
Unexpected halide anion binding modes in meso-bis-ethynyl picket-calix[4]pyrroles: effects of meso-π (ethynyl) extension†
Ranjan
Dutta
a,
Dikhi
Firmansyah
a,
Jihoon
Kim
a,
Hongil
Jo
b,
Kang Min
Ok
 b and
Chang-Hee
Lee
*a
b and
Chang-Hee
Lee
*a
aDepartment of Chemistry, Kangwon National University, Chun Cheon 24341, Korea. E-mail: chhlee@kangwon.ac.kr
bDepartment of Chemistry, Chung-Ang University, Seoul, 06974, Korea
First published on 21st June 2018
Abstract
meso-Ethynyl extended aryl-picket calix[4]pyrroles 2 and 3 are designed and synthesized by directly anchoring arylethynyl groups at diametrically opposed meso-positions. The critical roles of direct ethynyl linkers are manifested through the isolation of unexpected host–anion conformers of meso-arylethynyl calix[4]pyrroles and a significant enhancement in halide binding affinities.
A ‘Picket Calix[4]pyrrole’ represents a calix[4]pyrrole bearing aryl groups at diametrical meso-positions in a cis-fashion when it becomes a cone conformation. The cavity created by the tetrapyrrolic core along with the axial aryl groups is suitable for inclusion of mono and polyatomic anions. In recent years, a variety of picket calix[4]pyrroles possessing various aryl groups at diametrical meso-positions have been developed and explored for anion recognition, sensing and transport in some cases.1 For instance, Ballester and co-workers have extensively studied energetics between differently substituted aromatic moieties of picket calix[4]pyrroles and anions.2 We have investigated anion recognition properties of these classes of hosts using a fluorescent dye displacement assay technique.3 As a result, a fluorescence ‘Turn-on’ type of recognition and selective sensing of different anions using various meso-aryl picket calix[4]pyrroles have been reported. For example, high fluoride anion selectivity was seen upon introduction of 4-fluorophenyl groups as a meso-picket.3a Exceptionally high affinity toward dihydrogen pyrophosphate anions was observed when 4-methyl pyridinium groups were introduced as a picket component.3b Most recently, high selectivity toward bicarbonate anions was reported when 2-benzimidazolium groups were introduced as a picket component.3c Due to the conformationally flexible natural and low energy barrier between different conformers, calix[4]pyrroles adopt many different conformations depending on the solvents and the meso-substituents. For instance, octamethylcalix[4]pyrrole 1 exists as a 1,3-alternate conformer in the solid state wherein the adjacent pyrrole rings are oriented in the opposite direction.4 Theoretical calculation reveals that the stability order between the different conformers in 1 is: 1,3-alternate > partial cone > 1,2-alternate > cone conformation.5 The macrocycle usually adopts a cone conformation upon complexation with anions. In the case of the meso-aryl picket calix[4]pyrroles, the anions usually occupy the deep pocket created by two axially positioned meso-aryl groups. The tetrapyrrole moiety and the meso-aryl substituents undergo dramatic changes in spatial orientation during the complexation process. This type of pocket-side binding along with the conformational changes of the host molecules has been reported with a wide variety of meso-aryl substituted calix[4]pyrroles.6 The trend of deep pocket binding holds even in the case of the hexyl armed calix[4]pyrrole in chloride anion complexation.7 DFT calculations also show this binding conformation to be the most stable. In order to clarify the binding modes of the meso-picket calix[4]pyrroles upon anion binding, we design and synthesize meso-picket calix[4]pyrroles 2, 3, 4 and 5 as shown in Fig. 1. Indeed, the detailed binding studies showed unexpected anion binding modes depending on the nature of the meso-pickets. We found that the aryl ethynyl arms occupy the equatorial positions in the sold state and the anion binds to the opposite side of the pocket in cases of 2 and 3. Systematic binding studies were performed for an in-depth understanding of this unexpected binding modes in meso-ethynyl extended calix[4]pyrroles 2 and 3.
