. The results were expressed as mean values ± standard deviation (n = 6). **P < 0.01, ***p < 0.001. | |
As shown in Fig. 9B, with the increase in DOX concentration, these DOX-loaded nanoparticles showed the same decrease tendency, while the modified nanoparticles showed a higher cytotoxicity compared to the unmodified nanoparticles.
3.9. Cellular uptake of CRGDYC-6-O-carboxymethyl chitosan nanoparticles
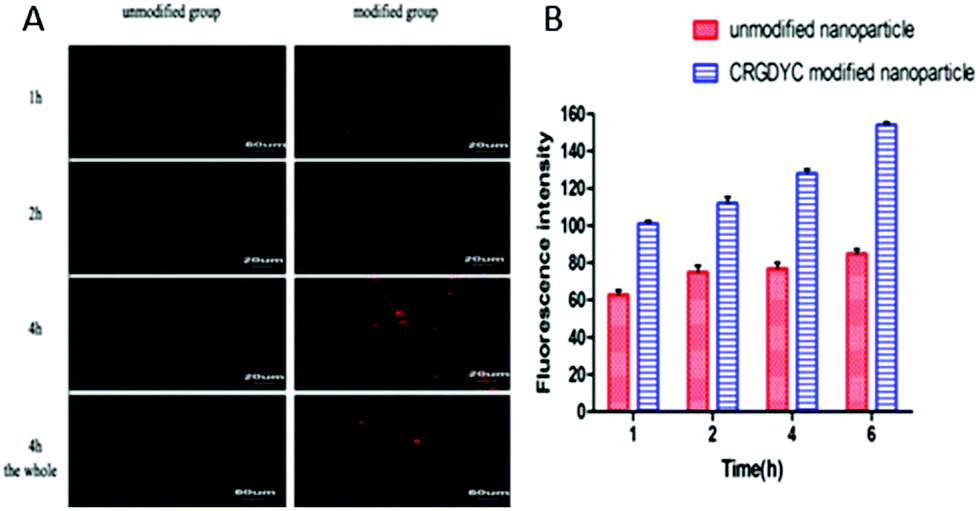
As shown in Fig. 10, after the co-incubation of MCF-7 and above preparations, the red fluorescence intensity of MCF-7 cells in the CRGDYC modified group is the strongest, apparently stronger than the unmodified one (Fig. 10A). In the flow cytometry experiment, the amount of the CRGDYC-modified group entering into the cells is twice as much as the unmodified one (Fig. 10B).
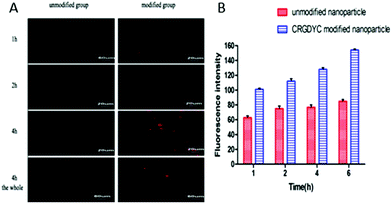 |
| | Fig. 10 Nanoparticle uptake by MCF-7cells. Confocal laser scanning microscopy of MCF-7 cells (1 × 105) after 1, 2 and 4 h of incubation (A). Flow cytometry of MCF-7 cells (1 × 105) after 1, 2, 4 and 6 h of incubation (B). | |
3.10.
In vivo imaging
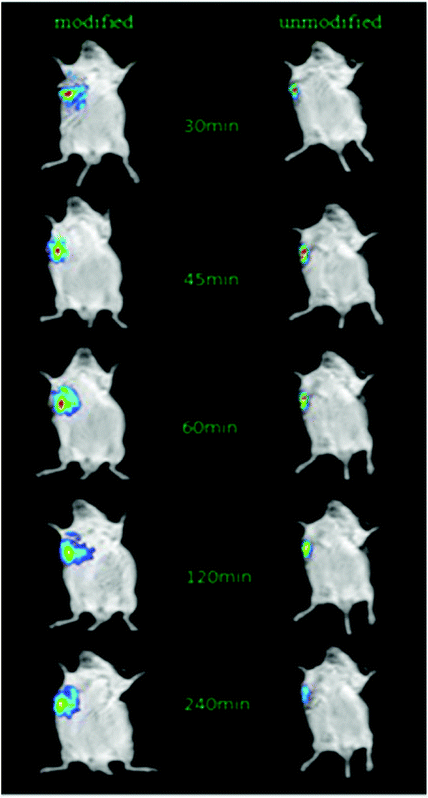
After the injection of NIRD-15-containing CRGDYC-6-O-carboxymethyl chitosan nanoparticle solution and 6-O-carboxymethyl chitosan nanoparticle solution, we obtained in vivo fluorescence images at different time points. From Fig. 11 we can see that the fluorescence intensity of modified nanoparticles is much higher than the unmodified nanoparticles. It may be due to the effect of the integrin receptor αvβ3 on the target polypeptide which can significantly improve the nanoparticle aggregation on the solid tumor area, to achieve its specific targeting. The results are also consistent with the results of cell experiments. It can be seen that RGD-modified 6-O-carboxymethyl chitosan nanoparticles showed specific tumor targeting in tumor-bearing mice, which can enhance the activity of anti-tumor drugs and reduce the side effects to normal tissues and organs.
