
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homologation of halostannanes with carbenoids: a convenient and straightforward one-step access to α-functionalized organotin reagents†
Saad
Touqeer
,
Laura
Castoldi
,
Thierry
Langer
,
Wolfgang
Holzer
and
Vittorio
Pace
 *
*
University of Vienna, Department of Pharmaceutical Chemistry, Althanstrasse, 14, A-1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at; Web: http://drugsynthesis.univie.ac.at/
First published on 9th August 2018
Abstract
A direct, single synthetic homologative transformation of halostannanes into mono- or di-substituted methyl analogues is documented. Critical for the success of the operation is the excellent nucleophilicity of carbenoid-like methyllithium reagents (LiCHXY, X, Y = halogen, OR, and CN): by simply individuating the reagents’ substitution pattern, the desired functionalized fragment is delivered to the electrophile. The wide scope of the protocol is evidenced also in the case of analogous halogermanium compounds. The tandem homologation–quenching with nucleophiles and the use of α-chloroallyllithium is also discussed.
The unique reactivity of the C–Sn bond together with the good stability makes organotin compounds highly versatile entities in chemistry.1 Accordingly, their use in preparative processes is not limited to pivotal synthetic operations (in primis the Stille coupling)2 but also encompasses fundamental organometallic techniques.3 In this context, α-halomethyl stannanes (R3Sn–CH2–X) represent privileged tin-containing reagents because of the formal analogy with metal carbenoids (M–CH2–X). In fact, their excellent stability overcomes important limitations of classical metal species (M = Li, MgY, and ZnY) such as α-elimination.4 As a consequence of this significant advantage, the reactivity portfolio of these stannanes has been considerably exploited in a series of synthetic processes ranging from the equivalence with alkoxymethyl anions5 to electrophilic carbon units suitable for the preparation of multifunctionalized organotin compounds via nucleophilic substitutions.6 For example, the corresponding α-alkoxy derivatives serve as placeholders for triggering Wittig rearrangements7 or Liebeskind-type couplings,8 whereas α-amino analogues could be employed in anionic (or nucleophilic) cyclizations (Scheme 1).9 Pd-catalyzed sp2–sp3 Stille cross-couplings with α-aminomethyl stannanes have been also recently developed by scientists at Merck for accessing anti-HIV molecules.10 Undoubtedly, the introduction of SnAP (tin amino