
Effect of steam and CO2 on ethane activation over Zn-ZSM-5†
Ali
Mehdad
 ,
Nicholas S.
Gould
,
Bingjun
Xu
and
Raul F.
Lobo
*
,
Nicholas S.
Gould
,
Bingjun
Xu
and
Raul F.
Lobo
*
Center for Catalytic Science and Technology, Department of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St., Newark, DE 19716, USA. E-mail: lobo@udel.edu
First published on 20th November 2017
Abstract
The impact of steam and CO2 on ethane activation over 5% (w/w) Zn-ZSM-5 catalysts was investigated. The main products in He, steam and CO2 were mixtures of ethene, methane, benzene and toluene. CO2 clearly increased the formation rate of ethene and CO, and steam increased the formation rate of ethene. In addition, the presence of CO2 decreased the formation rates of methane and aromatics by factors of ∼3 and 7, respectively. Hydrogen formed from ethane dehydrogenation reacted with CO2, via the reverse water gas shift reaction, to form CO and water. Under vacuum, FTIR spectra of adsorbed ethene on Zn-ZSM-5 showed stronger adsorption on zinc Lewis acid sites than on Brønsted acid sites. In the presence of steam, strong adsorption of ethene on the zinc Lewis acid sites (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 1595 cm−1) splits the spectra into two absorption bands (at 1650 and 1568 cm−1), implying the hydrolysis of Zn(II) sites to form Brønsted acid sites and Zn(OH)+ sites. Hydrolysis of Zn(II) sites suppresses oligomerization/aromatization reactions. The changes in selectivity are reversible and can be stopped by decreasing water vapor pressure. The results confirm that Zn(OH)+ sites are effective in ethane dehydrogenation, but the Zn(II) sites are necessary for aromatization. The absence of aromatization reactions in the presence of steam shows that Zn(II) sites catalyze aromatization at faster rates than Brønsted acid sites.
C at 1595 cm−1) splits the spectra into two absorption bands (at 1650 and 1568 cm−1), implying the hydrolysis of Zn(II) sites to form Brønsted acid sites and Zn(OH)+ sites. Hydrolysis of Zn(II) sites suppresses oligomerization/aromatization reactions. The changes in selectivity are reversible and can be stopped by decreasing water vapor pressure. The results confirm that Zn(OH)+ sites are effective in ethane dehydrogenation, but the Zn(II) sites are necessary for aromatization. The absence of aromatization reactions in the presence of steam shows that Zn(II) sites catalyze aromatization at faster rates than Brønsted acid sites.
1. Introduction
Production of light alkanes—ethane, propane, butanes—has increased due to the widespread application of hydraulic fracturing for the extraction of shale gas and oil. As a result, abundant and inexpensive feedstocks containing light alkanes have become increasingly available. This availability has made the transformation of these species into value-added olefins and aromatics increasingly attractive. Among the various catalysts considered for this reaction, metal-containing zeolites are particularly attractive as shape selectivity of zeolites decreases the rate of coke formation.Dehydroaromatization rates of light alkanes in ZSM-5 (MFI) zeolites can be increased by the introduction of zinc into the pores. Zn replaces protons in the zeolite resulting in lower rates of hydrocarbon cracking and higher rates of hydrogen desorption.1–4 In Zn-ZSM-5, heterolytic dissociative adsorption of methane, ethane and hydrogen has been reported.5–10 This is explained, in part, by the fact that zinc makes strong bonds with hydrogen and carbon, as shown by the stability of compounds such as ZnH2,11 Zn(Me)2 and Zn(Et)2.12
The non-oxidative dehydrogenation reaction requires high temperature because of thermodynamic limitations. To overcome the thermodynamic limitations of isothermal dehydroaromatization, co-feeding of hydrogen acceptor molecules has been investigated. Co-feeding of CO2, CO or O2, for example, to H- or metal-exchanged ZSM-5 increased the rate of propane aromatization, with CO2 being more selective for scavenging hydrogen than O2,13 which also enhanced combustion reactions. Depending on the reaction conditions and catalyst properties, CO2 can react with hydrogen or ethane through different reactions: CO2 can be reduced with hydrogen via the reverse water gas shift reaction (RWGS) to form CO and water (eqn (1)), and can react with ethane through C–H bond activation by oxidative dehydrogenation (ODH) (eqn (2)), or through C–C bond activation via the dry reforming reaction (eqn (3)).14,15
| CO2 + H2 → CO + H2O | (1) |
| CO2 + C2H6 → C2H4 + CO + H2O | (2) |
| 2CO2 + C2H6 → 4CO + 3H2 | (3) |
The identity of the metal incorporated into ZSM-5 affects the products formed by the reaction of ethane and CO2. At temperatures higher than 500 °C, the reaction of ethane and CO2 proceeds via reforming on Rh/ZSM-516 and via ODH on Cr/ZSM-5.17,18 The positive role of CO2 was attributed to the removal of deposited coke by the reverse Boudouard reaction. CO2 can also decrease the rate of coke formation by facilitating ethene desorption from Ga2O3/TiO2 during ethane dehydrogenation.19 Reduction of CO2 by propane versus H2 on Zn-ZSM-5 showed higher conversion and selectivity toward CO, as in the presence of hydrocarbons CO could also form by oxidation of hydrocarbons.20
In this report, we investigate the use CO2 as a hydrogen scavenger during ethane dehydroaromatization on Zn-ZSM-5. Coke formation is the main deactivation mechanism of Zn-ZSM-5 catalysts during ethane aromatization,4 and CO2 can improve catalyst lifetime by removing a fraction of the coke via the reverse Boudouard reaction. Further, CO2 can also serve as a mild oxidant to stabilize ZnO in ZSM-5,21 preventing the loss of zinc in a high temperature reducing atmosphere. The dehydrogenation of propane in CO2 on Zn/ZSM-5 (impregnated) revealed that the promoting effects of CO2 were related to the removal of H2 by the RWGS reaction and to a suppressed rate of coke formation.22 The dehydroaromatization of ethane diluted in He, CO2 and H2O on 5% Zn loaded silicalite-1 (MFI) and ZSM-5 with different Si/Al ratios was investigated. The change in product selectivity and the roles of different active sites responsible for this change during ethane dehydroaromatization in each gaseous environment are discussed. Our results indicated that water suppresses oligomerization/aromatization of ethane by hydrolysis of Zn(II) sites, while the newly formed Zn(OH)+ site can catalyze ethane dehydrogenation but not oligomerization/aromatization.
