
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-induced decarbonylation in diphosphine-ligated palladium acetates [CH3CO2Pd((PR2)2CH2)]+ (R = Me and Ph)†
Michael
Lesslie
a,
Yang
Yang
b,
Allan J.
Canty
 c,
Elettra
Piacentino
a,
Francis
Berthias
d,
Philippe
Maitre
d,
Victor
Ryzhov
*a and
Richard A. J.
O'Hair
*b
c,
Elettra
Piacentino
a,
Francis
Berthias
d,
Philippe
Maitre
d,
Victor
Ryzhov
*a and
Richard A. J.
O'Hair
*b
aDepartment of Chemistry and Biochemistry, and Center for Biochemical and Biophysical Sciences, Northern Illinois University, DeKalb, IL 60115, USA. E-mail: ryzhov@niu.edu
bSchool of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, The University of Melbourne, Victoria 3010, Australia. E-mail: rohair@unimelb.edu.au
cSchool of Physical Sciences, University of Tasmania, Private Bag 75, Hobart, Tasmania 7001, Australia
dLaboratoire de Chimie Physique, Bâtiment 349, Université Paris-Sud, CNRS, Université Paris-Saclay, F-91405 Orsay, France
First published on 11th December 2017
Abstract
A new decarbonylation reaction is observed for [(K2-acetate)Pd(K2-diphosphine)]+ complexes. Gas-phase IR experiments identify the product as [CH3Pd(OP(Ph2)CH2PPh2)]+. DFT calculations uncovered a plausible mechanism involving O atom abstraction by the diphosphine ligand within the coordination sphere to yield the acetyl complex, [CH3COPd(OP(Ph2)CH2PPh2)]+, which then undergoes decarbonylation.
Gas-phase studies utilising tandem mass spectrometry (MS/MS) methods in conjunction with DFT calculations provide valuable fundamental information on how the auxiliary ligands in metal complexes can tune the fragmentation chemistry of coordinated carboxylates under collision-induced dissociation (CID) conditions (Scheme 1).1 For example, the acetate ligand in Pd(II) cationic complexes can undergo decarboxylation to give an organometallic cation (eqn (1))2 or act as an intramolecular base to promote C–H bond activation (eqn (2)).3 Both of these modes of reactivity have been widely exploited in the condensed phase to generate organometallic precursors for use in organic synthesis.4,5 A recent study has used mechanistic insights from MS/MS experiments to develop a new decarboxylative protocol for the synthesis of thioamides,6 highlighting the value of gas-phase model systems. Here we report on a newly discovered mode of reactivity, ligand-induced decarbonylation (eqn (3)), one that has little precedence in the condensed phase7–9 and is thus of considerable mechanistic interest.
