
Palladium-catalyzed oxidative allylation of bis[(pinacolato)boryl]methane: synthesis of homoallylic boronic esters†
Chunsheng
Li
,
Meng
Li
,
Jianxiao
Li
,
Wanqing
Wu
* and
Huanfeng
Jiang
 *
*
Key Laboratory of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: cewuwq@scut.edu.cn; jianghf@scut.edu.cn; Fax: +86 20-87112906; Tel: +86 20-87112906
First published on 22nd November 2017
Abstract
A palladium-catalyzed oxidative allylation of bis[(pinacolato)boryl]methane to afford the corresponding homoallylic organoboronic esters with moderate to excellent yields is reported. This novel transformation provides an efficient strategy for the construction of homoallylic organoboronic esters in one step with a broad substrate scope. It is proposed that the palladium-catalyzed oxidative allylic C–H bond activation process may be involved in the catalytic cycle.
1,1-Bisborylalkanes have emerged as a valuable synthon in contemporary organic synthesis due to their easy access to highly functionalized, complicated organoboron molecules.1–3 Shibata and co-workers reported the pioneering work on the allylation of bis[(pinacolato)boryl]methane with allyl halides under mild conditions, giving highly valuable homoallylic boronic esters.4 Hoveyda et al. reported the Cu-catalyzed enantioselective allylic substitution (EAS) of allylic electrophiles with 1,1-bisborylalkanes.5 Niu's group developed the iridium-catalyzed asymmetric allylation strategy to prepare β-substituted chiral homoallyl boronic esters.6 Despite the significant progress that has been achieved along this line, all of these elegant developments employed the unique and high reactivity of 1,1-bisborylalkanes toward electrophiles. However, the application of 1,1-bisborylalkanes as soft nucleophiles in the allylic C–H activation process has not been investigated, and is still highly desirable.
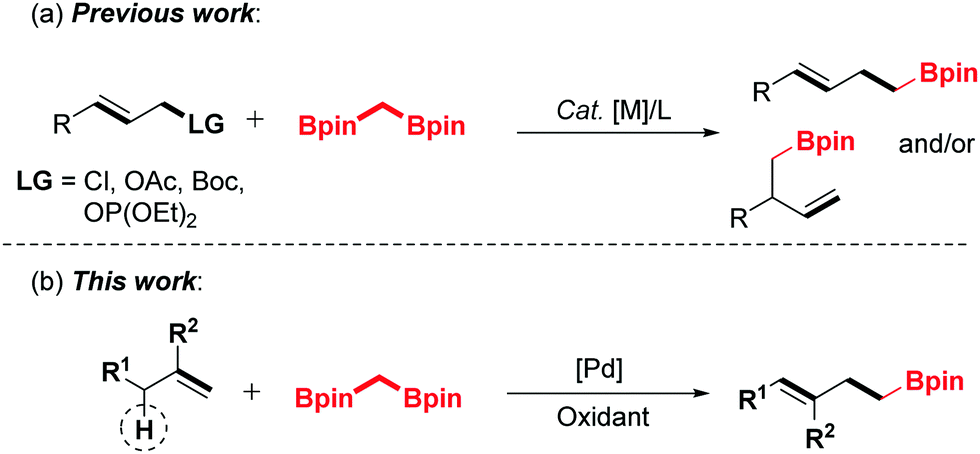
The transition metal-catalyzed oxidative functionalization of allylic C–H bonds has been recognized as a powerful method toward the construction of carbon–carbon or carbon–heteroatom bonds in a concise manner.7,8 Most notably, palladium-catalyzed oxidative allylic C–H bond activation has been defined as an expedient and atom-economic strategy for the synthesis of complex molecules.9 As a class of high-activity electrophiles, the key allylpalladium intermediate could undergo cross-coupling reactions with various nucleophiles.10–13 Recently, our group has realized the palladium-catalyzed direct coupling of terminal alkenes with water,11c amines12h and alcohols.10a,11g On the basis of the previous endeavours, we envision that the allyl-Pd intermediate could be captured by the 1,1-bisborylalkanes to afford the organoboron compounds by introducing an α-boroalkyl group. Herein, we disclose a strategically distinct approach to synthesize homoallylic boronic esters through palladium-catalyzed oxidative allylic C–H functionalization of terminal olefins (Scheme 1).
