
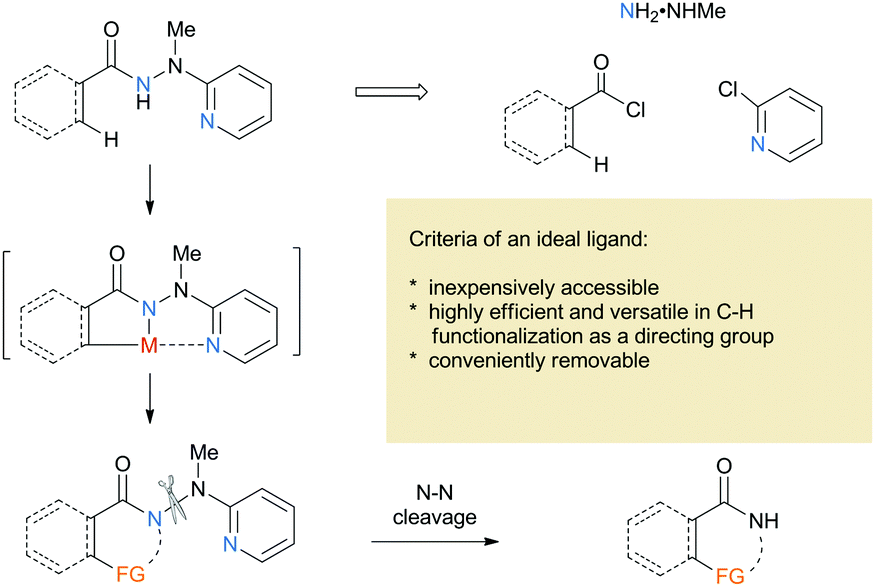
2-(1-Methylhydrazinyl)pyridine as a reductively removable directing group in a cobalt-catalyzed C(sp2)–H bond alkenylation/annulation cascade†
Shengxian
Zhai
a,
Shuxian
Qiu
b,
Xiaoming
Chen
b,
Jiang
Wu
a,
Hua
Zhao
b,
Cheng
Tao
a,
Yun
Li
 a,
Bin
Cheng
a,
Huifei
Wang
b and
Hongbin
Zhai
*abc
a,
Bin
Cheng
a,
Huifei
Wang
b and
Hongbin
Zhai
*abc
aThe State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
bLaboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China. E-mail: zhaihb@pkusz.edu.cn
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 24th November 2017
Abstract
We describe a new application of 2-(1-methylhydrazinyl)pyridine as a bidentate directing group to directing cobalt-catalyzed C(sp2)–H alkenylation/annulation of the corresponding benzoic hydrazides to form an isoquinoline backbone, via reacting with a terminal or internal alkyne followed by annulation. The reaction shows a broad substrate scope with the products obtained in good to excellent yields and high regioselectivity. Moreover, the directing group can be reductively removed in one step under mild conditions.
Research on transition metal-catalyzed C–H functionalization has increased significantly over the past decades1 and emerged as a powerful strategy for efficient and economic synthesis of complex natural products.2 The control of the site selectivity is challenging and the use of a directing group has proven to be a common, practical, and powerful strategy. In 2005, Daugulis and coworkers reported a groundbreaking palladium-catalyzed C(sp3)–H arylation using 8-aminoquinoline as a bidentate directing group with excellent regioselectivity.3 It was believed that bidentate auxiliaries could generate stable metallacycles and promote C–H activation. After their pioneering work, 8-aminoquinoline was widespreadly studied and proved to be the most successful and promising bidentate directing group,4 which set a stage for the further development of novel approaches toward selective C–H functionalization. Various new bidentate auxiliaries have been disclosed over the past few years,3,5 but most auxiliaries incorporate a stable C–N linkage that is relatively difficult to remove. Thus, development of novel removable bidentate directing groups remains highly desirable. To the best of our knowledge, the bidentate auxiliaries containing the N–X (X = S and N) linkage seem to be rare,6 although it is well known that the N–X linkage can be easily cleaved in a traceless fashion compared to the C–N counterpart. Therefore, it is essential to develop new N-hetero bond-containing directing groups that are both easily available and readily removable.