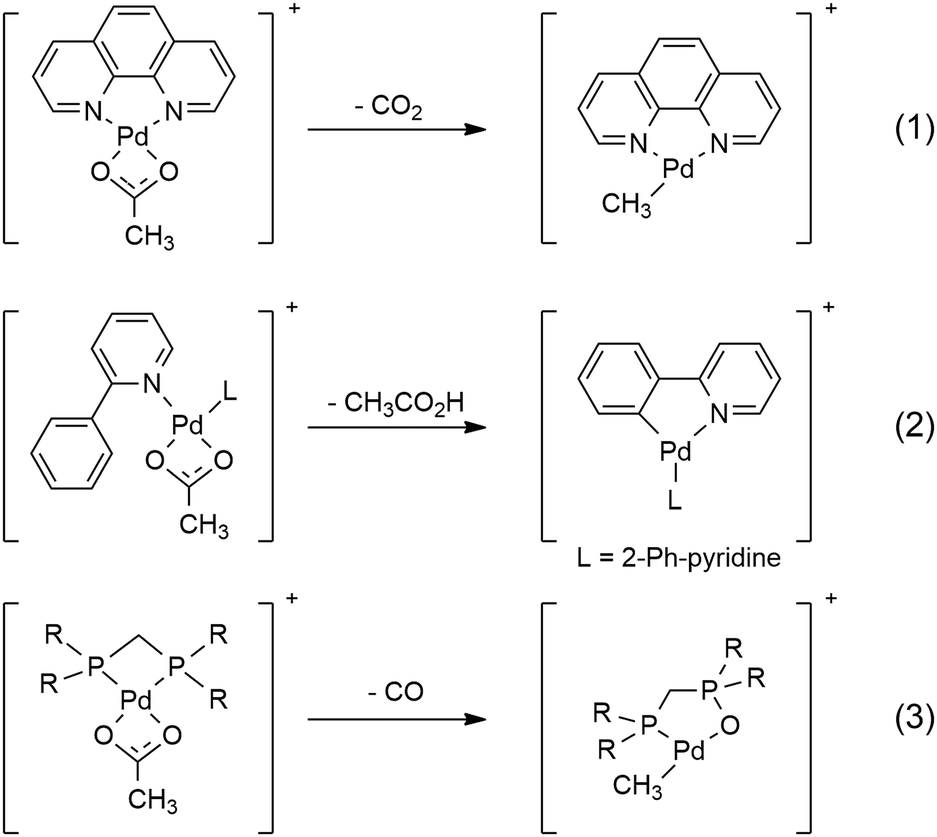 | ||
| Scheme 1 Modes of reactivity observed for coordinated acetates in Pd(II) cationic complexes: decarboxylation (eqn (1));2 as a base to facilitate C–H bond activation (eqn (2));3 decarbonylation (eqn (3), this work). | ||
Electrospray ionisation (ESI) of methanolic solutions containing palladium acetate and a diphosphine ligand, R2PCH2PR2 (R = Ph = dppm ligand; R = Me = dmpm ligand) gave rise to the desired diphosphine-ligated palladium acetates [CH3CO2Pd((PR2)2CH2)]+, 1a, R = Ph and 1b, R = Me, which were mass selected and subjected to low-energy CID in ion trap mass spectrometers. The unimolecular chemistry of 1a (m/z 549, Fig. 1a) is dominated by loss of CO to give an ion at m/z 521 (eqn (3), Scheme 1). Minor fragment ions at m/z 305 and 397 are assigned as [Ph2PCH2Pd]+ and [Ph2PCH2Pd(Me)(Ph)]+, respectively, and arise from subsequent C–P activation of the dppm ligand, as established by MS3 on m/z 521, which gave m/z 305 and 397 (Fig. S1, ESI†). CID of the labelled acetate complexes [CH313CO2Pd((PR2)2CH2)]+ (m/z 550, Fig. 1b) and [CD3CO2Pd((PR2)2CH2)]+ (m/z 550, Fig. 1c) confirmed these assignments. Complex 1b also underwent decarbonylation (Fig. S2, ESI†). Decarboxylation is only a very minor pathway in the CID spectra of 1a (<0.1%) and 1b (<1%), which is in stark contrast to CID on [CH3CO2Pd(phen)]+, where decarboxylation dominates (eqn (1), Scheme 1). This highlights the unique role of the diphosphine auxiliary ligand in promoting decarbonylation.
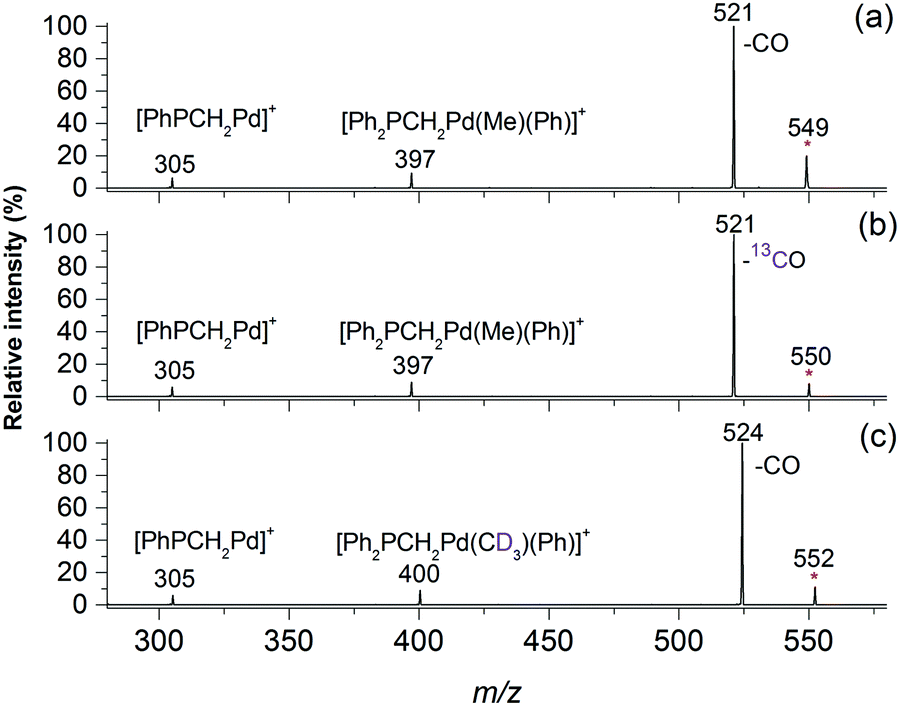 | ||
| Fig. 1 LTQ MS2 CID spectra of: (a) [CH3CO2106Pd((PPh2)2CH2)]+, m/z 549, 1a; (b) [CH313CO2106Pd((PPh2)2CH2)]+, m/z 550; (c) [CD3CO2106Pd((PPh2)2CH2)]+, m/z 552. Product ions are labeled according to the neutral fragment that was lost from the mass-selected precursor, which is marked with an asterisk. | ||