protocol) reagents by Bode in 2013 – as an elegant alternative to metal-catalyzed cross-couplings for preparing in one step saturated heterocycles from aldehydes – constitutes one of the most versatile uses of α-halomethyl stannanes as precursors of functionalized organometallics.11 Additionally, the C–X bond of α-halomethyl stannanes can undergo Pd-insertion, thus enabling the access to halopalladio-carbenoids as showcased by Fillion.12
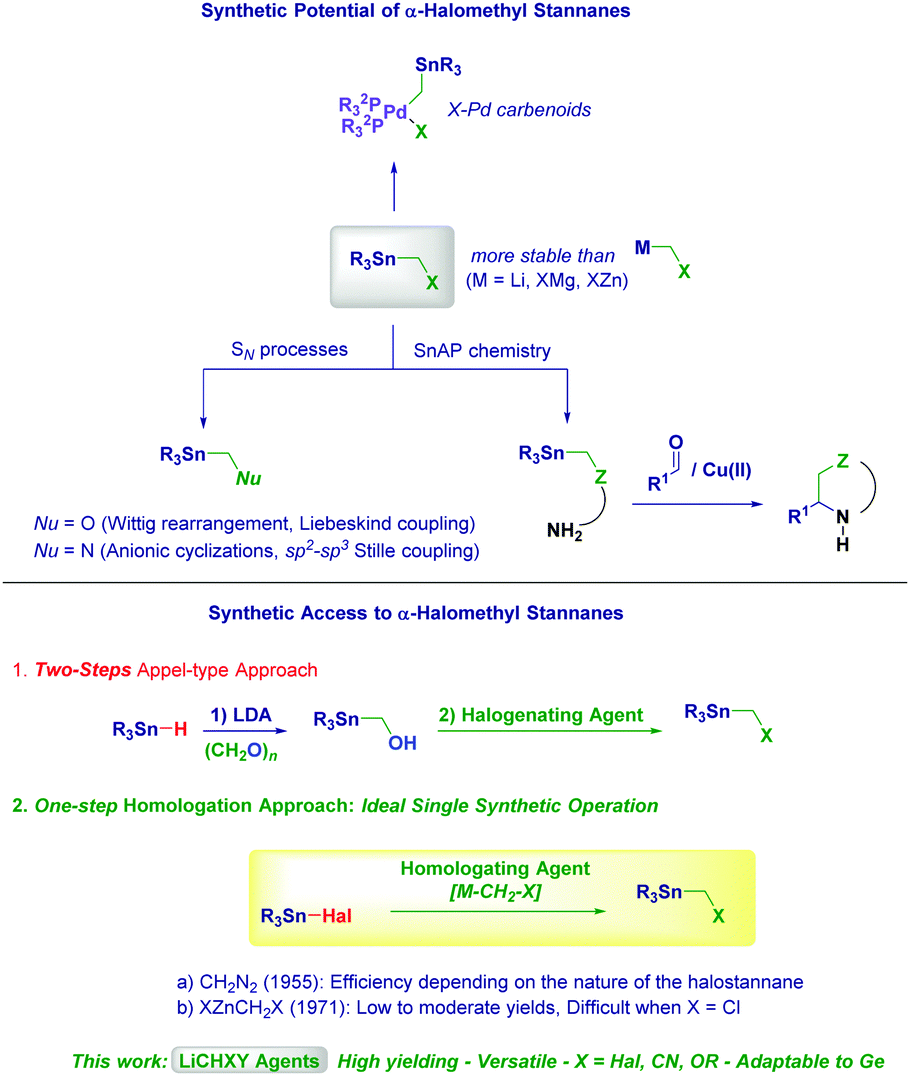 | ||
| Scheme 1 General context of the presented work. | ||
The synthetic potential of α-halomethyl stannanes is rather counterbalanced by the inherent difficulties in accessing them through simple and direct procedures. It seems nowadays that the two-step Appel-type protocol13 starting from a tin hydride derivative and subsequent halogenation is the preferred technique for these α-halo analogues.7,14 Cognizant of the strict safety limitations dealing with the use of toxic Sn compounds in chemical processes,15 we deemed a homologation process of a commercially available halostannane derivative (R3Sn–X) with a M–CH2–X reagent would be ideal for the purpose. Initial studies with diazomethane in the 1950s demonstrated the feasibility of the approach, although it was shown to be highly dependent on the substrate.16 Seyferth also introduced in the early 1970s the use of zinc carbenoids, observing a strict dependence of the success of the transformation on the nature of the carbenoid (i.e. the halogen).17 α-Iodo or α-bromo derivatives could be accessed in modest to moderate yields (30–66%), while α-chloro analogues could be prepared only via nucleophilic displacement (AgCl, seven days) starting from the corresponding iodinated compound.
In recent years, our group has documented a series of homologation techniques for introducing into a given carbon or heteroatom electrophile a CH2X fragment through a single synthetic operation, thus compressing the synthetic sequence to the only required transformation.18
Herein, we report the effectiveness of the nucleophilic substitution on halostannanes with lithium carbenoids and related reagents (LiCH2X, X = halogen, OR, and CN) for accomplishing a direct, one-step and straightforward formation of R3Sn–CH2–X type reagents. We anticipate the applicability of the protocol to the homologation of analogous organogermanium derivatives. Conceptually, the overall process can be regarded as a transmetallation of Li into Sn or Ge carbenoids, in which the products retain the α-halomethyl unit susceptible to late functionalization.