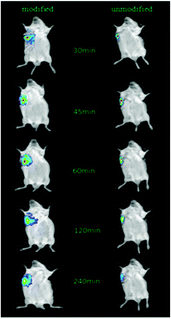 |
| | Fig. 11
In vivo imaging of tumor-bearing mice. | |
4. Discussion
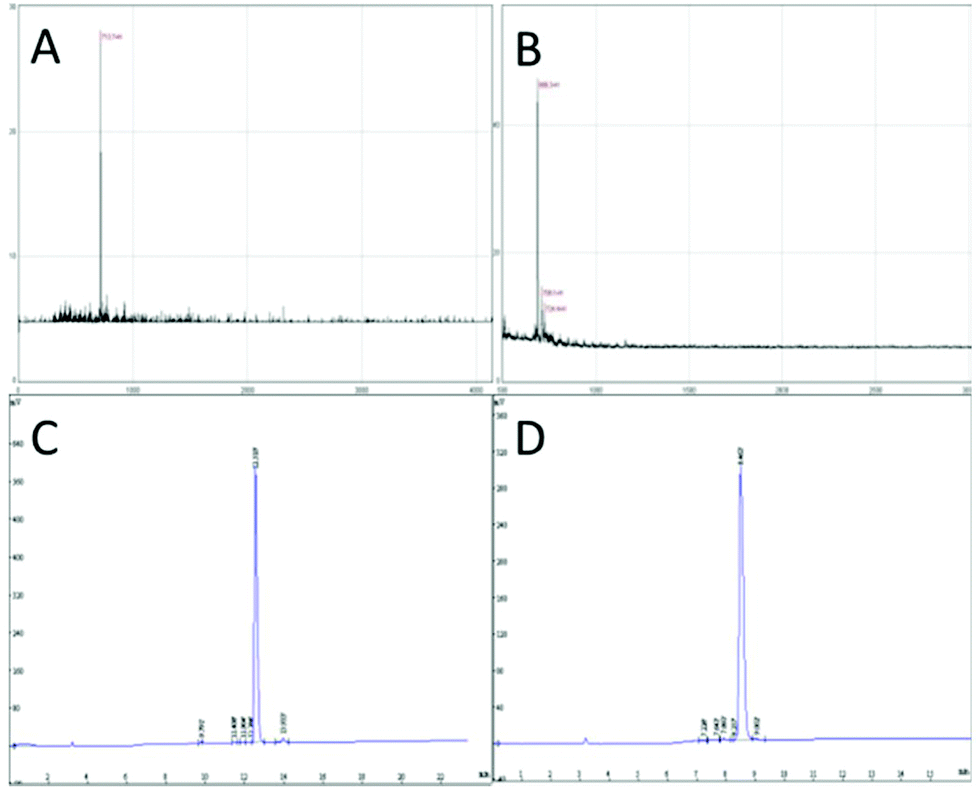
In this study, targeting peptides CRGDYC and CRGDHC were successfully synthesized by SPPS for the modification of 6-O-carboxymethyl chitosan. The sequence of the amino acid was synthesized strictly according to the order from C-terminal to N-terminal. Therefore, the composition and sequence of the amino acid totally satisfied the requirements. The peptides, after separation and purification, were certificated by mass spectra (Fig. 12) and analytical HPLC (Fig. 13). The results of the laser confocal experiment (Fig. 12) indicated that, after the co-incubation of MCF-7 with FITC-CRGDYC and FITC-CRGDHC, the green fluorescence intensity of MCF-7 cells in the FITC-CRGDYC group was stronger than that in the FITC-CRGDHC group. Besides, the results of the flow cytometry experiment (Fig. 13) also showed that the cellular uptake of FITC-CRGDYC was higher than FITC-CRGDHC. In consequence, we selected CRGDYC peptides as target peptides for modification in the next step.
