
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly and activation of supported cobalt nanocrystal catalysts for the Fischer–Tropsch synthesis†
T. W.
van Deelen
 a,
H.
Su
bc,
N. A. J. M.
Sommerdijk
bc and
K. P.
de Jong
*a
a,
H.
Su
bc,
N. A. J. M.
Sommerdijk
bc and
K. P.
de Jong
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: k.p.dejong@uu.nl
bLaboratory of Materials and Interface Chemistry & Center for Multiscale Electron Microscopy, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO box 513, 5600 MB, Eindhoven, The Netherlands
cInstitute of Complex Molecular Systems, Eindhoven University of Technology, PO box 513, 5600 MB, Eindhoven, The Netherlands
First published on 20th February 2018
Abstract
The effect of oxidative treatments on the depostion of cobalt nanocrystals (Co–NC) onto a support and subsequent ligand removal was investigated. Deposition of ε-cobalt NC led to extensive clustering of NC and low Fischer–Tropsch synthesis activity. Low-temperature oxidation of ε-cobalt NC resulted in a very uniform CoO–NC distribution and high activity whereas high-temperature oxidation to Co3O4 led to less uniform NC distributions and lower activity.
Nanosized metal particles dispersed on a support material are the workhorses of industrial catalysis. Hence, their synthesis has been studied extensively to optimize catalytic performance.1,2 The two most used techniques to synthesize supported metal catalysts are impregnation and precipitation.3 However, these only offer limited control over e.g. size and distribution of the metal particles. To enable rational catalyst design, new synthesis methods are developed such as melt-infiltration, atomic layer deposition and colloidal synthesis.3,4 Among these, especially colloidal synthesis of metal nanocrystals (M–NC) is promising to synthesize well-defined catalysts, since it provides control over the size, shape, and composition of M–NC. This makes M–NC-based catalysts very suitable to investigate structure–performance relationships.5,6
Two challenges arise in the catalyst assembly procedure when using colloids. Firstly, the M–NC have to be uniformly deposited onto a support. This is often achieved by mixing a M–NC suspension with the support followed by separation and washing. Such methods lead to uniform M–NC distributions only if the particle–support and particle–particle interactions are optimal.7–9 Secondly, the surfaces of M–NC are usually covered with stabilizing ligands, which limit accessibility of the metal surface for reactants and thereby hamper catalytic activity.10,11 Ligands are commonly removed through oxidative treatments at high temperatures, but such severe conditions can affect for example the M–NC size and shape.12
These challenges have been investigated and largely resolved for noble-metal NC.13 This even led to the introduction of the first commercial NC-based catalysts containing Pd– or Pt–NC.14 On the other hand, base-metal NC have been studied less for catalysis and are more challenging than noble-metal NC, because base metal NC are more susceptible to oxidation and magnetic interactions.15 Furthermore, common ligands, such as oleic acid, are hard Lewis bases and bind stronger to base metals, which are hard Lewis acids as compared to noble metals.16
As a showcase of base metal NC, this study focusses on cobalt catalysts for the Fischer–Tropsch (FT) synthesis. In FT, fuels and chemicals are produced from synthesis gas, a H2/CO mixture that can be derived from natural gas, coal or biomass.17 The understanding of FT could benefit from colloidal techniques to provide more precise structure–performance relationships that are not readily obtained otherwise.18,19 Supported cobalt catalysts for FT have been prepared using colloidal techniques.8,20–28 Most were activated through high-temperature (HT) oxidation followed by reduction to arrive at high intrinsic activities.8,20–25 When HT-oxidative treatments were omitted, significantly lower intrinsic activities were obtained.26–28 The development of a milder activation procedure would reduce the impact on the Co–NC and thereby increase the potential of Co–NC for future structure–performance studies.5,29 Here, we investigated the effect of oxidative treatments on the deposition and activation of Co–NC on carbon nanotubes (CNT) as FT catalysts (Fig. 1).
