
Fe–Zr–O catalyzed base-free aerobic oxidation of 5-HMF to 2,5-FDCA as a bio-based polyester monomer†
Dongxia
Yan
abc,
Jiayu
Xin
b,
Qiu
Zhao
b,
Kai
Gao
bc,
Xingmei
Lu
bc,
Gongying
Wang
*a and
Suojiang
Zhang
 *b
*b
aChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu, 610041, P. R. China. E-mail: gywang@cioc.ac.cn
bBeijing Key Laboratory of Ionic Liquids Clean Process, Key Laboratory of Green Process Engineering, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: sjzhang@ipe.ac.cn; Fax: +86 10 8262 7080; Tel: +86 10 8262 7080
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 2nd October 2017
Abstract
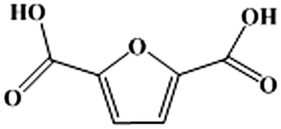
An environment-friendly and economical route for 5-hydroxymethylfurfural (HMF) aerobic oxidation to 2,5-furandicarboxylic acid (FDCA) in an ionic liquid (IL)-promoted base-free reaction system was reported using Fe–Zr–O as a catalyst. A series of FexZr1−xO2 catalysts were synthesized by a hydrothermal method and the catalytic performance was investigated. Among these catalysts, Fe0.6Zr0.4O2 exhibited excellent catalytic activity in the HMF oxidation. A 60.6% FDCA yield and 99.9% HMF conversion could be obtained after 24 h under 2 MPa O2 pressure and base-free conditions. The good performance could be attributed to the large amount of acidic and basic sites on the surface of the catalyst and its high reducibility and oxygen mobility. In addition, the formation of humins and the reaction pathways in the ILs were also investigated, which revealed parallel reactions between FDCA and humin formation. A plausible reaction mechanism was proposed based on the results of a series of designed experiments. Finally, the catalyst was used five times without obvious loss of activity. To the best of our knowledge, this is the best result for a non-noble metal catalyzed base-free oxidation of HMF to FDCA using molecular oxygen as an oxidant.
Introduction
Due to its desirable properties such as durability, low production cost, optical clarity and barrier properties, polyethylene terephthalate (PET) has been the dominant manmade polymer with applications ranging from beverage bottles, packaging materials and medical devices to textiles for the past four decades, of which about 56 million tons were produced worldwide in 2013 alone.1,2 However, the presence of a high ratio of aromatic components in its structure and the corresponding limited mobility of the polymer chains make PET resistant to microbial attack and non-degradable under normal environmental conditions, resulting in the accumulation of PET in the environment and thus creating environmental hazards.1,3 More importantly, the monomer of PET, terephthalic acid (TPA), is obtained from fossil resources, which are non-renewable. Therefore, much research was conducted to find bio-based alternatives to PET to reduce the pressure on the environment and resources.4,5 Bio-based 2,5-furandicarboxylic acid (FDCA) contains two carboxyl groups at the para position of the furan ring, which is very similar to fossil-based TPA and can be a green substitute to produce plastics. Meanwhile, as an analogue of PET, a FDCA-based polyethylene, furandicarboxylate (PEF), has been well prepared and investigated. It is a degradable bio-based polymer with superior properties to PET, such as improved thermal stability and gas barrier properties with low-temperature formability and reduced chain mobility.2,5 Therefore, as an important monomer for PEF production, 100% bio-based FDCA has attracted much attention recently.6–9Generally, FDCA could be prepared by oxidation of 5-hydroxymethylfurfural (HMF), and strong stoichiometric oxidation reagents, such as chromate (CrO42−), dichromate (Cr2O72−) and permanganate (MnO4−) salts were often employed to accomplish the reaction.10 However, these oxidants are high cost and environment-unfriendly. Thus, the conversion of HMF to FDCA with green molecular oxygen is more attractive. Recently, the aerobic oxidation of HMF to FDCA has been accomplished using a homogeneous catalyst (e.g. Co/Mn/Zr/Br) combined with acetic acid as solvent11 or a homogeneous catalyst (Co(OAc)2/Zn(OAc)2/NaBr) combined with trifluoroacetic acid as an additive;12 however, it is difficult to reuse the homogeneous catalysts. Therefore, the use of molecular oxygen and a heterogeneous catalyst is quite advantageous for HMF oxidation. Thus, a large number of reports focused on supported heterogeneous noble metal catalysts, such as Au,13 Pt,14 Pd,15 Ru (ref. 16) and their alloys.7 A high yield of FDCA can be obtained with the combined use of an additional base. However, the product obtained in these catalytic systems is in the form of FDCA salt, which cannot be used directly for polymer production and needs to be separated by adding a large amount of strong mineral acids, such as HCl or H2SO4. Therefore, the excessive use of base and the high cost of catalysts resulted in a less efficient and less sustainable process.
In order to reduce the cost of the catalyst, the heterogeneous non-noble metal catalyzed oxidation of HMF was investigated recently. Zhang et al.17,18 reported that moderate to high FDCA yields could be obtained over the nano-Fe3O4–CoOx catalyst or the Merrifield resin-supported Co(II)-meso-tetra(4-pyridyl)-porphyrin catalyst using t-BuOOH as oxidant. Recently, another group used MnxFey mixed oxide as a catalyst for the production of FDCA; however, the yield was very low by direct conversion of HMF.19 MnO2 and its mixed oxides MnOx–CeO2 were also used for the oxidation of HMF and showed excellent catalytic performance in the presence of a high concentration of NaHCO3 or KHCO3.20,21 Generally, even though the noble metal catalysts showed excellent performance for FDCA production, their high costs and excessive use of base imposed a restriction for the further use. Non-noble metals can reduce the cost of catalysts; however, catalytic systems often employ high cost and environment-unfriendly t-BuOOH as the oxidant as well as the use of excessive base.7 Thus, it is believed that the economical, environment-friendly and simple direct conversion of HMF to FDCA over non-noble metal catalysts under base-free conditions is highly desired.
Herein, with the purpose of developing an efficient and economic catalytic system for the green conversion of HMF, a series of FexZr1−xO2 catalysts were synthesized and employed as heterogeneous catalysts for the production of FDCA using 1-butyl-3-methylimidazolium chloride ([Bmim]Cl) ionic liquids (ILs) as solvents and molecular oxygen under base-free conditions. The catalytic activity and stability towards the oxidation of HMF were examined, which showed good stability and obtained the best result for the non-noble metal catalyzed base-free aerobic oxidation of HMF to FDCA to the best of our knowledge. Furthermore, a series of characterization methods were used to reveal the factors affecting the activity of the catalyst. Additionally, the formation of humins and the reaction pathways in the [Bmim]Cl ILs were also investigated, and a plausible mechanism was proposed based on the results of a series of designed experiments.
