
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble uranium oxo cations derived from uranyl by borane or silane reduction†
Bradley E.
Cowie
,
Gary S.
Nichol
 ,
Jason B.
Love
* and
Polly L.
Arnold
*
,
Jason B.
Love
* and
Polly L.
Arnold
*
EaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, The King's Buildings, Edinburgh, EH9 3FJ, UK. E-mail: Jason.Love@ed.ac.uk; Polly.Arnold@ed.ac.uk
First published on 28th March 2018
Abstract
A new type of double uranium oxo cation [O–U–O–U–O]4+ is prepared by selective oxygen-atom abstraction from macrocyclic uranyl complexes using either boranes or silanes. A significant degree of multiple U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding is evident throughout the U2O3 core, but either trans-,cis- or trans-,trans-OUOUO motifs can be isolated as boron- or silicon-capped oxo complexes. Further controlled deoxygenation of the borylated system is also possible.
O bonding is evident throughout the U2O3 core, but either trans-,cis- or trans-,trans-OUOUO motifs can be isolated as boron- or silicon-capped oxo complexes. Further controlled deoxygenation of the borylated system is also possible.
Until recently, the oxo groups of the uranyl(VI) dication, [UO2]2+, which has a linear geometry and short, strong U–O bonds were considered to be very inert.1 However, under anaerobic conditions, one electron reduction of uranyl compounds is now recognised to form uranyl(V) complexes that do not disproportionate, although the reactions rarely proceed further to lower UIV oxidation state.2 Reduction reactions increase the oxo basicity, generating oxo-donor interactions to other Lewis acidic ions. This makes reduced uranium oxo compounds better models for the heavier, highly radioactive transuranic metal actinyl cations [AnO2]n+ (An = Np, Pu; n = 1, 2) for which clustering behaviour is problematic in PUREX separation processes for civil nuclear waste treatment.3–6 Actinide oxo-bridges also facilitate electron-transfer reactions in environmental waste remediation,7–9 enrich the coordination chemistry of actinides in minerals,10 and can generate interesting electronic and magnetic structures.11–13
We originally reported reductive silylation as a route to the first covalent bond formation reaction of a molecular uranyl complex,14 and, with others, have developed generic systems and rules that govern UVI to UV oxo-metalation,15–19vs. oxo-silylation.20,21 Only very rarely does reduction to UIV occur, recently seen using oxophilic, highly reducing UIII cations to form [(UO2I4){UICl(py)4}2], for example.22 Also, exhaustive deoxygenation can convert simple uranyl salts to UIV halides when combinations of alkali metal and Group 1 alkyl reductants, and excesses of halosilanes are used.23,24
Here we show for the first time how simple diboranes are effective reagents for metal oxo-abstraction chemistry and how borane and silane-mediated uranyl reduction, oxo-functionalisation and abstraction, can afford a new U2O3 motif with trans,-cis- or trans,-trans-OUOUO linkages.
The dinuclear uranyl(VI) complex [{UO2(py)}2(LA)] (1; py = pyridine) reacts with two equivalents of the diborane B2pin2 (pin = pinacolate) at 80 °C in pyridine to yield the new paramagnetic complex [(py)(pinBO)UOU(OBpin)(py)(LA)] 2 (Scheme 1, and ESI†). Both [UVIO2]2+ ions in 1 have undergone UVI→UIV reduction and borylation, and a single oxo-atom abstraction, resulting in extrusion of O(Bpin)2. The O(Bpin)2 byproduct gives rise to singlets at 23 and 16 ppm in the 11B NMR spectrum of the reaction solution, in which the highest frequency chemical shift is attributed to the pyridine adduct of OB(pin)2 (see Fig. S16 and S18, ESI†), and accurately identified via an independent synthesis of O(Bpin)2 from B2pin2 and Me3NO in pyridine (see ESI†). In spite of the strength of B–O bonds, diboranes have only rarely been used to deoxygenate molecules, such as pyridine-oxides and phosphine-oxides,25,26o-nitrostyrenes,27 and CO2.28 To our knowledge, this is the first example of their use to deoxygenate a metal complex. Reactions of uranyl with hydrosilanes such as HSiEt3 can produce oxo-silylated UV-OSiR3 products such as [U(OSiR3)2(I)2(PDI)], (PDI is a redox non-innocent pyridinediimine),29 usually requiring a strong Lewis acid activator such as B(C6F5)3, e.g. to form the intermediate [U(OB{C6F5}3)(OSiR3)(dbm)2(THF)] (dbm = OC(Ph)CHC(Ph)O),30 and deoxygenation usually requires more aggressive reagents such as halosilanes.24,29 Complex 1 also reacts with HBpin, forming 2, H2 and O(Bpin)2. However, this reaction requires an excess of HBpin (10 equiv.) and elevated temperatures (125 °C).