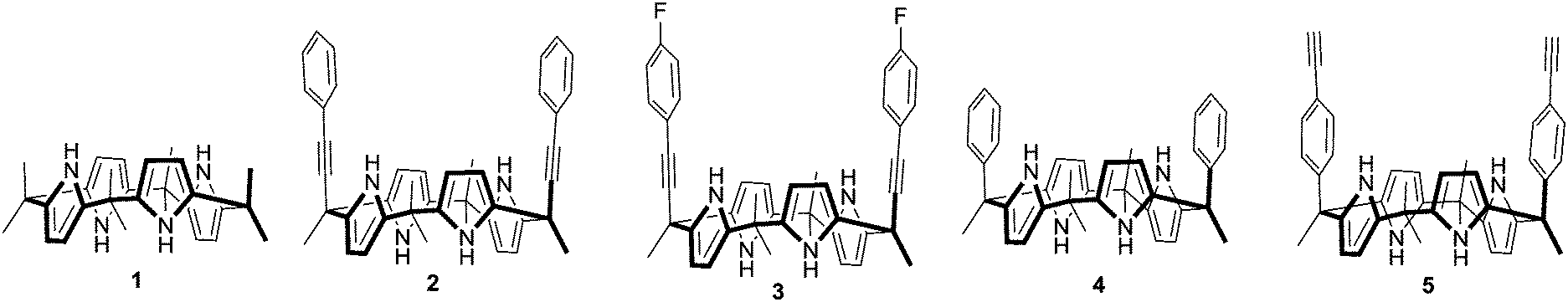 | ||
| Fig. 1 Chemical structures of calix[4]pyrroles 1, 2, 3, 4 and 5. | ||
The synthesis of receptor 2 is outlined in Scheme S1 (ESI†). Briefly, meso-[(2-ethynylphenyl)methyl]dipyrromethane (8) is synthesized in 52% yield by acid-catalyzed condensation of 4-phenyl-3-butyn-2-one, 6, with freshly distilled pyrrole at 0 °C. It is worth mentioning that a longer reaction time at elevated temperature (rt) led to the formation of meso-([2-phenyl]methyl)dipyrromethane 10 and 11 presumably via the cleavage of triple bonds thorough Meyer–Schuster rearrangement8 followed by retro Aldol type reaction (ESI†). Compounds 10 and 11 were isolated and verified. Mechanistic details of such a transformation are provided in the ESI.†
Condensation of 8 with excess of acetone in the presence of BF3·Et2O as a catalyst afforded cis- and trans-isomers as a mixture. Repeated column chromatography on silica gel yielded pure 2 in 7% yield, with characteristic meso-methyl protons as three distinct singlets in a 1/1/1 ratio in the 1H NMR spectrum (ESI†). Structural analysis of single crystals of compound 2 obtained from a solvent mixture (ethyl acetate/hexane) revealed that the calix[4]pyrrole core adopted a 1,3-alternate conformation in the solid state and the phenyl ethynyl pickets adopted pseudo-axial conformation, whereas a partial cone conformer with bound solvents was obtained when the crystals were grown from acetone/methanol (ESI†). In this case one of the meso-phenylethynyl groups occupied the axial position. Receptor 3 bearing a meso-parafluorophenylethynyl picket was synthesized in 12% yield and characterized following a similar protocol (ESI†). Hosts 42c and 59 were synthesized for a comparison study and will provide crucial information regarding the electronic and positional effects of ethynyl groups on anion binding.
