
Tuning protein assembly pathways through superfast amyloid-like aggregation†
Chen
Li
a,
Lu
Xu
b,
Yi Y.
Zuo
b and
Peng
Yang
 *a
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Xi'an 710119, China. E-mail: yangpeng@snnu.edu.cn
bDepartment of Mechanical Engineering, University of Hawaii at Manoa, 2540 Dole St, Holmes Hall 302, Honolulu, HI 96822, USA
First published on 7th February 2018
Abstract
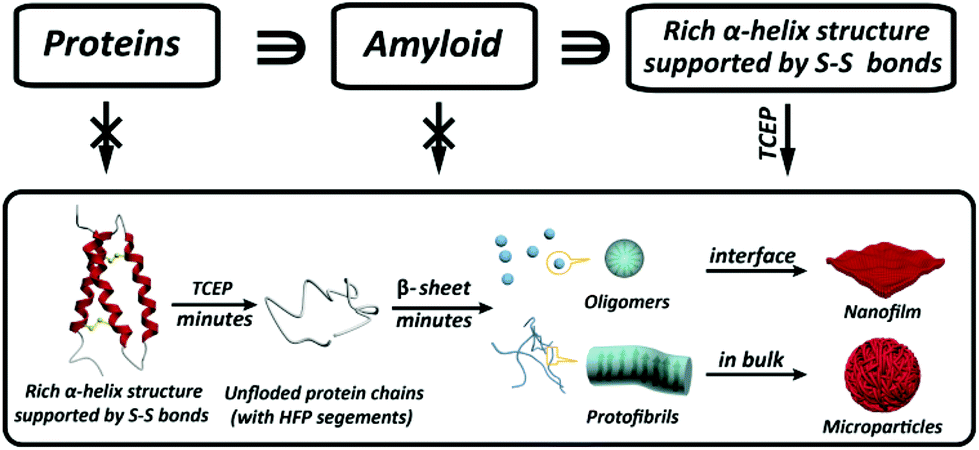
Amyloid formation of proteins is not only relevant for neurodegenerative diseases, but has recently emerged as a groundbreaking approach in materials science and biotechnology. However, amyloid aggregation of proteins in vitro generally requires a long incubation time under extremely harsh conditions, and the understanding of the structural motif to determine amyloid assembly is extremely limited. Herein we reveal that the integration of three important building blocks in typical globular proteins is crucial for superfast protein amyloid-like assembly including the segment required for high fibrillation propensity, abundant α-helix structures and intramolecular S–S bonds to lock the α-helix. With the reduction of the S–S bond by tris(2-carboxyethyl)phosphine (TCEP), the α-helix was rapidly unlocked from the protein chain, and the resultant unfolded monomer underwent a fast transition to β-sheet-rich amyloid oligomers and protofibrils in minutes, which further assembled into a macroscopic nanofilm at the air/water interface and microparticles in bulk solution, respectively.
Introduction
Protein assembly has demonstrated its marvellous value in the fields of supramolecular chemistry, biomimetics, materials, synthetic biology, biomedicine, and chemical biology.1–4 One unique and important protein assembly approach in nature is protein amyloidosis, which is closely related to neurological diseases5 and host defense,3 as well as recent breakthroughs to produce novel nanohybrids.6 However, the detailed amyloid-like assembly pathway and kinetics are poorly understood,7 and an overly slow amyloid assembly process is generally required for proteins, with a frequent need for stringent denaturing conditions to unfold and/or degrade the biopolymer chain. Accordingly, a methodology for superfast amyloid protein assembly, albeit technically challenging, is highly necessary and significantly valuable in various fields.Initially, amyloid-like protein assembly requires noticeable protein unfolding, followed by a structural transition from an unfolded monomeric conformation to an insoluble β-sheet-rich fibrillar structure.8 For the conventional amyloid assembly of stable globular proteins, attainment of the unfolded state requires a transition from the native state across an energy barrier (Ea1) for unfolding to occur (Scheme 1a).9 In such a process, slow nucleation from native proteins, typically including denaturation to form unfolded or even hydrolyzed polypeptide fragments (monomers) and the subsequent transition to β-sheet-rich oligomers and protofibrils (Ea2), plays a key role in the conventional amyloid aggregation,10 which typically relies on harsh unfolding conditions such as an organic solvent, extreme pH, high temperature and ionic strength. Then, the monomers join the nucleus end to grow slowly into a mature fibril, and the entire process is typically described as a sigmoidal curve in which a lag phase of nucleation is followed by a growth phase (Scheme 1b). Although a few oligomers and protofibrils could be observed by the conventional way on a time scale of minutes, effective assembly to generate tailored materials with statistical significance usually requires a time scale of hours and harsh denaturation conditions.11 Using trifluoroethanol as a mild denaturant, Plakoutsi, Chiti and Dobson suggested the formation of amyloid fibrils from some native-like globular proteins with high content of β-sheets in their folded state.12–15 Our present design finds a way to induce mild and fast protein unfolding (E′a1) in aqueous quasi-physiological buffer without the use of an organic solvent (Scheme 1c). Then the resultant unfolded monomers can rapidly transform into amyloid-like oligomers and protofibrils in a few minutes (E′a2), which consequently drives fast assembly under mild conditions to form novel a macroscopic nanofilm at the air/water interface and microparticles in bulk solution (Scheme 1d). This strategy thereby exhibits its capability (being different from existing systems) for the spontaneous and efficient formation of scalable amyloid-based biomaterials (e.g. nanofilms).