 | ||
| Scheme 1 Diborane or silane-mediated deoxygenation of co-axial uranyl dications to form the new tetracations [OUOUO]4+2, 3, and 5, with either trans,-cis- (for boron) or trans,-trans- (for silicon) geometry, and the further deoxygenation of 3 to afford the UOU-containing 4. | ||
Resonances in the 1H NMR spectra of 2 range from +29 to −41 ppm and a significant reduction in the asymmetric OUO stretching frequency is observed, from 912 cm−1 for the uranyl group in 131 to 566 cm−1 in 2. Complex 1 also reacts with the diborane B2cat2 (cat = catecholate) at 80 °C in pyridine, forming a catecholboroxy-analogue of 2, [(py){(py)catBO}UOU(OBcat)(py)(LA)] 3. Similarly to the formation of 2, 3 is the product of UVI→UIV reductive borylation of both U centres, and O-atom extrusion forming O(Bcat)2 which was identified by 11B NMR spectroscopy (singlets at 15 and 9 ppm, Fig. S23 and S25, ESI†); the highest frequency singlet is due to the pyridine adduct of O(Bcat)2 (verified via an independent synthesis from B2cat2 and Me3NO in pyridine, see ESI†). Whereas complex 2 can be isolated cleanly on a preparative scale, 3 transforms slowly into the new catecholate-bridged complex [(py)UOU(μ-O2C6H4)(py)(LA)] 4 which is the product of loss of both boroxy ligands and the addition of a catecholate ligand, [C6H4O2]2−, that bridges the two UIV centres (Scheme 1); upon the addition of a third equivalent of B2cat2 and heating for 48 h at 80 °C, 3 is converted exclusively into 4 in 77% yield. Only very small quantities of 3 have been isolated cleanly by fractional crystallisation. Complexes 3 and 4 may also be obtained from 1 and HBcat and, as with the formation of 2 from HBpin, these reactions require an excess of HBcat (10 equiv.).
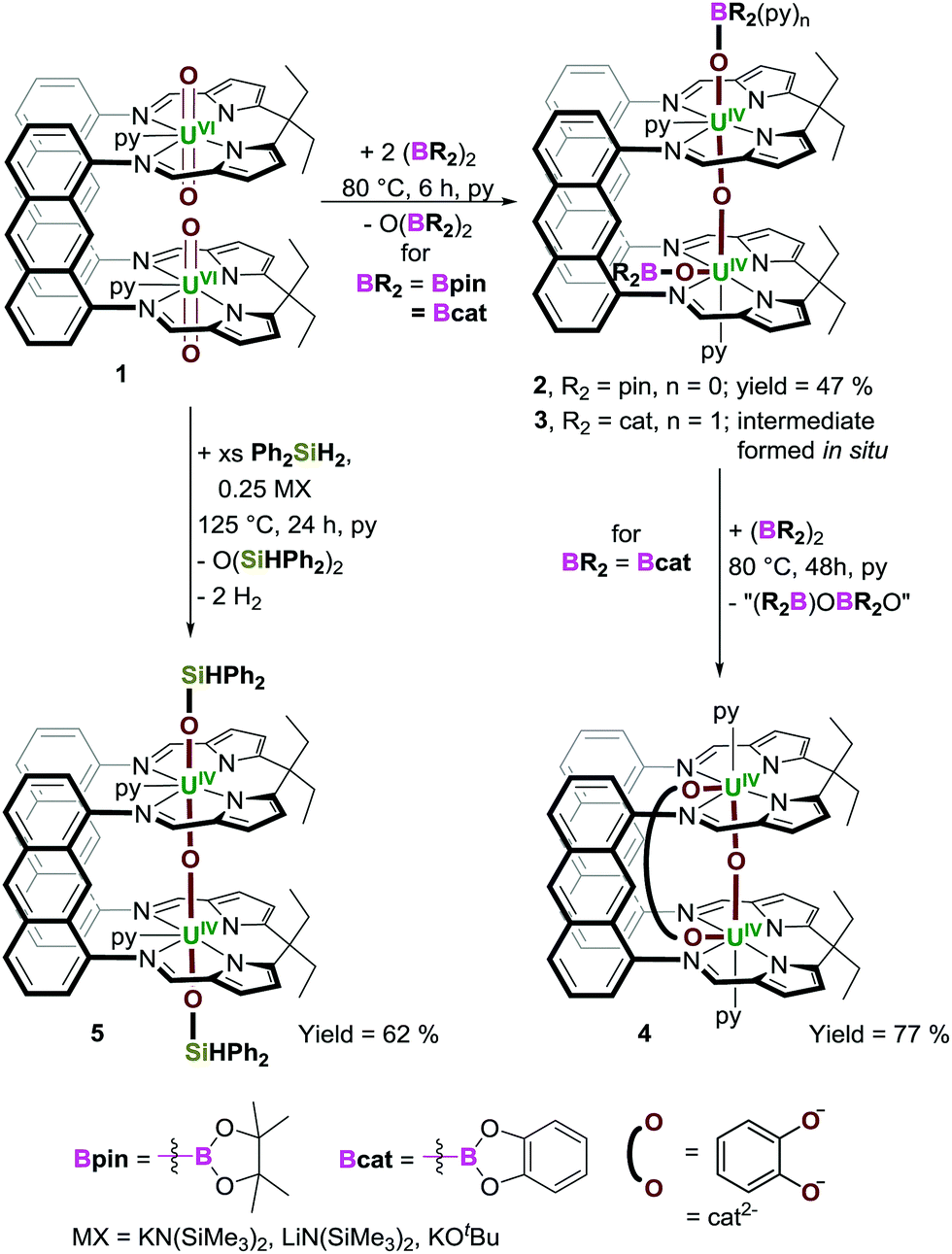