In general, transition metal-catalyzed C–H activation relies mainly on noble metal (such as Ru,7 Rh,8 or Pd9) catalysts. In spite of their high catalytic activity, these metals are less abundant in nature. So, it would be economically advantageous to achieve catalytic C–H activation using more abundant and lower atomic weight transition metals with comparable efficacy. Recently, cobalt-catalyzed C–H activation has attracted considerable attention in its low valence,10 cationic,11 or other forms.12 Because of its high reactivity, low cost, low toxicity, and abundant availability from the natural sources, cobalt was considered as an attractive catalyst for C–H activation.
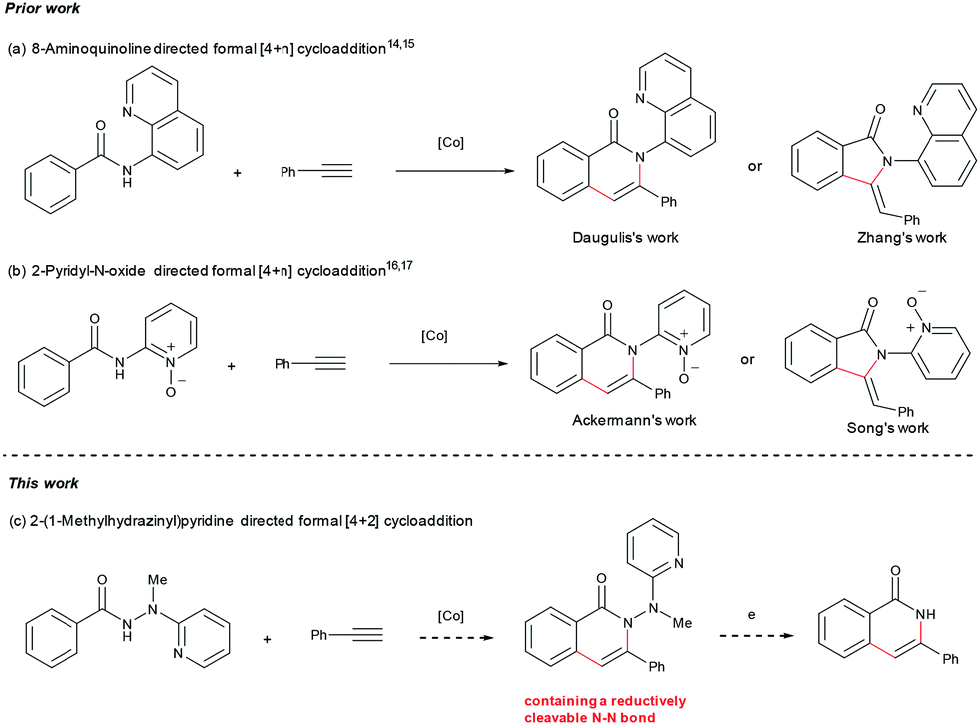
Based on the considerations mentioned above, we envisioned that 2-(1-methylhydrazinyl)pyridine might be a promising bidentate auxiliary candidate,13 since it can be easily obtained from 2-chloropyridine and methylhydrazine at low cost. Also, the N–N linkage can be much more easily cleaved compared to most other bidentate directing groups (Scheme 1). Cobalt-catalyzed C(sp2)–H alkenylation/annulation assisted by a bidentate directing group was pioneered by Daugulis and coworkers,14 and later flourished by the groups of Zhang,15 Song,16 and Ackermann17 (Scheme 2a and b). Inspired by the previous studies, we initiated our investigations (Scheme 2c) on the reaction of N′-methyl-N′-(pyridin-2-yl)benzohydrazide (1a) with phenylacetylene (2a), using the catalytic system reported by Daugulis.14 Gratifyingly, the reaction generated the desired product 3aa in moderate yield (Table 1, entry 1), and the structure of 3aa was determined by an X-ray diffraction analysis.18 Encouraged by the above results, we then screened some other bases for the reaction and found that Na2CO3 and K2CO3 were both slightly better than NaOPiv (entries 2 and 3). A significant increase of the reaction efficiency was observed when 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was used as the solvent (entry 4). The yield of 3aa was further increased by the incorporation of tetrabutylammonium iodide (TBAI) as a phase-transfer catalyst into the reaction system (entry 5).15 In addition, control experiments indicated that the C–H activation step was completely inhibited in the absence of either Co(OAc)2·4H2O or Mn(OAc)3·2H2O (entries 6 and 7). Nevertheless, the reaction could still proceed to some extent without a base (entry 8).