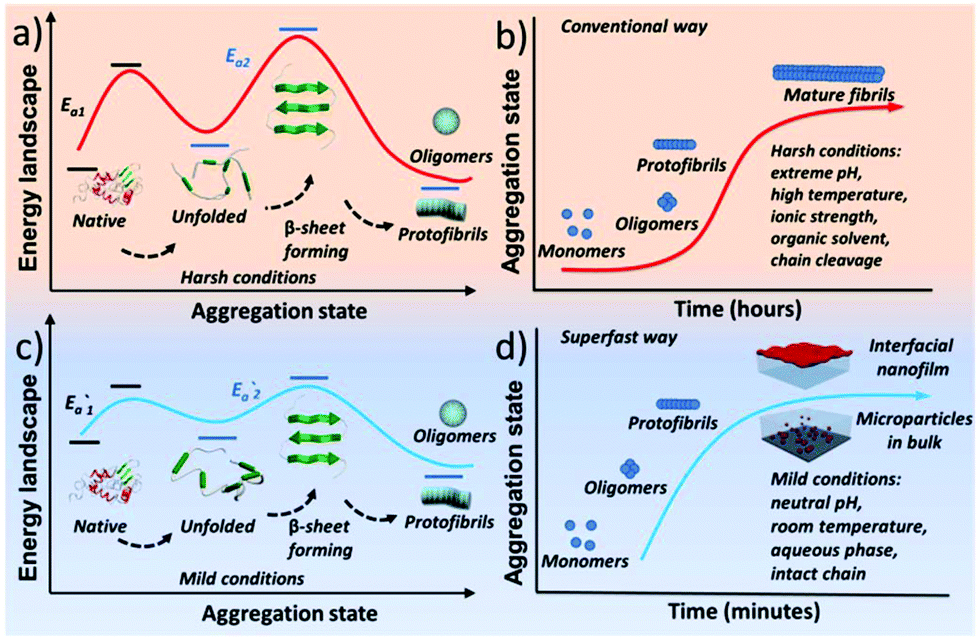 | ||
| Scheme 1 Schematic process for the conventional amyloid aggregation (a, b) and superfast amyloid-like assembly (c, d). | ||
Results and discussion
The unfolding approach in our system starts from the efficient reduction of the intramolecular disulfide bonds of proteins. The disulfide (S–S) bond is an important covalent bond in proteins, and a lack of S–S bonds is generally related to a decrease of protein stability and the consequent unfolding of its secondary structure.16 Nonetheless, the understanding of the S–S bond in the formation of amyloids is still limited.17 Herein, tris(2-carboxyethyl)phosphine (TCEP) was used as a reducing agent due to its very high reaction efficiency towards the S–S bond and stability at different pH values.18,19 In the Raman spectra of lysozyme as a model globular protein, the intramolecular S–S peak at 505 cm−1 significantly decreased over 6 minutes after the addition of TCEP, which reflected the high reaction efficiency towards the reduction of the S–S bond (Fig. 1a).20 The corresponding anilino-1-naphthalene sulfonate (ANS) assay presented a rapid enhancement of fluorescence at 470 nm after the addition of TCEP at different pH values, which suggested that the breakage of S–S bonds stimulated fast protein unfolding and subsequent aggregation of hydrophobic residues (Fig. 1b).21 Hydrophobic aggregation of misfolded species is a crucial and common feature of amyloid formation,21 and the latter was directly monitored by the thioflavin T (ThT) assay as a well-established method to characterize amyloid assembly.22 Different from the traditional sigmoidal curve,23 a rapid increase in ThT fluorescence was observed in our system after 2 min upon the addition of TCEP (Fig. 1c). Both ANS and ThT fluorescence intensified with increasing pH, and a plateau was observed in these curves at ∼10 min. The pH-sensitive behaviours indicated that hydrophobic aggregation was mediated by the net charge of protein colloids, and by increasing the pH to approach the pI of the protein (11 for lysozyme), the resultant amyloid assembly was enhanced through attenuation of electrostatic repulsion among the colloids.24 The transparent solution then became turbid after 4–10 min due to the formation of microparticles in bulk solution (Fig. S1†). This drastic phase separation of amyloid structures from the solution attenuated the ANS and ThT fluorescence to a plateau, followed by a drop in these fluorescence signals (Fig. S2a†). Such a phase separation could be largely attenuated at low pH (4) with a still effective amyloid assembly-induced ThT fluorescence enhancement (Fig. S2b†). In contrast, the conventional amyloid aggregation of lysozyme on a short time scale only exhibited an