An X-ray diffraction study on single crystals of 2 obtained from slow evaporation of a toluene solution at room temperature (Fig. 1a) shows it comprises two exogenous boroxide ligands and that one endo-oxo atom has been eliminated, with the remaining forming a fused UIV–O–UIV core that is essentially linear (U(1)–Oendo–U(2) = 176.2(1)°). The U–Oexo bond lengths are significantly longer than the U–Oendo bonds; the U(1)–O(1) and U(2)–O(2) bond lengths are 2.161(2) and 2.172(2) Å, respectively, whereas the U(1)–O(3) and U(2)–O(3) bond lengths are 2.139(2) and 2.112(2) Å, respectively. Of greatest interest is that one reduced OUO dication retains the trans-(pinB)OUO geometry (O(1)–U(1)–O(3) = 169.05(8)°), but the other has rearranged to a cis-OUO configuration with a O(2)–U(2)–O(3) angle of 96.51(7)°. The trans,-cis-configurations of 2 and 3 are retained in solution – the 11B NMR spectrum of 2 shows two resonances at 475 and 221 ppm (496 and 126 ppm for 3), and two sets of Bpin–CH3 resonances are seen in the 1H NMR spectra of 2.
 | ||
| Fig. 1 Solid-state structures of 2·2toluene (a) and 3·THF (b). Displacement ellipsoids are drawn with 50% probability, and carbon atoms of LA and U-coordinated solvent molecules drawn wireframe. For clarity, hydrogen atoms, lattice solvent, and lower-fractional occupancy disorder components of the OBpin ligand (B(1)), and LA ethyl groups of 2·2toluene are omitted. | ||
X-ray quality crystals of 3·THF were obtained by diffusion of hexanes vapour into a THF solution of a dried, crude reaction product mixture containing ca. 90% 3, Fig. 1b. The core is similar to 2, Fig. 1a, possessing axial and equatorial boroxides; the O(1)–U(1)–O(3) and O(2)–U(2)–O(3) bond angles are 170.7(1) and 99.2(1)°, respectively. However, the catBO-ligand that is axially coordinated to U(1) in 3 contains an additional pyridine donor, hampering direct comparison with 2 and resulting in a relative contraction of the U–Oexo bond length in the py-solvated half of the structure, (U(1)–O(1) = 2.092(2) Å; U(2)–O(2) = 2.219(2) Å), and elongation of the B–Oexo bond lengths (B(1)–O(1) = 1.400(5) Å; B(2)–O(2) = 1.315(5) Å) and of the U–Oendo (U(1)–O(3) = 2.176(2) Å; U(2)–O(3) = 2.068(2) Å), presumably as a result of the lower Lewis acidity of B(1) than B(2).