Solution state anion binding studies of receptor 2 with various halide anions (as their TBA salt) were performed using 1H NMR spectroscopy in CD3CN. Incremental addition of the fluoride anion (0.25 equiv.) to a solution of 2 in CD3CN (∼10 mM) induced the splitting of the pyrrolic N-Hs signals into two sets along with a large downfield shift (Δδ = 5.40 ppm). Upon the addition of ca. 1.0 equiv. of F−, the signals corresponding to the free host disappeared completely (ESI†). This result indicated slow complexation/decomplexation kinetics with high binding affinity. Noticeably, a 0.13 ppm downfield shift of the ortho-C–Hs of the meso-phenyl groups was observed while other phenyl–Hs remained unperturbed. These results ruled out the presence of any anion–π interactions which are generally associated with the up-field shift of aryl protons. All the meso-methyl protons endured a slight downfield shift during titration. These findings suggested an equatorial alignment of meso-phenylethynyl groups rather than an axial orientation which is unprecedented in the anion binding of the meso-aryl picket calix[4]pyrrole family. Fairly similar observations were noted during 1H NMR titration with the chloride anion (ESI†). The complexation induced chemical shift change was calculated to be 3.85 ppm (Δδ) for chloride with slow complexation–decomplexation kinetics. Moreover, the change of counter cations from tetrabutylammonium to tetraethylammonium did not alter the binding mode as clearly evident from the 1H NMR titration with tetraethylammonium chloride (ESI†). On the other hand, broadening of NH signals along with a concomitant downfield shift was observed during titration with the bromide anion, which was saturated after the addition of one equiv. of bromide anion (ESI†). The chemical shift changes of aryl and meso-methyl protons were consistent with the equatorial alignment of meso-phenylethynyl groups, while a small yet detectable downfield shift of NH protons of 2 was observed during titration with the iodide anion (ESI†). Only a few picket calix[4]pyrroles with highly electron withdrawing meso-aryl substituents were known to display a response to the iodide anion.2c This indicated that receptor 2 shows high affinity towards anions in general. Similar observations were noted during titration of receptor 3 with halides. The equatorial alignment of meso-parafluorophenylethynyl pickets was evident from the chemical shift changes of aryl-CH and meso-methyl protons. Details of titrations for 3 with different halides are provided in the ESI.†
In the case of meso-phenyl calix[4]pyrrole 4, upon incremental addition of the fluoride anion (as its TBA salt), the pyrrolic NH resonance first disappeared due to peak broadening and then reappeared upon addition of ∼1.0 equiv. of fluoride anion along with a concomitant downfield shift (ESI†). 0.25 and 0.11 ppm up-field shifts were observed for the proton signals corresponding to phenyl groups indicating the interaction of the fluoride anion with the face of the π-surface of the phenyl ring but not with the C–H groups involving CH·F− hydrogen bonds. The up-field shift observed for the signal corresponding to methyl protons (set a) provides additional support for the existence of anion–π interactions operating between the phenyl ring and the bound F− anion. Similar 1H NMR spectral changes were noted with the chloride anion indicating the pocket side binding of the anion (ESI†). Relatively smaller downfield changes in NH protons were observed in the case of bromide indicating weaker hydrogen bonding interactions than fluoride and chloride anions. However, pocket side binding of bromide in association with anion–π interaction was evident from the 1H NMR spectral changes of meso-aryl and meso-methyl protons. The 1H NMR titration spectral pattern of 5 with chloride and bromide anions was consistent with the trend observed in the case of 4 (ESI†). Pocket side binding of anions was established as the preferred binding mode in cases of meso-aryl picket calix[4]pyrrole receptors with a variety of aryl groups. Thus, the observed halide binding mode in the case of receptors 2 and 3 is unique in the meso-substituted calix[4]pyrrole family.