 | ||
| Fig. 1 Schematic overview of the oxidative treatments applied to the NC. The treatments include no oxidation of Co–NC, low-temperature oxidation of Co–NC to CoO–NC and high-temperature oxidation of CoO–NC/CNT to Co3O4–NC/CNT. | ||
Co–NC were synthesized using colloidal techniques and were imaged after synthesis using cryogenic transmission electron microscopy (cryo-TEM). The Co–NC had neither been washed nor exposed to air. The as-synthesized Co–NC were cubical and formed chains, indicating magnetic interactions between metallic Co–NC (Fig. 2A). Electron diffraction experiments using cryo-TEM showed that the crystal structure of the freshly synthesized NC was ε-cobalt (Fig. 2B and Table S1, ESI†), which is a crystal structure of Co that is accessible via wet-chemical synthesis of Co–NC.30
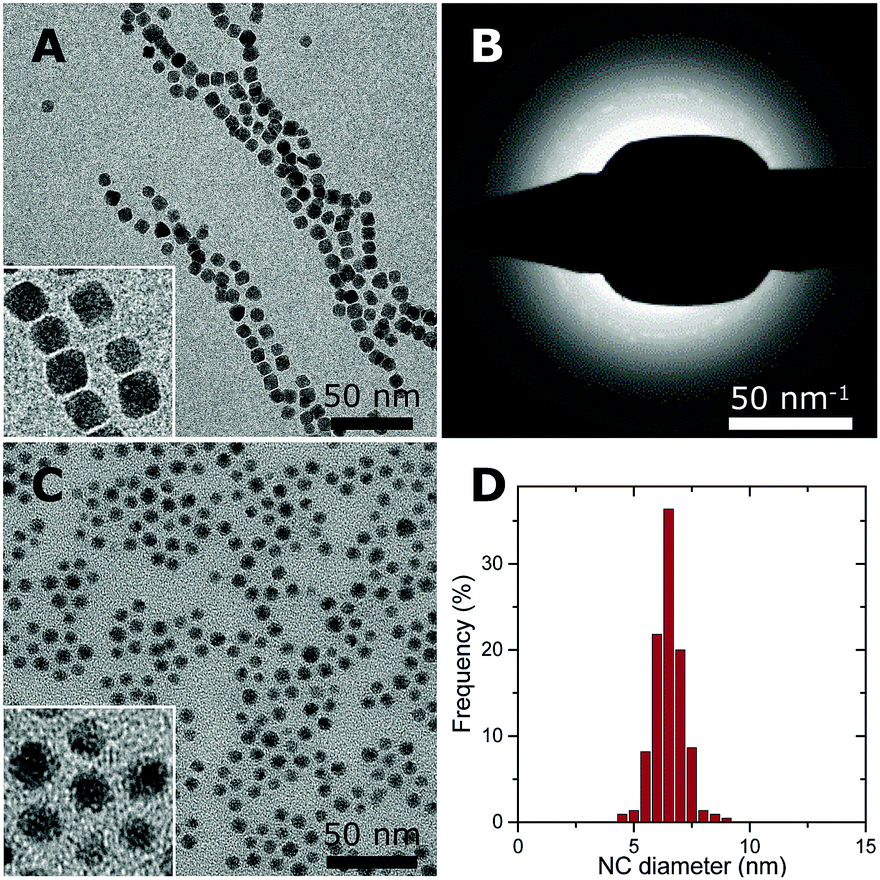 | ||
| Fig. 2 Overview of the Co- and CoO–NC. (A) Cryo-TEM of as-synthesized Co–NC before exposure to air with a higher magnification inset and (B) the corresponding electron diffraction pattern. (C) TEM of CoO–NC with a higher magnification inset and (D) size distribution of CoO–NC with a surface-averaged particle diameter of 6.8 nm. | ||
Low-temperature (LT) oxidation of as-synthesized Co–NC was pursued by air-exposure at ambient conditions during subsequent washing steps. This reproducibly resulted in particles of 6–7 nm with a narrow particle size distribution (Fig. 2C and D) according to transmission electron microscopy (TEM). In a typical NC synthesis, a cobalt yield of ∼50% was obtained. The final NC after the washing steps consisted of CoO, as inferred from electron diffraction (Fig. S1 and Table S1, ESI†), and had become irregularly spherical, owing to their polycrystalline nature. No remaining metallic cores were detected for the CoO–NC using either TEM or electron diffraction. Consequently, the magnetic interactions were reduced and the formation of chains on the TEM grid was no longer observed (Fig. 2C).
Both Co- and CoO–NC were deposited on untreated, hydrophobic CNT through mixing at elevated temperature. This method resulted in nearly quantitative deposition of the NC on the CNT with some variation of the final metal loading due to a slightly variable NC concentration in the suspension. The supported Co–NC and CoO–NC were designated Co1/CNT and Co2/CNT, respectively. In the case of Co1/CNT, oxidation through air-exposure was avoided by omitting the washing steps after the Co–NC synthesis. The Co–NC were exposed to air for the first time during drying after deposition on the CNT.