 | ||
| Scheme 1 Strategies for the synthesis of homoallylic boronic esters. | ||
Inspired by these advances and our previous work,6,11g,12h we investigated the oxidative α-boroalkylation reaction of allylbenzene (1a) and bisborylmethane (2a). At first, the reaction was initially carried out with the combination of Pd(OAc)2, AgOTf and tBuOK in 1,4-dioxane at 50 °C for 24 h under 1 atm of oxygen. Unfortunately, no desired product 3a was detected (Table 1, entry 1). Subsequently, a series of silver salts were screened, such as AgTFA, AgOAc and AgBF4 (Table 1, entries 2–4), among which AgBF4 was the optimal one and afforded the desired product 3a in trace yield (entry 4). Next, various bases were surveyed, and KH2PO4 was found to be the most effective base for this reaction (Table 1, entry 8). Further examination of oxidants revealed that NQ (1,4-naphthoquinone) increased the yield of 3a distinctly (Table 1, entry 15). After screening of different ligands (Table 1, entries 16–20), 1,2-bis(phenylsulfinyl)ethane was identified as the optimal ligand for the present transformation (Table 1, entry 20). Screening of various solvents indicated that 1,4-dioxane gave the best result for this reaction (see the ESI† for details). Thus, the standard conditions were obtained as Pd(OAc)2 (10 mol%), AgBF4 (20 mol%), KH2PO4 (2.0 equiv.), NQ (2 equiv.) and 1,2-bis(phenylsulfinyl)ethane (15 mol%) in 1,4-dioxane (2.0 mL) at 50 °C with stirring for 24 h.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Ligand | Base | Oxidant | Yieldb (%) |