 | ||
| Scheme 1 Novel bidentate directing group-assisted C–H activation. | ||
 | ||
| Scheme 2 Cobalt-catalyzed C–H activation. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (2.0 equiv.), Co(OAc)2·4H2O (20 mol%), air, 100 °C, 24 h. b Isolated yield. c TBAI (2.0 equiv.) was added. d No cobalt was added. | ||||
| 1 | Mn(OAc)3·2H2O | NaOPiv | TFE | 51 |
| 2 | Mn(OAc)3·2H2O | Na2CO3 | TFE | 61 |
| 3 | Mn(OAc)3·2H2O | K2CO3 | TFE | 61 |
| 4 | Mn(OAc)3·2H2O | Na2CO3 | HFIP | 84 |
| 5 | Mn(OAc) 3 ·2H 2 O | Na 2 CO 3 | HFIP | 90 |
| 6c,d | Mn(OAc)3·2H2O | Na2CO3 | HFIP | N.D. |
| 7c | — | Na2CO3 | HFIP | N.D. |
| 8c | Mn(OAc)3·2H2O | — | HFIP | 44 |
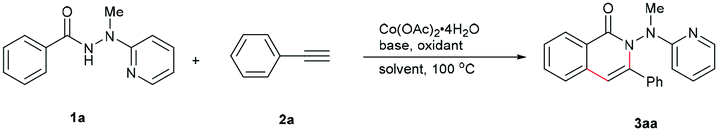
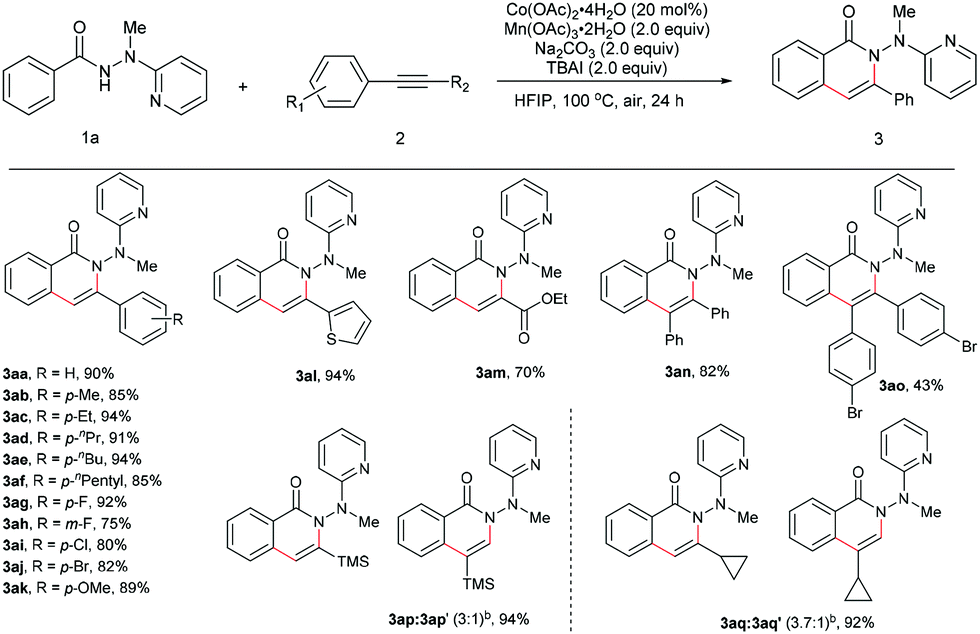
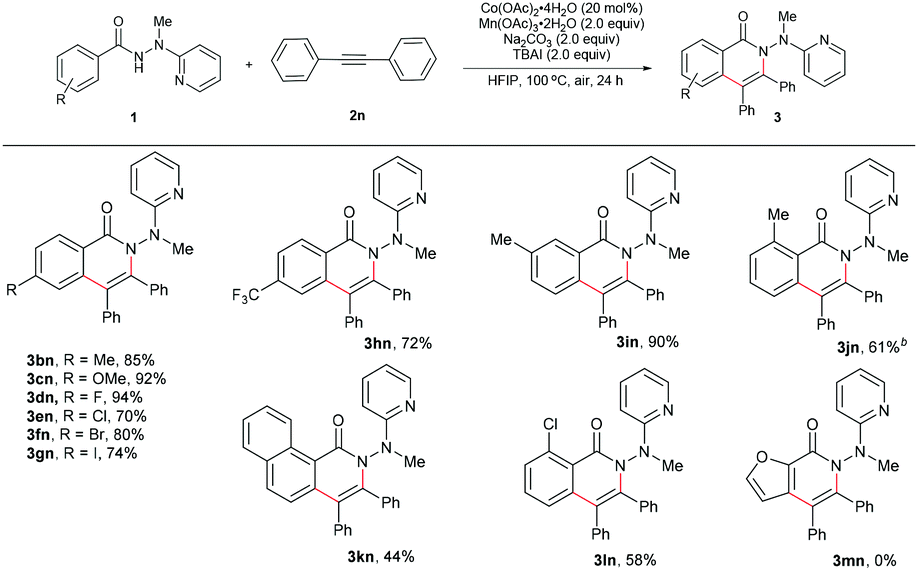
The scope for the alkynes was next investigated under the optimal reaction conditions obtained above (see Table 1, entry 5). In general, both electron-rich and electron-deficient alkynes were compatible with the reaction (Table 2, 3aa–3ak). For example, phenylacetylene and 1-ethynyl-4-pentylbenzene produced nearly equivalent yields (3aa and 3af). Phenylacetylenes with a substituent on the benzene ring such as fluoro (3ag and 3ah), chloro (3ai), bromo (3aj), and methoxy (3ak) groups were well tolerated, providing ample opportunities for further derivatization of the products. 