increase of ANS fluorescence and no increase of ThT fluorescence was observed, which indicated that the protein just aggregated randomly (to enhance the ANS fluorescence) without β-sheet stacking to produce the enhancement of ThT fluorescence (Fig. S3†). The protein assembly in our system was further supported by circular dichroism (CD) (Fig. S4†)25 and infrared (IR) spectra (Fig. 1d and Fig. S5†),26 which showed a successive helicity loss with a gradual increase of β-sheet structure after the addition of TCEP. Then, oligomers with a diameter of 20 nm and protofibrils that were hundreds of nanometers in length were directly visualized using TEM after 1–2 min of superfast amyloid-like aggregation (Fig. 1e and f).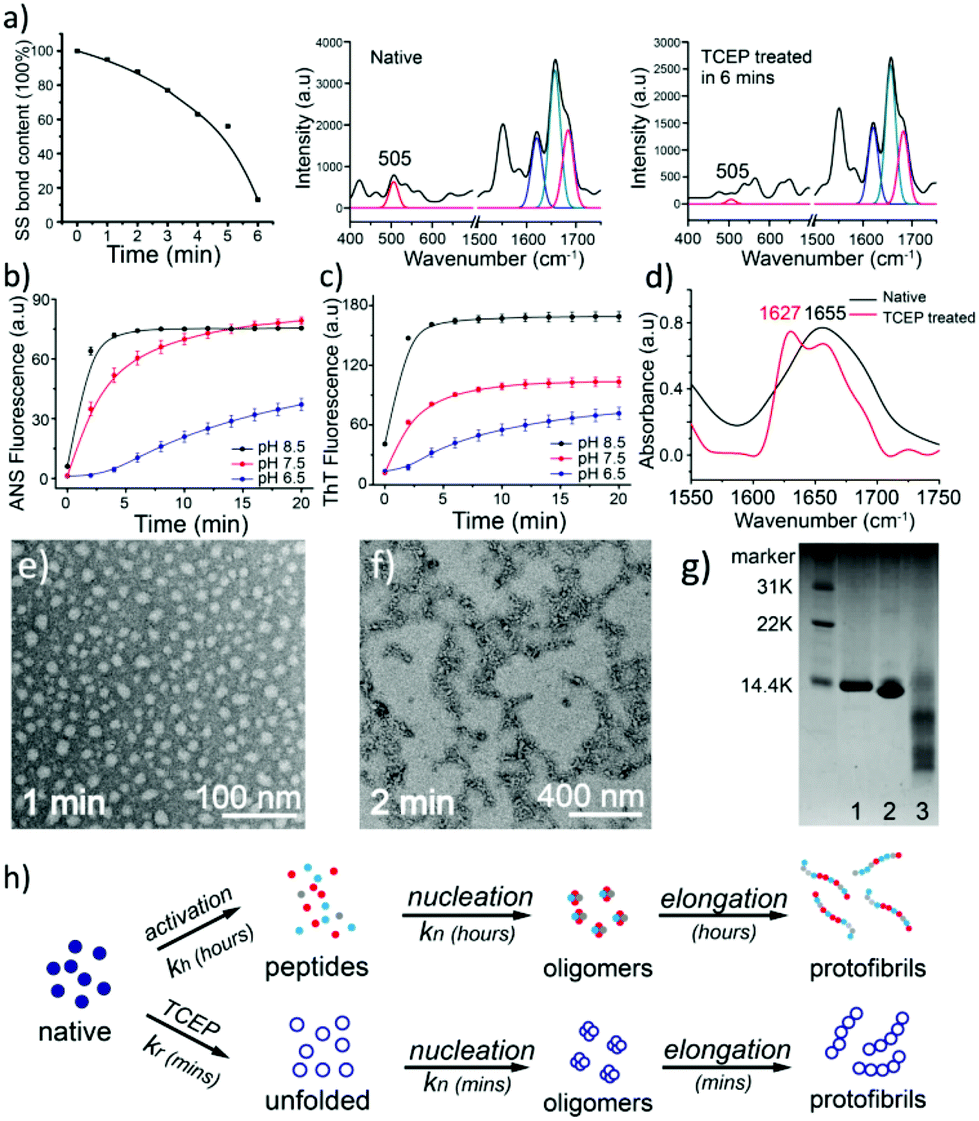 | ||
| Fig. 1 (a) Raman spectra of the reduction of S–S bonds of lysozyme by TCEP; (b, c) ANS (b) and ThT (c) fluorescence of lysozyme during the reduction of S–S bonds by TCEP at different pH values; (d) IR spectra of lysozyme after the reduction of S–S bonds; (e, f) TEM images of oligomers (e) and protofibrils (f) formed after the reduction of S–S bonds by TCEP at 1 and 2 min; (g) SDS-PAGE of native lysozyme (lane 1), TCEP-reduced lysozyme (lane 2), and conventional lysozyme amyloid fibrils (lane 3); (h) schematic for conventional and superfast amyloid aggregation. | ||
In one kind of classical in vitro amyloid aggregation of globular proteins, short polypeptide chains, which are cleaved by hydrolysis (Fig. 1g), are defined as monomers for the nucleation of building blocks.27 The monomer concentration for conventional amyloid aggregation then closely depends on the hydrolysis rate.28 The nucleation kinetics for conventional amyloid aggregation is commonly described as an nc-order reaction with a rate r1 (Fig. 1h and Appendix I†).28–30
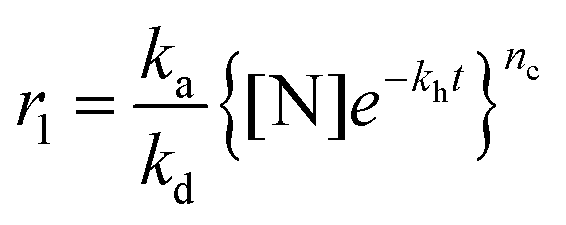
In conventional amyloid aggregation, long and rigid mature fibrils are generated after tens of hours under harsh conditions by oligomers and protofibrils. However, in our superfast protein process, an abundance of monomers was formed in a few minutes to facilitate assembly into substantial oligomers and protofibrils, which can proceed further via unique assembly pathways under mild conditions.32 First, oligomers, as charged colloidal particles that consist of a few protein monomers, tended to assemble at the air/water interface driven by the reduction of interfacial free energy.33 This process led to a 2D amyloid nanofilm at the air/water interface.22 As characterized via Constrained Drop Surfactometry (CDS) (Fig. 2a),34 the surface tension of the lysozyme solution droplet decreased to a minimum (56 mN m−1) before adding TCEP, due to the adsorption of amphipathic proteins at the air/water interface (Fig. 2b). With