Experimental
Materials
HMF (98%), 5-hydroxymethyl-2-furancarboxylic acid (HMFA) (95%), 2,5-diformylfuran (DFF) (98%), 5-formyl-2-furancarboxylic acid (FFCA) (98%) and FDCA (97%) were purchased from the J&K Chemical Co. Ltd. (Beijing, China). Zr(NO3)4·5H2O was supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Fe(NO3)3·9H2O was purchased from Xilong Chemical Co., Ltd. (Guangdong, China). Sulfuric acid (guaranteed reagent) and ammonia (25–28 wt%) were purchased from Beijing Chemical Co. Ltd. (Beijing, China). O2 (99.9%) was supplied by Beijing Beiwen Gas Factory. [Bmim]Cl was commercially available (purchased from Lanzhou Institute of Chemical Physics, CAS) and was dried under vacuum at 60 °C for 24 h before use. The commercial chemicals were of analytical grade and therefore were used as received.Catalyst preparation
A series of FexZr1−xO2 mixed oxides were prepared by a hydrothermal method, where x is the atomic ratio of Fe/(Fe + Zr) and varied from 0 to 1 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1.0). The required amounts of Fe(NO3)3·9H2O and Zr(NO3)4·5H2O were dissolved in deionized water and well mixed for 2 h at room temperature. The total concentration of Fe and Zr cations was 0.25 mol L−1. The solutions of Fe(NO3)3·9H2O and Zr(NO3)4·5H2O and their mixed solution were gradually dripped in an ammonia solution (5 mol L−1) while maintaining the pH at about 10 with stirring. Then, the reaction mixture was vigorously stirred for 2 h and allowed to settle for 2 h. Thereafter, 65 ml of slurry was transferred into 100 ml Teflon-lined stainless steel autoclaves. The autoclaves were tightly sealed and placed in an oven for hydrothermal treatment at 220 °C for 48 h. After cooling, the samples were washed with distilled water and ethanol until a pH of 7 was reached before air-drying at 110 °C for 12 h; thus, the FexZr1−xO2 mixed oxides were obtained.Catalyst characterization
The prepared catalysts were systematically investigated using different characterization techniques like X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), hydrogen temperature-programmed reduction (H2-TPR) analysis, temperature-programmed desorption (TPD) of ammonia and carbon dioxide (NH3 and CO2-TPD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), inductively coupled plasma optical emission spectroscopy (ICP-OES) and specific surface area analysis (BET). The details for the catalyst characterization can be found in the ESI.†Humin characterization
The humins formed during the aerobic oxidation of HMF were analyzed by Fourier transform infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), thermogravimetric (TG) analysis and gel permeation chromatography (GPC). The details for the humin characterization can be found in the ESI.†Catalytic reaction and product analysis
In a typical experiment, 1 g of [Bmim]Cl IL was added to a 50 ml batch-type Teflon-lined stainless-steel autoclave along with 0.1 mmol HMF and 0.01 g powdery catalyst. The autoclave was purged 3 times with O2 and then pressurized to 2 MPa at room temperature. Once the system reached the desired temperature (160 °C), the reaction was started by vigorous stirring with a magnetic stirrer for 24 h. Then, the reaction mixtures were diluted with deionized H2O, filtered using 0.2 μm PTFE filters, and subsequently analyzed using a Shimadzu LC-20A HPLC instrument equipped with refractive index (RI) and photo-diode array (PDA) detectors. A Bio-Rad Aminex HPX-87H column (300 mm × 7.8 mm) and a 5 mM H2SO4 aqueous solution were employed to perform the separation at 35 °C (0.6 ml min−1). The retention times and calibrations for the observed products were determined by injecting known concentrations of standard reference compounds and quantified using an external standard calibration curve method.Results and discussion
Characterization of catalyst
The XRD patterns of the catalysts are presented in Fig. 1a. The Fe2O3 and ZrO2 samples show the characteristic reflections of the rhombohedral phase of hematite (α-Fe2O3) (JCPDF 33-0664) and the tetragonal as well as the monoclinic phases of ZrO2 (JCPDF 50-1089 and 37-1484). For the FexZr1−xO2 mixed oxides (x = 0.2, 0.4, 0.6 and 0.8), both ZrO2 and Fe2O3 can be detected clearly. However, compared with ZrO2, a slight shift of ZrO2 peaks toward higher 2θ values was observed over the four samples (see Fig. 1a-2), revealing that a part of the solid solution was present in the mixed oxides with some Fe doped into the lattice of ZrO2.22,23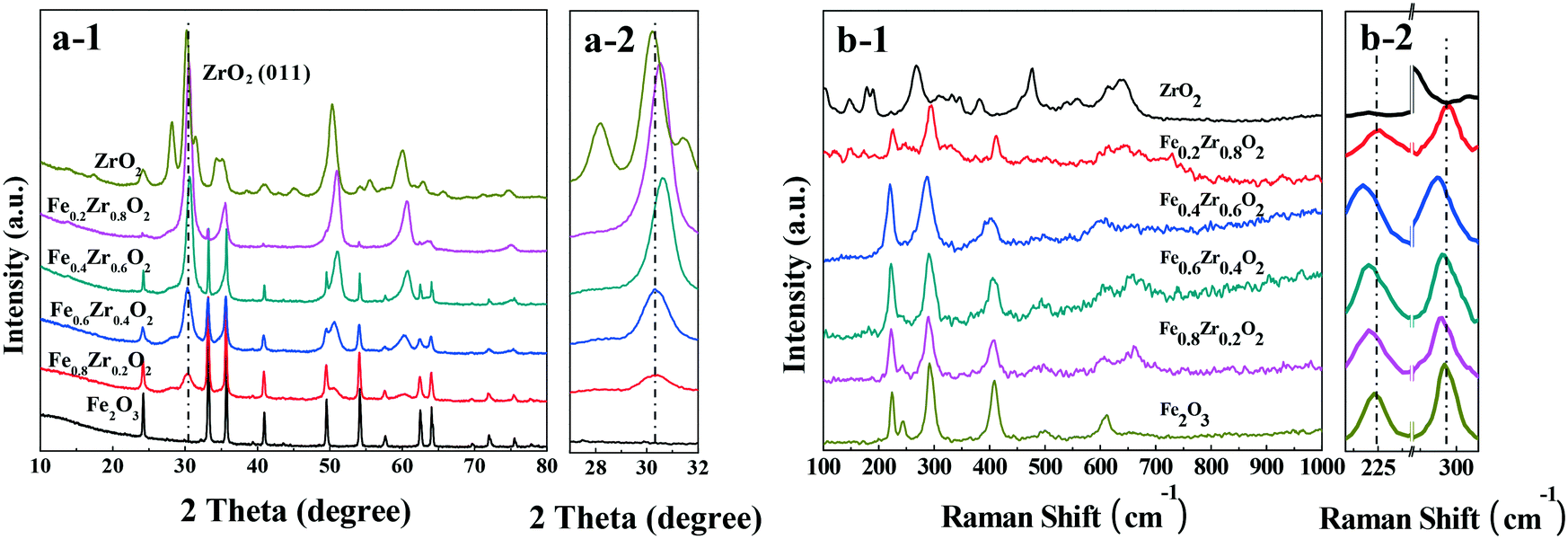 | ||
| Fig. 1 (a) XRD patterns (a-2: amplification of the (011) reflection for ZrO2) and (b) Raman spectra (b-2: amplification of the Raman spectra) of FexZr1−xO2 samples. | ||
The Raman spectra are displayed in Fig. 2b. For single ZrO2, five obvious peaks at 146, 267, 309, 460 and 641 cm−1 are indexed as the tetragonal phase of ZrO2.24 The Raman spectrum of ZrO2 also indicates the characteristic diffraction peaks at approximately 178, 189, 331, 345, 381, 479 and 623 cm−1, which correspond to the monoclinic phase of ZrO2.25 For the Raman spectrum of Fe2O3, the diffraction signals at approximately 223, 244, 291, 408, 492 and 610 cm−1 indicate the hematite structure of Fe2O3.26 It should be noted that for FexZr1−xO2 mixed oxides (x = 0.2, 0.4, 0.6 and 0.8), the main bands of Fe2O3, such as those at approximately 224 and 291 cm−1, are shifted to lower wavenumbers (red shift) relative to those of the Fe2O3 sample (see Fig. 1b-2). This phenomenon can be attributed to changes in the surface strain or defects due to the incorporation of Zr into the Fe2O3 crystal structure, because the strains and defects in the crystal structure or effects of particle size can lead to the shifts in Raman peaks,27 which was consistent with the XRD result that a certain amount of solid solution formed in the mixed oxides.
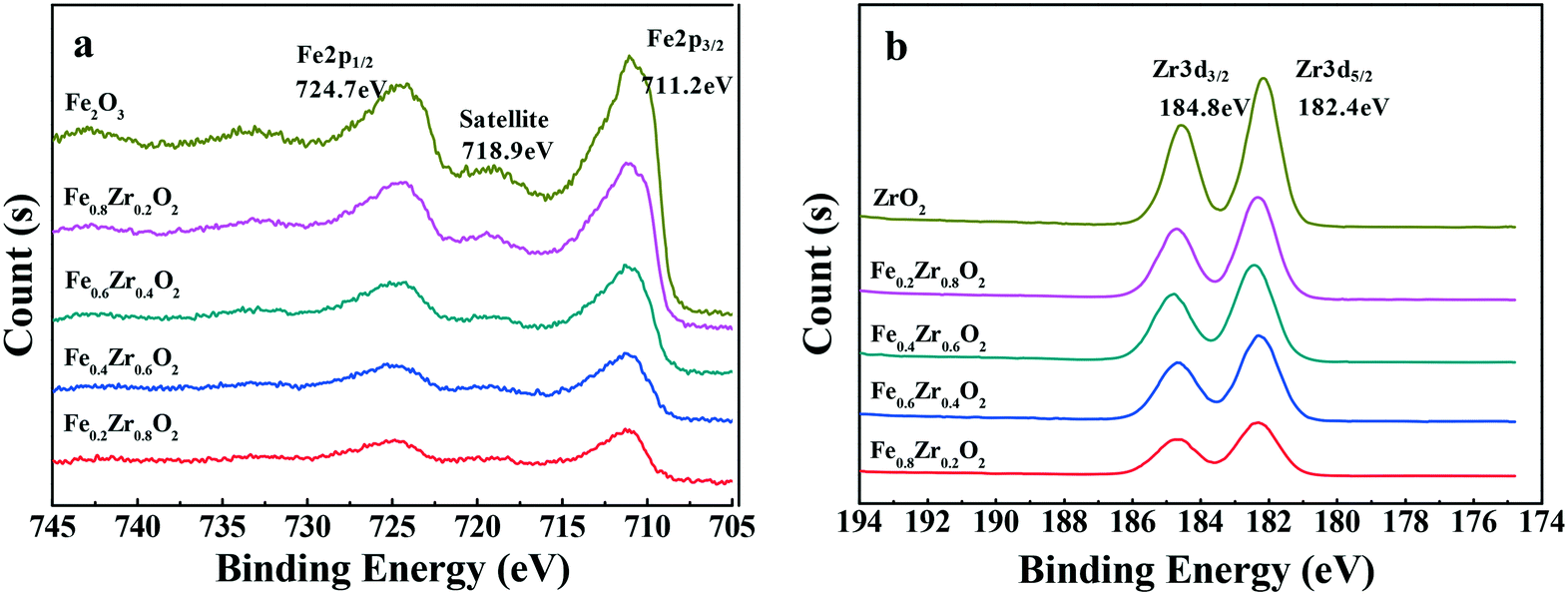 | ||
| Fig. 2 XPS spectra of FexZr1−xO2 samples: survey spectra of (a) Fe 2p and (b) Zr 3d. | ||
The samples were further analyzed by XPS to determine the distribution of the elements and degrees of oxidation on the surface of the compound oxide catalysts. As shown in Fig. 2a, the Fe 2p XPS spectra for the samples that contained iron revealed the presence of Fe2O3 on the surface of the catalysts due to the shape and position of the Fe 2p3/2 peak at a binding energy of 711.2 eV and the Fe 2p1/2 peak at a binding energy of 724.7 eV.28,29 Additionally, a shake-up satellite of the Fe 2p3/2 peak, approximately 7.7 eV below the main line, was also observed at approximately 718.9 eV, which was also characteristic of Fe3+ in Fe2O3.30 As for the samples that contained zirconium (see Fig. 2b), the Zr 3d XPS spectra showed the exiting of ZrO2 on the surface of the catalysts because Zr 3d3/2 and Zr 3d5/2 peaks were observed at a binding energy of 184.8 and 182.4 eV, respectively.28,31
Moreover, the types of surface oxygen species were also identified by the O 1s XPS spectra as displayed in Fig. 3. It can be seen that the O 1s curves of the FexZr1−xO2 mixed oxides (x = 0.2, 0.4, 0.6 and 0.8) can be fitted into three peaks, which were denoted as OI, OII and OIII, the relative fractions of which are listed in Table 1. According to the literature, the main peak OI at 529.6–530.2 eV is ascribed to lattice oxygen atoms (O22−); a shoulder peak OII at 531.0–531.8 eV is assigned to oxygen vacancies, surface adsorbed oxygen ions (O2− or O−) or OH groups; another shoulder peak OIII at 532.4–532.9 eV is likely to be associated with adsorbed molecular water.32,33 Importantly, surface oxygen species are considered to be more reactive than the others because they have higher mobility and are more easily activated. At the same time, it is also reported that the high amount of oxygen vacancies and HO− groups (denoted as OII) contributes to the high activity of the catalysts.34,35 As for the FexZr1−xO2 mixed oxides, a certain amount of OII and OIII species was observed. In combination with XRD, Raman spectroscopy and the following TEM and BET analysis, the oxygen vacancies could be ascribed to the formation of an Fe–Zr solid solution, and the adsorbed oxygen ions or OH groups as well as molecular water could be associated with the small particle size and high specific surface area of the samples (see Table 2).36 Additionally, there is no obvious difference in the relative fractions of surface oxygen species in all mixed oxides except the Fe0.6Zr0.4O2 sample (see Table 1), which showed the best catalytic performance (discussed in the section Catalytic performance for aerobic oxidation of HMF).