2-Ethynylthiophene (3al) and ethyl propiolate (3am) were also suitable for this reaction. Furthermore, internal alkynes such as diphenylacetylene and its derivatives did not appear to influence the reactions, and gave the products in moderate to good yields (3an19 and 3ao). In the cases of trimethylsilylacetylene (3ap) and cyclopropyl acetylene (3aq), the corresponding cyclization products were formed in high yields, yet with relatively low regioselectivities.
Subsequently, a wide range of hydrazides were explored in the cobalt-catalyzed annulation with alkyne 2n (Table 3). Benzoic hydrazides containing either an electron-donating or electron-withdrawing group (e.g., methyl, methoxy, fluoro, chloro, bromo, or iodo group, in the para-position) on the benzene ring (3bn–3gn) worked well in this reaction. Moreover, the hydrazide with a para trifluoromethyl group furnished the product in 72% yield (3hn), indicating that the reaction might not be very sensitive to the electronic effect. A single product was generated in an excellent yield of 90% with the meta-methyl-substituted benzoic hydrazide 3in used as the substrate, therefore the current reaction might have a strong steric effect. Indeed, ortho-substituted benzoic hydrazides with either an electron-donating or an electron-withdrawing group gave the corresponding annulation products in only moderate yields (3jn, 3kn, and 3ln). Moreover, for the hydrazide with a heteroaromatic moiety 1m, no desired product (3mn) was formed at all, even at an elevated reaction temperature.