the addition of TCEP, the surface tension decreased rapidly again within the first 10 min and reached 46 mN m−1 in 60 min (Fig. 2c). Such a decrease of surface tension was ascribed to the enrichment of oligomers at the air/water interface via superfast amyloid-like aggregation.35 This conclusion was supported using an AFM observation of the interfacial aggregate prepared via Langmuir–Blodgett (LB) transfer from the drop surface (Fig. S6†), after carefully exchanging the subphase inside the droplet with HEPES buffer under a constant surface tension of 46 mN m−1 (Fig. 2d). The internal structure of the resultant interfacial aggregate was further revealed by TEM and AFM images of the nanofilm formed at the air/water interface (Fig. 2e–h and Fig. S7†). This evidence demonstrated that oligomers that formed in the solution could preferentially assemble at the air/water interface to form a nanofilm. Alternatively, the oligomers in bulk solution would further propagate very quickly to protofibrils, which subsequently aggregated and fused into microparticles (Fig. S1†).24 In accordance with the pH-dependent effect on the ANS and ThT assays (Fig. 1b and c), the assembly process of the protofibrils also exhibited a pH-dependent morphology evolution from irregular to well-defined microparticles on increasing the pH towards the pI of the lysozyme (Fig. S8a–c†). By contrast, soluble protofibrils without further agglomeration were also observed by TEM at low pH (e.g., 4.5) (Fig. S8d†). According to the AFM image of the protofibrils formed at pH 4.5, the protofibrils were composed of a few oligomers (Fig. S8e†). This result supported the conclusion that the oligomers could assemble into one-dimensional fibrils in bulk solution (being in contrast to two dimensional nanofilms at the air/water interface). Moreover, protofibrils did not assemble into long and rigid mature fibrils with long time incubation (10 days, room temperature). We believe that unfolded lysozyme as the monomer for amyloid-like assembly was rapidly consumed for assembly into oligomers and protofibrils in a short time. Therefore, no monomers further took part in the elongation of amyloid fibrils.
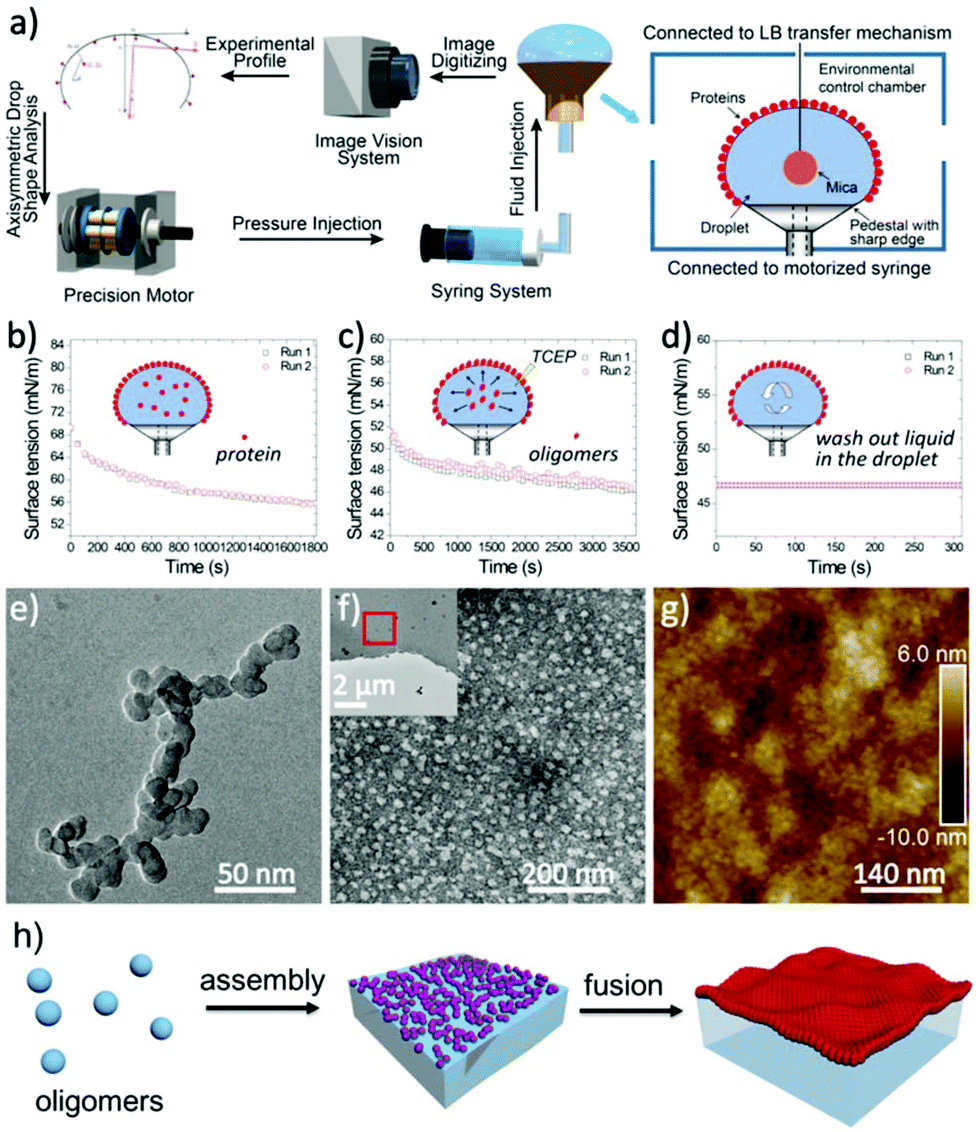 | ||
| Fig. 2 (a) Schematic of the CDS integrated with closed-loop axisymmetric drop shape analysis (ADSA) and in situ Langmuir–Blodgett (LB) transfer; (b) the surface tension decrease with the adsorption of native lysozyme at the air/water interface; (c) the surface tension decrease with the adsorption of the oligomers at the air/water interface; (d) constant surface tension during the exchange of the solution inside the droplet with HEPES buffer; (e–g) TEM images of the oligomers (e), the resultant negatively stained nanofilm (f) and the corresponding AFM image (g); (h) schematic process for the interfacial assembly of the oligomers at the air/water interface. | ||