2. Materials and methods
2.1. Catalyst synthesis and gases
NH4-ZSM-5 (SBV 2314 and CBV 5524G with Si/Al = 11.5 and 25) were obtained from Zeolyst International. In this work, ZSM-5 (x) indicates a zeolite with a Si/Al = x. Silicalite-1 (MFI) was prepared in-house following the protocol described elsewhere.4 5% Zn was loaded by incipient wetness impregnation (IWI). Zn(NO3)2·6H2O (Sigma-Aldrich, >99%) was dissolved in DI water to a final concentration of 2 M, and the solution was added dropwise to the NH4-ZSM-5 and pre-calcined silicalite-1 and mixed in a mortar and pestle. Samples were flash-frozen in liquid nitrogen, subsequently dried overnight in a freeze-drier and finally calcined in air at 500 °C. Ethane (99.99%, Matheson), CO2 (grade 5.0, Keen gas), He (grade 5.0, Keen gas) and air (grade 0.1, Keen gas) were used as received.2.2. Characterization
Zn and Al contents were measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) at Galbraith Laboratories (Knoxville, TN). Pore volumes were measured by using the t-plot technique in a 3Flex characterization apparatus (Micromeritics) and the density of BAS in the samples was measured at the University of Pennsylvania by simultaneous temperature-programmed-desorption and thermogravimetric analysis (TPD-TGA) of 2-propanamine. Detailed synthesis procedures and characterization results were reported in a previous report.42.3. Thermal treatment
2.4. Transmission FTIR spectroscopy
Transmission Fourier transform infrared (FTIR) spectra were obtained on an Agilent Cary 660 FTIR spectrometer equipped with an MCT detector (128 scans at a spectral resolution of 2 cm−1) with a homemade in situ transmission cell.23 A vacuum level of 0.01 mTorr in the transmission cell was reached through a vacuum manifold, which was connected to a mechanical pump and a diffusion pump. A self-standing zeolite wafer was loaded into a custom-made sample holder, followed by annealing at 450 °C under vacuum for 30 min after a heating ramp of 2.5 °C min−1 to completely remove adsorbed water. After cooling to the desired temperature, 100 mTorr of pyridine, water, benzene or ethene was introduced to the transmission cell via the vacuum manifold.2.5. Catalytic tests
The catalysts were pelletized and sieved to the mesh size of 40–60. About 95 mg of the catalyst was loaded between two quartz wool plugs in a 60 cm long, 6 mm (¼ inch) diameter quartz tube used as a flow micro-reactor. The catalysts were dehydrated in 50 ml min−1 of He to the reaction temperature of 500 °C with a temperature ramp of 10 °C min−1. Then, the flow was switched to the reactant flow of 50 mL min−1 of 16.5% C2H6 in He or in CO2. The gas phase products were analyzed using an online MS or every 30 min the reactor effluents were injected into an online GC (2014 Shimadzu) equipped with flame ionization detector (FID) and thermal conductivity detector (TCD). The GC was equipped with a GS-Al/KCL column (50 m, 0.535 mm) attached to a FID and a GS-CarbonPLOT column (30 m, 0.53 mm) attached to a TCD. The GC program was 5 min isothermal at 40 °C, then the temperature was increased with 20 °C min−1 to the final temperature of 200 °C, which was held isothermally for 10 min. The effluent lines from the reactor to the GC and MS were heat traced to prevent any condensation. Conversion was calculated based on the disappearance of ethane and selectivity was measured based on the mol% of products as reported in eqn (4) and (5): | (4) |
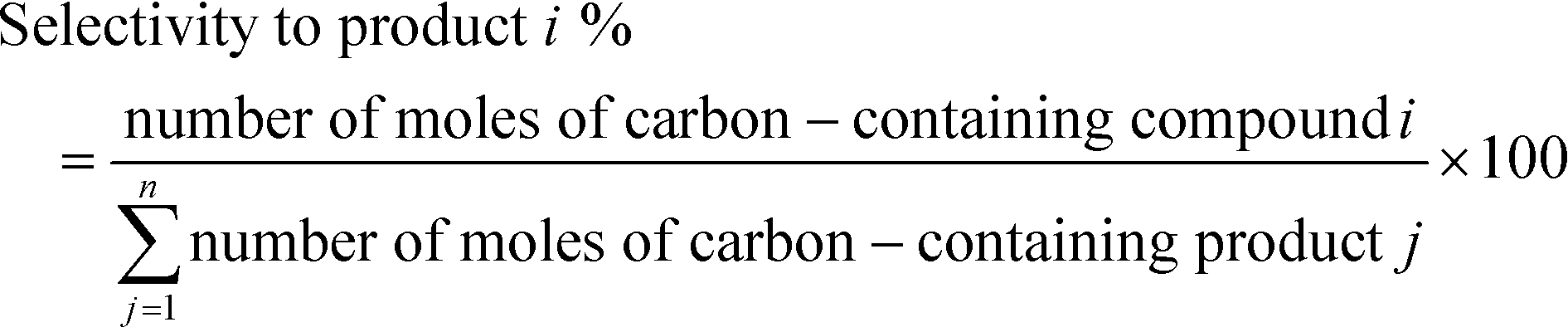 | (5) |
3. Results
3.1. Brønsted acid site density and physical properties of samples
Table 1 summarizes the Brønsted acid site (BAS) densities, measured via 2-propanamine decomposition, and the pore volumes, measured via nitrogen adsorption, of the samples investigated in this work.4 At constant Zn loading (5% w/w), decreasing the Al concentration from 1160 to 504 (μmol g−1) resulted in a reduction in the number of accessible BAS from 210 to 124 (μmol g−1) and increasing the Zn/BAS ratio from 3.6 to 6.2. The number of accessible BAS on silicalite-1 was below the detection limit. The samples pore volumes, at 5% Zn loading, increased only slightly from 0.119 to 0.135 cm3 g−1 by increasing the Si/Al from 11.5 to 25. Additional characterization details (XRD, UV-vis and SEM images) can be found in ref. 4.| Sample | Pore volume (cm3 g−1) | Zn (μmol g−1) | Al (μmol g−1) | BAS density (μmol g−1) |