The average NC sizes and distributions over CNT were investigated using TEM. Co1/CNT was non-uniform with isolated NC as well as 20–60 nm aggregates (Fig. S2, ESI†). The inhomogeneity was believed to result from magnetic interactions between NC consisting of superparamagnetic ε-cobalt,31 which prevented the Co–NC from distributing uniformly over the CNT. Werner et al.8 reported similar non-uniform distributions of metallic Co–NC over silica supports and ascribed this to magnetic interactions of the Co–NC.
For Co2/CNT, the CoO–NC retained their diameter of 6–7 nm during deposition (Fig. 3A). High-resolution transmission electron microscopy (HR-TEM) confirmed that the NC were polycrystalline and attached as rather spherical particles onto the CNT (Fig. 3B). Yang et al.32 obtained comparable polycrystalline CoO–NC through rapid oxidation at 473 K of isolated, monocrystalline NC of hcp-Co. Furthermore, the NC were distributed uniformly over the support, indicating that the magnetic interactions were limited and that diffusion of the NC into the open porous network of the CNT was facile, which is in line with previous observations.7,33 Clearly, oxidation of the Co–NC to CoO under air-exposure proved crucial for their uniform distribution over CNT.
 | ||
| Fig. 3 Overview of NC supported on CNT. (A) TEM and (B) HR-TEM of Co2/CNT, and (C) TEM and (D) HR-TEM of Co3/CNT. | ||
X-ray diffraction (XRD) pointed to ε-Co crystallites for Co1/CNT (Table 1 and Fig. S3, ESI†). The average crystallite size of ∼6 nm was in the range of the NC size prior to deposition. XRD analysis of Co2/CNT indicated 2–3 nm crystalline CoO domains (Table 1 and Fig. S3, ESI†), which was consistent with HR-TEM (Fig. 3B).
To investigate the effect of HT-oxidation, a commonly applied treatment to remove ligands, Co2/CNT was exposed to synthetic air (20 vol% O2) at 523 K. The obtained catalyst was labelled Co3/CNT. XRD measurements after the oxidative treatment showed that CoO was converted into Co3O4 (Table 1 and Fig. S3, ESI†). The treatment did not affect the particle size and distribution as both remained largely unchanged (Fig. 3C and Fig. S4, ESI†). However, at the local scale, HR-TEM showed that the NC now consisted of single crystalline domains although some clustering of NC and embedding into the CNT was apparent, possibly because of cobalt oxide locally catalyzing oxidation of CNT at high temperature (Fig. 3D).
The reduction of the catalysts was investigated using thermogravimetric analysis coupled with mass spectrometry (TGA-MS) in diluted H2. For Co2/CNT, the main weight loss around 923 K was associated with cobalt-catalyzed methanation of CNT (Fig. 4).34 At 623 K, CoO was reduced to Co0 as indicated by weight loss and water evolution. Interestingly, CH4, CO2 and the C4H7+ ion, a fragment ion of oleic acid, were produced as well. These signals were related to desorption and/or decomposition of oleic acid ligands simultaneously with reduction of CoO at 623 K. Quantification of the weight loss confirmed that, besides the loss of oxygen from CoO, also ligands were removed. When assuming full reduction of CoO, ∼35% of the amount of oleic acid added in the NC synthesis was removed during reduction. For Co3/CNT, neither the C4H7+ fragment nor CH4 was observed with TGA-MS and the observed weight loss of 3.8% matched the theoretical weight loss of 3.1% (Fig. S5, ESI†). The ligands had thus already been removed in the HT-oxidative treatment.