We selected the aliphatic tri-cyclohexyltin chloride 1 as the model substrate to rapidly evaluate the proposed homologation based strategy (Table 1). Moreover, the scarce commercial availability of the corresponding hydride required for applying the Appel-type protocol could be efficiently overcome. Generating chloromethyllithium (LiCH2Cl) via lithium/iodine exchange on ICH2Cl promoted by MeLi–LiBr under the usual Barbier-type conditions demonstrated the feasibility of the process. Although the reaction with 1.2 equiv. of LiCH2Cl reached 68% completion, as judged by GC-MS analysis of the crude, the subsequent chromatographic purification on silica gel was troublesome, finally leading to 56% isolated yield of the desired α-chloromethyl stannane 2 (entry 1). Switching to Et2O was detrimental (entry 2), while by progressively increasing the carbenoid loading up to 2.8 equiv. a quantitative conversion could be reached, thus significantly simplifying the purification procedure to a simple fast column chromatography – 93% isolated yield (entry 4). Attempting the homologation with a more stable (but less nucleophilic) magnesium carbenoid (ClMgCH2Cl–LiCl) resulted in no transformation (entry 5).19 It is worth observing the chemoselectivity of the process: no deleterious effect originated from the competitive Sn–Li exchange was noticed. As expected, changing ICH2Cl with ICH2Br – ceteris paribus – generated LiCH2Br,20 which accomplished the homologation in comparable efficiency (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Carbenoid (equiv.) | Solvent | Conversionb (%) | Yield of 2c (%) |
| a Otherwise stated, LiCH2Cl was generated from ICH2Cl (3.0 equiv.) and MeLi–LiBr (2.8 equiv.) in THF at −78 °C. b GC-MS calculated conversion. c Yields refer to isolated and purified compounds. d Formed from ICH2Cl (3.0 equiv.) and i-PrMgCl–LiCl (2.8 equiv.) in THF at −78 °C under non-Barbier conditions. e Formed from ICH2Br (3.0 equiv.) and MeLi–LiBr (2.8 equiv.) in THF at −78 °C under Barbier conditions. | ||||
| 1 | LiCH2Cl (1.2) | THF | 68 | 56 |
| 2 | LiCH2Cl (1.2) | Et2O | 55 | 31 |
| 3 | LiCH2Cl (2.2) | THF | 95 | 83 |
| 4 | LiCH2Cl (2.8) | THF | >99 | 93 |
| 5d | ClMgCH2Cl–LiCl (2.8) | THF | — | — |
| 6e | LiCH2Br (2.8) | THF | 95 | 90 |

Having disclosed the optimal conditions for the one-step homologation of halostannanes into α-halomethyl stannanes, we then investigated the scope of the reaction (Scheme 2). Aliphatic halostannanes proved to be excellent substrates for the transformation, enabling the formation of all the targeted α-chloro, α-bromo and α-iodo analogues in excellent yields through a single synthetic operation (2–9). Notably, a dihalo-stannane smoothly undergoes the substitution of both halogens, affording the bis-(chloromethyl) derivative 10 in 82% yield, previously obtained in 40% yield with the Appel method.21 Switching to aryl-stannanes was possible without compromising the efficiency of the process, as indicated by the reactions with LiCH2Cl, LiCH2Br and LiCH2I, respectively (11–13). It is worth mentioning the higher performance of the Li carbenoid approach compared to the Zn species which afforded 12 in 30% yield.22 By adopting a deprotonation strategy on dihalomethanes of type XCH2Y, the corresponding dihalolithium carbenoids were easily prepared23 and then reacted with the halostannanes giving the corresponding dihalomethyl tin compounds (14–18) whose synthesis proved to be challenging with other methodologies.24 The precise control of the chemoselectivity of the process is further documented by the one-step preparation of the not previously reported analogue 18 featuring two different halogens on the attacking carbon. Carbenoid-like reagents such as LiCH2CN (generated via MeCN deprotonation, 19)25 and LiCH2OEt (generated via the Yus’ reductive lithiation of ClCH2OEt, 20)26 could be advantageously employed in the process, thus expanding the synthetic potential of the methodology.
 | ||
| Scheme 2 Chemoselective homologative α- and α,α-difunctionalization of halostannanes. | ||
We were pleased to find that the protocol showcased its versatility in the homologation of halogermanium derivatives, as well (Scheme 3).27 Indeed, these starting materials afforded the corresponding halo- (21–23) or dihalo- (24) methyl derivatives in high yields. The fine tuning of the stoichiometry enables the addition of two different lithium reagents (LiCH2Cl and MeLi) giving the mixed aromatic–aliphatic halogermanium 25. Otherwise, double substitution with the same LiCH2Cl was noticed (26) in analogy to the above observed tin analogue.