Since the structure(s) of the decarbonylation product is unknown, we next: (1) used DFT calculations to optimise four chemically reasonable isomers, including the organopalladium complexes, [CH3Pd(OP(Ph2)CH2PPh2)]+2 and 3, containing the Ph2PCH2P(O)Ph2 (dppmO) ligand,10 the palladium methoxide complex, [CH3OPd((PPh2)2CH2)]+411 and the Pd(IV) complex, [CH3Pd(O)((PPh2)2CH2)]+5.12 (2) compared the theoretically predicted IR spectra of these isomers to the gas-phase infrared multiphoton dissociation (IRMPD) spectrum of 1a-CO acquired using a 3D ion-trap tandem mass spectrometer coupled with the CLIO free electron laser.13

Although 4a benefits from an agostic interaction between the methyl group and Pd (Fig. S3, ESI†), calculations at the M06/SDD-6-31G(d) level of theory show the following stability order: 2a (0 kcal mol−1) > 3a (+22 kcal mol−1) > 4a (+30 kcal mol−1). 5a is unstable, undergoing rearrangement to 4a. Although the experimental IRMPD spectrum of 1a-CO exhibits quite broad features, an examination of Fig. 2 reveals that the lowest-energy isomer 2 displays the best match (Fig. 2(a) and Table S1, ESI†). The most intense absorption band in the experimental spectrum (the broad band at 1070–1200 cm−1) matches the predicted P–O and P–C asymmetric stretches at 1134 cm−1 flanked by the shoulder peaks at 1076 cm−1 (Ph ligand C–H bend coupled with P–O stretch) and 1196 cm−1 (CH3 rocking mode). The experimental absorption between 1400 and 1450 cm−1 matches the aromatic ring deformation of the ligand, calculated to be at 1442 cm−1. The smaller experimental peaks at around 1300 and 1000 cm−1 can be assigned to the aromatic ligand C–H bending (1299 cm−1) and C–H/C–C stretching/bending (988 cm−1), respectively. In contrast, the higher-energy isomers 3a and 4a show substantially poorer matches to the experimental IRMPD spectrum (Fig. 2(b) and (c)). Isomer 3a is predicted to have a strong band at 1042 cm−1, corresponding to the P–O stretching and Pd–O bending motions. This band is absent from the experimental spectrum (Fig. 2b). Isomer 4a has the CH3 umbrella band calculated to be at 1580 cm−1, which is also absent from the experimental spectrum. Thus, the IRMPD data is consistent with structure 2a for the 1a-CO ion.