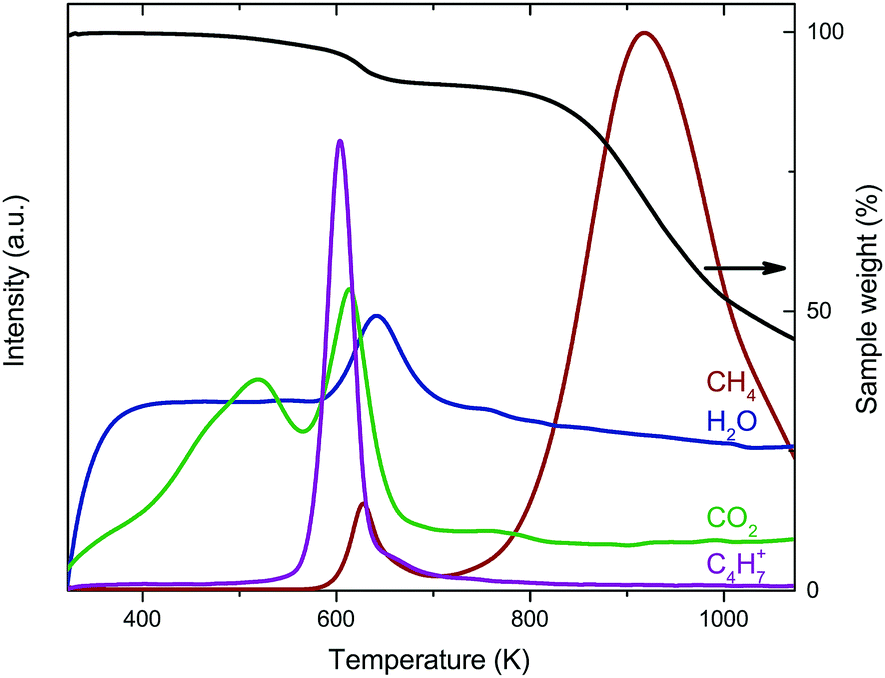 | ||
| Fig. 4 Combined TGA and MS results of Co2/CNT in 25 vol% H2 with 10 K min−1 heating. | ||
Co2/CNT was highly active in FT after in situ reduction (Table 2 and Fig. S6, ESI†). In fact, its cobalt time yield (CTY) was higher than that of a Co/CNT catalyst prepared by Eschemann et al.35via incipient wetness impregnation using a cobalt nitrate precursor solution. The high activity of our Co2/CNT implied that the surface of the cobalt particles was fully accessible to the synthesis gas reactants. Furthermore, during the first few hours of FT, substantial activation of the Co2/CNT could indicate incomplete reduction or residual ligands desorbing from the NC surface due to the formed FT products.
The activity of Co1/CNT was close to zero. This was most likely caused by a large decrease in metallic surface area due to the clustered particles growing together under reaction conditions (Fig. S7, ESI†). Additionally, the lack of washing steps between the synthesis and deposition of the Co–NC may have increased the ligand concentration, which in turn may have led to less binding of the Co–NC to the CNT. Casavola et al.7 described a similar effect of ligands on the binding of Fe–NC.
Co3/CNT was significantly less active than Co2/CNT. The HT-oxidative treatment promoted particle growth during in situ reduction and FT. TEM of the spent catalysts revealed a larger particle size for Co3/CNT than for Co2/CNT (Table 2 and Fig. S7, ESI†). An explanation for the adverse effect of HT-oxidation on the stability of the catalysts could be a different interaction of the NC with the support for Co2/CNT and Co3/CNT, as shown in Fig. 3. Furthermore, some NC clustering was apparent for the fresh Co3/CNT, which probably led to increased particle growth during reduction and catalysis.
The C5+-selectivity of both Co2/CNT and Co3/CNT was comparable, although Co3/CNT was slightly more selective to C5+ products than Co2/CNT (Table 2). For Co1/CNT, no selectivity could be determined owing to its low activity.
The turnover frequencies (TOF) of Co2/CNT and Co3/CNT were similar (Table 2), thereby proving that the higher CTY of Co2/CNT originated from a larger cobalt surface area instead of higher intrinsic activity. Because the surface of Co3/CNT was free of ligands, as shown before, the surface of the Co2/CNT must have been equally stripped to obtain a similar TOF. When comparing the TOF with literature values for LT- or non-oxidized NC catalysts,26,27 it is apparent that we obtained more active catalysts, which are now at the high end of the TOF range of conventional catalysts (Fig. 5). For the HT-oxidized Co3/CNT, full activation and a high TOF was expected, but this is the first time, to the best of our knowledge that only LT-oxidation followed by in situ reduction was sufficient to fully activate a NC-based cobalt catalyst for FT.