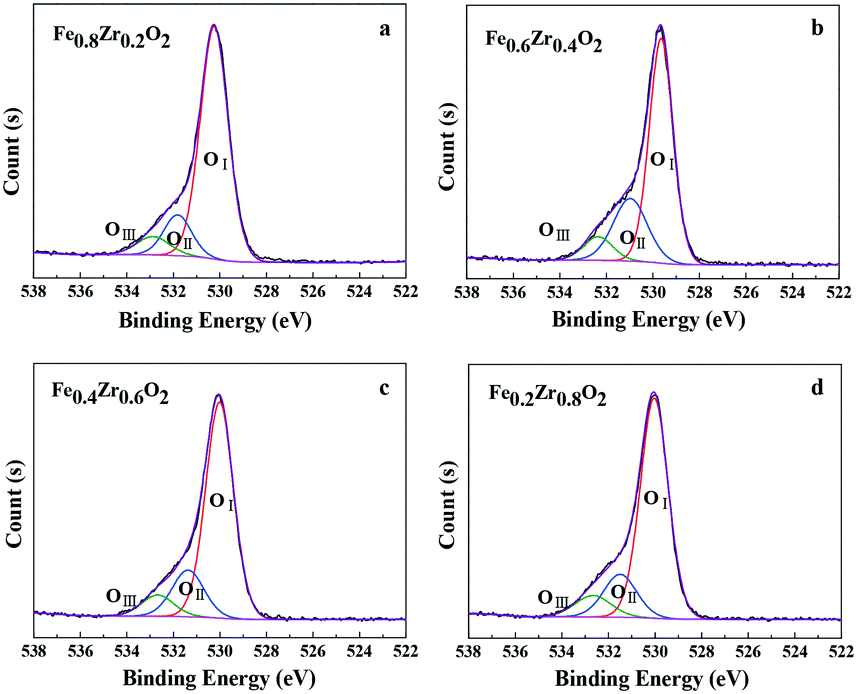 | ||
| Fig. 3 (a–d) O 1s XPS spectra of FexZr1−xO2 samples. | ||
| Catalyst | OI | OII | OIII | |||
|---|---|---|---|---|---|---|
| B.E. (eV) | Fractions (%) | B.E. (eV) | Fractions (%) | B.E. (eV) | Fractions (%) | |
| Fe0.8Zr0.2O2 | 530.2 | 78.8 | 531.8 | 13.8 | 532.9 | 7.4 |
| Fe0.6Zr0.4O2 | 529.6 | 65.4 | 531.0 | 26.2 | 532.4 | 8.4 |
| Fe0.4Zr0.6O2 | 530.0 | 73.5 | 531.4 | 18.2 | 532.7 | 8.3 |
| Fe0.2Zr0.8O2 | 530.0 | 73.7 | 531.5 | 17.0 | 532.7 | 9.3 |
| Catalyst | Mean diametera (nm) | X value of FexZr1−xO2b | Surface areac (m2 g−1) | Consumption of H2d (mmol g−1) | Total aciditye (mmol NH3 per g) | Total basicityf (mmol CO2 per g) |
|---|---|---|---|---|---|---|
| a Determined by TEM. b Based on ICP analysis. c BET surface area calculated from N2 adsorption isotherms at 77 K. d Consumption of H2 from hydrogen TPD measurements. e Acid properties from ammonia TPD measurements. f Base properties from carbon dioxide TPD measurements. g A mixture of two different sizes of particles. | ||||||
| Fe2O3 | 74.0 | — | 15 | 13.92 | 0.20 | 0.42 |
| Fe0.8Zr0.2O2 | 5.0/47.3g | 0.76 | 62 | 10.40 | 0.34 | 0.91 |
| Fe0.6Zr0.4O2 | 7.0 | 0.58 | 96 | 5.66 | 0.60 | 1.46 |
| Fe0.4Zr0.6O2 | 7.1 | 0.37 | 102 | 4.09 | 0.58 | 0.97 |
| Fe0.2Zr0.8O2 | 7.2 | 0.18 | 122 | 1.44 | 0.59 | 0.96 |
| ZrO2 | 8.2 | — | 113 | 0.05 | 0.43 | 0.71 |
The reducibility of FexZr1−xO2 samples was investigated by H2-TPR as shown in Fig. 4a, while the H2 consumption values are displayed in Table 2. The H2-TPR profiles of ZrO2 do not show any peaks attributed to H2 consumption due to its non-reductive property below 930 °C in hydrogen.37 For single Fe2O3 two distinct peaks are attributed to the stepwise reduction of Fe2O3 to Fe3O4 and the further reduction of Fe3O4 to Fe.38 However, the Fe–Zr mixed oxides represent three reduction peaks labeled α, β and γ. It is reported that because of the interaction between Fe species and other metals, the high-temperature reduction peak of pure Fe2O3 can be divided into two peaks due to the stepwise reduction of Fe2O3 to Fe through FeO as an intermediate.36 Therefore, it can be concluded that there should be an interaction between Fe and Zr species which affects the reduction process of Fe2O3 in the mixed oxides. Thus, the reduction of Fe–Zr mixed oxides may be separated into three steps which are the reduction of Fe2O3 to Fe3O4 (peak α), the reduction of Fe3O4 to FeO (peak β) and the reduction of FeO to Fe (peak γ). It is worth mentioning that hydrogen consumption during the TPR measurements increases with the increase of the Fe proportion in the Fe–Zr mixed oxides; the quantification results of H2-TPR experiments are included in Table 2. In addition, the higher temperature reduction peaks in mixed oxides are shifted to a lower temperature compared with those of single Fe2O3, indicating a chemical interaction between Fe and Zr species, which could enhance the reducibility and oxygen mobility of Fe–Zr mixed oxides.39,40 Importantly, the Fe0.6Zr0.4O2 sample has the lowest reduction peaks, which reveals the highest reducibility and oxygen mobility of the sample. This finding is in accordance with the O 1s XPS result that the Fe0.6Zr0.4O2 sample has the largest amount of OII fraction, which has a higher mobility and is more easily activated.