The stability constants and Gibbs free energy (ΔG) of binding for the 1/1 stoichiometric complexes of 2 and 3 with halide anions were determined by isothermal titration calorimetry (ITC) in acetonitrile at 298 K and are summarized in Tables S1 and S2 (ESI†). The highest binding affinity was observed for the fluoride anion, benefitting from a favorable entropic term. The binding affinity values followed the order F− > Cl−> Br− > I− as expected for typical calix[4]pyrroles. Interestingly, the binding affinity values of 2 were much higher than those of its meso-phenyl congener, 4 (Table 1). For example, sixteen fold enhancements in chloride binding affinity were calculated for 2 over 4. Similarly, nine fold increments in bromide binding affinity were calculated for 2 over 4. The measured halide binding affinities of receptor 3 also followed the trend observed for 2. For example, twenty two and fourteen fold increments in chloride and bromide binding affinity values were calculated respectively for 3 over 4. Noticeably, two fold enhancements in chloride and bromide binding affinities were measured respectively for 2 over meso-octamethylcalix[4]pyrrole 1. Differences in binding energies between picket calix[4]pyrroles and octamethylcalix[4]pyrrole (no picket) were the direct consequence of attractive or repulsive anion–π interactions between the anion and aromatic walls.2a–c Taking the chloride anion as a representative case, the higher affinity of 2 compared to 1 clearly indicated that no repulsive anion–π interaction was present between phenylethynyl groups and chloride, indeed no anion–π interaction was operating in the case of 2 (vide supra). Two other factors also contributed to the observed higher binding affinity of 3 for halides. In general, the binding energies of calix[4]pyrroles are the outcome of primary hydrogen bonding interactions between the anion and the tetrapyrrolic core. The complexation induced chemical shift value for 2 with chloride was found to be 3.85 ppm (Δδ) which is significantly higher than the values (Δδ = 3.0–3.23 ppm) found in cases of picket calix[4]pyrroles with varying degrees of meso-substitutions.2c We postulate that the tetrapyrrolic core of 2 offered much stronger hydrogen bond donors for anions compared to the picket calix[4]pyrroles reported thus far. Further evidence of such stronger hydrogen bonding interactions came from the relatively shorter N⋯Cl bond distances of the crystal structure of the chloride complex of 2. Moreover, the additional CH⋯Cl interactions in the case of 2 contributed to the enhanced binding affinity. However, the effect of the counter cation on the halide binding affinity value for 2 was minimal. For instance, a slightly higher binding affinity (∼1.3 fold) is calculated for 2 with chloride from ITC measurement when the counter cation was changed from TEA to TBA species (ESI†). On the other hand, meso-phenyl calix[4]pyrrole 4 displayed relatively lower binding affinity than 2 as a consequence of repulsive anion–π interactions between phenyl groups and halides (Table 1).2c Calix[4]pyrrole 5 bearing terminal ethynyl groups showed slightly higher binding affinity to chloride and bromide than 4 (Table 1). Thus, the higher anion binding affinity of 2, 3 and 5 compared to 4 indicated the electron withdrawing nature of ethynyl groups. However, the lower affinity of 5 compared to 1 suggested that repulsive anion–π interaction is still operational between ethynylphenyl groups and halides albeit to a lower extent.
