
Carbon nanofibres-supported KCoMo catalysts for syngas conversion into higher alcohols†
Ho Ting
Luk
a,
Tim
Forster
a,
Cecilia
Mondelli
*a,
Sebastian
Siol
 b,
Daniel
Curulla-Ferré
c,
Joseph A.
Stewart
c and
Javier
Pérez-Ramírez
*a
b,
Daniel
Curulla-Ferré
c,
Joseph A.
Stewart
c and
Javier
Pérez-Ramírez
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, HCI E125, Vladimir-Prelog-Weg 1, CH-8093 Zurich, Switzerland. E-mail: cecilia.mondelli@chem.ethz.ch; jpr@chem.ethz.ch
bEMPA, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
cTotal Research & Technology Feluy, Zone Industrielle Feluy C, B-7181 Seneffe, Belgium
First published on 6th November 2017
Abstract
The development of industrially-viable heterogeneous catalysts for higher alcohol (HA) synthesis via direct syngas conversion is hindered by the limited understanding of structural and electronic descriptors of their performance. Here, K-promoted CoMo(5 wt%)-based catalysts were investigated to shed light onto property–function relations. Evaluation of impregnated oxides and C-based materials identified a higher HA selectivity for the catalyst supported on carbon nanofibres obtained by the simultaneous addition of all metals. Using this carrier, additional solids were synthesised via ball milling as well as a sol–gel method with citric acid, which enhanced the CO conversion. After confirming the positive influence of K and a unitary Co/Mo ratio, the impact of temperature (573–773 K) and pressure (0.1–5 MPa) upon activation through reduction by hydrogen was investigated. A combination of two steps, one at 723 K and ambient pressure, fostering the HA selectivity (22%), and a second, more extended, at 573 K and 5 MPa, boosting the CO conversion (14%), maximised the space time yield of HA, which matches that of the best comparable CoMo system reported in the literature (ca. 0.12 gHA gcat−1 h−1) in spite of the 13-times lower metal loading. Characterisation by X-ray diffraction, temperature-programmed reduction by H2, X-ray photoelectron spectroscopy, operando infrared spectroscopy and electron microscopy uncovered that this originated from the atomic intermixing of Co and Mo in binary oxide phases and their high dispersion, enabling proximity and an effective reduction to low oxidation states of the metal sites (0 and +2 for Mo and 0 for Co) upon activation and reaction.
1. Introduction
Higher alcohols (HA) find a broad application in the (fine) chemical and polymer industries and in the energy sector.1–4 Their demand is expected to raise substantially in the next decade as a consequence of the growing needs of the increasing world population and, in particular, due to the phasing out of methyl tert-butyl ether as a fuel additive in some countries and the interest in developing safer and more efficient fuel cell technologies based on ethanol rather than methanol.5,6 This has revitalised interest in the direct conversion of syngas as a potentially more sustainable route to HA than conventional processes based on the hydration of oil-derived alkenes. Indeed, the reaction comprises a single step and could utilise syngas obtained from renewable (biomass) or unconventional (shale gas) sources.Various types of catalysts have been studied for this transformation since the 1930s, with Mo-containing systems receiving particular attention after the publication of patents on alkali-promoted MoS2 by Dow Chemicals and Union Carbides in the 1980s.7–9 In view of the restrictions relative to the emission of H2S and organic sulfonated compounds, research efforts soon switched to sulphur-free catalyst compositions, which typically still contained K to suppress the formation of methanol and enhance that of HA. Among these, interesting developments have been observed for materials in which Mo has been associated to the Fischer-Tropsch element Co to complement the ability of the first metal to molecularly adsorb CO with that of the second to bind it in a dissociative manner. Early studies reported catalysts in bulk form or systems supported on oxidic carriers with a typical CoMo loading of 40–50 wt%, which reached moderate CO conversion (10%) and HA selectivity (21%).10–12 In contrast, the use of carbon-based solids as a support or an additive led to more significant, though varied, catalytic performance.13–19 KCoMo/C catalysts exhibited a CO conversion of 47% and a HA selectivity of 16%.15 These two parameters reached values of 57 and 46%, respectively, over a composite containing the metals and Co-decorated carbon nanotubes in an 88/12 mass ratio but the alcohol distribution was considerably altered in favour of C4–C9 products.16 An even higher HA selectivity (65%) at a slightly lower CO conversion (42%) was reported for CoMo catalysts patented by SABIC, which surprisingly were K-free and exhibited 10-times lower CO2 selectivity than generally observed.20 It is worth noting that these last two materials stand among the top 10 catalysts for HA synthesis considering all families of systems investigated.4
As a quite common situation and challenge in direct HA synthesis due to the complexity and multicomponent nature of the catalysts, sound structure–performance relations have not been derived to rationalise the differences in the catalytic behaviour and guide the design of more industrially-relevant materials. While the relative placement of the metals, the support effect and the role of K have been addressed very infrequently, some efforts have been devoted to uncover the speciation and oxidation state of Co and Mo. Still, the majority of the analyses have been conducted ex situ.15,18,21 Exceptions are two studies characterising the catalyst through in situ X-ray photoelectron spectroscopy and diffuse reflectance infrared Fourier transform spectroscopy.17,19 The first highlighted Co2+ and Mo4+ as the likely active sites for the reaction, but was based on the analysis of freshly reduced and Mn-doped samples.19 The second reported the presence of Moδ+ (1 < δ < 4) species and of Co in metallic form and with oxidation states of 1 and 2 upon syngas adsorption at ambient temperature and pressure, indicating that Co favours the binding of CO to Mo and that the higher catalyst activity of a KCoMo/C sample with Co/Mo = 0.5 and reduced at 773 K was related to a higher concentration of Moδ+ sites at the surface.17
In this study, we gathered deeper insights into supported KCoMo catalysts with special attention on the influence of the carrier, the proximity of the metals and the chemical nature of Co and Mo. For this purpose, the focus was placed on materials with a low CoMo loading (5 wt%) to better control the metals' nanostructure and more easily detect support effects. At the synthesis level, we varied the type of carrier (oxidic and carbon-based materials), the preparation method (dry impregnation, a sol–gel route and ball milling), the order of introduction of the metals and the composition (distinct Co/Mo ratios and the presence/absence of K). With respect to catalyst activation by reduction, the impact of the temperature was evaluated along with that of the pressure, where ambient pressure and the same as employed upon testing were applied. Indications about performance descriptors were obtained combining catalytic data with information gathered on reduced and used samples by X-ray diffraction, monitoring the evolution of crystalline phases, temperature-programmed reduction by hydrogen, X-ray photoelectron spectroscopy and operando diffuse reflectance infrared Fourier transform spectroscopy, unravelling the oxidation states of Co and Mo, and electron microscopy, visualising the size of the phases and mapping the metals distribution.