Our present work proposed a three-in-one principle towards a rational and general design of superfast amyloid-like protein assembly including (1) a high fibrillation propensity (HFP) segment, (2) abundant alpha-helices and (3) the reduction of S–S bonds by TCEP (Fig. 3). First, lysozyme belongs to typical amyloid proteins that are widely used for amyloid studies.36,37 Previous studies have resolved this feature into specific functional segments with a HFP in the primary structure of the protein, allowing self-complementary β-sheets to form the spine of an amyloid structure.38,39 Second, the secondary structure of lysozyme is mainly composed of α-helices (54%). The α-helix in globular proteins generally represents one type of amphipathic structure, in which non-polar residues are mainly on one side of the α-helix, with polar and charged residues on the other side.40 Accordingly, plentiful α-helix structures in globular proteins provide many hydrophobic cores, which are exposed to the polar solvent and enhance hydrophobic aggregation of proteins during misfolding. In fact, this helical intermediate is an important structural block that promotes β-sheets stacking,41,42 and the resultant α to β transition has been associated with amyloid fibrillization.42,43 Third, the intramolecular S–S bond is a key element that initiates superfast amyloid-like aggregation. In previous studies, the effects of S–S bonds on conventional protein amyloid aggregation and helix destabilization were complicated by harsh denaturation conditions e.g. heating and extreme pHs.36 In some cases, harsh conditions unfolded and compacted protein chains, and the S–S bonds even remained intact in the mature fibrils, which revealed that the direct relationship between the S–S bonds and amyloid aggregation required more straightforward study.44 In our system, molecular dynamics simulations for 200 ns proved that a helical domain (residues 18–24) in a native lysozyme underwent a significant conformational change from α-helix to random coil and β-sheet after cleavage of S–S bonds, which indicated the rapid α to β transition (Fig. 4). The sulfur atoms of the Cys6–Cys127 S–S bond showed a significant separation, which indicated that the Cys6–Cys127 S–S bond probably played a role in the unfolding process (Fig. S9†). Previously, Wang et al. reported the formation of native-like lysozyme fibrils via reduction of a Cys64–Cys80 S–S bond under UV illumination.45 Therefore, it is interesting to experimentally study the location role of S–S bond on our superfast amyloid-like aggregation, and more global effects are needed to be considered in this context.