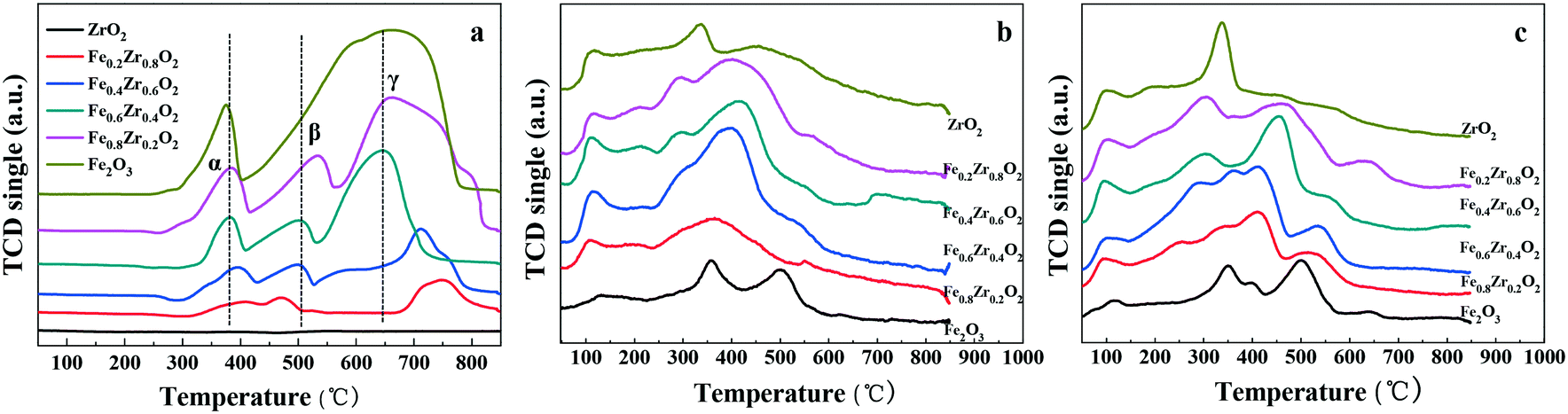 | ||
| Fig. 4 (a) H2-TPR, (b) NH3-TPD and (c) CO2-TPD profiles of FexZr1−xO2 samples. | ||
The acid–base properties of FexZr1−xO2 samples were revealed by NH3-TPD and CO2-TPD analysis, and the results are shown in Fig. 4b and c. The total acidity and basicity which has been estimated from the area of the TPD profiles are summarized in Table 2. The results showed that all the samples have both acidic and basic sites as well as the same acid and base types. The NH3-TPD curves (Fig. 4b) show that the amount of total acidity for the FexZr1−xO2 samples initially increased and then slightly decreased with the increase of Zr proportion in the Fe–Zr mixed oxides. Moreover, the amount of total acidic sites on the FexZr1−xO2 samples ranged from 0.20 mmol (Fe2O3 sample) to 0.60 mmol (Fe0.6Zr0.4O2 sample) of NH3 per gram of catalyst (see Table 2). There is no significant difference in the NH3-TPD curves except the relative acid strength (Table S1†), suggesting the same acid types.
The CO2-TPD profiles of all oxides in Fig. 4c present the basic properties of the oxides. Fe2O3 presents mainly medium and strong basic sites, while ZrO2 presents preponderant weak and medium basic sites. Additionally, as revealed in Fig. 4c and Table 2, both the total amount of basic sites and the base strength distribution are influenced by the Zr proportion in the Fe–Zr mixed oxides. The total amount of basic sites of FexZr1−xO2 samples initially increased and then decreased with the increase of Zr content in the Fe–Zr mixed oxides. The surface base concentrations increased from 0.91 mmol CO2 per g in Fe0.8Zr0.2O2 to 1.46 mmol CO2 per g in Fe0.6Zr0.4O2 and subsequently decreased to 0.96 mmol CO2 per g in Fe0.2Zr0.8O2. More importantly, as can be seen from the CO2-TPD profiles, sites of medium strength were the most abundant in Fe0.6Zr0.4O2 mixed oxide, with a content as high as 0.81 mmol CO2 per g (Table S2†).
TEM images and size distributions of the prepared catalysts are presented in Fig. 5, while the mean diameters of each catalyst are listed in Table 2. The results showed that a good degree of metal dispersion and homogeneity was achieved for all samples except Fe0.8Zr0.2O2. Fe2O3 and ZrO2 clearly have a much larger average particle size (74.0 and 8.2 nm, respectively) than the Fe–Zr mixed oxides, which revealed that the generation of mixed oxides can significantly reduce the particle size of the catalysts, especially for the Fe2O3 sample. It is worth mentioning that Fe0.6Zr0.4O2, in comparison with other mixed oxides, has a smaller particle size (7.0 nm). Additionally, because of the relatively high iron content in the Fe0.8Zr0.2O2 catalyst, there were lots of large Fe2O3 particles generated (about 47.3 nm, Fig. S1†) and small particles (about 5.0 nm), which were still much smaller than pure Fe2O3 particles (about 74.0 nm). The SEM images of the catalysts (Fig. S2†) also showed the smaller particle size of Fe–Zr mixed oxides than that of pure oxides.
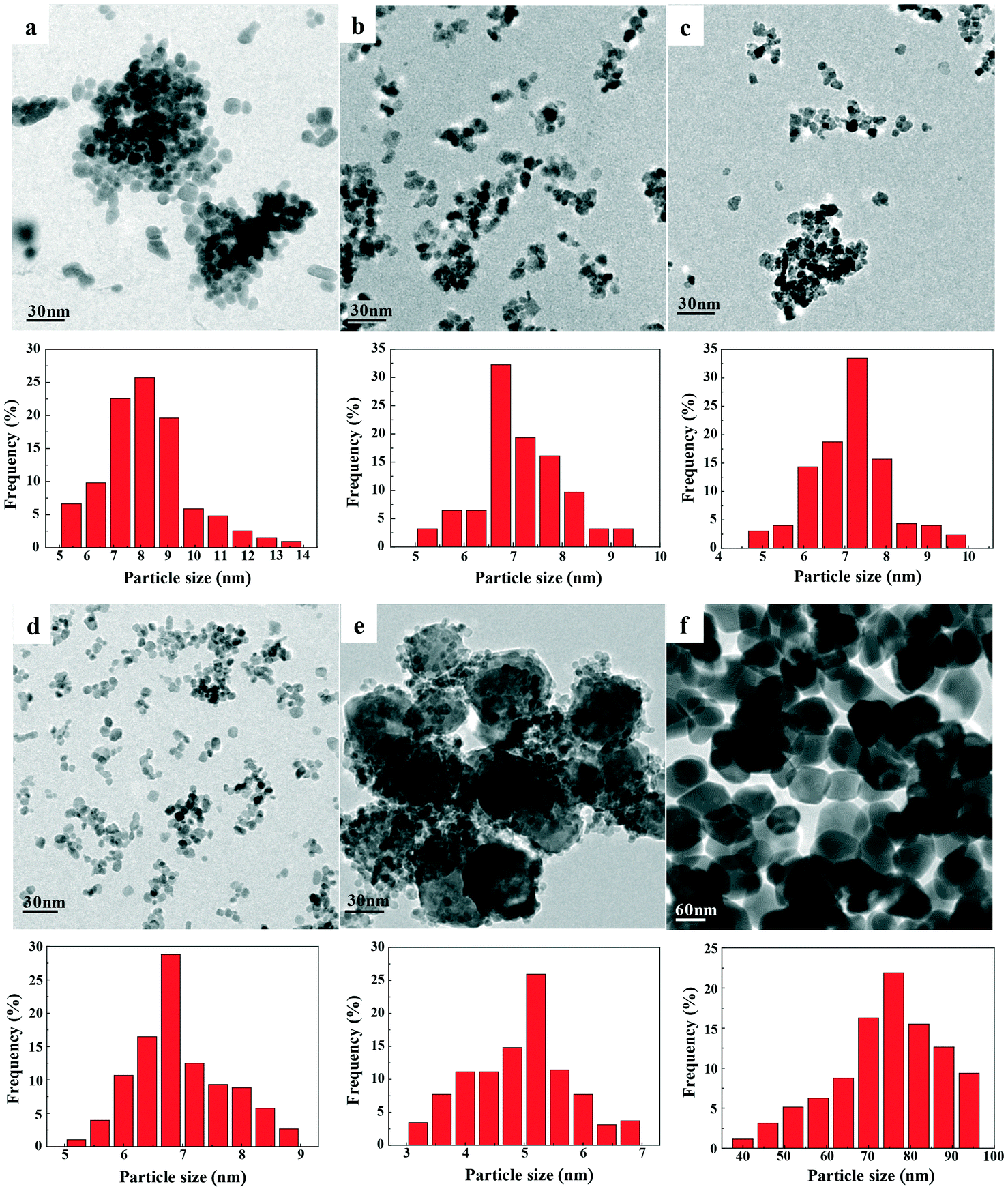 | ||
| Fig. 5 TEM images and histograms showing the size distributions of FexZr1−xO2 samples for (a) ZrO2, (b) Fe0.2Zr0.8O2, (c) Fe0.4Zr0.6O2, (d) Fe0.6Zr0.4O2, (e) Fe0.8Zr0.2O2 and (f) Fe2O3. | ||
The composition of the prepared catalysts was determined by ICP analysis (Table 2), and the results showed that the tested value was approximately consistent with the expected value. The specific surface areas of the catalysts are shown in Table 2. The Fe2O3 sample presented the lowest BET surface because of its largest crystallite size of 74.0 nm (as shown in TEM). Additionally, it is clear that the addition of Zr can significantly change the surface area of the catalysts, which is consistent with the previous report that the formation of mixed oxides could help to increase the surface area of the catalysts.41 With the increase of Zr content in Fe–Zr mixed oxides, the specific surface area of the catalyst increased from 62 m2 g−1 in Fe0.8Zr0.2O2 to 122 m2 g−1 in the Fe0.2Zr0.8O2 sample. It should be noted that due to the generation of large Fe2O3 particles (as shown in TEM), the Fe0.8Zr0.2O2 sample has a relatively low specific surface area compared with other mixed oxides.