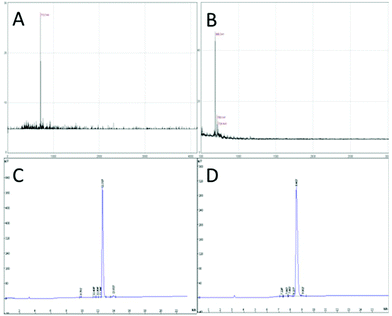 |
| | Fig. 12 Mass spectra of CRGDYC (A) and CRGDHC (B), analytical HPLC of CRGDYC (C) and CRGDHC (D). | |
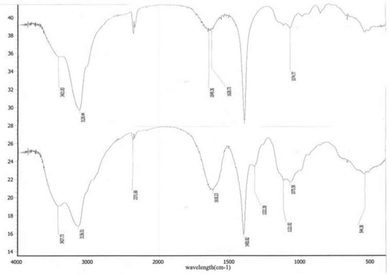 |
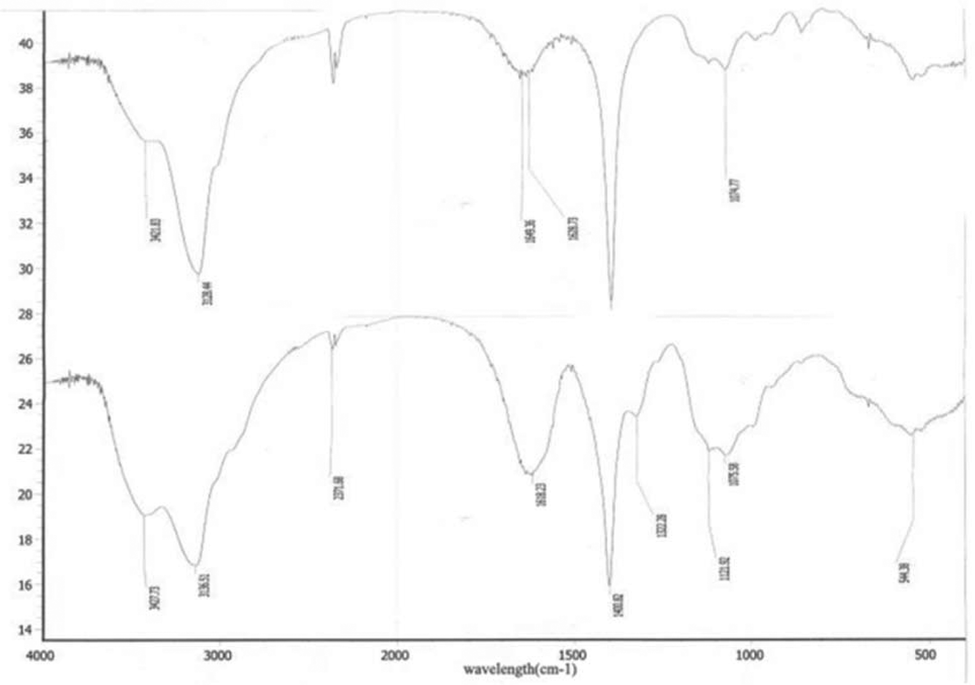
| | Fig. 13 IR of CRGDYC peptide modified 6-O-carboxymethyl chitosan. | |
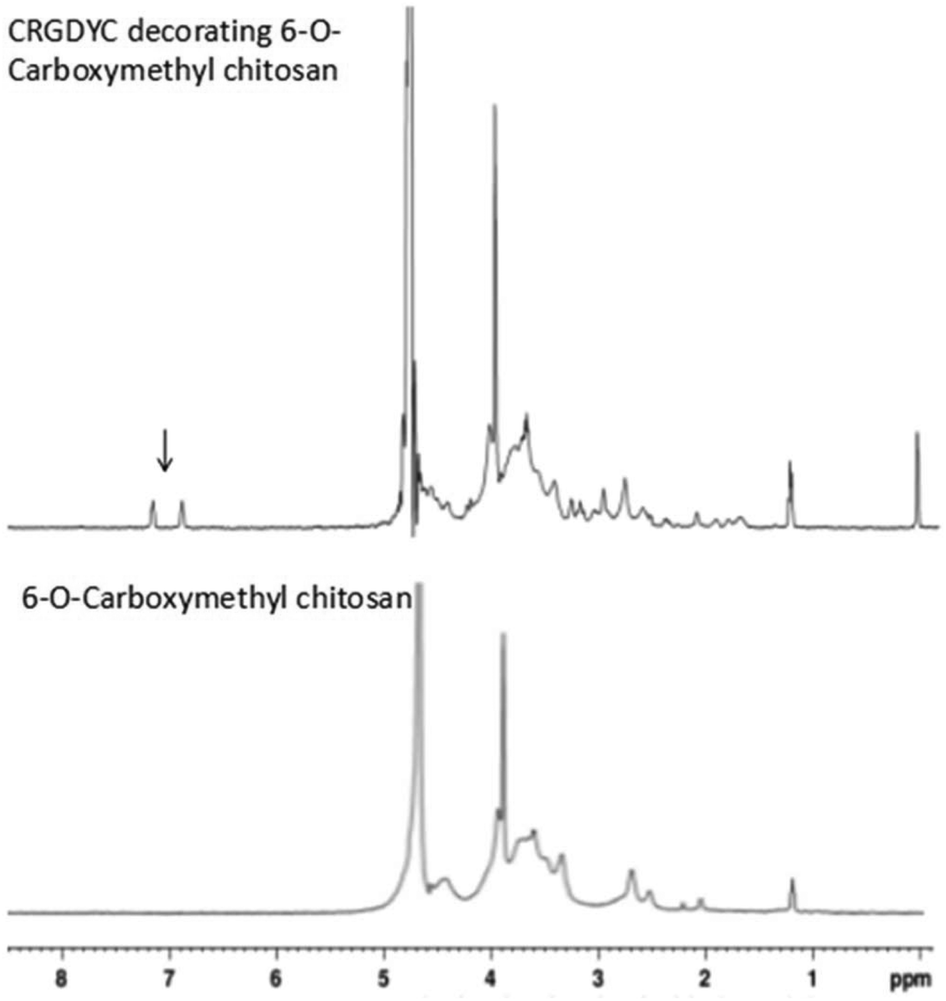
After the method of CRGDYC decorating 6-O-carboxymethyl chitosan had been optimized, we selected the best scheme. After 3 mg of EDC·HCl was added (Fig. 3), we spared 5 min for its activation (Fig. 4) and 6 h for agitation (Fig. 6) at the temperature of 30 °C (Fig. 5), and CRGDYC-modified 6-O-carboxymethyl chitosan was determined by IR (Fig. 13) and 1H-NMR (Fig. 14). As shown in Fig. 14, two characteristic absorption peaks appeared at 7.0 ppm, representing hydrogen atoms in the benzene ring, indicating that 6-O-carboxymethyl chitosan has been successfully modified with the CRGDYC peptide. The peptide modification rate was relatively higher when 3 mg of EDC·HCl was added.