Unambiguous confirmation of halide binding was obtained from the X-ray crystal structure analysis of the chloride complex of 2 (complex 1). The crystal structure analysis of complex 1 [(2)·(TEACl)] revealed that the tetrapyrrolic core adopted a cone conformation to encapsulate the chloride anion (Fig. 2). The tetrapyrrolic core offered four strong hydrogen bond donors for the chloride anion. The average N–Cl bond distances and N–H⋯Cl bond angles were calculated to be 3.26 Å and 174° respectively (ESI†). Notably, several CH⋯Cl interactions contributed to chloride binding. Intermolecular CH⋯Cl interactions were observed between the ortho-CH protons of two phenylethynyl groups and chloride anions which further ruled out the presence of anion–π interactions between the aryl groups and anions (vide infra). The TEA countercation occupied the π-rich cavity generated by four pyrrole groups. Further evidence of such a unique conformation is obtained from the crystal structure analysis of the fluoride complex of 3 [complex 2, (3·TBAF·H2O·CH3CN)]. Single crystals of complex 2 revealed the trapping of the monohydrated fluoride anion by 3 (Fig. 2). Notably, meso-parafluorophenylethynyl groups were oriented in pseudo-equatorial positions. Details of hydrogen bonding parameters for complex 2 are provided in the ESI.† Significant conformational differences were noted in the orientation of the meso-arylethylnyl groups compared to the trend observed for meso-aryl picket calix[4]pyrroles. Generally, the meso-aryl groups of picket calix[4]pyrroles adopt a face-to-face orientation to create a pocket for anions, while the meso-methyl groups occupy the equatorial positions. In cases of complexes 1 and 2, meso-methyl groups are axially oriented while the meso-arylethynyl groups are pointed equatorially. The binding of halide anions occurred in this cavity side rather than the pocket side created by meso-arylethynyl groups. A possible explanation for such a unique spatial arrangement of aryl groups could be obtained from one of the crystal structures of 2. Crystals of the partial cone conformer of 2 was obtained with a bound water molecule where one of the meso-phenylethynyl groups occupied the axial position (ESI†). Distance from the bound water (hydrogen bond acceptor) to the centroid of the phenyl group is clearly out of range for any weak interaction. In this analogy, anion–π interaction is ruled out between the phenyl group and chloride in this conformation. However, repulsive interaction between the anion and π-cloud of the triple bond is likely as evident from the contact distance (ESI†). Such a repulsive interaction could enforce the meso-phenylethynyl groups to orient towards the equatorial direction. The lesser steric repulsion could also contribute to the observed equatorial orientation of meso-arylethynyl groups in cases of 2 and 3. The proposed anion binding mechanisms for meso-aryl and meso-arylethynyl calix[4]pyrroles are given in the ESI.†
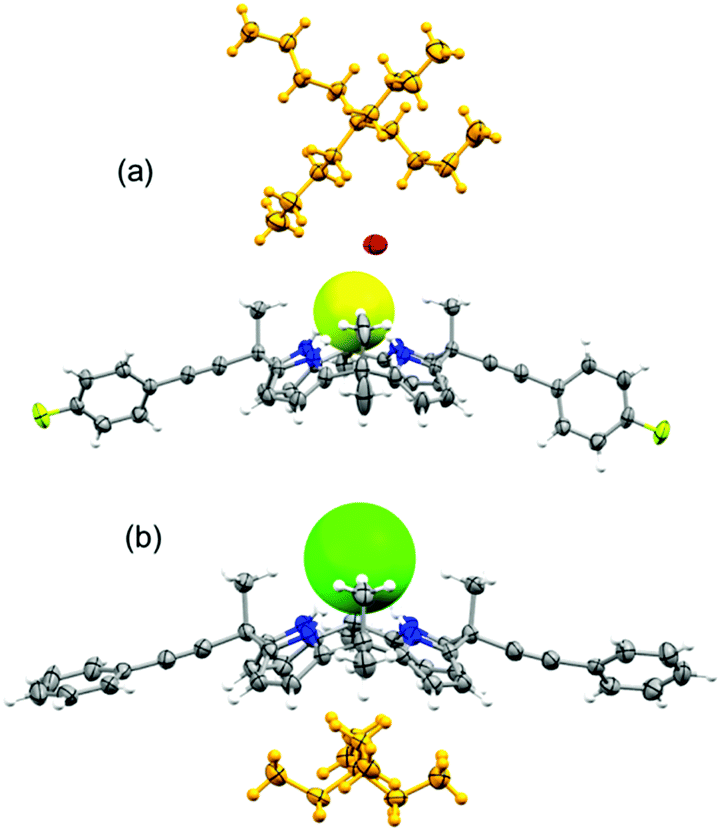 | ||
| Fig. 2 Crystal structures of the TBAF complex of 3 (a) and the TEACl complex of 2 (b). | ||
In summary, we have designed and synthesized new picket calix[4]pyrroles bearing arylethynyl groups at diametrically opposed meso-positions. These alkynyl extended calix[4]pyrroles, 2 and 3, are established as superior halide anion receptors to their analogous meso-aryl congener. Single crystal X-ray structure analysis and solution state binding studies of the halide–host complexes demonstrate a pseudo-equatorial alignment of the meso-arylethynyl groups, which are unprecedented and quite different from the meso-aryl calix[4]pyrroles reported thus far. These results indicate that the repulsive anion–alkyne interaction is stronger so that the equilibrium favours the unexpected conformer. Further exploration of the anion–triple bond interaction paradigm in conjunction with analogous receptors with varying substituents on the ethynylaryl groups is currently in progress.