 | ||
| Fig. 3 Schematic to show the three key elements in a protein structure leading to superfast amyloid-like assembly. | ||
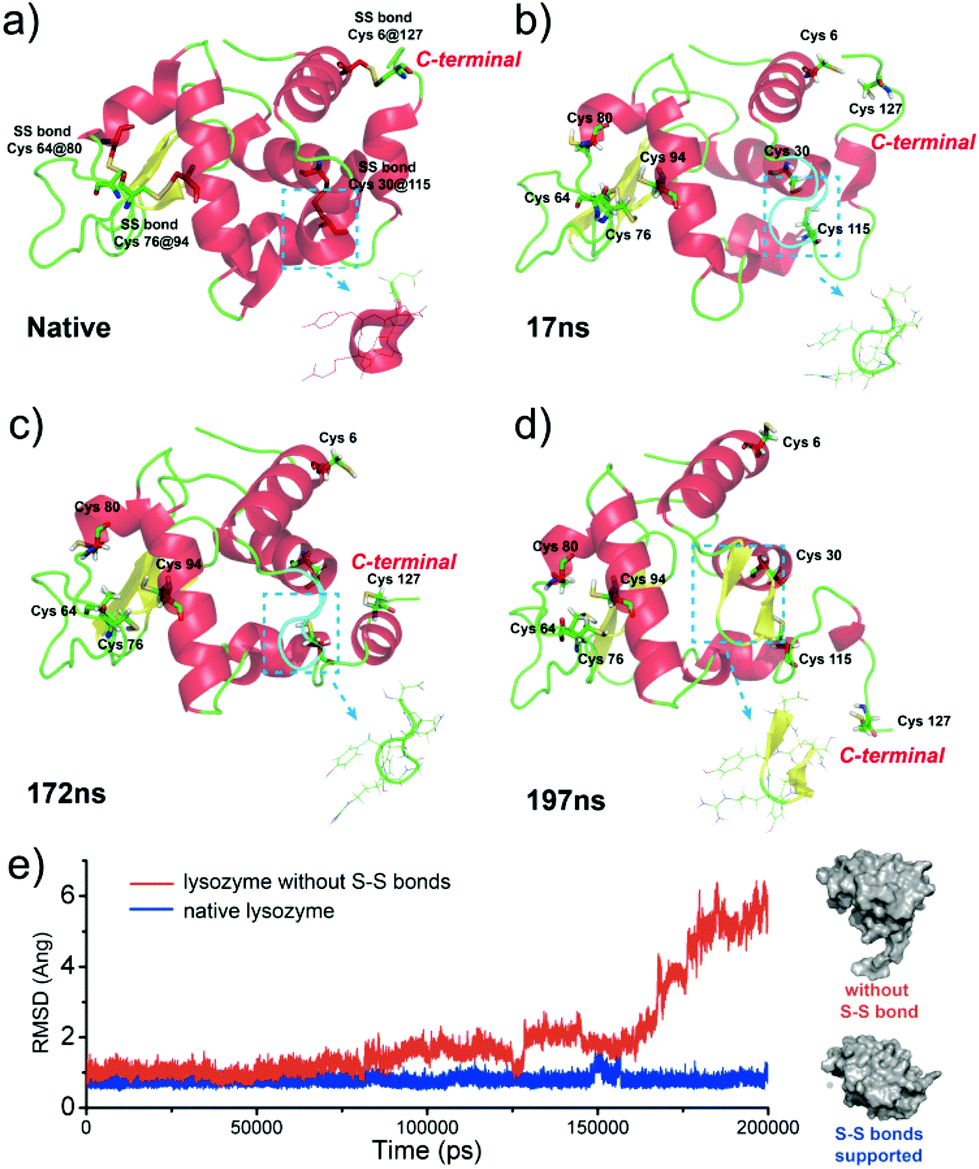 | ||
| Fig. 4 (a)–(d) The conformation change of lysozyme triggered by the breakage of S–S bonds; (e) the root mean square deviation (RMSD) of native lysozyme and lysozyme without S–S bonds from the initial structure as a function of time for the simulations. | ||
The proposed three factors were further supported by designing positive and negative controls (Table 1 and Table S1†). In commercially available samples, we just found that insulin, bovine serum albumin and α-lactalbumin could serve as positive controls, as they share all three factors with lysozyme. In the future, we would make use of gene engineering to deliberately express/design more positive control proteins. Similar to lysozyme, all of positive controls exhibited superfast amyloid-like assembly with the treatment of TCEP at room temperature, as supported by the ANS, ThT, CD and IR characterizations (Fig. S10–13†). The cleavage of protein chains was not observed in these processes except for insulin, because the A-chain and B-chain of insulin are linked by two interchain S–S bonds, resulting in molecular cleavage during the TCEP treatment (Fig. S14†). These processes at different pHs generated macroscopic nanofilms at air/water interface and microparticles in bulk solution with a morphology similar to those from lysozyme (Fig. S15–20†). By contrast, a series of proteins with partial three factors or without all three factors was selected as negative controls (e.g. β-lactoglobulin, ribonuclease A, pepsin, horseradish peroxidase, myoglobin), which showed no interfacial nanofilm or microparticles after the treatment by TCEP, and the clear solution did not show any obvious amyloid transition, as reflected by ANS, ThT fluorescence assay (Fig. S21†) and AFM characterization (Fig. S22†).