2. Experimental
Catalyst synthesis
Supported K-promoted CoMo catalysts with a nominal molar K/Co/Mo ratio of 0.1/1/1 and a CoMo loading of 5 wt% were prepared by simultaneous dry impregnation (DI) using SiO2 (Sigma-Aldrich, 99.5%), Al2O3 (Sasol Puralox SCFa, 98%), MgO (Strem Chemicals, 99%), activated carbon (AC, NORIT CN1), carbon black (CB, Cabot Vulcan XC-72) and carbon nanofibres (CNF, Sigma-Aldrich, iron content <100 ppm) as carriers. 0.197 g of Co(NO3)2·6H2O (ABCR, 99%) and 0.120 g of (NH4)6Mo7O24·4H2O (ABCR, 99.98%) were dissolved in deionised water. The subsequent addition of 0.0047 g of K2CO3 (Sigma-Aldrich, 99.99%) led to the formation of a purple precipitate, which was dissolved by adding a few drops of diluted (0.2–0.3 M) HNO3 (Fisher Chemicals, 65 wt% aqueous solution). The resulting mixture was incorporated drop-wise into 2 g of the support under magnetic stirring. The impregnated solid was stirred for 3 h at room temperature, dried at 338 K in static air overnight and activated by reduction in a 10 vol% H2/He flow of 20 cm3 min−1 for 4 h at 673 K (ramp rate = 3 K min−1), which is referred to as reduction under the standard conditions in this manuscript. These catalysts are identified by the formula or the abbreviation of the support.Additional catalysts with the same molar metals ratio and CoMo loading were synthesised using CNF by sequential DI, ball milling (BM) and a sol–gel (SG) route described by Bao et al.22 The latter method was also used to produce materials with the same CoMo loading but nominal molar K/Co/Mo ratios of 0/1/1, 0.1/0.5/1 and 0.1/2/1. For sequential DI, an aqueous solution of K2CO3 and Co(NO3)2·6H2O or of K2CO3 and (NH4)6Mo7O24·4H2O was added to 2 g of CNF and the mixture was stirred and dried as described above. Prior to impregnation with the third metal, the solid was treated in a 10 vol% H2/He flow of 20 cm3 min−1 for 4 h at 533 or 673 K (3 K min−1, after Co and K or after Mo and K addition, respectively). For BM, the three metal precursors and the CNF were loaded into a stainless-steel 12 cm3 crucible equipped with stainless-steel balls of 5 mm in diameter in the same amounts as mentioned above. A water volume equal to that used in DI was added and the crucible was sealed under air and operated at 500 rpm for 2 h. The solid retrieved was dried at 338 K in static air overnight. For SG synthesis, a Co(NO3)2·6H2O solution (0.611–1.433 g in 5 cm3 of water) and a (NH4)6Mo7O24·4H2O solution (0.435–0.741 g in 6.6 cm3 of water) were prepared separately and mixed under magnetic stirring. With the addition of 4–5 drops of NH4OH (Acros Organics, 25 wt% aqueous solution), a purple precipitate was formed. A citric acid (Sigma-Aldrich, ≥95%) solution (0.484–0.567 g in 1.5 cm3 of water) was then added under magnetic stirring to dissolve the solid. A K2CO3 solution (0.017–0.029 g in 1.3 cm3 of water) was subsequently incorporated, if desired. The pH of the sol was adjusted to 3.5 using a few drops of HCOOH (Merck product, 98–100%) and NH4OH. An aliquot of this solution was added to 2.0 g of CNF to achieve the desired CoMo loading. The mixture was magnetically stirred for 6 h at 338 K and dried in air at the same temperature overnight. All DI-, BM- and SG-prepared catalysts were activated under standard conditions. Their names comprise the abbreviation of the method, ‘673’ to indicate the temperature of reduction, and additional codes to highlight changes in the synthesis parameters, i.e., ‘Co/KMo’ or ‘MoK/Co’ for catalysts prepared by sequential DI, ‘noK’ for the catalyst without K and ‘0.5’ or ‘2’ for the catalysts with a Co/Mo ratio equal to these values. The SG-prepared sample with K/Co/Mo = 0.1/1/1 and 5 wt% CoMo was also alternatively activated in (i) a 10 vol% H2/He flow of 20 cm3 min−1 for 4 h at 723 or 773 K (3 K min−1), (ii) a H2 flow of 20 cm3 min−1 for 4 or 18 h at 5 MPa and 573 K or (iii) in a 10 vol% H2/He flow of 20 cm3 min−1 for 4 h at 723 (3 K min−1) and, after re-activation in the catalytic reactor (vide infra), in a H2 flow of 20 cm3 min−1 for 18 h at 5 MPa and 573 K. The latter multi-step activation was also applied to a portion of the BM-prepared catalyst and alternatively conducted on the SG-prepared solid using CO or syngas with a molar H2/CO ratio of 2. In these cases, the catalyst name contained the abbreviation of the method, ‘673’, ‘723’ or ‘773’ to indicate the reduction at ambient pressure, ‘573(4 or 18)’ to indicate the reduction at high pressure and/or ‘sg’ or ‘CO’ to indicate the reduction in syngas or CO, respectively. Table S1 in the ESI† provides an explanation of the codes of all CNF-containing catalysts in relation to their synthesis and activation protocols.
Finally, the SG route was applied to prepare CNF-supported K-promoted CoO and MoO2, which were physically mixed to yield a reference catalyst with a K/Co/Mo ratio of 0.1/1/1 and a CoMo loading of 5 wt%. To attain CoO, the dried catalyst was heated in an Ar flow of 20 cm3 min−1 for 4 h at 573 K (3 K min−1), while a reduction in a 10 vol% H2/He flow of 20 cm3 min−1 for 4 h at 773 K (3 K min−1) was applied to produce MoO2. The composite material was labelled KCoO–MoO2.