Catalytic performance for aerobic oxidation of HMF
The reaction conditions were selected on the basis of our previous work regarding the base-free conversion of HMF to FDCA in ILs.42 We have already shown that the formation of FDCA nearly cannot take place below 120 °C over non-noble metal catalysts, and 160 °C is the most suitable temperature for the reaction. Therefore, the measurement of the catalytic performance of the synthesized Fe–Zr–O catalyst and the determination of the reaction pathways for the production of FDCA were performed with batch experiments at 160 °C and 2 MPa of O2. The choice of [Bmim]Cl ILs as solvent not only can avoid the excessive use of base due to the unique dissolving abilities of ILs but also can effectively promote the oxidation of HMF into FDCA with a non-noble metal catalyst, a reaction which cannot take place when other solvents are used instead of ILs, such as water, toluene, CH3CN, 1,4-dioxane, methanol, GVL or CH3COOH.42Table 3 presents the catalytic results of mixed oxides with different Fe/Zr ratios and pure Fe2O3 and ZrO2 oxides for the aerobic oxidation of HMF to FDCA in ILs. As reported,42 because of the strong hydrogen bond formation ability and the unique dissolving ability of FDCA, [Bmim]Cl played a favorable role as a co-catalyst and solvent in HMF oxidation, and a 21.6% yield of FDCA is achieved at 160 °C without any catalysts (Table 3, entry 1). Importantly, although over 98% conversion of HMF is achieved with all the catalysts, the FDCA yield is strongly influenced by the Fe/Zr ratio. Compared with the blank experiment, ZrO2 did not exhibit any catalytic activity for HMF oxidation, while the Fe2O3 catalyst showed 31.6% FDCA yield (Table 3, entries 2 and 3). Additionally, a strong synergetic effect was observed when the Fe–Zr mixed oxides were generated by a hydrothermal method. For the bimetallic oxides, with the increase in Zr content, the FDCA yield first sharply increased to 60.6%, and then decreased rapidly to 27.1% (Table 3, entries 5–7). Although all catalysts show similar HMF conversion (>98%), the employment of the Fe0.6Zr0.4O2 catalyst results in the highest FDCA yield of 60.6%. It is well known that the acid–base properties of catalyst have a great effect on catalysis. According to recent reports,16 the acidic active sites on the surface of the catalysts played an important role in enhancing the yield of FDCA. Recent literature reports also emphasized that the surface basicity of bimetallic oxides improves the oxidant strength of the catalyst;43 in particular, the medium strength basic sites favor the oxidation of HMF to FDCA.19 For the Fe0.6Zr0.4O2 catalyst, as we mentioned above (Fig. 4b and c and Table 2), a higher total number of acidic sites as well as basic sites were generated compared with pure oxides or other mixed oxides. Importantly, basic sites of medium strength were the most abundant in the Fe0.6Zr0.4O2 catalyst (Table S2†), which resulted in the best catalytic performance.
| Entry | Catalysts |
|
|
|
|
|---|---|---|---|---|---|
| FDCA yield (%) | FFCA yield (%) | HMFA yield (%) | HMF conversion (%) | ||
| a Reaction conditions: HMF (0.1 mmol), catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C, 24 h. b Physical mixture of Fe2O3 and ZrO2 with the atomic ratio of Fe0.6Zr0.4O2. | |||||
| 1 | Blank | 21.6 ± 0.9 | 2.4 ± 0.2 | 0.5 ± 0.1 | 99.1 ± 0.5 |
| 2 | ZrO2 | 16.7 ± 0.8 | 11.4 ± 0.8 | 0.6 ± 0.1 | 99.1 ± 0.8 |
| 3 | Fe2O3 | 31.6 ± 1.7 | 5.3 ± 0.4 | 1.7 ± 0.2 | 98.1 ± 0.9 |
| 4 | Fe0.8Zr0.2O2 | 53.5 ± 2.1 | 2.4 ± 0.3 | 3.5 ± 0.3 | 98.5 ± 1.2 |
| 5 | Fe0.6Zr0.4O2 | 60.6 ± 2.4 | 1.3 ± 0.2 | 0.2 ± 0.0 | 99.7 ± 0.2 |
| 6 | Fe0.4Zr0.6O2 | 35.6 ± 1.6 | 2.8 ± 0.4 | 3.2 ± 0.3 | 98.7 ± 0.9 |
| 7 | Fe0.2Zr0.8O2 | 27.1 ± 1.3 | 3.6 ± 0.4 | 1.8 ± 0.2 | 99.2 ± 0.5 |
| 8 | Fe2O3 + ZrO2b | 36.7 ± 1.4 | 4.2 ± 0.3 | 0.1 ± 0.0 | 98.2 ± 1.0 |
It is worth noting that, in contrast, the physical mixture of Fe2O3 and ZrO2 with a corresponding mole fraction as Fe0.6Zr0.4O2 was added to the reaction (Table 3, entry 8); however, a much lower FDCA (36.7%) yield was obtained from it. The significant difference between the Fe0.6Zr0.4O2 catalyst prepared by the hydrothermal and that prepared by the physical mixing method indicated the importance of the solid solution structure. As we mentioned above, the XRD and Raman measurements along with O 1s XPS revealed the formation of solid solutions and oxygen vacancies. As reported previously,26,36 more oxygen vacancies on the material could be created with the formation of a solid solution. Therefore, the redox properties and activity of the catalyst, which are beneficial to the reaction, could be improved as well. In the Fe–Zr mixed oxides, the Fe0.6Zr0.4O2 sample has relatively low reduction peaks (see Fig. 3a), revealing the highest reducibility and oxygen mobility of the sample, which is in accordance with the O 1s XPS finding that the largest amount of reactive oxygen species was observed in the Fe0.6Zr0.4O2 catalyst. It is worth stressing that the by-products during the oxidation of HMF are small amounts of FFCA and HMFA, which could be detected by HPLC, and a relatively large amount of humins, which could be observed clearly after the reaction (Fig. S4†) and will be discussed in the following.
Formation of humins
As reported previously,44 HMF was not stable and degraded into humins significantly at high temperature. Therefore, the FDCA yield was restrained (60.6% under optimal conditions) by the parallel formation of insoluble humins even though the HMF conversion reached over 99%. In our work, some yellowish-brown humins were observed together with the used catalyst after the oxidation of HMF (Fig. S4†). In order to obtain more insights into their formation mechanisms, the solid residues (containing insoluble humins and used catalyst) were washed with ethanol three times, collected by centrifugation and then oven-dried overnight at 70 °C for FT-IR, SEM, TG and GPC analyses.As shown in Fig. 6, the FT-IR spectrum of humins shows a broad peak at 3378 cm−1 attributed to O–H stretching vibrations, suggesting the existence of hydroxyl groups.45 The peaks at 2959–2872 cm−1 were due to stretching vibrations of methyl and methylene groups, and the peak at 1712 cm−1 was ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations in aldehydes.46 Importantly, peaks corresponding to furanic rings exist in the IR spectrum of the humins. As reported in the literature, peaks at 1611 and 1577 cm−1 correspond to CC stretching in furan rings, and peaks at 1407, 1364, 779 and 740 cm−1 were ascribed to C–O–C stretching and aromatic C–H out-of-plane bending vibrations in furan rings, respectively.47 Additionally, the peak at 1018 cm−1, which is attributed to primary alcohols, is also observed in the IR spectrum of the humins. In conclusion, the formation of humins may be through the polymerization and cross-polymerization of HMF and intermediates as reported in the literature.48,49 Understanding and controlling the formation of humins are still challenging; some work is ongoing in our laboratory to reduce the formation of humins and improve the yield of FDCA.
O stretching vibrations in aldehydes.46 Importantly, peaks corresponding to furanic rings exist in the IR spectrum of the humins. As reported in the literature, peaks at 1611 and 1577 cm−1 correspond to CC stretching in furan rings, and peaks at 1407, 1364, 779 and 740 cm−1 were ascribed to C–O–C stretching and aromatic C–H out-of-plane bending vibrations in furan rings, respectively.47 Additionally, the peak at 1018 cm−1, which is attributed to primary alcohols, is also observed in the IR spectrum of the humins. In conclusion, the formation of humins may be through the polymerization and cross-polymerization of HMF and intermediates as reported in the literature.48,49 Understanding and controlling the formation of humins are still challenging; some work is ongoing in our laboratory to reduce the formation of humins and improve the yield of FDCA.