Support from the Basic Science Research Program (2015R1A2A1A10052586) funded by the National Research Foundation (NRF) under the Ministry of Science, ICT & Future Planning of Korea is acknowledged. The Central Laboratory at KNU and the University-Industry Cooperation Foundation at KNU (D1000436-01-01) are also acknowledged.
Conflicts of interest
There are no conflicts to declare.References
- (a) D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC; (b) I. Saha, J. T. Lee and C.-H. Lee, Eur. J. Org. Chem., 2015, 3859–3885 CrossRef; (c) C.-H. Lee, Bull. Korean Chem. Soc., 2011, 32, 768–778 CrossRef.
- (a) G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Angew. Chem., Int. Ed., 2008, 47, 4114–4118 CrossRef PubMed; (b) L. Adriaenssens, C. Estarellas, A. V. Jentzsch, M. M. Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef PubMed; (c) L. Adriaenssens, G. Gil-Ramírez, A. Frontera, D. Quiñonero, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2014, 136, 3208–3218 CrossRef PubMed.
- (a) P. Sokkalingam, J. Yoo, H. Hwang, P. H. Lee, Y. M. Jung and C.-H. Lee, Eur. J. Org. Chem., 2011, 2911–2915 CrossRef; (b) P. Sokkalingam, D. S. Kim, H. Hwang, J. L. Sessler and C.-H. Lee, Chem. Sci., 2012, 3, 1819–1824 RSC; (c) D. Sareen, J. H. Lee, H. Hwang, S. Yoo and C.-H. Lee, Chem. Commun., 2016, 52, 5852–5855 RSC; (d) E. Mulugeta, Q. He, D. Sareen, S.-J. Hong, J. H. Oh, V. M. Lynch, J. L. Sessler, S. K. Kim and C.-H. Lee, Chem, 2017, 3, 1008–1020 CrossRef.
- (a) A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184–2185 CrossRef; (b) P. A. Gale, J. L. Sessler, V. Kral and V. M. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef.
- Y. D. Wu, D. F. Wang and J. L. Sessler, J. Org. Chem., 2001, 66, 3739–3746 CrossRef PubMed.
- (a) G. Bruno, G. Cafeo, F. H. Kohnke and F. Nicolo, Tetrahedron, 2007, 63, 10003–10010 CrossRef; (b) K.-C. Chang, T. Minami, P. Koutnik, P. Y. Savechenkov, Y. Liu and P. Anzenbacher, Jr., J. Am. Chem. Soc., 2014, 136, 1520–1525 CrossRef PubMed; (c) A. Kim, R. Ali, S. H. Park, Y.-H. Kim and J. S. Park, Chem. Commun., 2016, 52, 11139–11142 RSC; (d) E. Mulugeta, R. Dutta, Q. He, V. M. Lynch, J. L. Sessler and C.-H. Lee, Eur. J. Org. Chem., 2017, 4891–4895 CrossRef.
- N. J. Williams, V. S. Bryanstev, R. Custelcean, C. A. Seipp and B. A. Moyer, Supramol. Chem., 2016, 28, 176–187 CrossRef.
- K. H. Meyer and K. Schuster, Chem. Ber., 1922, 55, 819 CrossRef.
- V. Valderrey, E. C. Escudero-Adán and P. Ballester, Angew. Chem., Int. Ed., 2013, 52, 6898–6902 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, NMR and ITC titration profiles, and crystal structures. CCDC 1827178–1827181 and 1846661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc04393k |
| This journal is © The Royal Society of Chemistry 2018 |