The formation of 4 could occur via the extrusion of two equivalents of [OBcat], which would presumably form an insoluble boroxide polymer. Both complexes 3 and 4 have paramagnetically shifted 1H NMR spectra (resonances ranging from ca. +70 to −60 ppm), and the FTIR spectrum of reaction solutions that contain predominantly 3 has bands at 580 and 531 cm−1, which are tentatively assigned as OUO stretches by comparison with 2.
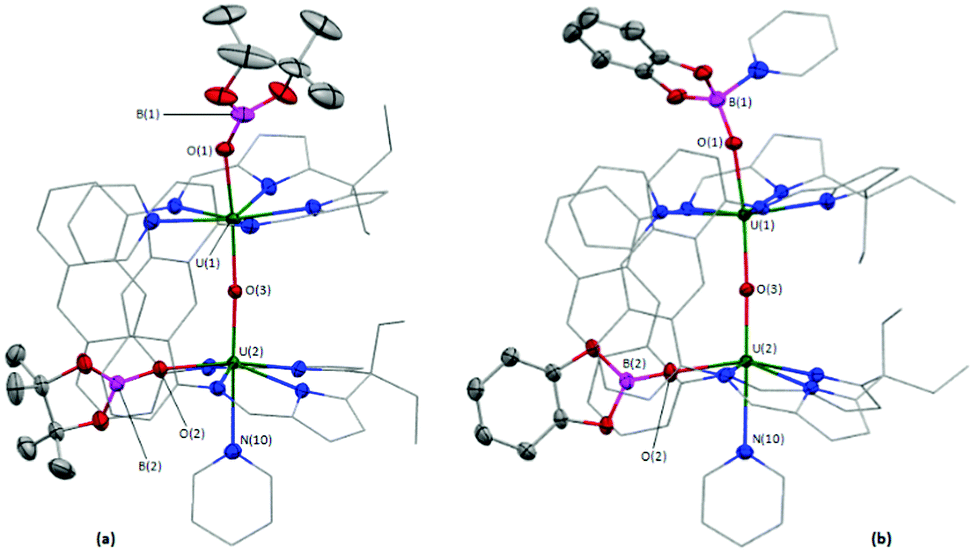
In the solid-state (Fig. 2a), 4 has crystallographically imposed mirror symmetry, with the central oxygen atom of the complex (O(3)) positioned on the mirror plane. It possesses a short U(1)–O(3) bond length of 2.090(2) Å and U(1)–O(1)/O(1′) bonds to the bridging catecholate ligand of 2.128(3) Å which, combined with the C(64)–O(1) bond length of 1.340(6) Å, indicate two UIV centres and a dianionic catecholate ligand.32 The U–Oendo–U angle in 4 (142.3(3)°) is significantly more acute than that in 2 and 3, resulting in a close approach of the two U centres (3.956 Å in 4versus 4.248 and 4.243 Å in 2 and 3, respectively).