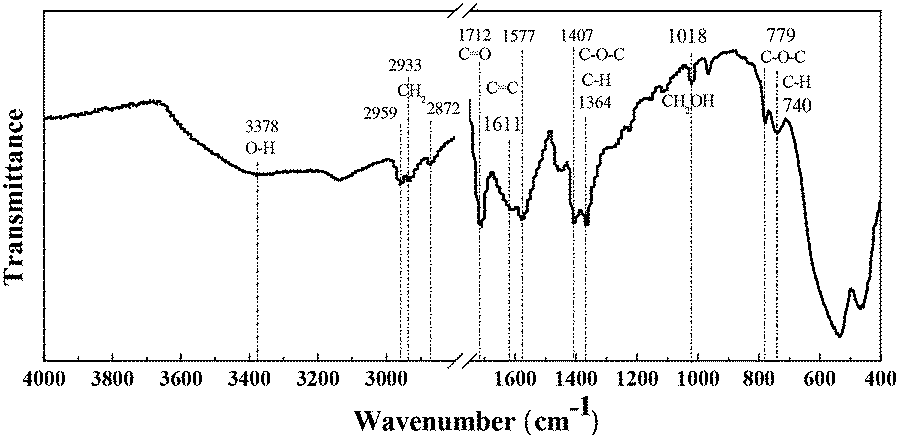 | ||
| Fig. 6 FT-IR spectrum of humins after aerobic oxidation of HMF. Reaction conditions: HMF (0.1 mmol), Fe0.6Zr0.4O2 catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C, 24 h. | ||
Further evidence for the formation of humins was obtained from SEM and TG analysis. Typical SEM images of the catalyst and humins after aerobic oxidation of HMF are given in Fig. 7. It is shown that the humins consisted of a lamellar membrane structure, which was very different from spherical particles as reported in the literature due to the use of a very different reaction solvent, [Bmim]Cl.50 TG analysis of the solid residues (containing insoluble humins and catalyst) showed a significant weight loss (Fig. S5†) corresponding to the decomposition of humins, which is also evidence for the formation of humins.
 | ||
| Fig. 7 SEM images of (a) fresh Fe0.6Zr0.4O2 catalyst and (b and c) mixtures of used catalyst and humins after aerobic oxidation of HMF. Reaction conditions: HMF (0.1 mmol), Fe0.6Zr0.4O2 catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C, 24 h. | ||
To gain insight into the average molecular weight distribution of humins, the solid residues were dissolved in THF and then subjected to GPC analysis (Fig. S6†). The GPC result demonstrates that the humins comprise a mixture of components with approximate weight-average molecular weights (Mw) of 277, 513 and 1065, which are two ranges of molecular weights of HMF. It should be noted that part of the humins is insoluble or partially soluble in THF, which exceeds the detection limit of instruments, and their molecular weights cannot be detected; as such, the molecular weights of the humins cannot be established unequivocally. However, it is evident that polymerization or cross-polymerization occurs to a significant extent because GPC showed obvious peaks. Coupling the TG behavior and the SEM characterization, we concluded that the main by-product was humins, whereas coupling the FT-IR and GPC analysis, we concluded that polymerization or cross-polymerization occurs with two ranges of molecular weights of HMF.
Reaction pathways for aerobic oxidation of HMF to FDCA
The reaction pathways for aerobic oxidation of HMF in [Bmim]Cl over Fe0.6Zr0.4O2 catalyst are investigated by recording the distributions and yields of the products at different reaction times. As shown in Fig. 8, with the increase of reaction time, HMF was progressively consumed, reaching a conversion of 88.8% at 4 h and almost complete conversion after 8 h. During the course of the reaction, FDCA was detected as a major product, with HMFA and FFCA being the main intermediates, while no DFF was detected. As the reaction progressed, the yield of FDCA increased, while the yield of FFCA first increased and then decreased, and the yield of HMFA was always low. When the reaction time was prolonged to 24 h, the FDCA yield achieved 60.6% while FFCA and HMFA yields declined to 1.3% and 0.2%, respectively.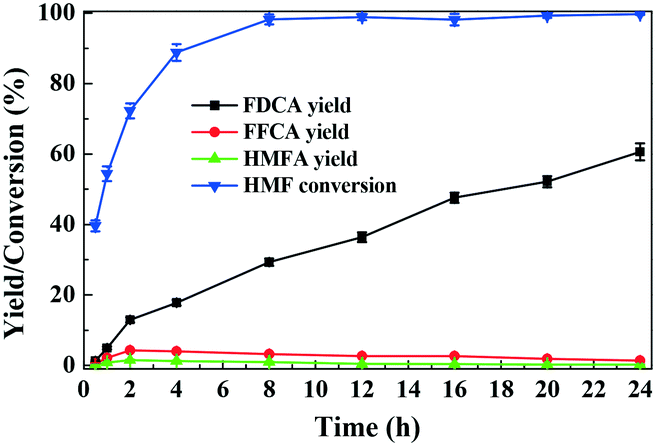 | ||
| Fig. 8 Time evolution of HMF conversion and yields of products over Fe0.6Zr0.4O2 catalyst. Reaction conditions: HMF (0.1 mmol), catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C. | ||
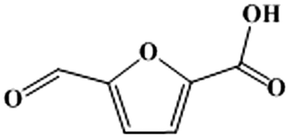
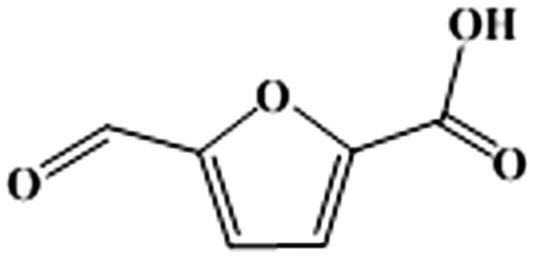
It should be underlined that there was no DFF generated during the reaction, suggesting that the oxidation of the aldehyde group to the carboxyl group was much easier than the oxidation of the hydroxymethyl group to the aldehyde group in [Bmim]Cl ILs with Fe0.6Zr0.4O2 catalyst. In order to confirm this, further oxidation experiments of commercial HMFA (which contains the hydroxymethyl group), DFF and FFCA (which contains the aldehyde group) in [Bmim]Cl over Fe0.6Zr0.4O2 catalyst were performed. As shown in Table 4, the hydroxymethyl group of HMFA was slowly converted into the aldehyde group of FFCA and further converted into the carboxyl group of FDCA, resulting in a 90.8% conversion of HMFA with 0.6% and 12.9% yields of FFCA and FDCA after 8 h, respectively, while the aldehyde groups in DFF and FFCA were quickly converted into the carboxyl group of FFCA or FDCA. In particular, for the DFF, almost complete conversion and 1.2% and 44.8% yields of FFCA and FDCA were attained in as short as 2 h. When the reaction time was prolonged to 6 h, a FDCA yield of 65.3% was achieved while the FFCA conversion increased to 98.8%.
| Entry | Reactant | Reaction time (h) |
|
|
Conversion (%) |
|---|---|---|---|---|---|
| FFCA yield (%) | FDCA yield (%) | ||||
| Reaction conditions: HMFA/DFF/FFCA (0.1 mmol), Fe0.6Zr0.4O2 catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C. | |||||
| 1 | HMFA | 0.5 | 3.2 ± 0.2 | 1.2 ± 0.1 | 27.1 ± 1.2 |
| 2 | HMFA | 1 | 4.0 ± 0.3 | 1.3 ± 0.1 | 32.2 ± 1.8 |
| 3 | HMFA | 4 | 1.3 ± 0.1 | 12.2 ± 0.7 | 70.1 ± 2.3 |
| 4 | HMFA | 8 | 0.6 ± 0.1 | 12.9 ± 0.6 | 90.8 ± 0.9 |
| 5 | DFF | 0.2 | 3.4 ± 0.4 | 0.8 ± 0.1 | 65.1 ± 2.0 |
| 6 | DFF | 0.5 | 25.6 ± 1.3 | 8.8 ± 0.5 | 97.6 ± 1.1 |
| 7 | DFF | 1 | 8.5 ± 0.4 | 36.0 ± 1.7 | 100.0 ± 0.0 |
| 8 | DFF | 2 | 1.2 ± 0.1 | 44.8 ± 2.0 | 100.0 ± 0.0 |
| 9 | FFCA | 0.5 | — | 9.5 ± 0.5 | 60.0 ± 2.2 |
| 10 | FFCA | 1 | — | 39.9 ± 1.8 | 86.4 ± 1.0 |
| 11 | FFCA | 4 | — | 58.9 ± 2.0 | 98.2 ± 0.8 |
| 12 | FFCA | 6 | — | 65.3 ± 2.3 | 98.8 ± 0.7 |
Accordingly, the reaction pathways for the aerobic oxidation of HMF to FDCA in [Bmim]Cl ILs over Fe0.6Zr0.4O2 catalyst could be obtained as shown in Scheme 1, which proceeded via the formation of a HMFA intermediate rather than the DFF intermediate and was consistent with previous reports.13,51,52 Importantly, the yield of HMFA was relatively low when compared to the yields of FDCA and FFCA, and the FFCA yield was much higher than the HMFA yield. This phenomenon revealed that the conversion of the aldehyde group in furan rings to the carboxyl group over Fe0.6Zr0.4O2 catalyst is still a challenge under base-free conditions, although it is very fast in the presence of a base.53 Therefore, conversions of HMF to HMFA and humins occur in parallel depending on the Fe0.6Zr0.4O2 catalyst employed, resulting in the high HMF conversion but limited FDCA yield, with humins as the main by-product.