Catalyst characterisation
X-ray fluorescence spectroscopy (XRF) was performed using an Orbis Micro-EDXRF spectrometer equipped with a Rh source operated at 35 kV and 500 μA and a silicon drift detector to obtain the bulk metal ratio in the catalysts. The bulk K, Co and Mo contents were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Horiba Ultra 2 instrument equipped with a photomultiplier tube detector. Prior to analysis, the samples were alternatively treated in H2SO4 (Fisher Chemicals, >95 wt% aqueous solution) or HCl (Merck product, 37 wt% aqueous solution) to dissolve Mo or Co, respectively. Thermogravimetric analysis (TGA) was performed in a Mettler Toledo TGA/DSC1 instrument equipped with a quadrupole mass spectrometer in the 298–1173 K range (10 K min−1) applying a synthetic air flow of 60 cm3 min−1. N2 sorption at 77 K was measured in a Micromeritics TriStar II instrument after degassing the samples at 573 K under vacuum for 3 h. The surface area of catalysts and supports was determined by applying the BET method. Powder X-ray diffraction (XRD) was conducted using a PANalytical X'Pert Pro-MPD diffractometer with Ni-filtered Cu Kα radiation (λ = 0.1541 nm), acquiring data in the 10–70° 2θ range with an angular step size of 0.033 and a counting time of 8 s per step. For selected samples, a semi-quantitative analysis of the patterns was conducted to estimate the amounts of the K-, Co- and/or Mo-based crystalline phases identified. For this purpose, the peaks specific to each compound were integrated and the sum of the areas was divided by the sum of all reflections in the pattern except for those related to the CNF. In the case of overlapping reflections, an approximation of the reflections' area was made based on the relative peaks intensity in reference patterns available in the database of the International Centre for Diffraction Data (ICDD). Temperature-programmed reduction with hydrogen (H2-TPR) was carried out using a Micromeritics Autochem II 2920 unit equipped with a thermal conductivity detector and coupled to an MKS Cirrus 2 quadrupole mass spectrometer. Samples (0.040 g) were dried in He (20 cm3 STP min−1) at 573 K for 1.5 h and cooled to room temperature before the temperature was ramped at 10 K min−1 up to 1173 K in a 5 vol% H2/N2 flow of 20 cm3 min−1 for the analysis. Samples reduced under 5 MPa of H2 and used materials were extracted from the reactors and loaded into the analysis tube in an Ar-conditioned glove box. Catalysts reduced under ambient pressure were handled in air but were reduced in a 5 vol% H2/N2 flow of 20 cm3 min−1 at 573 K for 3 h and cooled down to room temperature prior to the analysis to simulate the re-activation step performed in situ in the catalytic reactor before use in HAS (vide infra). The amount of H2 consumed upon H2-TPR (μmol H2 gcat−1) was quantified based on integration of the curve, followed by calibration based on a curve obtained reducing CuO (Alfa Aesar, 99.9999% on a metal basis). For the used samples, the area of their negative peak was excluded from the integration, which might lead to a slight underestimation of the H2 consumption. X-ray photoelectron spectroscopy (XPS) was conducted using a Physical Electronics (PHI) Quantum 2000 X-ray photoelectron spectrometer featuring monochromatic Al Kα radiation, generated from an electron beam operated at 15 kV and 32.3 W, and a hemispherical capacitor electron-energy analyser, equipped with a channel plate and a position-sensitive detector. The energy scale of the instrument was calibrated using Au and Cu reference samples. The samples were firmly pressed onto indium foil patches, which were then mounted onto a sample plate and introduced into the spectrometer. The analysis was conducted at 1 × 10−6 Pa, with an electron take off angle of 45° and a pass energy of 46.95 eV. Charge compensation during the measurement was achieved using a low energy electron source. The total X-ray exposure of all specimens was ca. 500 min. No changes were observed in the spectra acquired after 120 and 360 min, indicating that sample degradation, if occurring, was confined to the initial exposure of the catalysts to the beam. This might have comprised a partial reduction of Mo6+ species. The spectra were aligned using the main component of the C1s core level spectra. Surface elemental concentrations were determined in atomic percent using the measured photoelectron peak areas after Shirley background subtraction and the built-in sensitivity factors for calculation. The relative amount of Mo species in different oxidation states was semi-quantitatively obtained by fitting the Mo3d core spectra. A symmetric shape (GL 30) was assumed for the peaks, thus screening effects were not considered. Additionally, a splitting of ΔE = 3.15 eV and an area ratio of 0.6 between the two components of the 3d doublets were assumed. Binding energy ranges and boundary conditions for the fits were chosen according to the literature.23 High-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy coupled to energy dispersive X-ray spectroscopy (STEM-EDX) were conducted in an FEI Talos F200A instrument equipped with a high brightness field emission gun, a high-angle annular dark-field (HAADF) and a large collection angle EDX detector and operated at 200 kV. Catalyst powders were dispersed on copper grids coated with a continuous carbon film. Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) investigations were performed using a Bruker Equinox 55 spectrometer, equipped with a liquid-N2-cooled MCT detector and a high-pressure diffuse reflectance cell (Harrick) and coupled to the gas chromatograph (GC) of the reactor set-up (vide infra) analysing the gas outlet stream. The sample holder was filled with the catalyst sample (10 mg) diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 with Si (Acros Organics, 99%). After in situ reduction based on the best activation protocol determined (vide infra), a catalytic run was conducted at 5 MPa and 543 K feeding a flow of 200 cm3 min−1 of H2, CO and Ar with a molar ratio of 0.5/1/98.5. Spectra were recorded by accumulating 200 scans in the range of 4000–600 cm−1 with a resolution of 4 cm−1 and were corrected by subtracting the gas-phase contribution of CO, using a spectrum collected upon flowing this gas under the reaction conditions over KBr.
:4 with Si (Acros Organics, 99%). After in situ reduction based on the best activation protocol determined (vide infra), a catalytic run was conducted at 5 MPa and 543 K feeding a flow of 200 cm3 min−1 of H2, CO and Ar with a molar ratio of 0.5/1/98.5. Spectra were recorded by accumulating 200 scans in the range of 4000–600 cm−1 with a resolution of 4 cm−1 and were corrected by subtracting the gas-phase contribution of CO, using a spectrum collected upon flowing this gas under the reaction conditions over KBr.