a A mixture of 1a (0.25 mmol, 2.5 equiv.), 2a (0.1 mmol), base (0.2 mmol, 2 equiv.), Pd(OAc)2 (10 mol%), additive (20 mol%), ligand (15 mol%), oxidant (2 equiv.) and anhydrous 1,4-dioxane (2 mL) was sealed in a 25 mL Schlenk tube at 50 °C for 24 h. N.D. = not detected. L1: PPh3. L2: dppf. L3: 4,4′-bipyridine. L4: 1,2-bis(phenylsulphonyl)ethane. L5: 1,2-bis(phenylsulfinyl)ethane.
b Isolated yield based on 2a.
c BQ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DDQ = 2:1.
d BQ:DDQ = 4:1. :DDQ = 2:1.
d BQ:DDQ = 4:1.
|
|||||
| 1 | AgOTf | — | t BuOK | O2 | N.D. |
| 2 | AgTFA | — | t BuOK | O2 | N.D. |
| 3 | AgOAc | — | t BuOK | O2 | N.D. |
| 4 | AgBF4 | — | t BuOK | O2 | Trace |
| 5 | AgBF4 | — | Cs2CO3 | O2 | Trace |
| 6 | AgBF4 | — | CF3COONa | O2 | 12 |
| 7 | AgBF4 | — | KPF6 | O2 | 23 |
| 8 | AgBF4 | — | KH2PO4 | O2 | 30 |
| 9 | AgBF4 | — | KH2PO4 | PhI(OAc)2 | N.D. |
| 10 | AgBF4 | — | KH2PO4 | BQ | Trace |
| 11 | AgBF4 | — | KH2PO4 | DDQ | Trace |
| 12 | AgBF4 | — | KH2PO4 | DMBQ | Trace |
| 13c | AgBF4 | — | KH2PO4 | BQ/DDQ | 35 |
| 14d | AgBF4 | — | KH2PO4 | BQ/DDQ | 55 |
| 15 | AgBF4 | — | KH2PO4 | NQ | 60 |
| 16 | AgBF4 | L1 | KH2PO4 | NQ | N.D. |
| 17 | AgBF4 | L2 | KH2PO4 | NQ | N.D. |
| 18 | AgBF4 | L3 | KH2PO4 | NQ | N.D. |
| 19 | AgBF4 | L4 | KH2PO4 | NQ | 43 |
| 20 | AgBF 4 | L5 | KH 2 PO 4 | NQ | 83 |
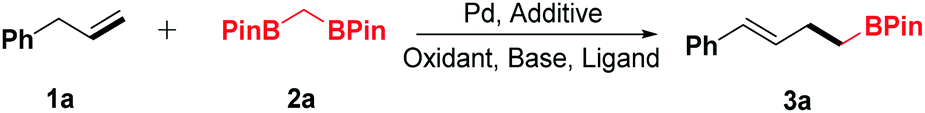
The generality of the new protocol was next tested as the optimized conditions established. Several structurally diverse allylbenzenes were initially explored, and the representative results are summarized in Table 2. Generally, both electron-donating (Me, Et, OMe) and electron-withdrawing groups (F, Cl, CF3) were compatible with this