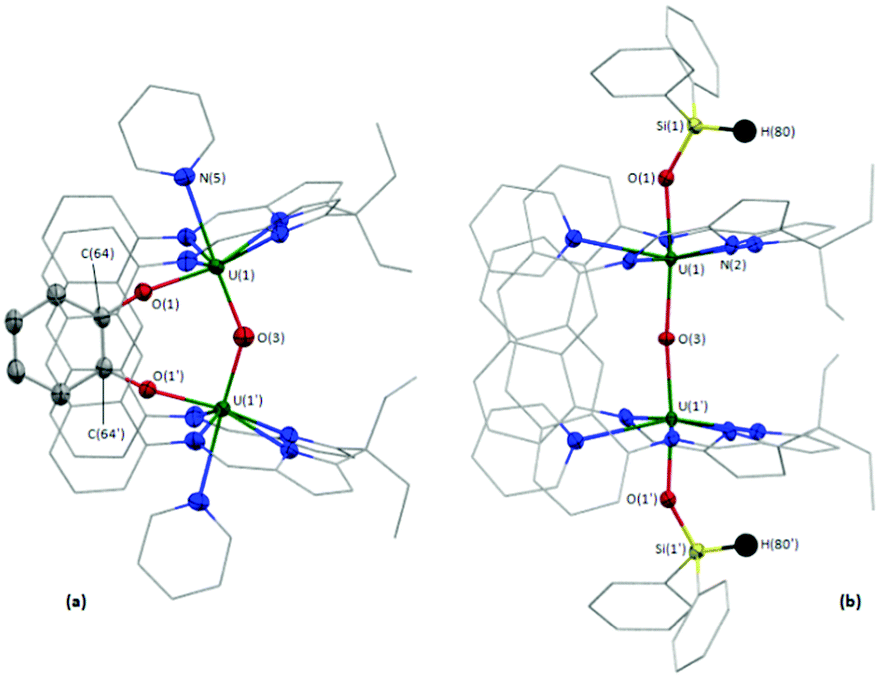 | ||
| Fig. 2 Solid-state structures of 4·5.5THF (a) and 5·py (b). Displacement ellipsoids are drawn at 50% probability, and carbon atoms of LA, U-coordinated solvent and SiPh2 groups drawn wireframe. For clarity, hydrogen atoms (except for H(80) and H(80′) of 5·py), the 50% occupancy disorder of the methyl carbon atom C(8) of 4·5.5THF, and lattice solvent are omitted. Atoms with a prime (′) character in the atom labels are located at equivalent positions: (x, 1/2 − y, z) for 4·5.5THF (a) and (1 − x, y, 3/2 − z) for 5·py (b). | ||
It was envisaged that 1 could react with other p-block reactants aside from diboranes, driven by the formation of strong new O–E bonds (E = p-block element). While 1 does not react with phosphines and stannanes (HPPh2, P2Ph4, HSnPh3, Sn2Me6, Sn2Ph6), it reacts with the silane Ph2SiH2 over 24 h at 125 °C to form [(py)(HPh2SiO)UOU(OSiPh2H)(py)(LA)] (5; Scheme 1).33 Complex 1 also reacts with PhSiH3, Si2Me6, Ph3SiH and Si2Ph2Me4. Reactions with the former two silanes lead to the formation of unidentified, insoluble precipitates, whereas the latter two resulted in decomposition into [UO2(py)(H2LA)] and unidentified uranyl-containing species.
Complex 5 is a siloxy-analogue of 2 and 3 and is only formed in the presence of a catalytic amount (25 mol%) of an alkali-metal salt such as KN(SiMe3)2, LiN(SiMe3)2 or KOtBu, which is suggestive of a hypervalent silicate facilitating bond homolysis. The simple Lewis acids such as BF3(OEt2) or B(C6F5)3 do not catalyse these reactions (see ESI†). The formation of 5 likely occurs in an analogous fashion to 2 and 3, but with Si–H bond homolysis driven by the formation of strong Si–O bonds, and release of H2 and O(SiHPh2)2 as reaction by-products. Indeed, when monitoring the reaction by 1H NMR spectroscopy, H2 was observed (4.31 ppm in d5-pyridine) although it was not possible to identify Si-containing by-products in the 29Si NMR spectrum, so these may be undergoing further condensation/catenation reactions.
The chemical shifts in the 1H NMR spectrum of paramagnetic 5 span +64 to −41 ppm, and no obvious high-frequency asymmetric OUO stretch is found in the FTIR spectrum, consistent with a UIV formal oxidation state. The trans-,trans-symmetry of this silyl-capped ion is retained in the solution, but may be enforced by an inability of the large silyl-groups to fit between the anthracenyl spacers of LA.