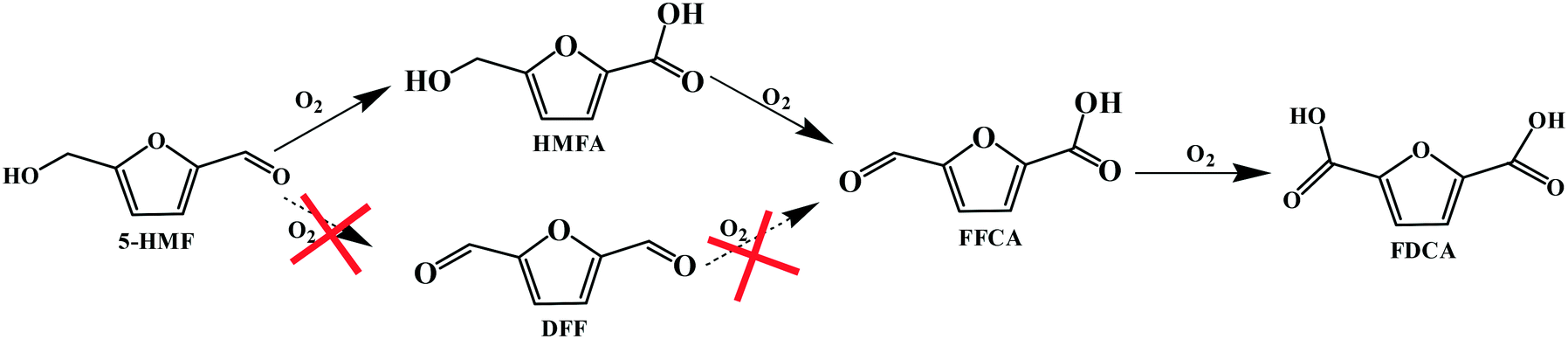 | ||
| Scheme 1 Reaction pathways for the aerobic oxidation of HMF to FDCA in [Bmim]Cl over Fe0.6Zr0.4O2 catalyst. | ||
Proposed reaction mechanism
According to the above experimental and catalyst characterization results as well as the reported work in the literature,13,54,55 the plausible reaction mechanism for the aerobic oxidation of HMF over Fe0.6Zr0.4O2 catalyst is proposed and shown in Scheme 2. In the initial stage of the reaction, the aldehyde groups of HMF were oxidized to the corresponding monocarboxylic acid (HMFA) through the formation of the intermediate hemiacetal. In this stage, both the acidity and the basicity of the catalyst could facilitate the formation of hemiacetal.56,57 The acidic sites can favor the adsorption of the aldehyde group on the surface of the catalyst, while the basic sites can further transform it into hemiacetal. During the transformation of hemiacetal into HMFA, a dehydrogenation reaction occurred and two hydrogen atoms were left on the surface of the catalyst, which were oxidized into water by active oxygen ions (O2− or O−) on the surface of the catalyst; thus, the catalyst is recovered. In the second stage, the hydroxymethyl group in HMF first adsorbed onto the surface of the catalyst to form a metal alkoxide intermediate with the help of acidic sites, and then is further converted to an aldehyde to form FFCA through β-hydride elimination with the aid of basic sites,55 also leaving two hydrogen atoms on the catalyst surface. As for the third step, the oxidation of aldehyde in FFCA undergoes the same oxidation process as the first step and finally generates FDCA. It is worth noting that both the acidity and the basicity as well as the active oxygen ions (O2− or O−) on the surface of the catalyst, which could supply molecular oxygen (O2), play important roles in the oxidation of HMF. In addition, as reported, the polar molecules or those containing a strong proton donor functionality (such as carboxylic acids, phenols and diols) can be dissolved in ILs, especially when the ILs have a H-bond acceptor capability, such as chloride-containing ILs.58 We also found that the solubility of FDCA in [Bmim]Cl was as high as 47.93%;42 therefore, the obtained HMFA, FFCA and FDCA molecules could be desorbed from the Fe0.6Zr0.4O2 catalyst with the help of [Bmim]Cl, and go into the IL solvent easily; thus, the catalyst was recovered.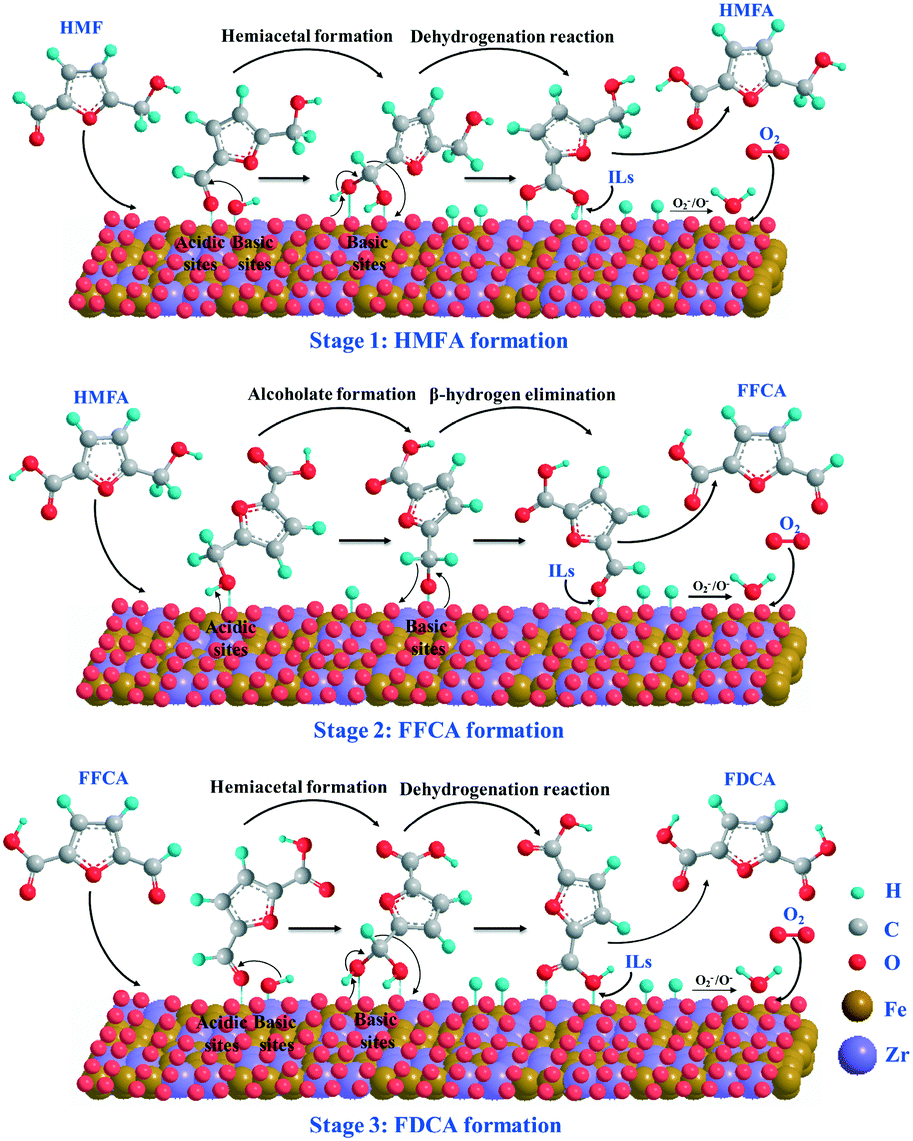 | ||
| Scheme 2 Plausible reaction mechanism of aerobic oxidation of HMF to FDCA over Fe0.6Zr0.4O2 catalyst under base-free conditions. | ||
Catalyst recycling
To test the reusability of the catalyst, Fe0.6Zr0.4O2 was successively reused five times under optimal conditions for the oxidation of HMF to FDCA. After the reaction, the catalyst was thoroughly washed with ethanol three times, dried in a vacuum oven at 70 °C overnight and then reused. As shown in Fig. 9, there was no significant change in the HMF conversion and FDCA yield, suggesting the stability of the catalyst. In the first run, HMF conversion and FDCA yield were 99.7% and 60.6%, respectively, while in the fifth run, they were still maintained at 98.1% and 50.1%, respectively, indicating that the catalyst was used five times without significant loss of activity. It is noteworthy that as our previous study pointed out,42 in the recycle experiments, some insoluble humins also formed and mixed together with the catalyst. However, they could be dissolved completely by [Bmim]Cl IL due to its unique dissolving abilities for polar compounds (Fig. S7†). Therefore, the adsorption of FDCA and humins on the active sites of the catalyst could be avoided, and thus the activity of the catalyst could be maintained.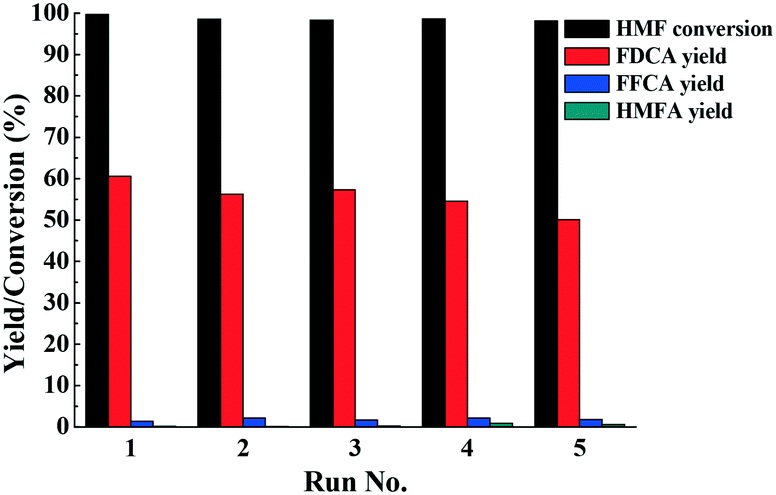 | ||
| Fig. 9 Recycle experiments of the catalyst. Reaction conditions: HMF (0.1 mmol), Fe0.6Zr0.4O2 catalyst (0.01 g), [Bmim]Cl (1 g), O2 (2 MPa), 160 °C, 24 h. | ||
Conclusions
In summary, a series of FexZr1−xO2 non-noble metal catalysts were synthesized and employed as catalysts in the aerobic oxidation of HMF to FDCA under base-free conditions. The catalysts demonstrated very different activities for FDCA production due to the different acidity and basicity as well as reducibility and oxygen mobility. A series of measurements revealed that the Fe0.6Zr0.4O2 mixed oxides have the highest degrees of acidity and basicity because of the catalyst composition and small particle size as well as the high specific surface area, which could improve the oxidant strength of the catalyst and favor the oxidation of HMF to FDCA. Additionally, the XRD, Raman and O 1s XPS revealed the generation of solid solutions and oxygen vacancies in the Fe0.6Zr0.4O2 mixed oxides, which are beneficial to the redox properties and activity of the catalyst and thus favor the oxidation of HMF. Thus, a favorable performance of the Fe0.6Zr0.4O2 catalyst for HMF oxidation was observed with a 99.9% HMF conversion and 60.6% FDCA yield under optimal reaction conditions, and the catalyst could be used five times without obvious loss of activity.Importantly, the formation of humins and the aerobic oxidation pathways of HMF to FDCA in the [Bmim]Cl ILs have also been investigated, which revealed a parallel reaction between FDCA and humin formation from HMF. Additionally, the reaction mechanism over the Fe0.6Zr0.4O2 catalyst was proposed based on the understanding of the acidity and basicity as well as the reducibility of the catalyst. Furthermore, given the cost and environmental advantage of non-noble metal catalysts used in base-free reaction systems, more research could be further performed in the development of highly active catalysts for FDCA production, which is ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, 2015CB251401) and the National Natural Science Foundation of China (No. 21576269, 91434203, 21210006, 21506231).Notes and references
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017 DOI:10.1111/1751-7915.12710.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- N. Mei, B. Liu, J. D. Zheng, K. L. Lv, D. G. Tang and Z. H. Zhang, Catal. Sci. Technol., 2015, 5, 3194–3202 CAS.