method (3a–3k). In addition, when (2-methylallyl)benzene (1l) was treated with 2a under the optimized conditions, two regioisomeric products 3l and 3l′ were obtained (dr = 1.2:1, determined using the NMR spectrum). Disubstituted allylbenzenes could also undergo this transformation, furnishing the corresponding boronic ester derivatives in good yields (3m–3q). Notably, functionalized allylbenzene derivatives, such as 1-allyl-2,3,4,5,6-pentafluorobenzene (3r), were transformed to the desired products with excellent yields. Only a trace amount of the desired product was detected by GC-MS when but-3-en-1-ylbenzene (1u) was surveyed.
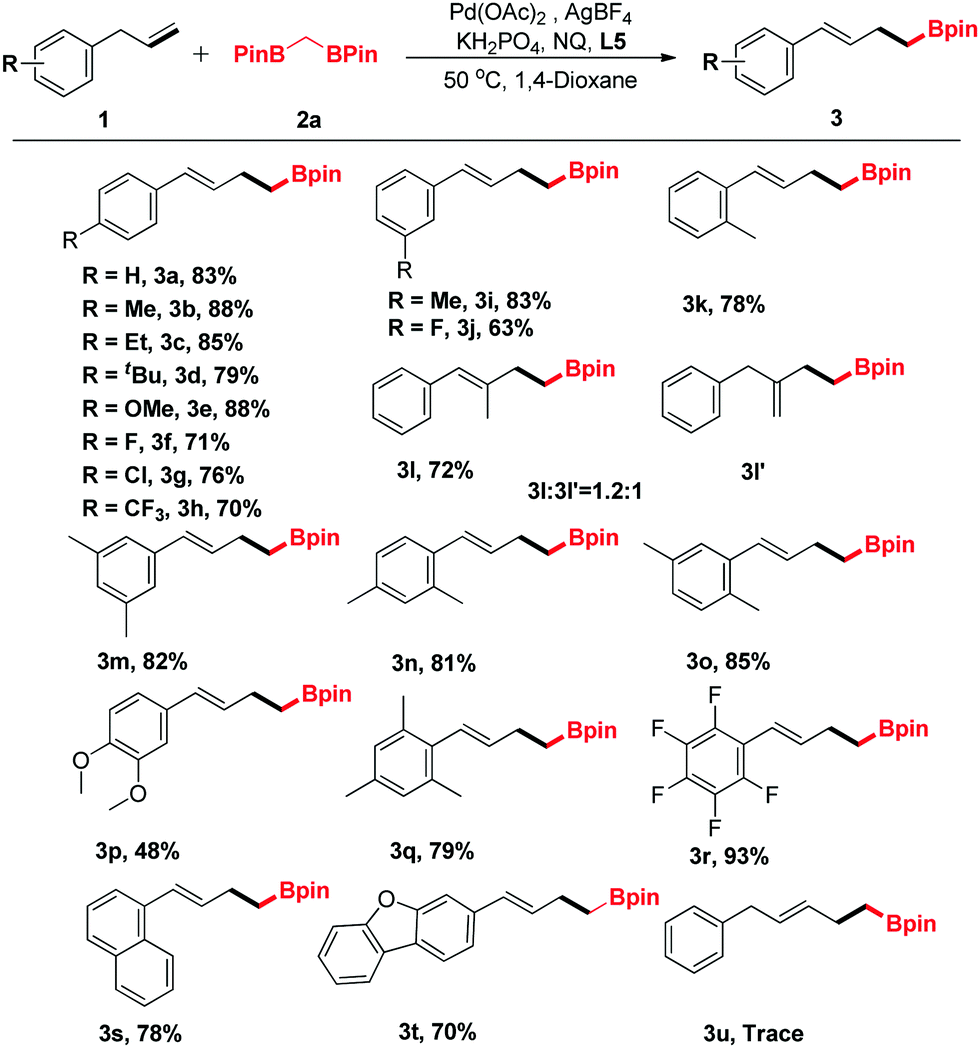
In view of the further applications of this transformation, the substrate scope was expanded to different substituted α-methylstyrenes (Table 3). α-Methylstyrene was first surveyed, which delivered a good yield of product 5a under our optimized conditions. Moreover, α-methylstyrenes bearing different functional groups such as F, Cl and Br at the para- or ortho-positions of the phenyl ring could also react smoothly to give the corresponding products 5c–5e in good yields ranging from 62% to 83%. Pleasingly, the reactions of 1-(prop-1-en-2-yl)naphthalene and 2-(prop-1-en-2-yl)naphthalene proceeded efficiently to give the corresponding products 5g and 5h in 82% and 78% yields, respectively.
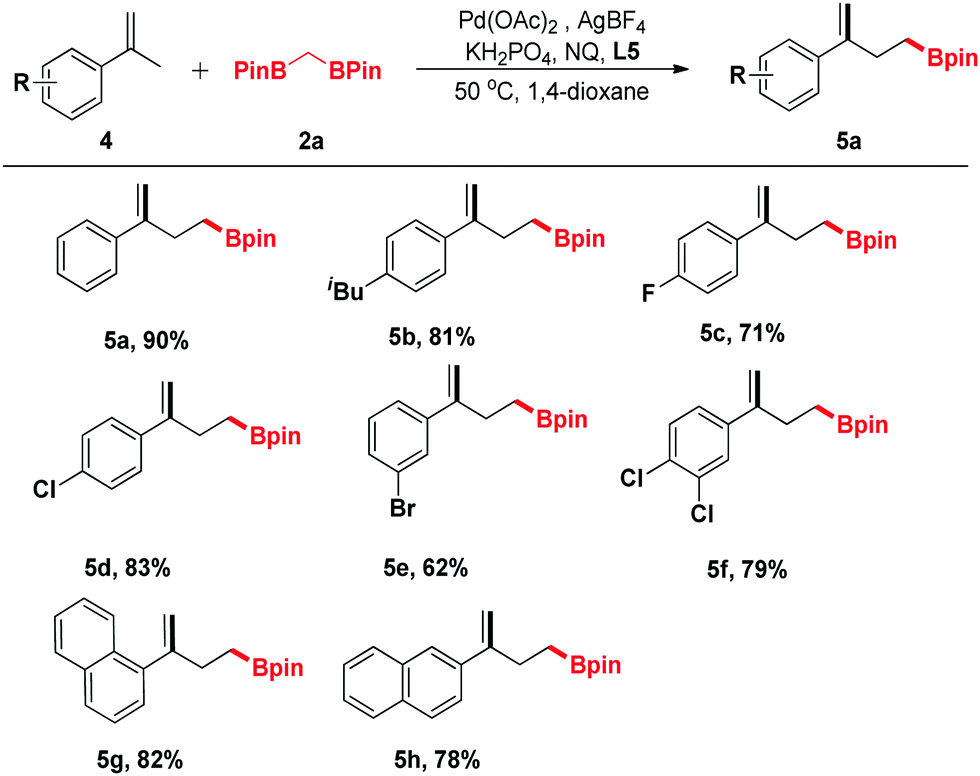
trans-β-Methylstyrene derivatives could be recognized as a precursor of allylic electrophiles. Thus, 6a and 6b were applied in this process and converted into the organoboronic esters in moderate yields (Scheme 2).