 | ||
| Scheme 3 Chemoselective homologative α- and α,α-difunctionalization of halogermanes. | ||
With the aim to gain full insight into the potential of these easily synthesized building blocks featuring a reactive electrophilic carbon, we designed an effective in situ functionalization (Scheme 4). Upon quenching the homologation reaction with acidic water, a solution of N-, O- or S-nucleophilic species was added to the reaction mixture directly leading to α-azidomethyl (27), α-aryloxymethyl (28) and α-arylthiomethyl stannanes, respectively (29). Finally, we were intrigued by the use of the more complex α-chloroallyl lithium as the homologating reagent.28 This nucleophilic reagent prepared via the deprotonation of allyl chloride with LDA may in principle display a dual reactivity towards a given electrophilic partner (α- or γ-attack). The homologation process occurring in a genuine regioselective γ-fashion did not stop at the expected chlorovinyl product. It further underwent lithiation giving an alkenyl-type carbenoid,29 which could realize an SN process on the allyl chloride, finally affording the bis-olefinic structures 30 and 31. Overall, through a single synthetic operation the full control of the inserting six carbon units can be achieved.
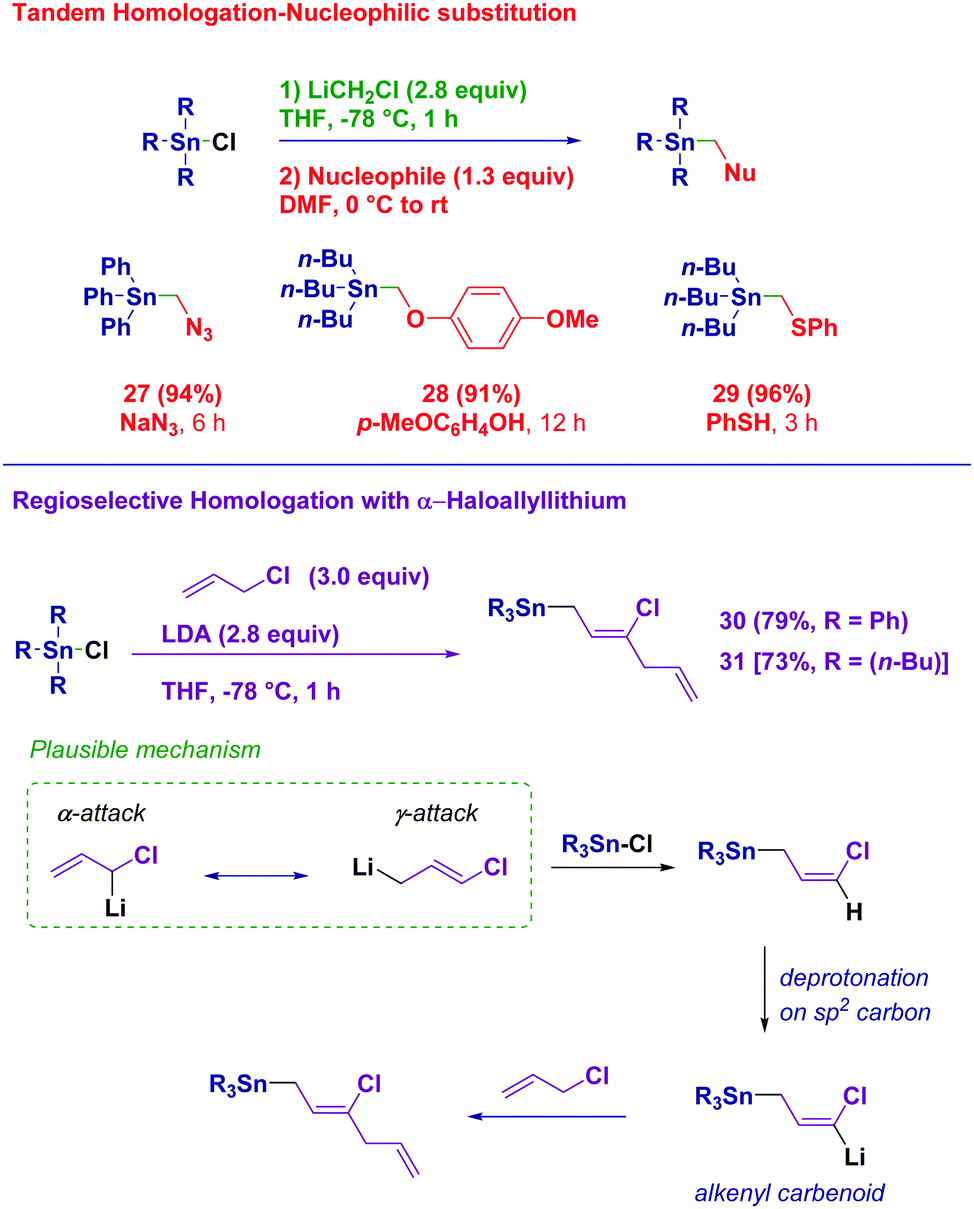 | ||
| Scheme 4 Immediate derivatization with added nucleophiles and use of α-chloroallyllithium as the homologation reagent. | ||
In summary, we report a direct nucleophilic substitution of halostannanes and halogermanes with lithium carbenoid-like reagents including LiCH2Hal, LiCHHalHal’, LiCH2OR and LiCH2CN to obtain through a single operation highly valuable α- and α,α-disubstituted methyl derivatives. The method represents a significant advancement compared to the commonly used two-step Appel-type protocol or the previously known low efficient homologation strategies dealing with the use of diazomethane or zinc carbenoids. The derivatization of the formed products and the use of a more challenging carbenoid further highlight the synthetic potential.