The solid-state structure of the pyridine solvate of 5·py (see the ESI† for the THF solvate) was obtained. 5·py possesses crystallographically imposed two-fold symmetry, with the central oxygen O(3) positioned on the two-fold axis, and shows a near linear U–O–U bond angle of 173.1(2)° (Fig. 2b), similar to 2 and 3. However, in contrast to the B-capped compounds, the exo-oxo-siloxides both remain trans with nearly linear O(1)–U(1)–O(3) and O(1′)–U(1′)–O(3) angles (172.09(9)°). The U(1)–O(1) and U(1)/U(1′)–O(3) bond lengths are 2.142(2) and 2.1486(3) Å, in good agreement with the R2BO–U bond lengths in 2 and 3, and with the U–O bond lengths in 4.
The U–O bond lengths in 2–5 range from 2.068(2)–2.219(2) Å, which are significantly elongated relative to the U–O bond length anticipated for [UVIO2]2+ (shorter than 1.80 Å) and [UVO2]+ complexes (∼1.85–1.95 Å),34,35 and are similar to those in the [UIVO2] complexes [(Ph2HSiO)2UCl2(OPPh3)2] (2.120(5) Å),23 [(Me3SiO)2UI2(bipy)2] (2.084(4) Å; bipy = 4,4′-bipyridine),36 [Cp2Co][{(C6F5)3BO}{Me3SiO}U(Aracnac)2] (U–O(siloxy) = 2.173(8) Å; Ar = C6H3-3,5-tBu2),37 [(UO2I4){DyI(py)5}2] (2.058(3) and 2.068(3) Å), [(UO2I4){UICl(py)4}2] (2.166(5) Å)22 and [(Cp2ClTiO)2UCl(L)] (2.062(7) and 2.066(7) Å; L = a monoanionic acyclic diimino-dipyrrin ligand),2 all of which derive from UVI→UIV reductive functionalisation of the uranyl(VI) ion. Furthermore, the average U–Npyrrolide/imine bond length in complexes 2–5 is 2.548 Å, which is longer than those see in uranyl(V)– (2.525 Å)14–16,19,20 and uranyl(VI)–pacman complexes (2.487 Å).12,15
The reductive deoxygenation of 1 by the diborane is a new reaction type and a mechanism would likely involve reaction at the most accessible exo-oxo ligands, with B–B bond homolysis forming UV–OBR2 and releasing BR2 which can either abstract H atoms from solvent, or react with the other uranyl exo-oxo. This will result in a reduced, UV intermediate [R2BOUV(O)2UVOBR2]4+ ion with elongated UVOendo bonds that now have greater oxo-basicity, facilitating the electron transfer required for one endo-oxo to form a covalent μ-oxo-bridge between the two U centres. The proposed di(boroxide),di(μ-oxo) intermediate is an analogue of the [Me3SiOU(μ-O)2UOSiMe3]4+ core seen previously.21 The catecholate dianion in Bcat enables a further deoxygenation by the B atoms resulting in the conversion of 3 to 4. The reaction of 1 with Ph2SiH2 presumably involves activation of the oxo group as a Lewis base through hypervalent silicate formation.20,38
Significantly, the use of a large spacer in the compartmental macrocycle LA to enforce proximal co-linearity in uranyl(VI) coordination31 has enabled the first reductive fusion of two uranyl dications into a single, double-uranium containing cation, and the diboranes B2pin2 and B2cat2 have been shown for the first time to be capable oxo-atom abstraction reagents; this latter feature should have a widespread utility for the deoxygenation of d-block metal oxo complexes. Both borane and silane reagents have allowed an unusually high degree of uranyl reduction, with the [OUOUO]4+ core existing in either trans-,trans-linear, or trans-,cis-bent conformation. The reaction that transforms complex 3 into the catechol-bridged diuranium(IV) complex 4 suggests that further reaction chemistry of these dinuclear uranium complexes will be possible. Work is in progress to explore the level of electronic coupling between the metal centres in all of these complexes, and to explore whether analogous oxo-ion fusion chemistry is possible for the actinyl cations of neptunium and plutonium, [AnO2]n+.
The authors thank the EPSRC-UK grants EP/N022122/1 and EP/M010554/1, the European Commission Directorate General and Actinet JRC Userlab (ACTINET-I3-CP-CSA-JRP-232631) and the Natural Sciences and Engineering Research Council of Canada for an NSERC Post-Doctoral Fellowship (BEC). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 740311).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC.
- G. R. Choppin, J. Radioanal. Nucl. Chem., 2007, 273, 695–703 CrossRef CAS.