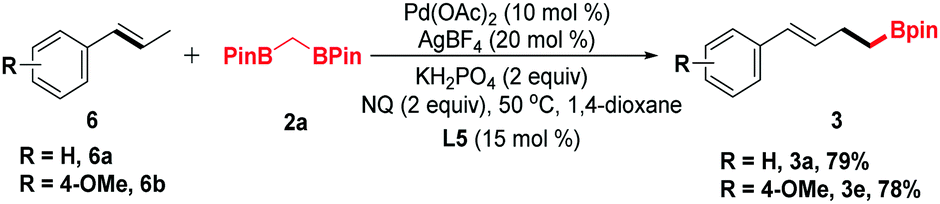 | ||
| Scheme 2 Substrate scope of trans-β-methylstyrenes. Reaction conditions: 6 (0.25 mmol, 2.5 equiv.), 2a (0.1 mmol), Pd(OAc)2 (10 mol%), AgBF4 (20 mol%), L5 (15 mol%), KH2PO4 (0.2 mmol, 2 equiv.), NQ (0.2 mmol, 2 equiv.) and 1,4-dioxane (2 mL) were sealed in a 25 mL Schlenk tube at 50 °C for 24 h. Isolated yields based on 2a. | ||
To demonstrate the synthetic utility of this protocol further, the homoallylic boronic esters were transferred into other desired synthons using the established chemistry (Scheme 3). For example, compound 3a could be oxidized by NaBO3·4H2O to homoallylic alcohol 8, which has been engaged in a wide range of chemical reactions to synthesize structurally complex products. Moreover, 3a could undergo amination to form homoallylic amine 9 in 78% yield.
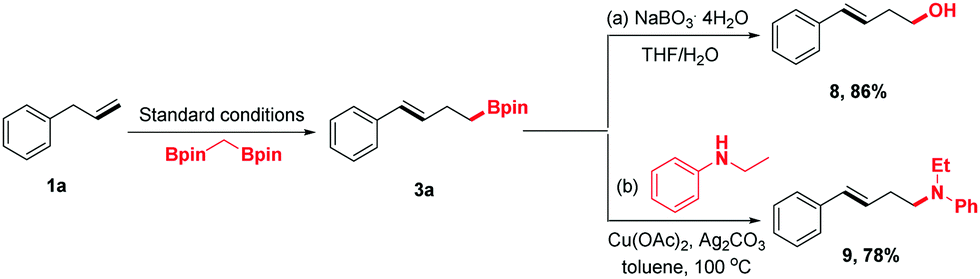 | ||
| Scheme 3 Elaboration of homoallyl boronates. Conditions: (a) 3a (0.2 mmol), NaBO3·4H2O (6.0 equiv.) in THF/H2O (2 mL/2 mL) at room temperature for 12 h. (b) 3a (0.2 mmol), N-ethylaniline (0.1 mL), Cu(OAc)2 (10 mol%), and Ag2CO3 (2.0 equiv.) in toluene at 100 °C with stirring for 20 h. | ||
In light of the previous literature, a plausible mechanism is outlined in Scheme 4. Initially, the catalytic cycle would begin with the coordination of palladium with olefins to generate the intermediate I. Next, the corresponding π-allylpalladium species II is formed by the electrophilic allylic C–H cleavage in the presence of ligand L5.10b,11a,14 With the addition of silver salt, 1,1-bis[(pinacolato)boryl]methane undergoes a deborylative transmetallation process to form an alkyl silver species III.6,15 Then, the π-allylpalladium intermediate II reacts with intermediate III by transmetallation to give a Pd-(α-boroalkyl) complex IV. The product homoallylic organoboronic ester would be obtained through the reductive elimination process. Finally, the reactive Pd(II) species is regenerated by the oxidation of NQ16 (see the ESI† for details).