|---|---|---|---|---|
| H-ZSM-5 (11.5) | 0.154 | 0 | 1223 | 989 |
| 5% Zn-ZSM-5 (11.5) | 0.119 | 765 | 1160 | 210 |
| 5% Zn-ZSM-5 (25) | 0.135 | 765 | 504 | 124 |
| 5% Zn-silicalite-1 | 0.123 | 765 | — | — |
3.2. Temperature-programmed reaction (TPR and TPO)
Fig. 1-a shows a comparison of TPR profiles of ethane/He versus ethane/CO2 on 5% Zn-ZSM-5 (11.5). Compared to the ethane in CO2, dilution in He resulted in H2 formation at lower temperatures, in higher amounts and at faster rates reaching a maximum at about 50 °C lower temperature. In the TPR trace of ethane/CO2, water (m/z = 18) started to form at 500 °C and reached a plateau at 600 °C; no water formation was observed during the reaction of ethane in He. This is the result of the RWGS reaction in ethane/CO2 above 500 °C. In the TPR of ethane/He, methane and hydrogen reached their maximum formation rate at the same temperature (620 °C). Formation rates of both methane and hydrogen increased and decreased in parallel and started to decline above 620 °C, possibly caused by coke deposition on the catalyst at these high temperatures. In contrast, formation rates of methane and hydrogen in CO2 did not change in parallel; methane formation increased monotonically above a temperature of 500 °C while the formation of hydrogen declined at temperatures higher than 660 °C.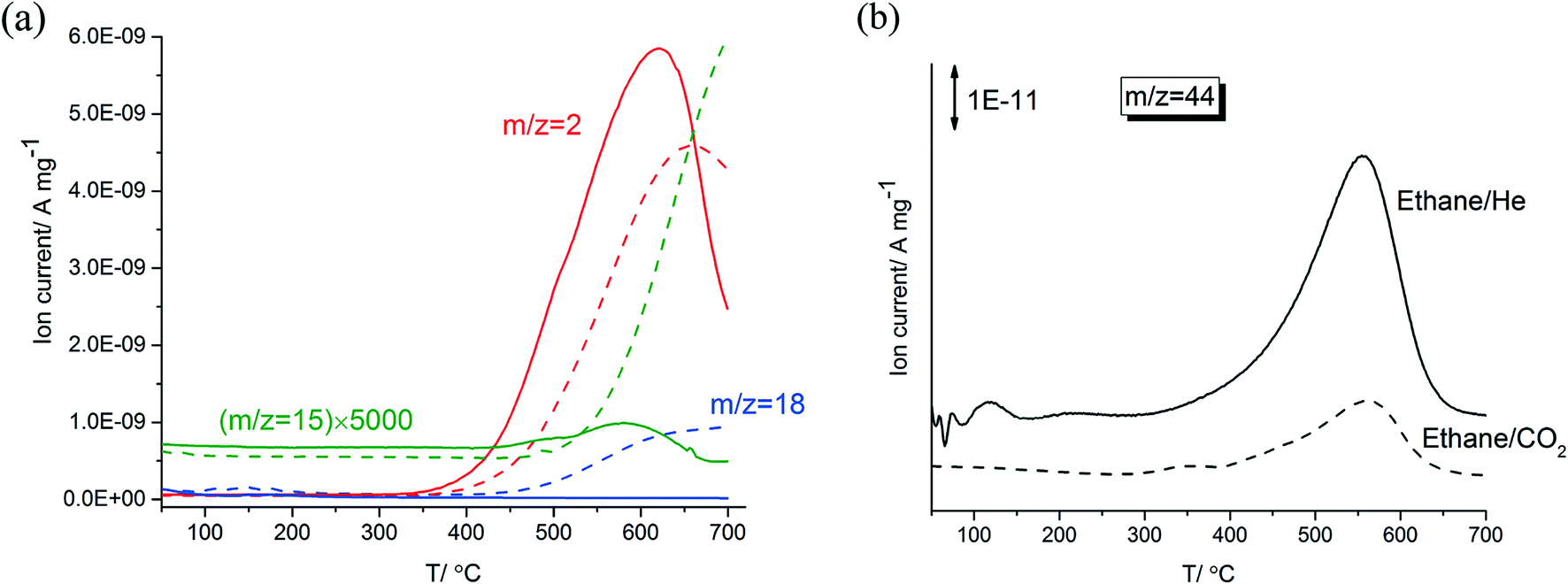 | ||
| Fig. 1 (a) TPR data for 16.5% C2H6 in He or in CO2 on 5% Zn-ZSM-5 (11.5). Reaction conditions: WHSV = 7.2 g gcat−1 h−1 and P = 1 atm. (b) CO2 formation from TPO of the spent 5% Zn-ZSM-5 (11.5) catalyst in 5% O2. Solid lines show ethane/He and dashed lines show ethane/CO2 with red (hydrogen, m/z = 2), green (methane, m/z = 15), blue (water, m/z = 18) and black (carbon dioxide, m/z = 44). | ||
After the dehydroaromatization reaction of ethane in CO2 or He, the spent samples of 5% Zn-ZSM-5 (11.5) were heated in a gas mixture containing 5% O2 (in He and N2). As shown in Fig. 1-b, both samples reached their maximum CO2 (m/z = 44) formation rate at 560 °C, implying that the deposited coke in the catalyst have similar reactivity. The spent sample (prepared in the ethane/He) formed more CO2, that is, this sample contained a larger amount of coke.
3.3. Catalytic tests
| Diluent gas | He | CO2 | He + 1% H2O | |||
|---|---|---|---|---|---|---|
| a Calculated based on CO formation. | ||||||
| Time (h) | 0.5 | 5 | 0.5 | 5 | 0.5 | 5 |
| Ethane conv. (%) | 19.8 | 12.1 | 16.4 | 15.9 | 17.2 | 15.7 |
| CO2 conv.a (%) | — | — | 1.4 | 1.2 | — | — |
| Selectivity (mol%) | ||||||
| CO | — | — | 41.6 | 39.4 | 3.2 | 3.1 |
| CO2 | — | — | — | — | 8.7 | 7.7 |
| Methane | 16.6 | 2.9 | 5.9 | 4.9 | 9.5 | 8.6 |
| Propane | 2.4 | 0.2 | — | — | 0.3 | 0.2 |
| Butane | 0.2 | — | — | — | 0.1 | — |
| Ethene | 57.3 | 87.9 | 47.8 | 51.3 | 74.2 | 76.7 |
| Propene | 3.4 | 2.7 | 1.2 | 1.2 | 1.4 | 1.3 |
| Butene | 0.9 | 1.4 | 0.5 | 0.5 | 0.8 | 0.8 |
| Benzene | 11.6 | 3.4 | 2.0 | 1.7 | 1.4 | 1.2 |
| Toluene | 7.6 | 1.4 | 0.9 | 0.8 | 0.5 | 0.5 |
While the initial ethane conversion was comparable in different atmospheres, selectivity was very different. For ethane in He, high selectivity toward aromatics (∼19%) and methane (∼17%) was observed. In CO2, the formation of methane and aromatics decreased by factors of ∼3 and 7, respectively. The production of CO was from 0 to ∼42% of products. Compared to CO2, in the wet atmosphere, the product contained a lower CO concentration (3.2 vs. 41.6%) and higher methane (9.5 vs. 5.9%) and ethene (74 vs. 48%) concentrations. The selectivity to saturated products (propane and butane) was about 3% in He while it was negligible in CO2 or wet atmospheres. In general, in the presence of CO2 or steam, oligomerization and aromatization of ethane were suppressed.