 | ||
| Fig. 2 IRMPD experimental (grey) spectrum of mass-selected [CH3CO2106Pd((PPh2)2CH2)–CO]+, m/z 521, vs. M06/SDD6-31G(d) calculated IR spectra (red) for structures: (a) 2a, (b) 3a, and (c) 4a. The experimental IR spectrum was obtained on a Bruker Esquire 3000+ quadrupole ion trap MS using the CLIO free-electron laser with a wavenumber step of 4.5 cm−1, an irradiation time of 1 s, and mean laser power was decreasing from 1500 to 700 mW when increasing the photon wavenumber. | ||
 | ||
| Fig. 3 DFT calculated competition between decarboxylation and decarbonylation of [CH3CO2Pd((PMe2)2CH2)]+, 1b at the M06/def2-TZVP//M06/SDD6-31G(d) level of theory. Energies are in kcal mol−1 and are given as ΔE. | ||
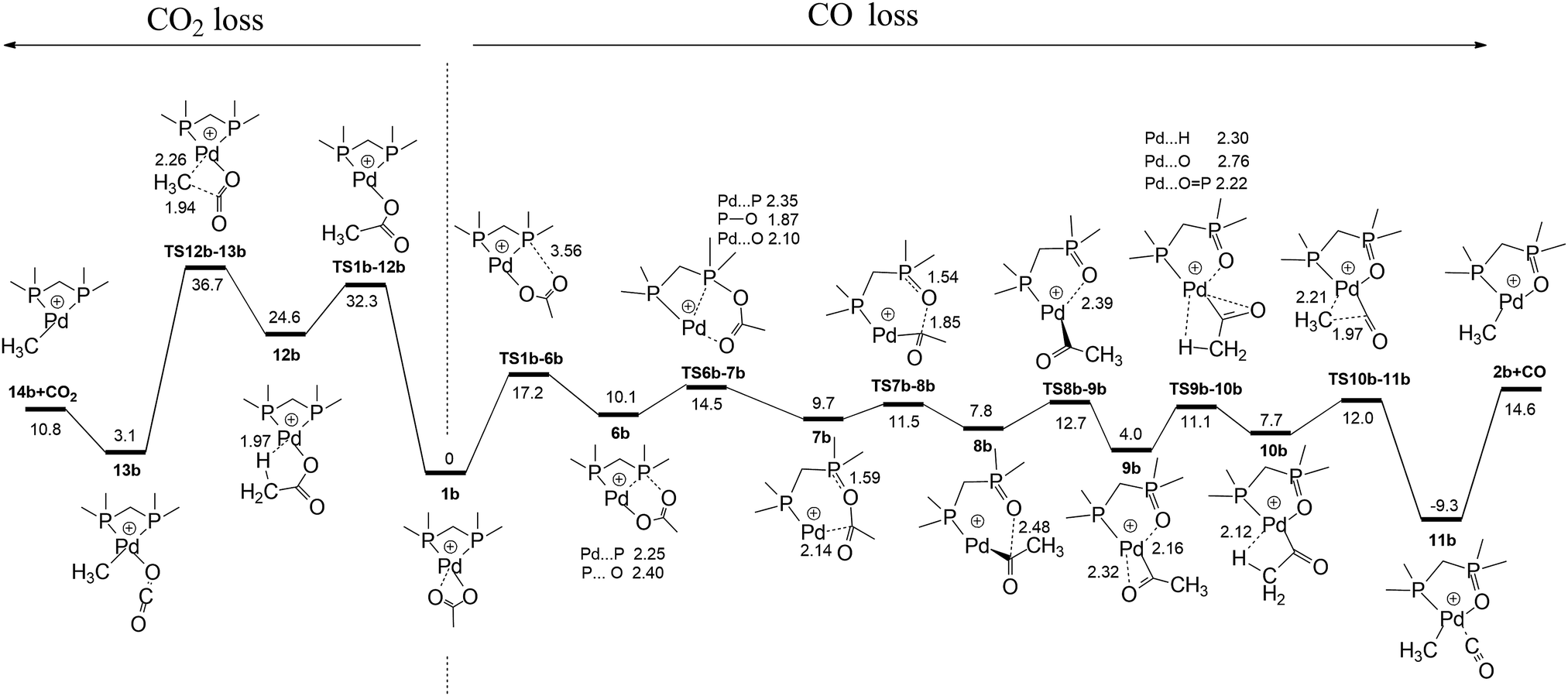
Having established the product structure as 2a, we next used DFT calculations to examine potential mechanisms for the decarboxylation of 1a and 1b and to establish that the barrier for decarbonylation is lower than that for decarboxylation, a requirement for formation of 2a and 2b as the major products under the low energy CID conditions used (Fig. 1 and Fig. S2, ESI†). The pathways for the decarbonylation and decarboxylation reactions are compared for 1b in Fig. 3 and Fig. S4 (ESI†), while the related reactivity for 1a is shown in Fig. S5 (ESI†). The decarboxylation pathway follows the same mechanism reported for [CH3CO2Pd(phen)]+.2b
Multiple decarbonylation pathways were considered. The lowest energy pathway connects 1b, 6b and 7bviaTS1b-6b and TS6b-7b. Cleavage of the C–O bond viaTS7b-8b gives the Pd-acetyl cation coordinated to a dmpmO ligand, 8b. Just as in the decarboxylation pathway, the coordinated acetyl cation needs to adopt reactive conformation 10b in order for decarbonylation to occur. This involves changing the conformation of the coordinated acetyl anion relative to the dmpmO ligand. Of the two pathways to connect 8b to 10b that were found (Fig. S4 and S5, ESI†), the lowest one involves traversing two transition structures TS8b-9b and TS9b-10b. Once formed, 10b decarbonylates viaTS10b-11b to give the CO coordinated organometallic cation, 11b, from which CO loss produces the experimentally observed cation, 2b. TS1b-6b. The most energetically demanding TS for decarbonylation (+17.2 kcal mol−1) is lower in energy than TS12b-13b (+36.7 kcal mol−1) for decarboxylation, consistent with the experimental observation that decarbonylation occurs predominantly. Related calculations were carried out for the competition between CO and CO2 loss for 1a (Fig. S6 and S7, ESI†). Similar reaction profiles were found, with TS6a-7a, the most energetically demanding TS for decarbonylation (+29.2 kcal mol−1) being lower in energy than TS12a-13a (+37.9 kcal mol−1) for decarboxylation, consistent with the experimental observation that decarbonylation occurs predominantly.