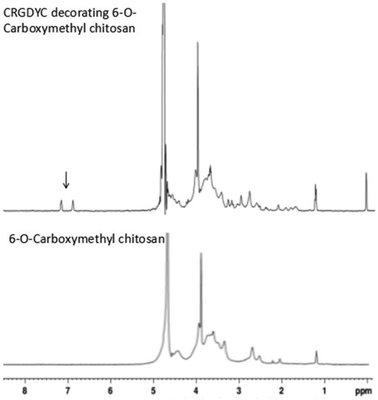 |
| | Fig. 14
1H-NMR of CRGDYC peptide modified 6-O-carboxymethyl chitosan. | |
Nanoparticles were prepared by the ionic gelation method, and the dosage of each ingredient in formulation was screened according to the particle size and DEE. The peptide modification rate was relatively higher when 3 mg of EDC·HCl was added (Fig. 3). As the main effect of EDC·HCl is to activate carboxyl, an excessive amount of EDC·HCl would cause the over-activation of 6-carboxyl of chitosan and even the amide bond formed by chitosan itself. So the amount of EDC·HCl was chosen as 3 mg. The peptide modification rate was the highest at 30 °C (Fig. 5). If the temperature is too low, EDC·HCl would not have been activated completely, leading to the low peptide modification rate. And if the temperature is too high, the amide bond between the amino group and activated 6-carboxyl of chitosan would form, leading to the low peptide modification rate. So the reaction temperature was chosen as 30 °C. The drug encapsulation efficiency (DEE) increased from 59.4% to 71.5% with the increase in the CaCl2/CRGDYC-6-O-carboxymethyl chitosan ratio from 2:3 to 2:1(Table 2). The reason is probably that the crosslinking degree of the calcium ion and carboxylate ion increases with the continuous addition of a cross-linking agent at a certain quantity of dosage. However, an increase in particle size was observed with the increase in crosslinking agent CaCl2. Taken together, the CaCl2/CRGDYC-6-O-carboxymethyl chitosan ratio was chosen as 4:3. The particle size of nanoparticles decreased from 203.9 nm to 193.4 mm, and DEE increased from 70.4% to 72.6% with the decrease in CaCl2 solution concentration from 5 mg ml−1 to 2 mg ml−1 (Table 3). The reason is probably that the crosslinking degree of the calcium ion and carboxylate ion increases with the decrease in the CaCl2 solution concentration and finally results in a decrease in particle size. However, if the CaCl2 solution concentration is too low, nanoparticles would form incompletely. So the optimal concentration of CaCl2 solution was chosen as 2 mg ml−1.
The results of the in vitro release experiment of CRGDTC-6-O-carboxymethyl chitosan nanoparticles and 6-O-carboxymethyl chitosan nanoparticles in the release medium with a pH of 6.8 showed that these two nanoparticles didn't have apparent difference in the release manner (Fig. 8). Their accumulated release amounts were both about 85% after 72 h, and the two curves were close to each other, abdicating that the modification of CRGDYC didn't affect the release of doxorubicin hydrochloride.
The MTT assay was used to determine the cytotoxicity of 6-O-chitosan nanoparticles and CRGDYC-6-O-carboxymethylchitosan nanoparticles against the human cancer cell line (MCF-7) in vitro. The pure carrier system of CRGDTC-6-O-carboxymethyl chitosan nanoparticles and 6-O-carboxymethyl chitosan nanoparticles had no effect on the activity of MCF-7. Even at the highest concentration, the cells’ survival rate was still higher than 85% (Fig. 9A). In conclusion, the nanoparticle delivery system with low cytotoxicity was safe. The doxorubicin hydrochloride nanoparticle drug delivery system was concentration dependent (Fig. 9B). The viability of MCF-7 cells decreased with the increase in the concentration of DOX-loaded nanoparticles. And with each concentration, the CRGDYC modified nanoparticle drug delivery system had higher inhibition efficiency to tumor cells than the unmodified one.
In the cellular uptake experiment, both confocal laser scanning microscopy and flow cytometry showed that the delivery of drug to the tumor cells was effectively facilitated by the RGD peptide modification for nanoparticle carriers, indicating that CRGDYC peptide modified nanoparticles can mediate the endocytosis of nanoparticles loaded with a fluorescence probe by specific binding of the CRGDYC peptide to integrins overexpressed on the surface of tumor cells.