As a function of time-on-stream, catalyst deactivation in ethane/He led to a decrease of the rate of aromatic and methane formation but to an increase in the formation rate of ethene. In ethane diluted in CO2 or in He + 1% H2O, there was only a small decrease in conversion with time-on-stream and the change in product selectivity was insignificant.
Table 2 shows that replacing He with CO2 affected the activity of the Zn-ZSM-5 catalyst. To investigate the reversibility of this process, ethane conversion was first monitored in the presence of CO2, then the flow was switched to pure He for 1 h and eventually a mixture of ethane in He was fed to the reactor at 500 °C. Water (m/z = 18) and hydrogen (m/z = 2) formation was monitored by online MS and the formation of all other products was monitored by online GC (Fig. 2).
 | ||
| Fig. 2 Change in the activity of 5% Zn-ZSM-5 (11.5) by changing the diluent gas. Reaction conditions: WHSV = 7.2 g gcat−1 h−1, T = 500 °C and P = 1 atm. | ||
The reaction of ethane/CO2 on 5% Zn-ZSM-5 formed water via the RWGS reaction as explained in section 3.4.1. The formation of hydrogen (m/z = 2) decreased slowly, possibly because of slow catalyst deactivation. After 1 h of isothermal treatment at 500 °C in pure He, only hydrogen desorption was observed via MS. When ethane/He was fed to the reactor, a concurrent increase in hydrogen and aromatic (∼ from 3 to 20%) formation was observed, as hydrogen is formed by the conversion of ethane to aromatics. However, increasing the time-on-stream resulted in rapid catalyst deactivation and decreased the formation of both hydrogen and aromatics. The results in Table 2 and Fig. 2 indicate that the reason for the change of activity in the presence of CO2 is water formation and this change in catalytic properties is reversible.
| Sample | 5% Zn-ZSM-5 (25) | 5% Zn-silicalite-1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Diluent gas | He | CO2 | He | CO2 | ||||
| a Calculated based on CO formation. | ||||||||
| Time (h) | 0.5 | 5 | 0.5 | 5 | 0.5 | 5 | 0.5 | 5 |
| Ethane conv. (%) | 20.0 | 16.0 | 10.5 | 10.2 | 2.8 | 2.4 | 1.8 | 2.0 |
| CO2 conv.a (%) | — | — | 0.6 | 0.6 | — | — | 0.13 | 0.14 |
| Selectivity (mol%) | ||||||||
| CO | — | — | 33.8 | 33.2 | — | — | 39.0 | 37.2 |
| Methane | 9.5 | 3.1 | 3.2 | 3.1 | — | — | — | — |
| Propane | 1.3 | 0.4 | — | — | — | — | — | — |
| Butane | 0.2 | 0.1 | — | — | — | — | — | — |
| Ethene | 69.7 | 84.7 | 59.3 | 60.1 | 100 | 100 | 61.0 | 62.8 |
| Propene | 3.3 | 2.8 | 1.0 | 1.0 | — | — | — | — |
| Butene | 1.0 | 1.2 | 0.5 | 0.5 | — | — | — | — |
| Benzene | 9.7 | 5.3 | 1.5 | 1.5 | — | — | — | — |
| Toluene | 5.2 | 2.1 | 0.5 | 0.5 | — | — | — | — |
The conversion of ethane on Zn-silicalite (in He or in CO2) is low (∼2%) and below the equilibrium conversion. Despite the low conversion, the selectivity in He was 100% ethene, while the selectivity in CO2 was 61% ethene and 39% CO. Consequently, the ZnO clusters present in this sample catalyze both dehydrogenation (although at a slower rate) and RWGS reactions.
3.4. FTIR spectroscopy
The results described in the previous sections show that the presence of water had a significant effect on the performance of the Zn-ZSM-5 catalysts. The role of BAS and Zn sites in these processes is still unclear and FTIR spectroscopy was used to investigate the effect of water on the sites formed in the dehydrated Zn samples. Fig. 3 shows the FTIR spectra collected at 25 °C for (i) ‘clean’ H-ZSM-5 (11.5), (ii) clean 5% Zn-ZSM-5 (11.5), (iii) pyridine adsorbed on Zn-ZSM-5 (11.5), and (iv) after exposing the adsorbed pyridine in (iii) to water.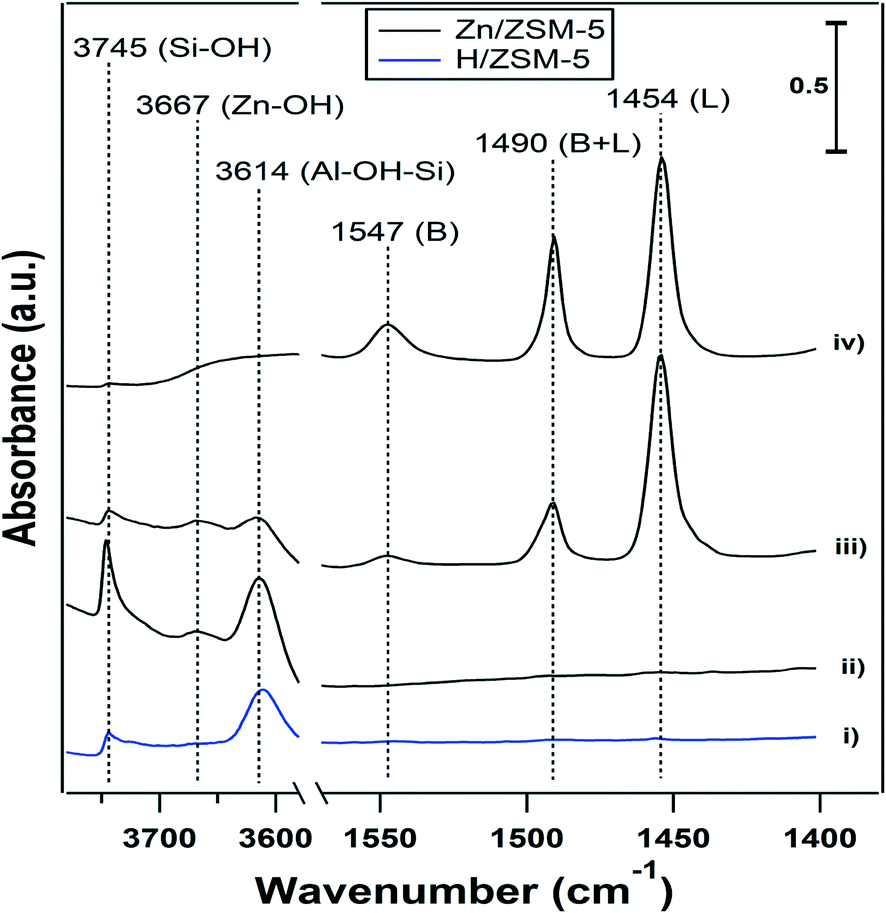 | ||
| Fig. 3 FTIR spectra of (i): clean H-ZSM-5, (ii): clean Zn-ZSM-5, (iii): Zn-ZSM-5 with adsorbed pyridine, and (iv): Zn-ZSM-5 with adsorbed pyridine and water. All spectra were collected at T = 25 °C. B refers to BAS and L refers to the Lewis acid site. | ||