One of the significant merits of the current strategy lies in the reductive removal of the methylaminopyridine moiety (originated from the ligand) under mild conditions. For instance, upon the N–N bond cleavage through treatment with SmI2 at 0 °C to room temperature overnight, compounds 3an, 3en, and 3fn were transformed smoothly into amides 5–7 in 88–94% yields (Scheme 3).20,21
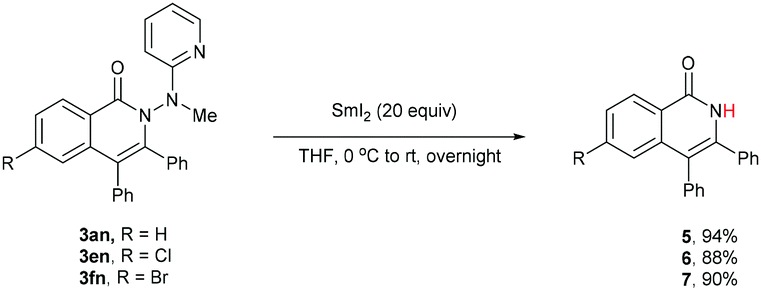 | ||
| Scheme 3 Reductive removal of the directing group. | ||
In order to gain further insight into the nature of the present reaction, the following experiments were performed to probe the reaction mechanism (Scheme 4). An intermolecular kinetic isotope effect (KIE) experiment was carried out between hydrazide 1a and isotopically labelled substrate [D5]-1a under standard reaction conditions (Scheme 4a), resulting in a K value of approximately 1.5, suggesting that the first step (for the formation of A) in the catalytic cycle was only slightly affected by the H/D isotope. Furthermore, no H/D exchange could be detected in both the reaction of isotopically labelled substrate [D5]-1a with 2n and that of 1a with 2n with CD3OD added as a co-solvent, indicating presumable irreversibility of the C–H cobaltation step (Scheme 4b).
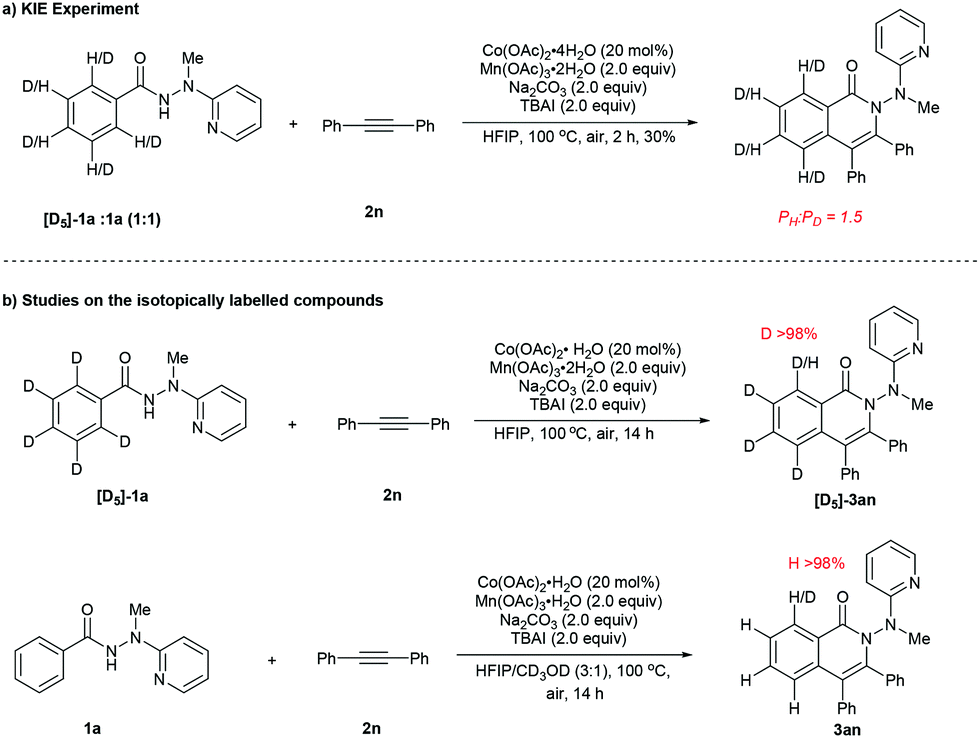 | ||
| Scheme 4 Preliminary mechanistic studies. | ||