Metal-mediated decarbonylation of carboxylic acid derivatives finds wide application in synthesis,7,14,15 although most processes show poor atom economy as they require an anhydride additive to facilitate formation of an acyl complex which then undergoes decarbonylation.8,9 Here we have shown that diphosphine and acetate ligands react with each other within the coordination sphere of complexes to trigger decarbonylation. While this mechanism appears to be unprecedented, there is a growing literature on ligand oxidation reactions occurring within the coordination sphere of metal complexes.16,17 Finally, the observed chemistry is not limited to acetate ligands, but occurs with a range of other coordinated carboxylates (e.g. benzoate and hydrocinnamate) as will be reported in due course.
We acknowledge support from the Australian Research Council (DP150101388) and the National Computing Infrastructure. Financial support from European Community's Framework Programme Horizon 2020 (CALIPSOplus, under grant agreement 730872) is gratefully acknowledged. ML thanks NIU graduate student travel award for funding his trip to conduct research at the University of Melbourne. We thank Victor Wan for carrying out preliminary DFT calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. A. J. O’Hair, “Gas Phase Ligand Fragmentation to Unmask Reactive Metallic Species”, in “Reactive Intermediates. MS Investigations in Solution”, ed. L. S. Santos, Wiley-VCH, Weinheim, 2010, ch. 6, p. 199, ISBN: 978-3-527-32351-7 Search PubMed; (b) R. A. J. O’Hair and N. J. Rijs, Acc. Chem. Res., 2015, 48, 329 CrossRef PubMed.
- (a) M. J. Woolley, G. N. Khairallah, G. da Silva, P. S. Donnelly, B. F. Yates and R. A. J. O’Hair, Organometallics, 2013, 32, 6931 CrossRef CAS; (b) M. J. Woolley, G. N. Khairallah, G. da Silva, P. S. Donnelly and R. A. J. O’Hair, Organometallics, 2014, 33, 5185 CrossRef CAS; (c) M. J. Woolley, A. Ariafard, G. N. Khairallah, K. H.-Y. Kwan, P. S. Donnelly, J. M. White, A. J. Canty, B. F. Yates and R. A. J. O’Hair, J. Org. Chem., 2014, 79, 12056 CrossRef CAS PubMed.
- A. Gray, A. Tsybizova and J. Roithová, Chem. Sci., 2015, 6, 5544 RSC.