Catalytic testing
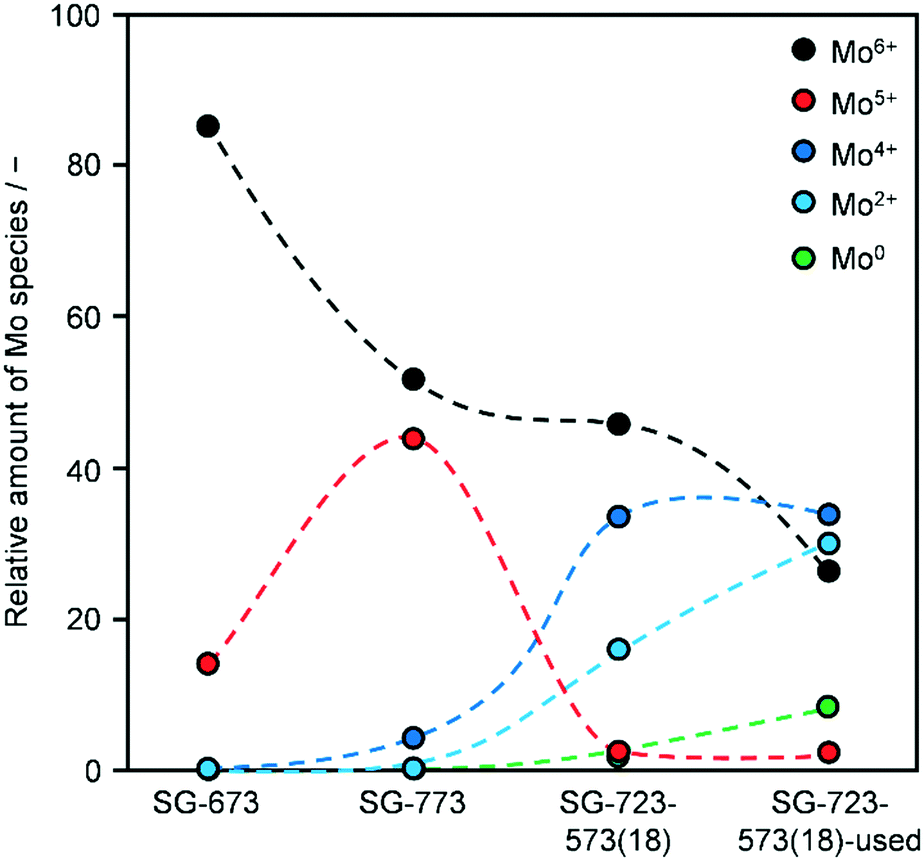
The performance of the catalysts was evaluated using a fixed-bed reactor setup (Fig. S1†) consisting of (i) mass flow controllers to feed H2, CO and Ar (Messer, ≥99.997%) and a liquid injection system based on a controlled evaporator unit (Bronkhorst) to feed alcohols and hydrocarbons for calibration, (ii) a nickel-lean steel reactor (0.65 cm i.d.) equipped with a thermocouple placed directly in the catalytic bed and heated by an oven, (iii) a hot trap kept at 453 K to condense alcoholic and aliphatic products with more than 6 and 10 carbon atoms, respectively, and (iv) an online GC (Agilent 6890A) comprising two columns (ShinCarbon ST and PoraPLOT Q PT), a thermal conductivity detector (TCD) and a flame ionisation detector (FID) to monitor the outlet gas composition. Typically, 0.5 g (sieve fraction = 0.125–0.300 mm) of the undiluted catalyst was loaded into the reactor and an Ar flow of 100 cm3 min−1 was applied for 0.5 h at ambient pressure. Under the same flow, the pressure was increased to 5 MPa and a leak test was conducted. Then, the pressure was lowered to 0.5 MPa and re-activation of the catalyst was performed in a 10 vol% H2/Ar flow of 100 cm3 min−1 at 573 K (3 K min−1) for 3 h for the catalysts reduced under ambient pressure at variable temperature. This step was replaced or followed by reduction under 5 MPa H2 at variable temperatures for different times for selected materials, as described above, or skipped in the case of the KCoO and CoMoO4 reference catalysts not to alter their chemical state. The reaction was carried out by feeding a mixture of H2, CO and Ar with a molar H2/CO/Ar ratio of 6/3/1 at 543 K, 5 MPa and a weight hourly space velocity (WHSV) of 4000 cm3 gcat−1 h−1. The CO conversion (XCO) was calculated using eqn (1): | (1) |
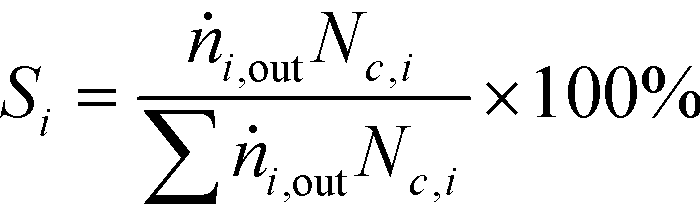 | (2) |
 | (3) |
 | (4) |
 | (5) |
3. Results and discussion
Comparison of supports
The oxides Al2O3, SiO2 and MgO and the C-based materials AC, CB and CNF were selected as supports to prepare K-promoted CoMo catalysts by simultaneous dry impregnation of the metal precursors, followed by reduction in diluted hydrogen at 673 K. The XRD patterns, surface areas and pore volumes of the carriers (Fig. 1 and Table 1) were comparable to literature data.24–26 The diffractograms of SiO2, AC and CB highlighted the amorphous or poorly crystalline nature of these materials and those of MgO and Al2O3 indicated the presence of a minor amount of Mg(OH)2 and AlO(OH) and Al(OH)3, respectively. The reflections specific to graphitic carbon were observed at 2θ = 24.4, 43.8 and 54.0° in the pattern of the CNF, which comprised rods with an internal diameter of 20–30 nm and a platelet-type structure, partly aggregated in coils of ca. 500 nm size with an opening of ca. 100 nm, as shown by TEM (inset of Fig. 1). Based on XRF and ICP-OES measurements, the molar K/Co/Mo ratio and CoMo loading of the KCoMo catalysts synthesised were close to the nominal values of 0.1/1/1 and 5 wt%, respectively. A deviation was evident for the Al2O3-supported catalyst, which had a ca. 50% higher CoMo loading and ca. 25% lower Co/Mo ratio. Likely due to the high dispersion of the metals, the XRD patterns of the reduced catalysts (Fig. S2†) did not show any additional metal phase to the support, except for the CNF- and SiO2-based solids, which contained weak reflections attributed to CoMoO4, CoMoO3 and CoO and to CoMoO4, respectively. The diffractograms of the systems supported on MgO and Al2O3 indicated that the catalyst preparation and activation led to the disappearance of the hydroxide phases. Such restructuring might explain the quite pronounced increase in the surface area upon metal deposition over these oxides. Testing of these systems in the conversion of syngas at 543 K and 5 MPa (Fig. 2) indicated that the AC-supported material was virtually inactive and the SiO2- and MgO-based systems had a very low activity (CO conversion <0.3%). This is in contrast to previous evidence for KCoMo systems supported on silica and carbon with 8- and 13-times higher metals loading, respectively.11,15 Among the other materials, the Al2O3-supported sample showed a CO conversion (5%) ca. double that of the CB- and CNF-based solids, which could be due to its higher CoMo content and Co/Mo ratio. All catalysts predominantly produced HC, especially the one containing alumina, possibly due to the acidity of this carrier that favours C–C coupling. Greatly superior conversion and HA selectivity over an Al2O3-supported CoMo catalyst was previously obtained upon promotion with Cs and application of a much higher pressure.27 The CNF-containing material exhibited a reasonable HA selectivity (18%), which was 3 times that of the other two active catalysts. One reason for this result could be the good interaction between Co and Mo, since the reduced catalyst mainly contained mixed oxides of Co and Mo among crystalline phases (vide infra). Still, intermixed metals might have been present also in the other solids and could not be detected by XRD due to a high dispersion. Another significant aspect is the high electrical conductivity of CNF (i.e., resistivity in the order of 10−4–10−5 Ω cm),28 which is only slightly inferior to that of carbon nanotubes (CNT, 10−5 Ω cm)29 and likely plays an important role in electron transfer processes between the active metals. Moreover, even if to a lower extent with respect to CNT,30–32 CNF have been found to enable hydrogen dissociation and spillover,16,29 which could support the reaction acting on hydrogenation steps and/or further reducing Co and Mo species. In the case of CNT-based materials, it has been reported that metal particles with different properties and performances are formed in the channels of the support due to confinement effects.33 Based on the much larger inner diameter of our CNF, the placement of active metals inside or outside the nanostructures is not envisaged to lead to a distinct catalytic behaviour. Indeed, STEM-EDX analysis (vide infra) indicates a similar size and composition for particles at the two positions. Additionally, diffusional disguises in the channels are unlikely in view of their large dimensions. While it would be interesting to confirm this experimentally, further assessment would require a separate study. | ||