Band assignments are reported in Table 4. The OH stretching region of 5% Zn-ZSM-5 (11.5) (see Fig. 3) displays three bands, assigned to silanols (3745 cm−1), BAS (3614 cm−1) and a new band at 3667 cm−1, which is not present in H-ZSM-5 (Fig. 3-i), and can be assigned to Zn-OH. The Zn(OH) in Zn-silicalite has been reported to have vibration at 3675 cm-1.24 This is consistent with MO–H (MCu, Fe and Ce) vibrations in zeolites that have vibration frequencies close to this value.26–28 The pyridine IR adsorption spectrum in Fig. 3-iii reveals the presence of both BAS (protonated pyridine at 1547 cm−1) and Lewis acid sites (1454 cm−1) on Zn-ZSM-5. The band at 1490 cm−1 contains contributions from both BAS and Lewis acid sites, which require deconvolution to characterize the relative contributions. For simplicity, the pure BAS band at 1547 cm−1 and the pure Lewis acid site band at 1454 cm−1 are used to analyze the two types of acid sites. Zn can replace the protons in H-ZSM-5 to form Lewis acid sites,4 and adding 5% Zn resulted in the loss of about 80% of the protons in H-ZSM-5 (11.5) (Table 1). Adsorption of water molecules in the pores of Zn-ZSM-5 resulted in a broad peak in the OH vibration region due to hydrogen bonding (Fig. 3-iv). But even in the presence of water, the sample containing adsorbed pyridine displayed the presence of both BAS and Lewis acid sites (Fig. 3-iv).
| IR band (cm−1) | Assignment |
|---|---|
| ν as: asymmetric stretching, νs: symmetric stretching, δas: asymmetric bending, δs: symmetric bending. | |
| 3745 | O–H stretching of external SiOH |
| 3610 | O–H stretching of Al–OH–Si (BAS) |
| 3675 | O–H stretching of Zn–OH in Zn/silicalite |
| 1546 | Pyridine adsorption on BAS |
| 1455 | Pyridine adsorption on the Lewis acid site |
| 2960 (νas), 2870 (νs), 1460 (δas), 1380 (δs) | –CH3 |
| 2925 (νas), 2850 (νs), 1470 (scissor) | –CH2 |
| 1650 and 1600 (νCC) |
Diene |
| 1650 and 1600 (νCC) |
Triene |
| Broad band 1650–1580 (νCC) |
Polyene |
Ethene had only weak interactions with H-ZSM-5, with weak features at 1470 δ(CH2), 2873 νs(CH3), 2932 νas(CH2) and 2960 νas(CH3) cm−1 (see Fig. 4-i). In another report,29 adsorption of ethene on BAS (forming OH–π complexes) allowed the formation of bands at 1340 δ(CH2), 1440 δ(CH2), 1612 ν(CC) and 2974 νs(CH2) cm−1. Ethene had a much stronger interaction with Zn-ZSM-5, with prominent bands in the bending region (1300–1500 cm−1) and weaker bands for the CH stretching regions (2800–3000 cm−1, Fig. 4-ii, note the scale bars). Adsorption of propene on Zn-MFI is reported to result in peaks at 1376, 1411, 1428, and 1451 cm−1 assigned to Zn-propyl groups and a band at 1587 cm−1 assigned to the stretching vibration of CC bonds coordinated to Zn cations.24 In Fig. 4-ii and -iii, a strong broad peak is observed in the range of 1550–1650 cm−1 with an apex at 1595 cm−1. These features are assigned to CC stretching modes. The broad band in the range of 1550–1650 cm−1 can be tentatively assigned to the CC bonds vibration of polyenes.25 In a separate measurement, adsorption of benzene on 5% Zn-ZSM-5 resulted in a band at 1470 cm−1 (Fig. S1†), suggesting that ethene has a stronger interaction than benzene with Zn-ZSM-5. In spectrum (ii), there is a small feature at 3090 cm−1, which is in the range for an alkene C–H stretch. It disappears at 150 °C (spectrum iii), suggesting that the ethene molecule with that stretch was weakly bound or rapidly reacts into different species. The absorption bands in the 2800–3000 cm−1 region (CH3 and CH2 stretches) can be observed for ethene adsorption on both H- and Zn-ZSM-5 at temperatures of 25 and 150 °C, as evidence of oligomerization. Oligomerization of ethene on H-ZSM-5 showed similar band intensities in the region of 2800–3000 and 1400–1600 cm−1.29 However, in this work, the bands in the 2800–3000 cm−1 region were significantly weaker than the bands in the 1300–1700 cm−1 region (see scale bars). The FTIR spectra, thus, reveal that ethene has a stronger interaction with Zn(II) sites (Lewis acid sites) than with BAS, resulting in the formation of polyenes on Zn-ZSM-5 and a small amount of oligomerization products on both H- and Zn-ZSM-5.