Based upon our preliminary mechanistic studies and the relevant literature reports,17,22 we proposed a mechanism for the cascade reaction sequence (Scheme 5). First, Co(II) is oxidized to Co(III) by Mn(OAc)3·2H2O. The chelation of Co(III) to hydrazide 1 followed by the subsequent C–H activation produces intermediate A. Alkyne 2 is then coordinated to the Co(III) center within intermediate A to provide intermediate B. Coordinative insertion of the carbon–carbon triple bond into the C–Co bond of intermediate B results in the seven-membered intermediate C, and the reductive elimination of which gives the desired product 3 as well as the low-valent Co(I). The active Co(III) species was regenerated through the oxidation of Co(I) with Mn(OAc)3·2H2O and O2, and the regenerated Co(III) species enters the next catalytic cycle.
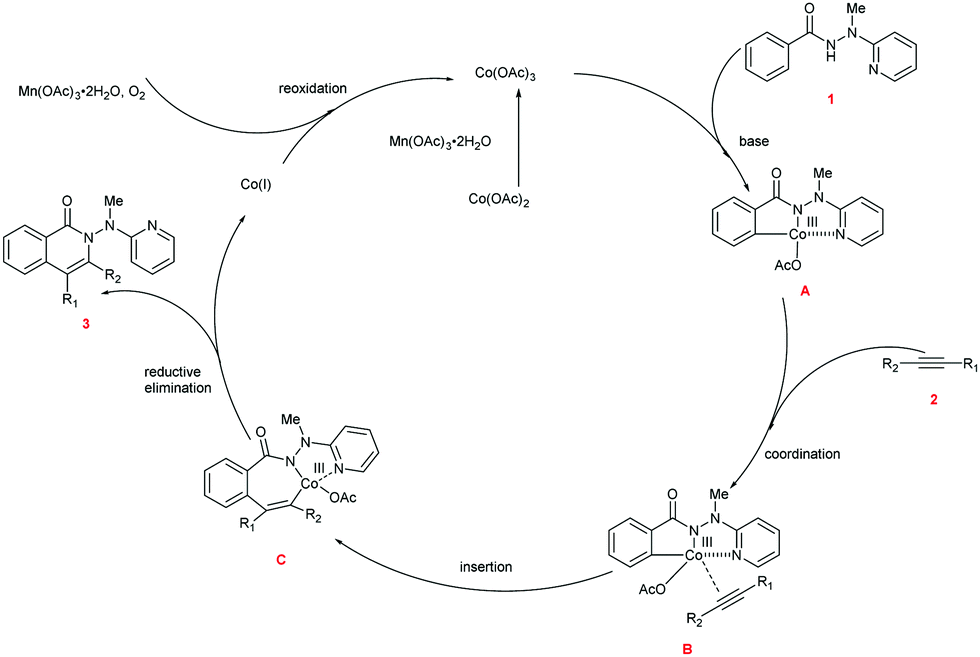 | ||
| Scheme 5 Proposed reaction mechanism. | ||
In conclusion, we have developed an efficient cobalt-catalyzed C(sp2)–H bond alkenylation/annulation cascade reaction of benzoic hydrazides with various terminal or internal alkynes via a novel 2-(1-methylhydrazinyl)pyridine-assisted C–H activation, providing a new rapid access to a series of isoquinoline derivatives. Moreover, the methylaminopyridine moiety originated from the ligand can be reductively removed under mild conditions. Further applications of 2-(1-methylhydrazinyl)pyridine as a bidentate directing group in other related types of C–H functionalization and detailed understanding of the mechanisms are currently being investigated in our laboratory.