5. Conclusion
In this study, on the basis of 6-O-carboxymethyl chitosan, the specific binding site of the RGD peptide and integrin αvβ3 was successfully found by MOE, and micromolecule targeted cyclopeptide c(CRGDYC) and c(CRGDHC) were successfully synthesized by SPPS. Taking the cell-uptake amount as the index, CRGDYC was chosen for the following experiment. Synthesis of the CRGDYC-6-O-carboxymethyl chitosan was confirmed by IR and 1H NMR analyses. DOX-loaded CRGDYC-6-O-carboxymethyl chitosan nanoparticles with a ∼193 nm mean diameter were prepared, and a high drug loading efficiency of up to 69.5% could be obtained. An in vitro cytotoxicity study showed that the pure carrier system of CRGDTC-6-O-carboxymethyl chitosan nanoparticles and 6-O-carboxymethyl chitosan nanoparticles with low cytotoxicity were safe. However, compared with DOX-loaded 6-O-carboxymethyl chitosan nanoparticles, DOX-loaded CRGDYC-6-O-carboxymethyl chitosan nanoparticles showed stronger destruction to tumor cells. The cellular uptake experiment showed that the delivery of drug to the tumor cells in vitro was effectively facilitated by the RGD peptide modification for nanoparticle carriers. And in vivo imaging also indicated that CRGDYC modified 6-O-carboxymethyl chitosan nanoparticles had strong tumor targets in tumor bearing mice. Taken together, these results suggest that CRGDYC-6-O-carboxymethyl chitosan may be a promising cancer targeting carrier which can enhance the intracellular uptake and cytotoxicity of the drug-loaded nanoparticles.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 81572978 & 81673299 & 21704104), the Natural Science Foundation of Jiangsu Province (no. BK2012355) and the Top-notch Academic Programs Project of Jiangsu Higher Education, the Thousand Talents Plan for Young Professionals, Guangdong Innovative and Entrepreneurial Research Team Program (2013S086), the Guangdong Natural Science Foundation (2014A030312018), and the Science and Technology Planning Project of Guangdong Province (2016A010103015). Meanwhile, we also want to sincerely acknowledge the funding from the Science and Technology Program of Guangzhou (201707010094).
References
- R. Siegel, J. Ma, Z. Zou and A. Jemal, CA-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- M. J. S. Su-Mei Cao and C.-N. Qian, Chin. J. Cancer, 2011, 30, 5 CrossRef.
-
H. M. Sandler and A. J. Mirhadi, in Seminars in oncology, Elsevier, 2012, pp. 583–587 Search PubMed.
- W. Wu, S. Coffelt, C. Cho, X. Wang, C. Lee, F. Chan, J. Yu and J. Sung, Oncogene, 2012, 31, 939–953 CrossRef CAS PubMed.
- Y. Wang, C. Li, C. Yao, L. Ding, Z. Lei and M. Wu, J. Biomed. Nanotechnol., 2016, 12, 1115–1135 CrossRef CAS PubMed.
- X. Liang and A. Aszalos, Curr. Drug Targets, 2006, 7, 911–921 CrossRef CAS PubMed.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- M. Yu, J. Wu, J. Shi and O. C. Farokhzad, J. Controlled Release, 2016, 240, 24–37 CrossRef CAS PubMed.
- F.-Y. Cheng, C.-H. Su, P.-C. Wu and C.-S. Yeh, Chem. Commun., 2010, 46, 3167–3169 RSC.
- S. Parveen and S. K. Sahoo, J. Drug Targeting, 2008, 16, 108–123 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- Q. Li, J. Liang, X. You, X. Liu, H. Wu, W. Gao, Y. Wen, J. Wu, X. Xu and F. Zhang, J. Biomed. Nanotechnol., 2017, 13, 513–521 CrossRef CAS.
- R. van der Meel, M. H. Fens, P. Vader, W. W. van Solinge, O. Eniola-Adefeso and R. M. Schiffelers, J. Controlled Release, 2014, 195, 72–85 CrossRef CAS PubMed.
- J. Wu and C.-C. Chu, J. Mater. Chem. B, 2013, 1, 353–360 RSC.
- M. A. Elgadir, M. S. Uddin, S. Ferdosh, A. Adam, A. J. K. Chowdhury and M. Z. I. Sarker, J. Food Drug Anal., 2015, 23(4), 619–629 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- H. Zhao, Z. Y. Lin, L. Yildirimer, A. Dhinakar, X. Zhao and J. Wu, J. Mater. Chem. B, 2016, 4, 4060–4071 RSC.