| Proteins | Fibrillation propensity? | α-Helix rich? | Intramolecular S–S bonds? | |
|---|---|---|---|---|
| Positive | Insulin | High | Yes | 3 |
| α-La | High | Yes | 4 | |
| BSA | High | Yes | 17 | |
| Negative | β-Lg | High | No | 2 |
| RNAse A | High | No | 4 | |
| Myoglobin | High | Yes | 0 | |
| Cyt c | High | Yes | 0 | |
| α-Amylase | Low | Yes | 4 | |
| HRP | Low | Yes | 4 | |
| Pepsin | Low | No | 3 |
Conclusion
In conclusion, cumulative evidences herein demonstrate that a class of proteins with at least three building blocks can induce superfast preparation of amyloid-based biomaterials. We propose that globular proteins with a high fibrillation propensity (HFP) and abundant α-helix structures locked by intramolecular S–S bonds can undergo rapid amyloid-like assembly after unlocking the S–S bonds by a reducing agent TCEP. The oligomers and protofibrils are generated in a few minutes after triggering fast amyloid-like aggregation, which further proceeds to produce macroscopic nanofilms at the air/water interface and microparticles in bulk solution. This controlled, unique pathway may offer a rational design towards protein assembly and functional materials. Future work is expected to focus on a remarkably diverse set of roles to capitalize on the unique properties and rich chemistry of proteins.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Y. thanks the National Natural Science Foundation of China (no. 51673112) and the 111 Project (no. B14041) for funding. C. L. thanks the Fundamental Research Funds for the Central Universities (2016CBZ005).Notes and references
- B. Pieters, M. B. van Eldijk, R. J. M. Nolte and J. Mecinović, Chem. Soc. Rev., 2016, 45, 24 RSC.
- Y. Bai, Q. Luo and J. Liu, Chem. Soc. Rev., 2016, 45, 2756 RSC.
- D. K. V. Kumar, S. H. Choi, K. J. Washicosky, W. A. Eimer, S. Tucker, J. Ghofrani, A. Lefkowitz, G. McColl, L. E. Goldstein, R. E. Tanzi and R. D. Moir, Sci. Transl. Med., 2016, 8, 340ra72 CrossRef PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188 CrossRef CAS PubMed.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661 RSC.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185 CrossRef CAS PubMed.
- B. H. Toyama and J. S. Weissman, Annu. Rev. Biochem., 2011, 80, 557 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Nat. Chem. Biol., 2009, 5, 15 CrossRef CAS PubMed.
- P. Arosio, T. P. J. Knowles and S. Linse, Phys. Chem. Chem. Phys., 2015, 17, 7606 RSC.
- O. G. Jones and R. Mezzenga, Soft Matter, 2012, 8, 876 RSC.
- G. Plakoutsi, F. Bemporad, M. Calamai, N. Taddei, C. M. Dobson and F. Chiti, J. Mol. Biol., 2005, 351, 910 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 35 CrossRef PubMed.
- G. Soldi, F. Bemporad, S. Torrassa, A. Relini, M. Ramazzotti, N. Taddei and F. Chiti, Biophys. J., 2005, 89, 4234 CrossRef CAS PubMed.
- G. Plakoutsi, F. Bemporad, M. Monti, D. Pagnozzi, P. Pucci and F. Chiti, Structure, 2006, 14, 993 CrossRef CAS PubMed.
- R. Silvers, F. Sziegat, H. Tachibana, S. Segawa, S. Whittaker, U. L. Günther, F. Gabel, J. Huang, M. Blackledge, J. Wirmer-Bartoschek and H. Schwalbe, J. Am. Chem. Soc., 2012, 134, 6846 CrossRef CAS PubMed.
- M. F. Mossuto, B. Bolognesi, B. Guixer, A. Dhulesia, F. Agostini, J. R. Kumita, G. G. Tartaglia, M. Dumoulin, C. M. Dobson and X. Salvatella, Angew. Chem., Int. Ed., 2011, 50, 7048 CrossRef CAS PubMed.
- J. C. Han and G. Y. Han, Anal. Biochem., 1994, 220, 5 CrossRef CAS PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648 CrossRef CAS.
- (a) H. E. Van Wart, A. Lewis, H. A. Scheraga and F. D. Saeva, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2619 CrossRef CAS PubMed; (b) D. Kurouski, R. P. Van Duyne and I. K. Lednev, Analyst, 2015, 140, 4967 RSC.
- B. Bolognesi, J. R. Kumita, T. P. Barros, E. K. Esbjorner, L. M. Luheshi, D. C. Crowther, M. R. Wilson, C. M. Dobson, G. Favrin and J. J. Yerbury, ACS Chem. Biol., 2010, 5, 735 CrossRef CAS PubMed.
- D. Wang, Y. Ha, J. Gu, Q. Li, L. Zhang and P. Yang, Adv. Mater., 2016, 28, 7414 CrossRef CAS PubMed.