- S. D. Reilly and M. P. Neu, Inorg. Chem., 2006, 45, 1839–1846 CrossRef CAS PubMed.
- S. Skanthakumar, M. R. Antonio and L. Soderholm, Inorg. Chem., 2008, 47, 4591–4595 CrossRef CAS PubMed.
- S. M. Cornet, L. J. L. Haller, M. J. Sarsfield, D. Collison, M. Helliwell, I. May and N. Kaltsoyannis, Chem. Commun., 2009, 917–919 RSC.
- M. Bühl and G. Schreckenbach, Inorg. Chem., 2010, 49, 3821–3827 CrossRef PubMed.
- C. Noubactep, G. Meinrath, P. Dietrich and B. Merkel, Environ. Sci. Technol., 2003, 37, 4304–4308 CrossRef CAS PubMed.
- V. Vallet, U. Wahlgren, B. Schimmelpfennig, Z. Szabo and I. Grenthe, J. Am. Chem. Soc., 2001, 123, 11999–12008 CrossRef CAS PubMed.
- M. J. Wilkins, F. R. Livens, D. J. Vaughan and J. R. Lloyd, Biogeochemistry, 2006, 78, 125–150 CrossRef CAS.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–317 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A.-F. Pécharman, N. Magnani, J.-C. Griveau, E. Colineau, R. Caciuffo, X. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12797–12801 CrossRef CAS PubMed.
- M. Zegke, G. S. Nichol, P. L. Arnold and J. B. Love, Chem. Commun., 2015, 51, 5876–5879 RSC.
- L. Natrajan, F. Burdet, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2006, 128, 7152–7153 CrossRef CAS PubMed.
- J.-C. Berthet, G. Siffredi, P. Thuery and M. Ephritikhine, Chem. Commun., 2006, 3184–3186 RSC.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J.-C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman, R. M. Lord, G. M. Jones, E. Hollis, G. S. Nichol, L. Maron, J. Fang, T. Davin and J. B. Love, Inorg. Chem., 2015, 54, 3702–3710 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- P. L. Arnold, B. E. Cowie, M. Suvova, M. Zegke, N. Magnani, E. Colineau, J.-C. Griveau, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2017, 56, 10775–10779 CrossRef CAS PubMed.
- J. J. Kiernicki, M. Zeller and S. C. Bart, Angew. Chem., Int. Ed., 2017, 56, 1097–1100 CrossRef CAS PubMed.
- J.-C. Berthet, G. Siffredi, P. Thuéry and M. Ephritikhine, Eur. J. Inorg. Chem., 2007, 4017–4020 CrossRef CAS.
- S. Bae and M. K. Lakshman, J. Org. Chem., 2008, 73, 1311–1319 CrossRef CAS PubMed.
- S. Hawkeswood and D. W. Stephan, Dalton Trans., 2005, 2182–2187 RSC.
- K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088–4091 CrossRef CAS PubMed.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- J. J. Kiernicki, D. P. Cladis, P. E. Fanwick, M. Zeller and S. C. Bart, J. Am. Chem. Soc., 2015, 137, 11115–11125 CrossRef CAS PubMed.
- E. A. Pedrick, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 12237–12239 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, Q. J. Pan, G. Schreckenbach and J. B. Love, Dalton Trans., 2012, 41, 6595–6597 RSC.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251–1260 CrossRef CAS PubMed.
- Complex 1 also reacts with PhSiH3 or Si2Me6 to yield unidentified, insoluble precipitates. Reactions with either Ph3SiH, Et3SiH or Si2Ph2Me4 result in the loss of one of the uranyl ions from 1, affording poor yields of the known mono-substituted [UO2(py)(H2LA)] reported previously by us.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- J. L. Brown, C. C. Mokhtarzadeh, J. M. Lever, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 5105–5112 CrossRef CAS PubMed.
- D. D. Schnaars, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 4695–4697 CrossRef CAS PubMed.
- E. A. Pedrick, G. Wu and T. W. Hayton, Inorg. Chem., 2015, 54, 7038–7044 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and characterisation details, and further IR spectroscopic and X-ray crystallographic data. CCDC 1812761–1812765. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc00341f |
| This journal is © The Royal Society of Chemistry 2018 |