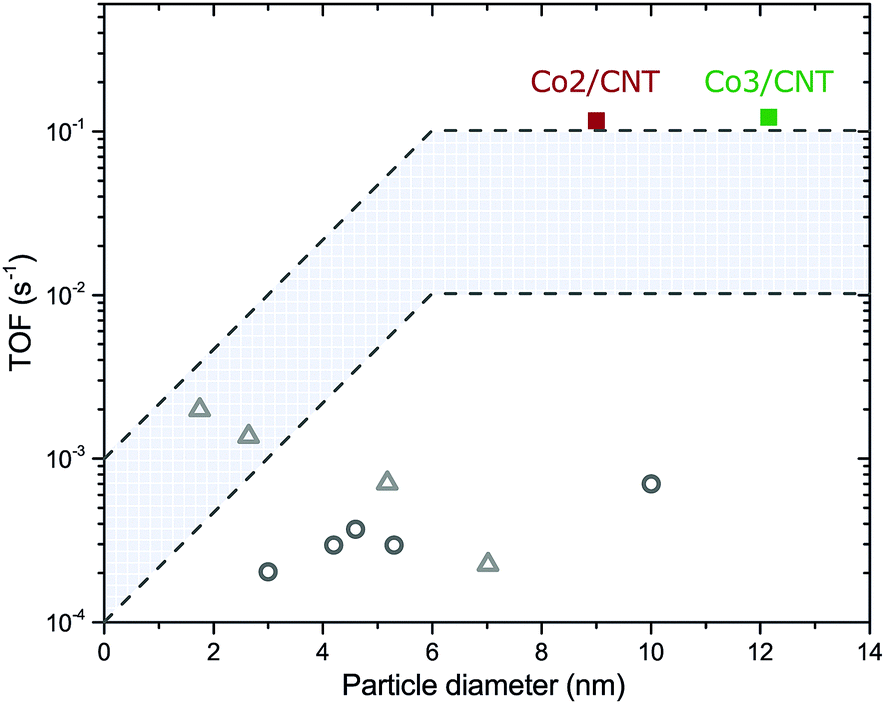 | ||
| Fig. 5 Turnover frequencies of Co2/CNT and Co3/CNT as a function of the average cobalt particle diameter. Recalculated TOF values of NC-based catalysts for LT-oxidized Co/SiO226 (grey open circles) and non-oxidized Co/TiO227 (grey open triangles) are given as comparison. Details of the recalculation can be found in the ESI.† The highlighted area is the typical TOF range of conventional catalysts. | ||
In this work, we investigated how oxidative treatments of cobalt nanocrystals (NC) affect their deposition on carbon nanotubes and activation for the Fischer–Tropsch (FT) synthesis. Deposition of ε-cobalt NC resulted in clustering of the NC (probably due to magnetic interactions between the NC) and low activity. After low-temperature oxidation of the NC to CoO, the NC were deposited uniformly. The ligands were readily removed during in situ reduction and high activity was obtained. In fact, it was possible to fully activate a NC-based cobalt catalyst for FT using this mild procedure. High-temperature (HT) oxidation to Co3O4 led to more pronounced particle growth during in situ reduction or FT and therefore the catalytic activity was lower. An HT-oxidative treatment was thus unnecessary and even counter-productive. These findings will facilitate the application of colloid-based catalysts to investigate structure–performance relationships in catalysis.
The authors acknowledge Shell Global Solutions and the Netherlands Association for Scientific Research (NWO) for funding through the CHIPP program. K. P. d. J. acknowledges the European Research Council, EU FP7 ERC Advanced Grant no. 338846. We thank Peter Munnik for helpful discussions and Paul Bomans (cryo-TEM), Lars van der Wal (HR-TEM), Marjan Versluijs-Helder (TGA-MS) and Helen de Waard (ICP-OES) for the measurements indicated.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Zečević, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245 CrossRef PubMed.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi and Y. Dai, et al. , Nature, 2016, 538, 84 CrossRef CAS PubMed.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746 RSC.
- C.-J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457 RSC.
- A. Gual, C. Godard, S. Castillón, D. Curulla-Ferré and C. Claver, Catal. Today, 2012, 183, 154 CrossRef CAS.
- M. Casavola, J. Hermannsdörfer, N. de Jonge, A. I. Dugulan and K. P. de Jong, Adv. Funct. Mater., 2015, 25, 5309 CrossRef CAS.
- S. Werner, G. R. Johnson and A. T. Bell, ChemCatChem, 2014, 6, 2881 CrossRef CAS.