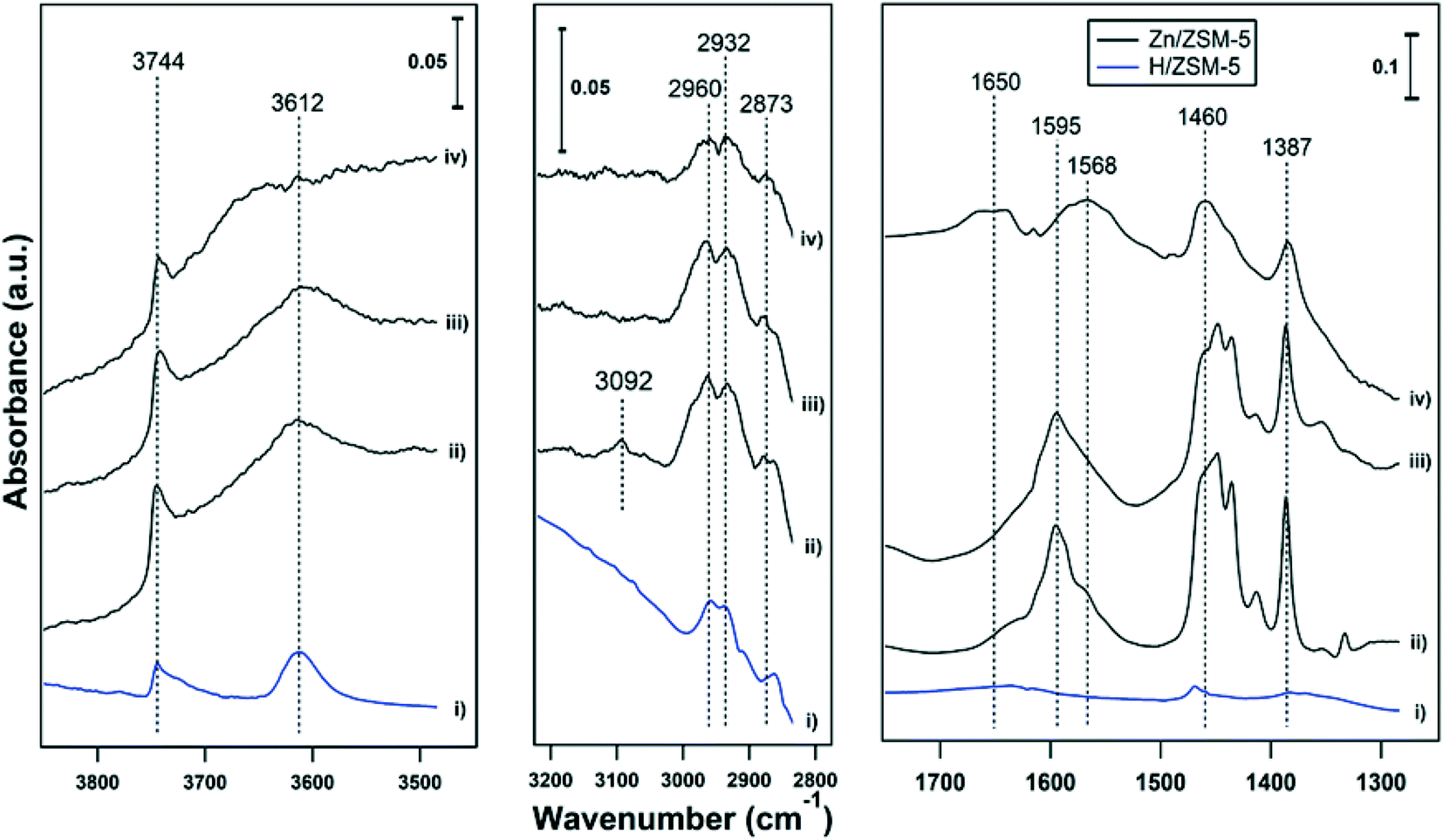 | ||
| Fig. 4 FTIR spectra of ethene adsorbed on (i) H-ZSM-5 at 25 °C, (ii) 5% Zn-ZSM-5 at 25 °C, (iii) 5% Zn-ZSM-5 at 150 °C, and (iv) is spectrum (iii) after exposure to water at 150 °C. | ||
At a temperature of 150 °C, a broad peak is observed in range of 1550–1700 cm−1 with a maximum at 1650 cm−1 in the presence of water (Fig. 4-iv). After subtraction of the water peak, a clear change in spectra can be observed compared to the spectra under the same conditions without water (Fig. 4-iii): most notably the band at 1595 cm−1 is split into two bands, one at 1650 cm−1 and another one at 1568 cm−1. The frequency of CC stretching depends on the adsorption site; in the case of propene adsorption, a band at 1632 cm−1 has been assigned to adsorption on BAS30 (ethene adsorption on H-ZSM-5 had ν(CC) = 1612 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29) and a band at 1585 cm−1 has been assigned to adsorption on Zn cations.24,31 As a result, formation of the two new bands in the presence of water can be attributed to the CC adsorption on BAS and Zn(II). Comparison of the spectra at 150 °C with (Fig. 4-iv) and without water (Fig. 4-iii) showed the intensity of bands decreased in the presence of water, implying ethene adsorption decreased in the presence of water.
29) and a band at 1585 cm−1 has been assigned to adsorption on Zn cations.24,31 As a result, formation of the two new bands in the presence of water can be attributed to the CC adsorption on BAS and Zn(II). Comparison of the spectra at 150 °C with (Fig. 4-iv) and without water (Fig. 4-iii) showed the intensity of bands decreased in the presence of water, implying ethene adsorption decreased in the presence of water.
4. Discussion
The ethane reactivity on Zn-ZSM-5 changed significantly when the He atmosphere was replaced with CO2. In the presence of CO2, product selectivity changed because of the suppression of oligomerization/aromatization and because of the formation of CO and water. The effect of pretreatment of Zn-ZSM-5 (11.5) with pure CO2 at 500 °C for 1 h and then switching the gas mixture to ethane/He was minimal, as we observed similar results using pretreatment in He. We also fed CO2 over Zn-ZSM-5 at 500 °C in the TGA and observed no weight change (Fig. S2†), implying carbonate species (CO2 adsorption on Zn-ZSM-5) did not form at 500 °C. These observations indicate that the change in selectivity of the catalyst is not because of the reaction of zinc sites with CO2. However, water was also formed in a CO2 atmosphere (Fig. 1-a), and the reactivity of the catalyst in 1% water + He was similar to the reactivity in the presence of CO2 (Table 2). This is an indication that the change in activity can be attributed to water.Zn-ZSM-5 catalyzes the RWGS reaction, resulting in water formation in a CO2 atmosphere. On ZnO, the RWGS reaction proceeds through intermediate formate species.32 However, the mechanisms and roles of different zinc sites in ZSM-5 for catalyzing RWGS reaction need further investigation.