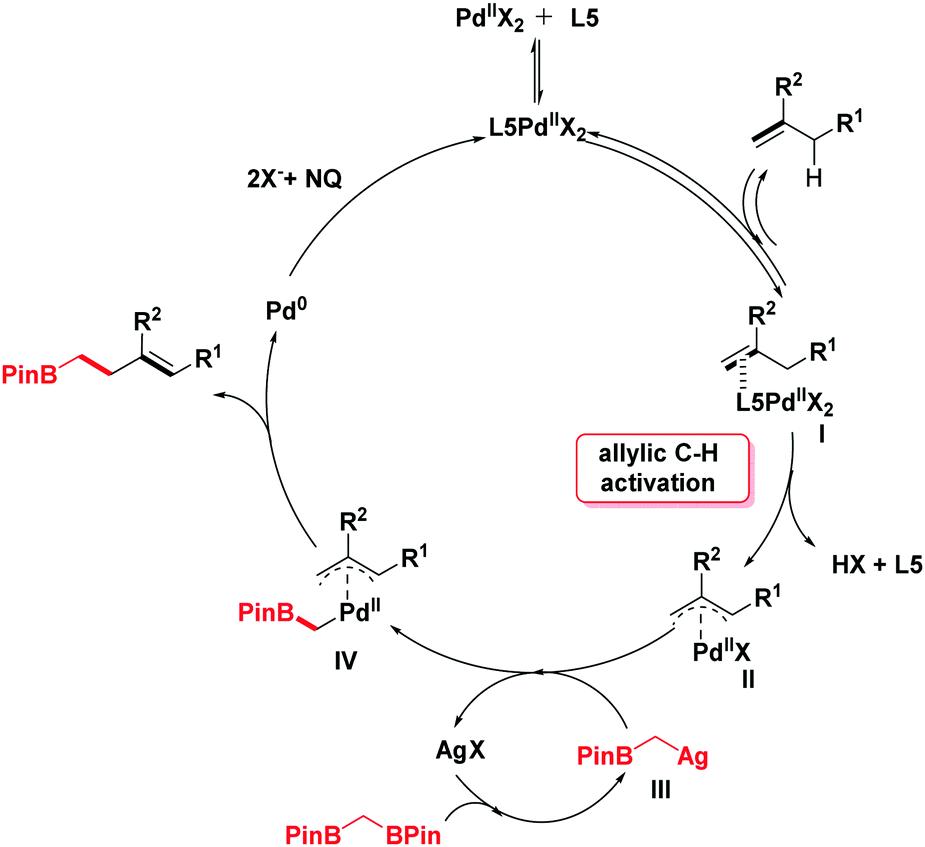 | ||
| Scheme 4 Possible reaction mechanism. | ||
In summary, a practical Pd-catalyzed oxidative α-boroalkylation reaction of simple olefins with 1,1-bis[(pinacolato)boryl]methane is presented. This novel procedure provides an efficient and attractive protocol for the construction of functionalized homoallylic boronic esters in good to excellent yields from readily available olefins with a broad substrate scope and excellent functional group compatibility. Remarkably, an efficient construction of the Csp3–Csp3 bond has been realized via the Pd-catalyzed oxidative functionalization of the allylic C–H bond.
We are grateful to the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (21672072, 21420102003 and 21502055), and the Fundamental Research Funds for the Central Universities (2015ZY001 and 2017ZD062).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For pioneering work in this area, see: (a) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033 CrossRef CAS PubMed; (b) K. Endo, T. Ohkubo and T. Shibata, Org. Lett., 2011, 13, 3368 CrossRef CAS PubMed; (c) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894 CrossRef CAS PubMed; (d) K. Endo, T. Ohkubo, T. Ishioka and T. J. Shibata, Org. Chem., 2012, 77, 4826 CrossRef CAS PubMed; (e) X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989 CrossRef CAS PubMed; (f) M. V. Joannou, B. S. Moyer, M. J. Goldfogel and S. J. Meek, Angew. Chem., Int. Ed., 2015, 54, 14141 CrossRef CAS PubMed; (g) J. Kim, S. Park, J. Park and S. H. Cho, Angew. Chem., Int. Ed., 2016, 55, 1498 CrossRef CAS PubMed.