We thank the NSFC (21732001, 21672017, 21472072, and 21290183), the Shenzhen Science and Technology Innovation Committee (JCYJ20150529153646078 and JSGG20160229150510483), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT: IRT_15R28), and the “111” Program of MOE for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews on C–H activation, see: (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) B. Su, Z.-C. Cao and Z.-j. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed; (c) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1769 CrossRef CAS PubMed; (d) I. Mkhalid, J. Barnard, T. Marder, J. Murphy and J. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed . For selected examples on C–H activation, see: ; (e) C.-Z. Luo, P. Gandeepan, Y.-C. Wu, C.-H. Tsai and C.-H. Cheng, ACS Catal., 2015, 5, 4837–4841 CrossRef CAS; (f) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS; (g) L.-C. Liang, P.-S. Chien and Y.-L. Huang, J. Am. Chem. Soc., 2016, 128, 15562–15563 CrossRef PubMed; (h) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858–5859 CrossRef CAS PubMed; (i) J. Park, J. Lee and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 4256–4260 CrossRef CAS PubMed.
- For selected examples, see: (a) J. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900–6903 CrossRef CAS PubMed; (b) S. O’Malley, K. Tan, A. Watzke, R. Bergman and J. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef PubMed; (c) H. Davies, X. Dai and M. Long, J. Am. Chem. Soc., 2006, 128, 2485–2490 CrossRef CAS PubMed; (d) W. Gutekunst and P. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed; (e) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS PubMed; (f) B. Wang, Y. Liu, R. Jiao, Y. Feng, Q. Li, C. Chen, L. Liu, G. He and G. Chen, J. Am. Chem. Soc., 2016, 138, 3926–3932 CrossRef CAS PubMed.
- V. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- For selected examples, see: (a) Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261–11270 CrossRef CAS PubMed; (b) H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909–9920 CrossRef CAS PubMed; (c) K. Shibata, S. Natsui and N. Chatani, Org. Lett., 2017, 19, 2234–2237 CrossRef CAS PubMed; (d) L. Ilies, Y. Itabashi, R. Shang and E. Nakamura, ACS Catal., 2017, 7, 89–92 CrossRef CAS; (e) B. Wang, C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 6260–6263 CrossRef CAS PubMed; (f) K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 5353–5357 CrossRef CAS PubMed; (g) N. THrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem. Int. Ed., 2016, 55, 12361–12365 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (b) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–8073 CrossRef CAS PubMed; (c) M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152–12155 CrossRef CAS PubMed; (d) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187–4192 RSC; (e) Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem. Int. Ed., 2014, 53, 3868–3871 CrossRef CAS PubMed; (f) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354–3357 CrossRef CAS PubMed; (g) Y.-F. Zhang, H.-W. Zhao, H. Wang, J.-B. Wei and Z.-J. Shi, Angew. Chem. Int. Ed., 2015, 54, 13686–13690 CrossRef CAS PubMed; (h) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem. Int. Ed., 2015, 54, 272–275 CrossRef CAS PubMed; (i) H. M.-F. Viart, A. Bachmann, W. Kayitare and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 1325–1329 CrossRef CAS PubMed; (j) T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem. Int. Ed., 2017, 56, 10924–10927 CrossRef CAS PubMed; (k) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 12394–12397 CrossRef CAS PubMed.