The authors thank the University of Vienna for financial support. S. T. acknowledges the OEAD for a predoctoral grant. We thank D. Dobusch for GC-MS and HRMS measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase and M. Oestreich, Wiley-VCH, Weinheim, 2014 Search PubMed.
- For reviews, see: (a) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 Search PubMed; (b) C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040 CrossRef.
- (a) M. S. Jensen, C. Yang, Y. Hsiao, N. Rivera, K. M. Wells, J. Y. L. Chung, N. Yasuda, D. L. Hughes and P. J. Reider, Org. Lett., 2000, 2, 1081 CrossRef PubMed; (b) M. Cormier, M. Ahmad, J. Maddaluno and M. De Paolis, Organometallics, 2017, 36, 4920 CrossRef.
- (a) V. Pace, L. Castoldi, S. Monticelli, M. Rui and S. Collina, Synlett, 2017, 879 CrossRef; (b) V. Pace, W. Holzer and N. De Kimpe, Chem. Rec., 2016, 16, 2061 CrossRef PubMed.
- J. Clayden, M. N. Kenworthy and M. Helliwell, Org. Lett., 2003, 5, 831 CrossRef PubMed.
- (a) A. Wieczorek and F. Hammerschmidt, J. Org. Chem., 2012, 77, 10021 CrossRef PubMed; (b) H. Kobayashi, J. A. Eickhoff and A. Zakarian, J. Org. Chem., 2015, 80, 9989 CrossRef PubMed.
- L. F. Nina and E. M. Carreira, Chem. – Eur. J., 2009, 15, 12065 CrossRef PubMed.
- H. Li, A. He, J. R. Falck and L. S. Liebeskind, Org. Lett., 2011, 13, 3682 CrossRef PubMed.
- (a) I. Coldham, R. Hufton and R. E. Rathmell, Tetrahedron Lett., 1997, 38, 7617 CrossRef; (b) G. Bélanger, V. Darsigny, M. Doré and F. Lévesque, Org. Lett., 2010, 12, 1396 CrossRef PubMed.
- A. ElMarrouni, M. Campbell, J. J. Perkins and A. Converso, Org. Lett., 2017, 19, 3071 CrossRef PubMed.
- (a) C.-V. T. Vo, M. Gediminas and J. W. Bode, Angew. Chem., Int. Ed., 2013, 52, 1705 CrossRef PubMed; (b) C.-V. T. Vo, M. U. Luescher and J. W. Bode, Nat. Chem., 2014, 6, 310 CrossRef PubMed; (c) M. U. Luescher, C.-V. T. Vo and J. W. Bode, Org. Lett., 2014, 16, 1236 CrossRef PubMed; (d) W.-Y. Siau and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 17726 CrossRef PubMed; (e) M. U. Luescher and J. W. Bode, Angew. Chem., Int. Ed., 2015, 54, 10884 CrossRef PubMed; (f) M. U. Luescher, K. Geoghegan, P. L. Nichols and J. W. Bode, Aldrichimica Acta, 2015, 48, 43 Search PubMed.