- (a) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed; (b) S. Xu, X. Shangguan, H. Li, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 7779 CrossRef CAS PubMed; (c) Z.-Q. Zhang, C.-T. Yang, L.-J. Liang, B. Xiao, X. Lu, J.-H. Liu, Y.-Y. Sun, T. B. Marder and Y. Fu, Org. Lett., 2014, 16, 6342 CrossRef CAS PubMed; (d) Z.-Q. Zhang, B. Zhang, X. Lu, J.-H. Liu, X.-Y. Lu, B. Xiao and Y. Fu, Org. Lett., 2016, 18, 952 CrossRef CAS PubMed; (e) J. Park, Y. Lee, J. Kim and S. H. Cho, Org. Lett., 2016, 18, 1210 CrossRef CAS PubMed; (f) A. Ebrahim-Alkhalil, Z.-Q. Zhang, T.-J. Gong, W. Su, X.-Y. Lu, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 4891 RSC.
- (a) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS PubMed; (b) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918 CrossRef CAS PubMed.
- K. Endo, T. Ohkubo, T. Ishioka and T. J. Shibata, Org. Chem., 2012, 77, 4826 CrossRef CAS PubMed.
- Y. Shi and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 3455 CrossRef CAS PubMed.
- M. Zhan, R.-Z. Li, Z.-D. Mou, C.-G. Cao, J. Liu, Y.-W. Chen and D. Niu, ACS Catal., 2016, 6, 3381 CrossRef CAS.
- For selected reviews, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (b) J. Thomas and F. Peter, Chem. – Eur. J., 2009, 15, 9632 CrossRef PubMed; (c) C. J. Engelin and P. Fristrup, Molecules, 2011, 16, 951 CrossRef CAS PubMed; (d) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC; (e) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863 CrossRef CAS.
- For selected examples, see: (a) H. M. L. Davies, P. Ren and Q. H. Jin, Org. Lett., 2001, 3, 3587 CrossRef CAS PubMed; (b) B. X. Yu, T. Jiang, J. P. Li, Y. P. Su, X. F. Pan and X. G. She, Org. Lett., 2009, 11, 3442 CrossRef CAS PubMed; (c) D. Huang, H. Wang, H. Guan, H. Huang and Y. Shi, Org. Lett., 2009, 13, 1548 CrossRef PubMed; (d) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120 CrossRef CAS PubMed; (e) M. E. Harvey, D. G. Musaev and J. DuBois, J. Am. Chem. Soc., 2011, 133, 17207 CrossRef CAS PubMed; (f) M. Sekine, L. Ilies and E. Nakamura, Org. Lett., 2013, 15, 714 CrossRef CAS PubMed; (g) Y. Shibata, E. Kudo, H. Sugiyama, H. Uekusa and K. Tanaka, Organometallics, 2016, 35, 1547 CrossRef CAS.
- For selected examples, see: (a) B. M. Trost, S. Mahapatra and M. Hansen, Angew. Chem., Int. Ed., 2015, 54, 6032 CrossRef CAS PubMed; (b) G. Hu, J. Xu and P. Li, Org. Lett., 2014, 16, 6036 CrossRef CAS PubMed; (c) Q. Yang, Y. Zhou, J. Chen, X. He, J. Xu, F. Kwong and B. Fan, Eur. J. Org. Chem., 2015, 5330 CrossRef CAS; (d) H.-P. Deng, L. Erikssonb and K. J. Szabó, Chem. Commun., 2014, 50, 9207 RSC; (e) B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176 CrossRef CAS PubMed; (f) K. Liu, M. T. Hovey and K. A. Scheidt, Chem. Sci., 2014, 5, 4026 RSC.
- For some examples of Pd-catalyzed allylic C–H alkylation: (a) H. Chen, C. Cai, X. Liu, X. Li and H. Jiang, Chem. Commun., 2011, 47, 12224 RSC; (b) L. Li, Q.-Y. Chen and Y. Guo, Chem. Commun., 2013, 49, 8764 RSC; (c) H. Jiang, W. Yang, H. Chen, J. Li, W. Wu and H. Jiang, Chem. Commun., 2014, 50, 7202 RSC; (d) S. Tang, X. Wu, W. Liao, K. Liu, C. Liu, S. Luo and A. Lei, Org. Lett., 2014, 16, 3584 CrossRef CAS PubMed; (e) Y. Nishikawa, S. Kimura, Y. Kato, N. Yamazaki and O. Hara, Org. Lett., 2015, 17, 888 CrossRef CAS PubMed; (f) C. C. Pattillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265 CrossRef CAS PubMed; (g) H.-C. Lin, P.-S. Wang, Z.-L. Tao, Y.-G. Chen, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2016, 138, 14354 CrossRef CAS PubMed; (h) L.-L. Li, Z.-L. Tao, Z.-Y. Han and L.-Z. Gong, Org. Lett., 2017, 19, 102 CrossRef CAS PubMed.