| Fig. 1 XRD patterns of the supports used in this study. The inset shows STEM and TEM images of the CNF visualising the organisation of the rods and the platelet structure. | ||
| Catalyst | CoMo loading (wt%) | Molar K/Co/Mo ratio (−) | S BET (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
| a Data for the supports in brackets. b XRF spectroscopy. c ICP-OES. d BET method. e t-plot method. | ||||
| Al2O3 | 8.3b | 0.15/1.0/1.3b | 187 (159) | 0.60 (0.49) |
| SiO2 | 4.8b | 0.09/1.0/1.0b | 143 (140) | 0.57 (0.56) |
| MgO | 4.9b | 0.08/1.0/0.8b | 44 (16) | 0.27 (0.07) |
| AC | 5.3c | 0.09/1.0/0.9c | 907 (1154) | 0.82 (1.05) |
| CB | 4.8c | 0.09/1.0/0.9c | 230 (233) | 0.50 (0.43) |
| CNF | 4.6c | 0.09/1.0/0.8c | 24 (22) | 0.07 (0.06) |
 | ||
| Fig. 2 Products yield over KCoMo catalysts supported on different carriers and reduced under the standard conditions. Reaction conditions: 543 K, 5 MPa, H2/CO = 2 and WHSV = 4000 cm3 gcat−1 h−1. | ||
Remarkably, after equilibration in the first 2 h on stream, the CNF-supported catalyst was quite stable for the whole duration of the run (14 h, Fig. S3a†). In contrast, the Al2O3-based catalyst continuously lost activity, reaching a value which was half of the original level. Since the catalyst powder turned black upon reaction, coke deposition was suspected as the main mechanism of deactivation. TGA data of the used Al2O3 catalyst (Fig. S3b†) confirmed that 4% of the weight of the used catalyst was due to C-based species that were burned off. In contrast, the TGA profiles of the reduced and used CNF-containing samples were practically identical. Based on its superior HA yield and durability, this system was selected for further investigations.
Impact of synthesis and activation on the state of Co and Mo and the performance of CNF-supported catalysts
In order to gather fundamental understanding of structural and electronic parameters relevant for HA production over CNF-supported KCoMo catalysts, various materials were prepared for testing and characterisation altering synthesis and activation variables.Firstly, distinct preparation methods were applied to tune the proximity of the metals as well as the size and distribution of the metal phases keeping the same metal ratio and loading. Two catalysts were again produced by DI, but Co and K together with Mo were added sequentially in this or the opposite order. Besides, the concomitant addition of all precursors to the CNF was assisted by BM in the presence of water or performed following a SG route using citric acid. These two methods were supposed to promote the intermixing of K, Co and Mo and the SG protocol was reported to boost the metal dispersion due to the formation of citrate–metal ion complexes.22 The experimental values derived for the metal ratio and CoMo loading were in good agreement with the expected figures. The porous properties of all catalysts were very comparable, except for the BM-prepared catalyst, which possessed a moderately higher surface area, likely due to particle disaggregation upon milling (Table 2). The XRD patterns of these materials (Fig. S4†) were analysed to derive semi-quantitative information on the Co- and Mo-containing phases. It is worth noting that, since single sites and small nanoparticles remain undetected by this technique, the findings might not be representative for the totality of metal species. As shown in Fig. 3a, the catalysts possessed a similar amount of crystalline compounds, but their composition varied strongly. DI-MoK/Co contained MoO2, CoO and CoMoO4 in a ratio of ca. 2/1/1. DI-673 (previously called CNF) mainly possessed CoMoO4 along with CoO and CoMoO3 in similar minor amounts and traces of MoO3. DI-Co/MoK comprised MoO3, MoO2 and CoMoO4 in comparable quantities and some additional KCoO2 phase. The presence of the latter might be due to the dissolution of a portion of previously deposited K upon impregnation of Co. SG-673 featured the highest relative amount of CoMoO4 and further contained CoO and MoO2 to a 4-times lower extent. In BM-673, CoO was the dominant phase, followed by CoMoO4, CoMoO3 and MoO2 in equal quantities. Oxides containing Mo4+ instead of Mo6+ species were more abundant in this catalyst compared to the other materials, hinting that the activation was more effective in reducing this metal, possibly due to the better dispersion achieved through BM. The catalytic data collected for these materials (Fig. 4) indicated the SG-derived system as the best performer in terms of HA yield, followed by the catalyst obtained by simultaneous DI of all metals and then by that produced by DI with the initial deposition of Mo and K and the subsequent incorporation of Co. The other two solids exhibited a very low CO conversion (<2%) and were mostly selective towards HC (up to 84%). The used samples had similar compositional and porous properties (Table 2), pointing to the absence of fouling and metal leaching. Still, based on XRD (Fig. 3b), redispersion and restructuring took place. The fraction of crystalline phases detected was ca. 10, 60 and 75% lower compared to the reduced solids for DI-derived catalysts, SG-673 and BM-673, respectively. The patterns of used DI-KMo/Co and DI-Co/KMo showed the presence of MoO2 and of CoO, in about two thirds of the amount of the former phase, while that of spent DI-673 pointed to comparable masses of CoO and CoMoO3. For SG-673, the compounds still detected were metallic Co and CoO in a 2/1 ratio. BM-673 after the reaction comprised CoO and CoMoO3 in equal amounts. Overall, XRD suggested that more reduced and intermixed Co and Mo species are relevant for HA synthesis, conditions which appeared maximised in the SG-prepared sample, likely due to the complexing effect of citric acid.