- E. Fillion and N. J. Taylor, J. Am. Chem. Soc., 2003, 125, 12700 CrossRef PubMed.
- R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801 CrossRef.
- J. Åhman and P. Somfai, Synth. Commun., 1994, 24, 1117 CrossRef.
- (a) E. Le Grognec, J.-M. Chrétien, F. Zammattio and J.-P. Quintard, Chem. Rev., 2015, 115, 10207 CrossRef PubMed; (b) D. C. Harrowven, D. P. Curran, S. L. Kostiuk, I. L. Wallis-Guy, S. Whiting, K. J. Stenning, B. Tang, E. Packard and L. Nanson, Chem. Commun., 2010, 46, 6335 RSC.
- (a) D. Seyferth and E. G. Rochow, J. Am. Chem. Soc., 1955, 77, 1302 CrossRef; (b) D. Seyferth, Chem. Rev., 1955, 55, 1155 CrossRef.
- (a) D. Seyferth and S. Brian Andrews, J. Organomet. Chem., 1971, 30, 151 CrossRef ; for a rare example of using an alkali metal carbenoid, see: ; (b) T. Kobayashi and K. H. Pannell, Organometallics, 1991, 10, 1960 CrossRef.
- (a) L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC; (b) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef PubMed; (c) G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef PubMed; (d) L. Castoldi, W. Holzer, T. Langer and V. Pace, Chem. Commun., 2017, 53, 9498 RSC; (e) S. Raffaele, I. Laura, U. Ernst, H. Wolfgang and P. Vittorio, Eur. J. Org. Chem., 2018, 2466 Search PubMed; (f) R. Senatore, L. Castoldi, L. Ielo, W. Holzer and V. Pace, Org. Lett., 2018, 20, 2685 CrossRef PubMed; (g) V. Pace, L. Castoldi and W. Holzer, Chem. Commun., 2013, 49, 8383 RSC.
- R. H. V. Nishimura, F. T. Toledo, J. L. C. Lopes and G. C. Clososki, Tetrahedron Lett., 2013, 54, 287 CrossRef.
- V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC.
- M. F. Connil, B. Jousseaume, N. Noiret and M. Pereyre, Organometallics, 1994, 13, 24 CrossRef.
- R. Altmann, K. Jurkschat, M. Schürmann, D. Dakternieks and A. Duthie, Organometallics, 1997, 16, 5716 CrossRef.
- (a) V. Pace, L. Castoldi, A. D. Mamuye, T. Langer and W. Holzer, Adv. Synth. Catal., 2016, 358, 172 CrossRef; (b) V. Pace, L. Castoldi, A. D. Mamuye and W. Holzer, Synthesis, 2014, 2897 CrossRefSee also: (c) D. M. Hodgson, Tetrahedron Lett., 1992, 33, 5603 CrossRef.
- (a) A. G. Davies and T. N. Mitchell, J. Chem. Soc. C, 1896, 1969 Search PubMed; (b) C. M. Warner and J. G. Noltes, J. Chem. Soc. D, 1970, 694 RSC.
- A. D. Mamuye, L. Castoldi, U. Azzena, W. Holzer and V. Pace, Org. Biomol. Chem., 2015, 13, 1969 RSC.
- (a) A. Guijarro, B. Mancheño, J. Ortiz and M. Yus, Tetrahedron, 1996, 52, 1643 CrossRef; (b) V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC.
- T. Boddaert, C. François, L. Mistico, O. Querolle, L. Meerpoel, P. Angibaud, M. Durandetti and J. Maddaluno, Chem. – Eur. J., 2014, 20, 10131 CrossRef PubMed.
- T. L. Macdonald, B. A. Narayanan and D. E. O'Dell, J. Org. Chem., 1981, 46, 1504 CrossRef.
- R. Lhermet, M. Ahmad, C. Fressigné, B. Silvi, M. Durandetti and J. Maddaluno, Chem. – Eur. J., 2014, 20, 10249 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04786c |
| This journal is © The Royal Society of Chemistry 2018 |