The ZnO species in Zn-silicalite catalyzed ethane dehydrogenation and RWGS (∼2% conversion, Table 3). However, unlike Zn-ZSM-5, CO2 had only a small effect on Zn-silicalite. Different zinc sites have been reported to be present in ZSM-5 such as Zn2+, Zn(OH)+, [ZnOZn]2+ and ZnO clusters.33 The Lewis acid sites are more effective than ZnO for dehydrogenation and also (unlike ZnO) can catalyze ethane aromatization.4 The suppression of ethane oligomerization/aromatization in the presence of water showed that the active sites responsible for oligomerization/aromatization have changed. This change was reversible since when CO2 was replaced with He, oligomerization/aromatization resumed (Fig. 2).
The water formation via the RWGS reaction can cause the hydrolysis of Zn2+ and [ZnOZn]2+ sites. Water has stronger interactions with isolated Zn2+ sites because of its stronger Lewis acidity than [ZnOZn]2+.34 Hydrolysis of the Zn(II) sites results in the formation of a different site (Zn(OH)+) and regeneration of a BAS as depicted in eqn (6) and (7) (where Z refers to the anionic oxygen attached to Al in the ZSM-5 structure).
| Z−(Zn2+)Z− + H2O → Z−(ZnOH)+ + Z−(H+) | (6) |
| Z−(ZnOZn)2+Z− + H2O → Z−(ZnOH)+ + Z−(ZnOH)+ | (7) |
The IR spectra (Fig. 3) illustrate the hydrolysis of Zn sites in Zn-ZSM-5. Comparing the spectra in Fig. 3-iii and -iv, it can be seen that the band area assigned to BAS increased by approximately a factor of four (from 0.45 to 1.84), while the area of the Lewis acid band was reduced approximately by 10% (from 7.02 to 6.44). It is possible that water displaced pyridine from the Zn(II) site to form BAS to a small extent. However, comparing the magnitude of the changes in the area of the two bands, and using the ratio of extinction coefficients for pyridine adsorbed on BAS and Lewis acid sites,35 it is concluded that the majority of the newly protonated pyridine (approximately 70%) in Fig. 3-iv did not originate from competitive adsorption on the Lewis acid sites. It is then likely that the water reacting with Zn2+ sites leads to the formation of Zn(OH)+ and BAS.
Ethene interaction with Zn(II) sites was much stronger than with BAS at both temperatures of 25 and 150 °C (Fig. 4). Because DFT calculations have shown that ethene has a stronger binding energy on Ag(I) than on BAS in ZSM-5,36 we suggest, that ethene adsorption is even stronger on Zn(II) in agreement with our IR results. Strong adsorption of ethene on Zn sites and the formation of polyenes implies a more important role for Zn(II) than BAS for oligomerization of ethene. At higher temperatures, cyclization and aromatization can also occur, as cations can induce electrostatic acceleration of cyclization.37
In the presence of water, the interaction of ethene with Zn2+ sites is weakened and the broad peak at 1595 cm−1 (ethene adsorption on Zn2+) splits into two bands: one at 1650 cm−1 and another one at 1568 cm−1 which can be assigned to ethene adsorption on BAS and Zn(OH)+ (eqn (6)). The ethene adsorption shifted to lower frequencies from Zn2+ to Zn(OH)+. Zn(OH)+ can activate the C–H bond.38 The catalytic activity of Zn(OH)+ is different from Zn2+, as Zn(OH)+ (in Zn/Na-ZSM-5, for example) is more efficient for dehydrogenation and hydrogen desorption than Zn2+ (in Zn/H-ZSM-5).39
According to eqn (4), BAS can be generated via hydrolysis of the Zn2+ site. The hydrolysis generated BAS (in Zn-ZSM-5) has been reported to be weaker than the original BAS in H-ZSM-5.40 However, in the case of hydrolyzed zinc sites (Zn(OH)+), the aromatization rate was much lower (Table 2). This observation suggests that Zn(OH)+ can catalyze dehydrogenation but not oligomerization/aromatization, perhaps due to the steric effects of the new OH group on the Zn cations. In contrast to the case of ethane, where water lowers the selectivity to aromatics on Zn-ZSM-5, water increases the selectivity toward aromatics for propane dehydroaromatization on Zn-41 and Ga-42 containing ZSM-5. The promoting effect of water has been ascribed to stabilization of oxygenated Zn sites and the creation of partially hydrolyzed Ga sites. The difference between aromatization of ethane and propane on Zn-ZSM-5 is that it is more difficult to activate ethane than propane on BAS. Therefore, unlike ethane, regeneration of BAS from hydrolysis can be helpful for the initial activation of propane.
The presence of water had a greater effect on the catalyst activity of higher Al content samples, with the least change in activity exhibited for Zn-silicalite samples (Tables 2 and 3). This implies that water (via hydrolysis) has a greater impact on the samples with higher concentrations of Lewis acid sites, while water has a minor effect on the catalytic activity of ZnO.
The Zn-ZSM-5 catalysts displayed improved stability in the presence of CO2 than in He, as the catalysts were shown to form less coke (Fig. 1-b). This is probably due to the lower formation rates of aromatic products, which are precursors for coke. However, the catalytic rates were more stable in CO2 than in wet He (Table 2).
Dehydrogenation products, such as olefins and aromatics, are more reactive than ethane, and their reaction with CO2 is likely. Therefore, we cannot rule out the possibility of product oxidation in the presence of CO2 and water, as co-feeding of water resulted in formation of CO and CO2 (Table 2).
5. Conclusion
We have shown that Zn-ZSM-5 catalyzes the RWGS reaction, and the produced water hydrolyzed the zinc sites in ZSM-5, forming Zn(OH)+ and a new BAS. Although BAS were generated during hydrolysis, aromatization decreased in favor of the formation of CO and the dehydrogenation of ethane to ethene. In fact, even with a small amount of water (1%), the product selectivity for ethane conversion changed significantly from aromatization to dehydrogenation. Zn(OH)+ sites catalyze the ethane dehydrogenation reaction but not aromatization. Lower formation rates of aromatics and possibly the reverse Boudouard reaction lead to lower coke formation rates on Zn-ZSM-5, which resulted in an increase in catalyst stability.The different Zn sites in ZSM-5 play varying roles in the dehydrogenation/aromatization of ethane. Zn-ZSM-5 with high Zn/BAS ratios showed high selectivity toward aromatics,4 implying a key role of Zn(II) compared to BAS for the aromatization of ethane. This conclusion is supported by the reported IR spectra, which revealed stronger adsorption of ethene on Lewis acid sites than on BAS and the formation of polyenes on Zn(II) sites. Zn in the form of Lewis acid sites (Zn2+ or (ZnOZn)2+) can catalyze ethane aromatization. Although ZnO and Zn(OH)+ can catalyze the ethane dehydrogenation but not aromatization, Zn(OH)+ catalyzes the reaction at faster rates than ZnO clusters in the zeolite pores.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This report is based upon work supported by the Department of Energy, Basic Energy Sciences, under Award Number DE-SC0014436.References
- J. A. Biscardi and E. Iglesia, Catal. Today, 1996, 31, 207–231 CrossRef CAS.