| Catalyst | CoMo loadingb (wt%) | Molar K/Co/Mo ratiob (−) | S BET (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|---|
| a Data for used catalysts in brackets. b ICP-OES. c BET method. d t-plot method. | ||||
| DI-Co/MoK-673 | 4.2 | 0.10/1.0/0.7 | 26 | 0.08 |
| DI-MoK/Co-673 | 4.7 | 0.07/1.0/1.1 | 25 | 0.08 |
| DI-673 | 4.6 (4.6) | 0.09/1.0/0.8 (0.09/1.0/0.8) | 24 (22) | 0.07 (0.07) |
| BM-673 | 4.7 (4.7) | 0.09/1.0/0.9 (0.09/1.0/1.0) | 28 (28) | 0.08 (0.08) |
| SG-673 | 5.0 (5.0) | 0.09/1.0/1.0 (0.09/1.0/1.0) | 22 (21) | 0.08 (0.08) |
| SG-2-673 | 4.8 | 0.09/2.2/1.0 | 21 | 0.08 |
| SG-0.5-673 | 4.3 | 0.10/0.5/1.0 | 16 | 0.06 |
| SG-noK-673 | 5.1 | 1.2/1.0 | 22 | 0.08 |
| SG-723 | 5.0 | 0.10/1.0/1.0 | 20 | 0.08 |
| SG-773 | 5.0 | 0.09/1.0/1.0 | 26 | 0.09 |
| SG-573(4) | 5.0 | 0.09/1.0/1.0 | 27 | 0.09 |
| SG-573(18) | 5.0 | 0.09/1.0/1.0 | 26 | 0.09 |
| SG-723-573(18) | 5.0 (4.6) | 0.09/1.0/1.0 (0.11/1.1/1.0) | 24 (11) | 0.1 (0.04) |
 | ||
| Fig. 3 Evolution of the crystalline phases in (a) as-reduced and (b) used KCoMo/CNF catalysts prepared by different synthesis routes and by the sol–gel method with variable Co/Mo ratios or without K and activated under the standard conditions and prepared by the sol–gel method and ball milling and reduced in one or two steps at variable temperatures and pressures for a distinct time, as determined from the XRD analysis. The total amount of phases in each sample is normalised to the total amount of phases estimated for the most crystalline material (SG-773). | ||
 | ||
| Fig. 4 Product yield over KCoMo/CNF catalysts prepared by different synthesis routes and by the sol–gel method with variable Co/Mo ratios or without K and reduced under the standard conditions. Reaction conditions: 543 K, 5 MPa, H2/CO = 2 and WHSV = 4000 cm3 gcat−1 h−1. | ||
Using the superior SG method, the effect of the relative amount of active metals and of the alkali promoter was explored by investigating catalysts with a nominal Co/Mo ratio of 0.5 and 2 and without K. Their basic characterisation was in line with the expectations, besides a moderately lower CoMo loading for SG-0.5-673 (Table 2). For this catalyst, very small amounts of MoO2 and metallic Co were identified by XRD (Fig. 3a and S4†), suggesting that the majority of the metals were highly dispersed. Only MoO2 was observed by Bao et al. for KCoMo/C catalysts with the same Co/Mo ratio reduced at identical temperature (673 K), possibly due to the even finer dispersion enabled by the high-surface area carrier.17,34 In the case of the solid with a higher Co content, CoMoO4 and MoO2 were mainly detected along with traces of CoO. Thus, this catalyst was more similar to the one with an equimolar amount of Co and Mo (SG-673), although it had a 40% lower fraction of crystalline species and only MoO2 instead of Mo(IV) and Mo(VI) oxides (Fig. 3a). Testing of these materials (Fig. 4) showed a 4-fold lower activity but a 50% higher HA selectivity for SG-0.5-673 compared to SG-673 and an opposite trend for SG-2-673 with a 20% higher CO conversion but a 30% lower HA selectivity with respect to the sample having a unitary Co/Mo ratio. The latter evidence is in line with the known relevance of Mo to boosting the production of C2+ alcohols. XRD analysis of the used catalysts indicated that metallic Co was formed in an equal amount to the existing MoO2 in SG-0.5-673. The metal phases in SG-2-673 underwent evident redispersion, so that the only detectable diffraction line in the pattern was that of metallic Co, which accounted for ca. 20% of the initially visible phases (Fig. 3b). Concerning the K-free catalyst (SG-noK-673), the only phases shown by XRD were CoO and CoMoO4 in a 2/1 ratio and their total amount was less than half compared to the K-containing analogue. Its CO conversion was slightly inferior to SG-673, which is in contrast with previous observations of a detrimental effect of alkali metals on MoS2-based systems due to blocking of a portion of the catalytic sites.35 Its methanol selectivity was 4.5-times higher than for the K-promoted catalyst, which supports the typical role of alkali to shift the product selectivity away from the Anderson–Schulz–Flory distribution. Specifically, the C2+/C1 alcohols ratio was 0.9 over SG-noK-673 instead of 1.6 over SG-673. Nevertheless, the HA selectivity was surprisingly high. Based on structural analysis, a Co2Mo3 phase formed upon reaction, hinting to a beneficial role of high proximity and reduction degree of the metals, which was likely favoured by the absence of K. Overall, the catalytic data for SG-noK-673 are in striking contrast with those published on K-free CoMo/AC catalysts by SABIC.20 It is worth mentioning that, intrigued by their results, we tried to reproduce their systems, but found contrasting indications in the patent about the metal loading and also realised that the solvent and pH value applied in their deposition–precipitation method are unsuitable to dissolve the metal precursors and fully precipitate Mo, respectively.