- Z. H. Zhang and K. J. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- Z. Z. Miao, Y. B. Zhang, X. Q. Pan, T. X. Wu, B. Zhang, J. W. Li, T. Yi, Z. D. Zhang and X. G. Yang, Catal. Sci. Technol., 2015, 5, 1314–1322 CAS.
- J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef CAS PubMed.
- T. S. Hansen, I. Sádaba, E. J. García-Suárez and A. Riisager, Appl. Catal., A, 2013, 456, 44–50 CrossRef CAS.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102–111 CrossRef CAS.
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81 CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- S. Siankevich, G. Savoglidis, Z. F. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- Z. H. Zhang, J. D. Zhen, B. Liu, K. L. Lv and K. J. Deng, Green Chem., 2015, 17, 1308–1317 RSC.
- D. K. Mishra, H. J. Lee, J. Kim, H.-S. Lee, J. K. Cho, Y.-W. Suh, Y. J. Yi and Y. J. Kim, Green Chem., 2017, 19, 1619–1623 RSC.
- S. G. Wang, Z. H. Zhang and B. Liu, ACS Sustainable Chem. Eng., 2015, 3, 406–412 CrossRef CAS.
- L. C. Gao, K. J. Deng, J. D. Zheng, B. Liu and Z. H. Zhang, Chem. Eng. J., 2015, 270, 444–449 CrossRef CAS.
- F. Neaţu, R. S. Marin, M. Florea, N. Petrea, O. D. Pavel and V. I. Pârvulescu, Appl. Catal., B, 2016, 180, 751–757 CrossRef.
- E. Hayashi, T. Komanoya, K. Kamata and M. Hara, ChemSusChem, 2017, 10, 654–658 CrossRef CAS PubMed.
- X. W. Han, C. Q. Li, X. H. Liu, Q. N. Xia and Y. Q. Wang, Green Chem., 2017, 19, 996–1004 RSC.
- Z. H. Gu, X. L. Sang, H. Wang and K. Z. Li, J. Rare Earths, 2014, 32, 817–823 CrossRef CAS.
- K. C. Soni, S. Chandra Shekar, B. Singh and T. Gopi, J. Colloid Interface Sci., 2015, 446, 226–236 CrossRef CAS PubMed.
- S. Kumar, S. Bhunia, J. Singh and A. K. Ojha, J. Alloys Compd., 2015, 649, 348–356 CrossRef CAS.
- P. E. Quintard, P. Barbéris, A. P. Mirgorodsky and T. Merle-Méjean, J. Am. Ceram. Soc., 2002, 85, 1745–1749 CrossRef CAS.
- Z. H. Gu, K. Z. Li, S. Qing, X. Zhu, Y. G. Wei, Y. T. Li and H. Wang, RSC Adv., 2014, 4, 47191–47199 RSC.
- G. Litt and C. Almquist, Appl. Catal., B, 2009, 90, 10–17 CrossRef CAS.
- A. Subramanian, A. Annamalai, H. H. Lee, S. H. Choi, J. Ryu, J. H. Park and J. S. Jang, ACS Appl. Mater. Interfaces, 2016, 8, 19428–19437 CAS.
- C. H. Miao, S. L. Ji, G. P. Xu, G. D. Liu, L. D. Zhang and C. H. Ye, ACS Appl. Mater. Interfaces, 2012, 4, 4428–4433 CAS.
- G. M. Wang, Y. C. Ling, D. A. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503–3509 CrossRef CAS PubMed.
- J. M. Luo, X. B. Luo, C. Z. Hu, J. C. Crittenden and J. C. Qu, ACS Appl. Mater. Interfaces, 2016, 8, 18912–18921 CAS.
- P. M. Kouotou, H. Vieker, Z. Y. Tian, P. H. T. Ngamou, A. El Kasmi, A. Beyer, A. Golzhauser and K. Kohse-Hoinghaus, Catal. Sci. Technol., 2014, 4, 3359–3367 Search PubMed.
- J. F. Nie and H. C. Liu, J. Catal., 2014, 316, 57–66 CrossRef CAS.
- V. P. Santos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 353–363 CrossRef CAS.
- X. Y. Wang, Q. Kang and D. Li, Appl. Catal., B, 2009, 86, 166–175 CrossRef CAS.
- K. Z. Li, H. Wang, Y. G. Wei and D. X. Yan, Chem. Eng. J., 2011, 173, 574–582 CrossRef CAS.
- E. Moretti, L. Storaro, A. Talon, P. Riello, A. I. Molina and E. Rodríguez-Castellón, Appl. Catal., B, 2015, 168–169, 385–395 CrossRef CAS.
- J. Zieliński, I. Zglinicka, L. Znak and Z. Kaszkur, Appl. Catal., A, 2010, 381, 191–196 CrossRef.
- K. Z. Li, H. Wang, Y. G. Wei and D. X. Yan, Appl. Catal., B, 2010, 97, 361–372 CrossRef CAS.
- E. J. Baran, Catal. Today, 1990, 8, 133–151 CrossRef CAS.
- J. Wang, C. Y. Han, X. Y. Gao, J. C. Lu, G. P. Wan, D. D. He, R. Chen, K. Z. Chen, S. F. He and Y. M. Luo, J. Power Sources, 2017, 343, 437–445 CrossRef CAS.
- D. X. Yan, J. Y. Xin, C. Y. Shi, X. M. Lu, L. L. Ni, G. Y. Wang and S. J. Zhang, Chem. Eng. J., 2017, 323, 473–482 CrossRef CAS.
- J. B. Branco, A. C. Ferreira, T. A. Gasche, G. Pimenta and J. P. Leal, Adv. Synth. Catal., 2014, 356, 3048–3058 CrossRef CAS.
- B. Liu, Y. S. Ren and Z. H. Zhang, Green Chem., 2015, 17, 1610–1617 RSC.
- S. R. Wang, B. Ru, H. Z. Lin and W. X. Sun, Fuel, 2015, 150, 243–251 CrossRef CAS.
- C. B. Rasrendra, M. Windt, Y. Wang, S. Adisasmito, I. G. B. N. Makertihartha, E. R. H. van Eck, D. Meier and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2013, 104, 299–307 CrossRef CAS.
- T. M. C. Hoang, E. R. H. van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- B. Liu, Z. H. Zhang and Z. K. Zhao, Chem. Eng. J., 2013, 215–216, 517–521 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 4745–4755 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- G. S. Yi, S. P. Teong and Y. G. Zhang, Green Chem., 2016, 18, 979–983 RSC.
- A. Abad, A. Corma and H. García, Chem. – Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed.
- Y. B. Wang, K. Yu, D. Lei, W. Si, Y. J. Feng, L.-L. Lou and S. X. Liu, ACS Sustainable Chem. Eng., 2016, 4, 4752–4761 CrossRef CAS.
- Y. C. Li, L. Wang, R. Y. Yan, J. X. Han and S. J. Zhang, Catal. Sci. Technol., 2015, 5, 3682–3692 CAS.
- F. Z. Su, J. Ni, H. Sun, Y. Cao, H. Y. He and K. N. Fan, Chem. – Eur. J., 2008, 14, 7131–7135 CrossRef CAS PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Germany, 2nd edn, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy01704a |
| This journal is © The Royal Society of Chemistry 2018 |