- N. Hondow and R. O. Fuller, J. Colloid Interface Sci., 2014, 417, 396 CrossRef CAS PubMed.
- J. Y. Park, C. Aliaga, J. R. Renzas, H. Lee and G. A. Somorjai, Catal. Lett., 2009, 129, 1 CrossRef CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358 CrossRef CAS.
- I. Lee, R. Morales, M. A. Albiter and F. Zaera, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15241 CrossRef CAS PubMed.
- V. Iablokov, Y. Xiang, A. Meffre, P.-F. Fazzini, B. Chaudret and N. Kruse, ACS Catal., 2016, 6, 2496 CrossRef CAS.
- P. T. Witte, P. H. Berben, S. Boland, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505 CrossRef CAS.
- E. Zacharaki, P. Beato, R. R. Tiruvalam, K. J. Andersson, H. Fjellvåg and A. O. Sjåstad, Langmuir, 2017, 33, 9836 CrossRef CAS PubMed.
- C. de Mello Donegá, Chem. Soc. Rev., 2011, 40, 1512 RSC.
- J. P. den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Frøseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197 CrossRef CAS PubMed.
- S. K. Beaumont, Phys. Chem. Chem. Phys., 2014, 16, 5034 RSC.
- S. Liu, Y. Li and W. Shen, Chin. J. Catal., 2015, 36, 1409 CrossRef CAS.
- G. Prieto, A. Martínez, P. Concepción and R. Moreno-Tost, J. Catal., 2009, 266, 129 CrossRef CAS.
- N. Fischer, E. van Steen and M. Claeys, J. Catal., 2013, 299, 67 CrossRef CAS.
- S. Lögdberg, M. Boutonnet, J. C. Walmsley, S. Järås, A. Holmen and E. A. Blekkan, Appl. Catal., A, 2011, 393, 109 CrossRef.
- J.-Y. Park, Y.-J. Lee, P. R. Karandikar, K.-W. Jun, K.-S. Ha and H.-G. Park, Appl. Catal., A, 2012, 411–412, 15 CrossRef CAS.
- Y.-J. Lee, J.-Y. Park, K.-W. Jun, J. Wook Bae and P. S. S. Prasad, Catal. Lett., 2009, 130, 198 CrossRef CAS.
- W. T. Ralston, G. Melaet, T. Saephan and G. A. Somorjai, Angew. Chem., Int. Ed., 2017, 56, 7415 CrossRef CAS PubMed.
- T. Herranz, X. Deng, A. Cabot, J. Guo and M. Salmeron, J. Phys. Chem. B, 2009, 113, 10721 CrossRef CAS PubMed.
- J. A. Delgado, C. Claver, S. Castillón, D. Curulla-Ferré, V. V. Ordomsky and C. Godard, Appl. Catal., A, 2016, 513, 39 CrossRef CAS.
- J. A. Delgado, C. Claver, S. Castillón, D. Curulla-Ferré, V. V. Ordomsky and C. Godard, J. Mol. Catal. A: Chem., 2016, 417, 43 CrossRef CAS.
- P. Sonström and M. Bäumer, Phys. Chem. Chem. Phys., 2011, 13, 19270 RSC.
- D. P. Dinega and M. G. Bawendi, Angew. Chem., Int. Ed., 1999, 38, 1788 CrossRef CAS.
- A. F. Gross, M. R. Diehl, K. C. Beverly, E. K. Richman and S. H. Tolbert, J. Phys. Chem. B, 2003, 107, 5475 CrossRef CAS.
- Z. Yang, I. Lisiecki, M. Walls and M.-P. Pileni, ACS Nano, 2013, 7, 1342 CrossRef CAS PubMed.
- B. Seo, Y. J. Sa, J. Woo, K. Kwon, J. Park, T. J. Shin, H. Y. Jeong and S. H. Joo, ACS Catal., 2016, 6, 4347 CrossRef CAS.
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, Catal. Today, 2013, 214, 50 CrossRef CAS.
- T. O. Eschemann, W. S. Lamme, R. L. Manchester, T. E. Parmentier, A. Cognigni, M. Rønning and K. P. de Jong, J. Catal., 2015, 328, 130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization (electron diffraction, TEM, XRD, TGA-MS), activity data and analysis of spent catalyst and turnover frequencies. See DOI: 10.1039/c7cc07741f |
| This journal is © The Royal Society of Chemistry 2018 |