Since the SG synthesis and a Co/Mo ratio of 1 led to a K-promoted material showing a greater HA yield and high Co–Mo intermixing, this combination of parameters was further applied to prepare catalysts to be activated under variable conditions to enhance the reduction degree of Co and Mo. Indeed, the XRD analysis (Fig. 3b and S5†) generally pointed to a higher abundance of low oxidation states for the two metals after the reaction. At first, the ambient pressure reduction was alternatively conducted at 723 and 773 K. This did not affect the porous properties of the catalyst significantly (Table 2), but had a remarkable impact on the nature and crystallinity of the Co- and Mo-based phases. 25% more crystalline compounds were detected in SG-723 and SG-773 compared to SG-673. The catalyst reduced at the intermediate temperature was almost exclusively composed of CoMoO4, with a very minor portion of CoO. In the case of SG-773, the 50-K higher temperature was sufficient to produce CoMoO3, i.e., to reduce Mo6+ to Mo4+. This phase was accompanied by CoO in a moderately higher amount with respect to the SG-723 catalyst (ca. 20% of CoMoO3). Assessment in HA synthesis indicated a halved CO conversion over both catalysts compared to SG-673 (ca. 4 versus 8%) but a greatly increased HA selectivity, which raised from 11% over SG-673 to 25 and 27% over SG-723 and SG-773, respectively (Fig. 5). Accordingly, the C2+ alcohol yield was almost double. In line with previous results, the metal phases in the two samples were strongly redispersed. The crystalline species in the used materials accounted for ca. 40–45% of their original amounts and mostly comprised Co and CoMoO3, with traces of CoO. These findings corroborate lower oxidation states of both Co and Mo and their proximity as important for the reaction.
 | ||
| Fig. 5 Product yield over KCoMo/CNF catalysts prepared by the sol–gel method and reduced in one or two steps at variable temperatures and pressures for a distinct time. Reaction conditions: 543 K, 5 MPa, H2/CO = 2 and WHSV = 4000 cm3 gcat−1 h−1. | ||
Aiming at boosting the HA selectivity without compromising the activity level, it was conceived to treat an as-prepared material at a lower temperature but at higher H2 concentration and total pressure. Hence, the reduction was conducted in pure H2 at 5 MPa, i.e., the same pressure as that applied during the reaction, at 573 K. While the porous properties were affected to a minor extent due to the stability of CNF and the low metals loading (Table 2), no Co- and/or Mo-based crystalline phases were formed (Fig. 3b and S5†), which proves the suppression of sintering but prevents gaining indications about the oxidation state of the metals. SG-573(4) exhibited a similar catalytic behaviour to the samples activated at 723 and 773 K. As expected from the XRD data of used samples previously investigated, no reflections were visible in the pattern recorded after the reaction for this material (Fig. 3b and S5†).
Based on these findings, the duration of the reduction at 573 K and 5 MPa in pure H2 was extended from 4 to 18 h. This treatment led to a material (SG-573(18)) with similar surface area and porous volume to SG-573(4), which also did not contain any crystalline phase. Interestingly, the catalyst activity increased (CO conversion = 8%), while the HA selectivity remained at 23%. Thus, this sample behaved as originally expected when lowering the activation temperature and increasing the reducing potential of the gaseous environment. XRD analysis of the used material unveiled the formation of a little amount of Co2Mo3. Thus, the harsher reduction conditions were able to overcome the negative influence of K on the reducibility of the metals. However, since the HA selectivity reached by SG-723 and SG-773 was not matched, a high-temperature activation step was combined with the extended activation at high pressure. Hence, an SG-prepared catalyst was reduced firstly at 723 K and ambient pressure for 4 h and then, after exposure of the catalyst to air and in situ reactivation in the reactor, at 573 K and 5 MPa for 18 h. Porous and structural properties were analogous to the other systems activated at 5 MPa. Still, while the C2+ alcohols selectivity was not enhanced (22%), the CO conversion increased remarkably, reaching 14%. Another difference was the enhancement of the water gas shift reaction at the expense of Fischer-Tropsch synthesis. Since the use of CO and syngas as reducing agents for CoCu catalysts has led to the generation of metal phases with distinct electronic properties and morphology and thus variable catalytic performance,36 these gases were alternatively applied in the most suitable two-step activation of the SG-prepared catalyst. The catalyst reduced in syngas (SG-723-573(18)-sg) showed substantially inferior CO conversion (3 versus 14%) and moderately reduced HA selectivity (20 versus 23%). The catalyst obtained using CO upon activation was almost inactive (CO conversion = 0.6%) and greatly less selective towards HA (15%). Based on XRD, no crystalline phases were detected in the two samples after the first activation step and after the reaction, indicating a high metal dispersion (Fig. S5†). TGA of the sample obtained upon the first ex situ activation step in CO showed the formation of a considerable amount of hard coke (Fig. S8†). The quantity of such a deposit was only slightly higher after the second activation step and the reaction (used SG-723-573(18)-CO), likely due to the minimal activity of the solid. This finding is in line with the ability of CoMo to disproportionate CO forming carbon nanotubes at 973–1123 K.37 Thus, it is likely that carbonaceous species block the active sites, suppressing the CO conversion. It is assumed that the presence of H2 in syngas partially counteracts the action of CO, hindering fouling and enabling an intermediate CO conversion.
Based on these results, the SG-723-573(18) material offered the highest HA yield in this study. Notably, its STY of HA based on the mass of active metals was 5.5-times higher than the best CoMo-based catalyst meaningful for comparison (38 versus 7 mmolHA gCoMo−1 h−1).15 Indeed, the SABIC systems and the catalysts containing Co-decorated CNT were excluded, in view of their irreproducible synthesis and drastically different, non-rationalised product distribution, respectively. It should be noted that the metal content used for the calculation was that of the fresh rather than the used catalyst. The CoMo loading derived by ICP-OES after the reaction was moderately inferior, but in view of the retained metal content for SG-673, BM-673 and DI-673 (Table 2) and of XPS and TGA data introduced later on indicating the occurrence of substantial coking, we believe that this decrease was due to the dilution of the metal by the additional carbon rather than to an effective leaching of Co and Mo upon reaction. It should be noted that the catalyst, after an equilibration phase of ca. 5 h, exhibited a substantially stable performance for 15 h on stream (Fig. S6†). Remarkably, when considering the STY of HA per mass of the catalyst, the SG-derived material almost matched the benchmark system15 (0.11 versus 0.12 gHA gcat−1 h−1) in spite of the 13-times lower CoMo loading. A reflection specific to Co2Mo3 was visualised in the pattern of the used material, which had a higher intensity than that for used SG-573(18). This is not surprising based on the overall longer contact of the catalyst with reducing gases upon activation and reaction.