- For some examples of Pd-catalyzed allylic C–H oxygenation: (a) K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032 CrossRef CAS PubMed; (b) A. N. Campbell, P. B. White, L. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15116 CrossRef CAS PubMed; (c) H. Chen, H. Jiang, C. Cai, J. Dong and W. Fu, Org. Lett., 2011, 13, 992 CrossRef CAS PubMed; (d) C. Le, K. Kunchithapatham, W. H. Henderson, C. T. Check and J. P. Stambuli, Chem. – Eur. J., 2013, 19, 11153 CrossRef CAS PubMed; (e) R. Tomita, K. Mantani, A. Hamasaki, T. Ishida and M. Tokunaga, Chem. – Eur. J., 2014, 20, 9914 CrossRef CAS PubMed; (f) S. E. Ammann, G. T. Rice and M. C. White, J. Am. Chem. Soc., 2014, 136, 10834 CrossRef CAS PubMed; (g) W. Yang, H. Chen, J. Li, C. Li, W. Wu and H. Jiang, Chem. Commun., 2015, 51, 9575 RSC; (h) P.-S. Wang, P. Liu, Y.-J. Zhai, H.-C. Lin, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 12732 CrossRef CAS PubMed; (i) S. E. Ammann, W. Liu and M. C. White, Angew. Chem., Int. Ed., 2016, 55, 9571 CrossRef CAS PubMed.
- For some examples of Pd-catalyzed allylic C–H amination: (a) F. Nahra, F. Liron, G. Prestat, C. Mealli, A. Messaoudi and G. Poli, Chem. – Eur. J., 2009, 15, 11078 CrossRef CAS PubMed; (b) G. Yin, Y. Wu and G. Liu, J. Am. Chem. Soc., 2010, 132, 11978 CrossRef CAS PubMed; (c) Y. Shimizu, Y. Obora and Y. Ishii, Org. Lett., 2010, 12, 1372 CrossRef CAS PubMed; (d) C. Jiang, D. J. Covell, A. F. Stepan, M. S. Plummer and M. C. White, Org. Lett., 2012, 14, 1386 CrossRef CAS PubMed; (e) H. Chen, W. Yang, W. Wu and H. Jiang, Org. Biomol. Chem., 2014, 12, 3340 RSC; (f) Y. Nishikawa, S. Kimura, Y. Kato, N. Yamazaki and O. Hara, Org. Lett., 2015, 17, 888 CrossRef CAS PubMed; (g) C. C. Pattillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265 CrossRef CAS PubMed; (h) C. Li, J. Li, Y. An, J. Peng, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 12189 CrossRef CAS PubMed.
- For some examples of Pd-catalyzed allylic C–H silylation: J. M. Larsson, T. S. N. Zhao and K. J. Szabó, Org. Lett., 2011, 13, 1888 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Engelin, T. Jensen, S. Rodriguez and P. Fristrup, ACS Catal., 2013, 3, 294 CrossRef CAS; (b) F. J. S. Duarte, G. Poli and M. J. Calhorda, ACS Catal., 2016, 6, 1772 CrossRef CAS.
- K. Endo, F. Kurosawa and Y. Ukaji, Chem. Lett., 2013, 42, 1363 CrossRef CAS.
- G. Yin, Y. Wu and G. Liu, J. Am. Chem. Soc., 2010, 132, 11978 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, copies of 1H and 13C NMR spectra for selected compounds. See DOI: 10.1039/c7cc07788b |
| This journal is © The Royal Society of Chemistry 2018 |