- (a) N. Rodriguez and L. Goossen, J. Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670 CrossRef CAS; (c) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS; (d) W. I. Dzik, P. P. Lange and L. J. Goossen, Chem. Sci., 2012, 3, 2671 RSC; (e) L. J. Gooßen and K. Gooßen, Top. Organomet. Chem, 2013, 44, 121 CrossRef.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- A. Noor, J. Li, G. N. Khairallah, Z. Li, H. Ghari, A. J. Canty, A. Ariafard, P. S. Donnelly and R. A. J. O’Hair, Chem. Commun., 2017, 53, 3854 RSC.
- Transition metal decarbonylative C–C bond coupling reactions of aldehydes, ketones and carboxylic acid derivatives have been widely studied: A. Dermenci and G. B. Dong, Sci. China: Chem., 2013, 56, 685 CrossRef CAS.
- For transition metal mediated decarbonylation reactions of carboxylic acids that utilize activating anhydride reagents, see: (a) A. John, M. O. Miranda, K. Ding, B. Dereli, M. A. Ortuño, A. M. LaPointe, G. W. Coates, C. J. Cramer and W. B. Tolman, Organometallics, 2016, 35, 2391 CrossRef CAS; (b) M. A. Ortuño, B. Dereli and C. J. Cramer, Inorg. Chem., 2016, 55, 4124 CrossRef PubMed.
- Transition metal mediated decarbonylation reactions of carboxylic acids that do not require activating anhydride reagents have only rarely been described, and it has typically been assumed that they proceed via the coordinated carboxylate: (a) R. H. Prince and K. A. Raspin, Chem. Commun., 1966, 156 RSC; (b) D. Fenton, US Pat., 3530198, 1970 Search PubMed; (c) T. A. Foglia and P. A. Barr, J. Am. Oil Chem. Soc., 1976, 53, 737 CrossRef CAS; (d) A. John, M. A. Hillmyer and W. B. Tolman, Organometallics, 2017, 36, 506 CrossRef CAS.
- For an excellent review of mixed phosphine−phosphine oxide ligands and their metal complexes, see: V. V. Grushin, Chem. Rev., 2004, 104, 1629 CrossRef CAS PubMed.
- For examples of palladium alkoxides, see: G. M. Kapteijn, A. Dervisi, D. M. Grove, H. Kooijman, M. T. Lakin, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1995, 117, 10939 CrossRef CAS.
- For a review on Pd(IV) complexes, see: P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS PubMed.
- L. MacAleese and P. Maître, Mass Spectrom. Rev., 2007, 26, 583 CrossRef CAS PubMed.
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231 RSC.
- Metal-mediated decarbonylation reactions play important roles in the activation of a range of other substrates including the greenhouse gas CO2. For lead references, see: (a) M. Firouzbakht, N. J. Rijs, P. González-Navarrete, M. Schlangen, M. Kaupp and H. Schwarz, Chem. – Eur. J., 2016, 22, 10581 CrossRef CAS PubMed; (b) M. Firouzbakht, M. Schlangen, M. Kaupp and H. Schwarz, J. Catal., 2016, 343, 68 CrossRef CAS; (c) S.-Y. Tang, N. J. Rijs, J. Li, M. Schlangen and H. Schwarz, Chem. – Eur. J., 2015, 21, 8483 CrossRef CAS PubMed.
- (a) B. L. Conley, S. K. Ganesh, J. M. Gonzales, W. J. Tenn, K. J. H. Young, J. Oxgaard, W. A. Goddard and R. A. Periana, J. Am. Chem. Soc., 2006, 128, 9018 CrossRef CAS PubMed; (b) J. Mei, K. M. Carsch, C. R. Freitag, T. B. Gunnoe and T. R. Cundari, J. Am. Chem. Soc., 2013, 135, 424 CrossRef CAS PubMed; (c) J. R. Webb, T. M. Figg, B. M. Otten, T. R. Cundari, T. B. Gunnoe and M. Sabat, Eur. J. Inorg. Chem., 2013, 4515 CrossRef CAS.
- For an example on the use of ESI-MS to monitor the palladium-catalyzed oxidation of phosphines, see: A. V. Hesketh, S. Nowicki, K. Baxter, R. L. Stoddard and J. S. McIndoe, Organometallics, 2015, 34, 3816 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of mass spectrometry experiments and DFT calculations; additional mass spectra and DFT calculated energy diagrams; Cartesian coordinates of all structures. See DOI: 10.1039/c7cc08944a |
| This journal is © The Royal Society of Chemistry 2018 |