- F. S. Ruggeri, G. Longo, S. Faggiano, E. Lipiec, A. Pastor and G. Dietler, Angew. Chem., Int. Ed., 2015, 54, 2462 CrossRef CAS PubMed.
- (a) Z. Wu and P. Yang, Adv. Mater. Interfaces, 2015, 2, 1400401 CrossRef; (b) A. Gao, Q. Wu, D. Wang, Y. Ha, Z. Chen and P. Yang, Adv. Mater., 2016, 28, 579 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain and S. Mukhopadhyay, J. Phys. Chem. B, 2011, 115, 4195 CrossRef CAS PubMed.
- F. S. Ruggeri, G. Longo, S. Faggiano, E. Lipiec, A. Pastor and G. Dietler, Nat. Commun., 2015, 6, 7831 CrossRef CAS PubMed.
- R. Mishra, K. Sörgjerd, S. Nyström, A. Nordigården, Y. Yu and P. Hammarström, J. Mol. Biol., 2007, 366, 1029 CrossRef CAS PubMed.
- A. Kroes-Nijboer, P. Venema, J. Bouman and E. van der Linden, Langmuir, 2011, 27, 5753 CrossRef CAS PubMed.
- A. Šarić, T. C. T. Michaels, A. Zaccone, T. P. J. Knowles and D. Frenkel, J. Chem. Phys., 2016, 145, 211926 CrossRef PubMed.
- T. C. T. Michaels, L. X. Liu, G. Meisl and T. P. J. Knowles, J. Phys.: Condens. Matter, 2017, 29, 153002 CrossRef PubMed.
- I. Gitlin, J. D. Carbeck and G. M. Whitesides, Angew. Chem., Int. Ed., 2006, 45, 3022 CrossRef CAS PubMed.
- K. Yuyama, M. Ueda, S. Nagao, S. Hirota, T. Sugiyama and H. Masuhara, Angew. Chem., Int. Ed., 2017, 129, 6843 CrossRef.
- E. D. Ruiz, M. Almada, M. G. Burboa, P. Taboada, V. Mosquera, M. A. Valdez and J. Juárez, Colloids Surf., B, 2015, 126, 335 CrossRef CAS PubMed.
- R. P. Valle, T. Wu and Y. Y. Zuo, ACS Nano, 2015, 9, 5413 CrossRef CAS PubMed.
- Y. Song, U. Shimanovich, T. C. T. Michaels, Q. Ma, J. Li, T. P. J. Knowles and H. C. Shum, Nat. Commun., 2016, 7, 12934 CrossRef CAS PubMed.
- (a) A. Cao, D. Hu and L. Lai, Protein Sci., 2004, 13, 319 CrossRef CAS PubMed; (b) S. S. S. Wang, K. N. Liu and Y. C. Lu, Biochem. Biophys. Res. Commun., 2009, 381, 639 CrossRef CAS PubMed.
- M. Mulaj, J. Foley and M. Muschol, J. Am. Chem. Soc., 2014, 136, 8947 CrossRef CAS PubMed.
- (a) L. Liu, L. Zhang, X. Mao, L. Niu, Y. Yang and C. Wang, Nano Lett., 2009, 9, 4066 CrossRef CAS PubMed; (b) L. Liu, L. Zhang, L. Niu, M. Xu, X. Mao, Y. Yang and C. Wang, ACS Nano, 2011, 5, 6001 CrossRef CAS PubMed; (c) L. Liu, L. Niu, M. Xu, Q. Han, H. Duan, M. Dong, F. Besenbacher, C. Wang and Y. Yang, ACS Nano, 2014, 8, 9503 CrossRef CAS PubMed.
- L. Goldschmidt, P. K. Teng, R. Riek and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3487 CrossRef CAS PubMed.
- T. C. Terwilliger, Nature, 1982, 299, 371 CrossRef.
- D. M. Ridgley, E. C. Claunch, P. W. Lee and J. R. Barone, Biomacromolecules, 2014, 15, 1240 CrossRef CAS PubMed.
- B. Kim, T. D. Do, E. Y. Hayden, D. B. Teplow, M. T. Bower and J. Shea, J. Phys. Chem. B, 2016, 120, 5874 CrossRef CAS PubMed.
- V. L. Anderson, T. F. Ramlall, C. C. Rospigliosi, W. W. Webb and D. Eliezer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18850 CrossRef CAS PubMed.
- D. Kurouski, J. Washington, M. Ozbi, R. Prabhakar, A. Shekhtman and I. K. Lednev, PLoS One, 2012, 7, e36989 CAS.
- J. B. Xie, Y. Cao, H. Pan, M. Qin, Z. Q. Yan, X. Xiong and W. Wang, Proteins: Struct., Funct., Bioinf., 2012, 80, 2501 CrossRef CAS PubMed.
- T. P. J. Knowles and R. Mezzenga, Adv. Mater., 2016, 28, 6546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; Fig. S1–S22, Table S1 and Appendix I. See DOI: 10.1039/c8bm00066b |
| This journal is © The Royal Society of Chemistry 2018 |