- J.-M. Williford, J. L. Santos, R. Shyam and H.-Q. Mao, Biomater. Sci., 2015, 3, 894–907 RSC.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- A. Agarwal, S. Saraf, A. Asthana, U. Gupta, V. Gajbhiye and N. K. Jain, Int. J. Pharm., 2008, 350, 3–13 CrossRef CAS PubMed.
- X.-D. Xu, Y.-J. Cheng, J. Wu, H. Cheng, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, Biomaterials, 2016, 76, 238–249 CrossRef CAS PubMed.
- J. Zong, S. L. Cobb and N. R. Cameron, Biomater. Sci., 2017, 5, 872–886 RSC.
- S. K. Sweeney, Y. Luo, M. A. O'Donnell and J. G. Assouline, J. Biomed. Nanotechnol., 2017, 13, 232–242 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
-
R. Merrifield, Advances in Enzymology and Related Areas of Molecular Biology, 2006, vol. 32, pp. 221–296 Search PubMed.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- CPBOL Group, D. Z. Li and L. M. Gao,
et al.
, Proc. Natl. Acad. Sci., 2011, 108(49), 19641–19646 CrossRef PubMed.
- E. Garanger, D. Boturyn and P. Dumy, Anti-Cancer Agents Med. Chem., 2007, 7, 552–558 CrossRef CAS PubMed.
- Y. Zhong, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2014, 15, 1955–1969 CrossRef CAS PubMed.
- Y. Hu, J. Li, J. Yang, P. Wei, Y. Luo, L. Ding, W. Sun, G. Zhang, X. Shi and M. Shen, Biomater. Sci., 2015, 3, 721–732 RSC.
- H. Kumagai, M. Tajima, Y. Ueno, Y. Giga-Hama and M. Ohba, Biochem. Biophys. Res. Commun., 1991, 177, 74–82 CrossRef CAS PubMed.
- R. Pasqualini, E. Koivunen and E. Ruoslahti, J. Cell Biol., 1995, 130, 1189–1196 CrossRef CAS PubMed.
- L. Upadhyaya, J. Singh, V. Agarwal and R. P. Tewari, J. Controlled Release, 2014, 186, 54–87 CrossRef CAS PubMed.
- X. Fei Liu, Y. Lin Guan, D. Zhi Yang, Z. Li and K. De Yao, J. Appl. Polym. Sci., 2001, 79, 1324–1335 CrossRef.
- Y. Luo, Z. Teng, X. Wang and Q. Wang, Food Hydrocolloids, 2013, 31, 332–339 CrossRef CAS.
- E. Bajelan, A. Haeri, A. M. Vali, S. N. Ostad and S. Dadashzadeh, J. Pharm. Pharm. Sci., 2012, 15, 568–582 CAS.
- H. Wang, F. Li, C. Du, H. Wang, R. I. Mahato and Y. Huang, Mol. Pharm., 2014, 11, 2600–2611 CrossRef CAS PubMed.
- E. Saltiel and W. McGuire, West. J. Med., 1983, 139, 332 CAS.
- J.-P. Xiong, T. Stehle, R. Zhang, A. Joachimiak, M. Frech, S. L. Goodman and M. A. Arnaout, Science, 2002, 296, 151–155 CrossRef CAS PubMed.
- A. Anitha, S. Maya, N. Deepa, K. P. Chennazhi, S. V. Nair, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2011, 83, 452–461 CrossRef CAS.
- A. Anitha, V. V. Divya Rani, R. Krishna, V. Sreeja, N. Selvamurugan, S. V. Nair, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2009, 78, 672–677 CrossRef CAS.
- X. Wang, Z. Teng, H. Wang, C. Wang, Y. Liu, Y. Tang, J. Wu, J. Sun, H. Wang and J. Wang, Int. J. Clin. Exp. Pathol., 2014, 7, 1337 Search PubMed.
- P.-Y. Li, P.-S. Lai, W.-C. Hung and W.-J. Syu, Biomacromolecules, 2010, 11, 2576–2582 CrossRef CAS PubMed.
Footnote |
| † These authors contribute equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yuan
Ren
a,
Yuan
Zhang
*e,
Jun
Wu
a,
Yuan
Ren
a,
Yuan
Zhang
*e,
Jun
Wu