- (a) N. Rodríguez, J. Romero-Revilla, M. Fernández-Ibáñez and J. Carretero, Chem. Sci., 2013, 4, 175–179 RSC; (b) R. Rit, R. Yadav and A. Sahoo, Org. Lett., 2012, 14, 3724–3727 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (b) P. Arockiam, C. Bruneau and P. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) S. Ko, H. Han and S. Chang, Org. Lett., 2003, 5, 2687–2690 CrossRef CAS PubMed; (d) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2012, 14, 4262–4265 CrossRef CAS PubMed.
- For selected reviews on Rh-catalyzed C–H activations, see: (a) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (c) D. Colby, A. Tsai, R. Bergman and J. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (d) J. Lewis, R. Bergman and J. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed; (e) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (f) D. Colby, R. Bergman and J. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y. Minami and T. Hiyama, Acc. Chem. Res., 2016, 49, 67–77 CrossRef CAS PubMed; (b) N. Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (c) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114–1123 CrossRef CAS PubMed; (d) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- (a) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed; (b) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (c) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251–8254 CrossRef CAS PubMed; (d) X. Cong, S. Zhai and X. Zeng, Org. Chem. Front., 2016, 3, 673–677 RSC.
- (a) J. Hummel and J. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef CAS PubMed; (b) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef CAS PubMed; (c) R. Mei, J. Loup and L. Ackermann, ACS. Catal., 2016, 6, 793–797 CrossRef CAS; (d) Q. Lu, S. Vásquez-Céspedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352–2356 CrossRef CAS.
- For selected examples, see: (a) P. Gandeepan, P. Rajamalli and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 4308–4311 CrossRef CAS PubMed; (b) W. Ma and L. Ackermann, ACS Catal., 2015, 5, 2822–2825 Search PubMed; (c) D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118–6121 CrossRef CAS PubMed; (d) O. Planas, C. Whiteoak, A. Company and X. Ribas, Adv. Synth. Catal., 2015, 357, 4003–4012 CrossRef CAS.
- For 2-hydrazinylpyridine applied to direct C–H activation in hydrazones, see: (a) S. Zhou, M. Wang, L. Wang, K. Chen, J. Wang, C. Song and J. Zhu, Org. Lett., 2016, 18, 5632–5635 CrossRef CAS PubMed; (b) Y. Xu, M. Young and G. Dong, J. Am. Chem. Soc., 2017, 139, 5716–5719 CrossRef CAS PubMed.
- (a) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed; (b) T. Nguyen, L. Grigorjeva and O. Daugulis, ACS Catal., 2016, 6, 551–554 CrossRef CAS PubMed.
- J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990–12996 CrossRef CAS PubMed.
- L.-B. Zhang, X.-Q. Hao, Z.-J. Liu, X.-X. Zheng, S.-K. Zhang, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 10012–10015 CrossRef CAS PubMed.
- R. Mei, H. Wang, S. Warratz, S. Macgregor and L. Ackermann, Chem. – Eur. J., 2016, 22, 6759–6763 CrossRef CAS PubMed.
- The structure of compound 3aa was confirmed by X-ray crystallography, CCDC 1547832†.
- The structure of compound 3an was confirmed by X-ray crystallography, CCDC 1548078†.
- C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed.
- Application of a lower amount of SmI2 resulted in a much lower yield of the product although the unreacted starting material could be recovered.
- (a) N. Thrimurtulu, A. Dey, D. Maiti and C. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361–12365 CrossRef CAS PubMed; (b) W. Ma and L. Ackermann, ACS. Catal., 2015, 5, 2822–2825 CrossRef CAS; (c) C. Kuai, L. Wang, B. Li, Z. Yang and X. Cui, Org. Lett., 2017, 19, 2102–2105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1547832 and 1548078. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08533h |
| This journal is © The Royal Society of Chemistry 2018 |