- J. A. Biscardi and E. Iglesia, J. Catal., 1999, 182, 117–128 CrossRef CAS.
- Y. Ono, Catal. Rev.: Sci. Eng., 1992, 34, 179–226 CAS.
- A. Mehdad and R. F. Lobo, Catal. Sci. Technol., 2017, 7, 3562–3572 CAS.
- E. A. Pidko and R. A. van Santen, J. Phys. Chem. C, 2007, 111, 2643–2655 CAS.
- A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi and Y. Kuroda, Angew. Chem., Int. Ed., 2012, 51, 7719–7723 CrossRef CAS PubMed.
- A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi and Y. Kuroda, J. Phys. Chem. C, 2013, 117, 19525–19534 CAS.
- V. B. Kazansky and E. A. Pidko, J. Phys. Chem. B, 2005, 109, 2103–2108 CrossRef CAS PubMed.
- V. B. Kazansky, A. I. Serykh and E. A. Pidko, J. Catal., 2004, 225, 369–373 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, N. Rane, R. A. van Santen and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2005, 7, 3088–3092 RSC.
- A.-K. Wiegand, A. Rit and J. Okuda, Coord. Chem. Rev., 2016, 314, 71–82 CrossRef CAS.
- J. Bacsa, F. Hanke, S. Hindley, R. Odedra, G. R. Darling, A. C. Jones and A. Steiner, Angew. Chem., Int. Ed., 2011, 50, 11685–11687 CrossRef CAS PubMed.
- E. Iglesia and J. E. Baumgartner, Catal. Lett., 1993, 21, 55–70 CrossRef CAS.
- M. D. Porosoff, M. N. Z. Myint, S. Kattel, Z. Xie, E. Gomez, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2015, 54, 15501–15505 CrossRef CAS PubMed.
- M. Myint, B. Yan, J. Wan, S. Zhao and J. G. Chen, J. Catal., 2016, 343, 168–177 CrossRef CAS.
- F. Solymosi, A. Szõke and L. Ovári, J. Catal., 1999, 186, 269–278 CrossRef CAS.
- N. Mimura, I. Takahara, M. Inaba, M. Okamoto and K. Murata, Catal. Commun., 2002, 3, 257–262 CrossRef CAS.
- N. Mimura, M. Okamoto, H. Yamashita, S. T. Oyama and K. Murata, J. Phys. Chem. B, 2006, 110, 21764–21770 CrossRef CAS PubMed.
- K. Nakagawa, C. Kajita, K. Okumura, N.-O. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Catal., 2001, 203, 87–93 CrossRef CAS.
- S.-K. Ihm, Y.-K. Park and S.-W. Lee, Appl. Organomet. Chem., 2000, 14, 778–782 CrossRef CAS.
- V. Abdelsayed, M. W. Smith and D. Shekhawat, Appl. Catal., A, 2015, 505, 365–374 CrossRef CAS.
- Y. Ren, F. Zhang, W. Hua, Y. Yue and Z. Gao, Catal. Today, 2009, 148, 316–322 CrossRef CAS.
- B. Murphy, M. E. Davis and B. Xu, Top. Catal., 2015, 58, 393–404 CrossRef CAS.
- Y. G. Kolyagin, V. V. Ordomsky, Y. Z. Khimyak, A. I. Rebrov, F. Fajula and I. I. Ivanova, J. Catal., 2006, 238, 122–133 CrossRef CAS.
- K. Nakanishi and P. H. Solomon, Infrared Absorption Spectroscopy, 2nd edn, 1978 Search PubMed.
- M. Iwasaki and H. Shinjoh, J. Catal., 2010, 273, 29–38 CrossRef CAS.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- E. Ito, Y. J. Mergler, B. E. Nieuwenhuys, H. P. A. Calis, H. van Bekkum and C. M. van den Bleek, J. Chem. Soc., Faraday Trans., 1996, 92, 1799–1806 RSC.
- G. Spoto, S. Bordiga, G. Ricchiardi, D. Scarano, A. Zecchina and E. Borello, J. Chem. Soc., Faraday Trans., 1994, 90, 2827–2835 RSC.
- A. K. Ghosh and R. A. Kydd, J. Catal., 1986, 100, 185–195 CrossRef CAS.
- I. I. Ivanova, Y. G. Kolyagin, V. V. Ordomsky, E. V. Asachenko, E. M. Pasynkova and Y. A. Pirogov, J. Mol. Catal. A: Chem., 2009, 305, 47–53 CrossRef CAS.
- J. Strunk, K. Kähler, X. Xia and M. Muhler, Surf. Sci., 2009, 603, 1776–1783 CrossRef CAS.
- J. A. Biscardi, G. D. Meitzner and E. Iglesia, J. Catal., 1998, 179, 192–202 CrossRef CAS.
- A. L. Yakovlev, A. A. Shubin, G. M. Zhidomirov and R. A. van Santen, Catal. Lett., 2000, 70, 175–181 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- M.-F. Hsieh, Y. Zhou, H. Thirumalai, L. C. Grabow and J. D. Rimer, ChemCatChem, 2017, 9, 1675–1682 CrossRef CAS.
- H. Jiao and P. V. R. Schleyer, J. Am. Chem. Soc., 1995, 117, 11529–11535 CrossRef CAS.
- X.-N. Wu, H.-T. Zhao, J. Li, M. Schlangen and H. Schwarz, Phys. Chem. Chem. Phys., 2014, 16, 26617–26623 RSC.
- J. A. Biscardi and E. Iglesia, Phys. Chem. Chem. Phys., 1999, 1, 5753–5759 RSC.
- E.-M. El-Malki, R. A. van Santen and W. M. H. Sachtler, J. Phys. Chem. B, 1999, 103, 4611–4622 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS.
- E. J. M. Hensen, E. A. Pidko, N. Rane and R. A. van Santen, Angew. Chem., Int. Ed., 2007, 46, 7273–7276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy01850a |
| This journal is © The Royal Society of Chemistry 2018 |