To verify whether the synthesis method or the reduction protocol was more crucial to attain a high HA yield, a catalyst produced by BM was subjected to the optimal two-step activation procedure applied to an SG-derived material. The negligible activity and scarce HA selectivity of BM-723-573(18) (Table S2†) clearly highlight the superiority of the SG preparation method.
With the purpose of getting insights into the importance of the proximity of Co and Mo, a reference material was prepared physically mixing two solids featuring only KCoO or KMoO2 attained according to the SG method. XRD analysis (Fig. S7†) confirmed the presence of the two desired phases in the catalyst, which displayed much inferior performance compared to the catalysts mostly comprising the mixed oxides CoMoO4 (SG-723) and CoMoO3 (SG-773)(Table S2†). Indeed, in spite of the comparatively high HA selectivity (33%), the CO conversion was limited to only 0.8%. XRD analysis of the used sample (Fig. S7†) revealed an enhancement of the CoO and MoO2 phases and the appearance of metallic Co.
Since XRD is a bulk technique unsuited to properly assessing low amounts of metals which are finely distributed on the support and was applied in ex situ mode, the most significant samples in as-reduced form and after use in the reaction were further characterised by complementary thermal, spectroscopic and imaging methods, under conditions more relevant to catalysis whenever possible, in order to gain a deeper understanding of the speciation and relative distribution of Co and Mo as well as the structure of their phases.
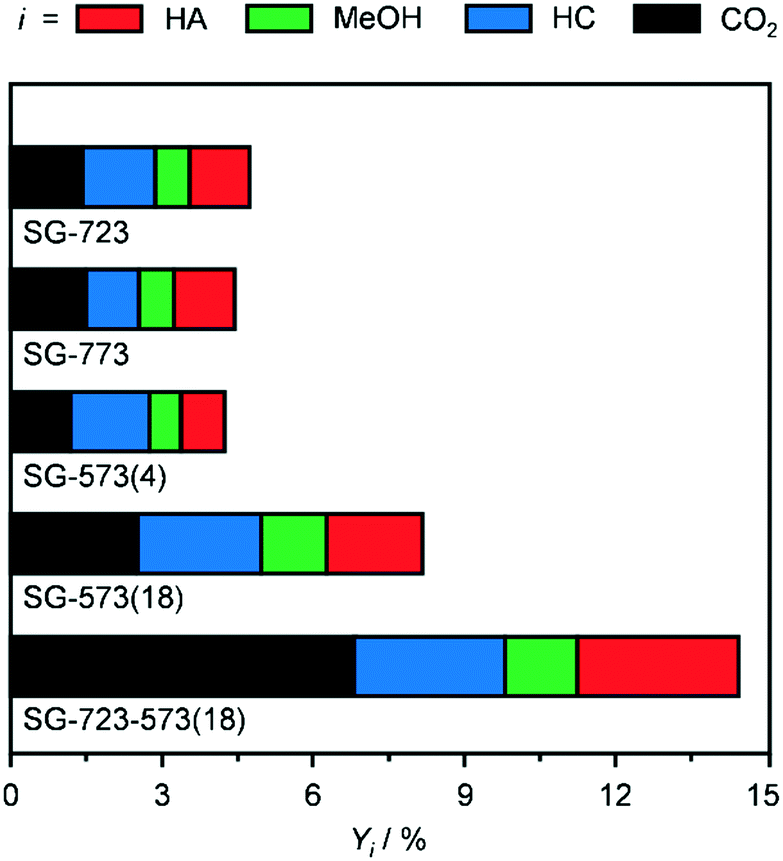
H2-TPR (Fig. 6) was performed on as-reduced and spent catalysts prepared by the three methods and activated under standard or high-temperature/-pressure conditions, which were not in contact with air prior to the analysis. The profiles of DI-673, SG-673, and BM-673 show the presence of two main reduction peaks. The low-temperature signal has a maximum at 773 (SG-673, BM-673) and 843 K (DI-673) and is attributed to the reduction of Mo6+ to Mo4+ species, while the high-temperature feature peaks at 963 K for all materials have been related to the reduction of Mo4+ atoms to species with an oxidation state of 1–3.17,18 In view of the asymmetry of these peaks and the catalyst composition, it is expected that a third reduction process at intermediate temperatures remained masked, which is typically related to the transformation of Co2+ species into metallic Co.17,18 Besides effects due to the size of the particles of individual and mixed Co- and Mo-based phases, the variable temperature associated with the reduction of Mo6+ to Mo4+ species might be linked to their distinct degree of interaction with Co, since the latter aids the reduction of strongly oxidised Mo species. Integration of the three curves revealed a similar overall hydrogen consumption for DI-673 and SG-673, while the use of half the hydrogen amount for the reduction of BM-673. The latter finding is in line with the lower oxidation state of Mo in the phases detected by XRD in this sample. The profiles of the SG-produced catalysts activated at higher temperature and/or pressure appear much broader and less intense and with contributions which cannot be properly defined in number and position between 463 and 973 K. This is likely the consequence of their more reduced state, and/or the higher dispersion of the metal phases, based on XRD. In line with the harsher reduction treatments, SG-723, SG-773, SG-573(4) and SG-723-573(18) consumed 20, 22, 25, respectively, and 60% less hydrogen in the analysis compared to SG-673. The curves of SG-773 and SG-723-573(18) in used forms also show a very wide temperature interval for reduction processes, with a more noticeable peak at 927 K for the former solid, and a generally low hydrogen consumption. However, a moderate to strong negative feature with a minimum at 723–743 K is present in both cases, which disables a more precise interpretation of the data. Based on the higher surface C content detected by XPS for the used SG-723-573(4) catalyst (vide infra), the negative peak is likely due to the evolution of adsorbed intermediates/products, which are not hydrogenated to methane according to the online MS analysis. The fact that the peak is larger for SG-723-573(18) seems in agreement with the higher dispersion of Co and Mo in this solid, which can bind more organic compounds. TGA analysis of the used sample confirmed a mass loss of 4 wt% for used SG-723-573(18) (Fig. S8†).
 | ||
| Fig. 6 H2-TPR curves of KCoMo/CNF catalysts prepared by different synthesis methods and reduced under the standard conditions and prepared by the sol–gel route and reduced in one or two steps at variable temperatures and pressures for a distinct time in as-reduced and used forms. | ||
Additional characterisation techniques were applied to SG-773 as well as SG-723-573(18) in as-reduced and used forms in view of their higher HA selectivity and yield, respectively, using SG-673 as a reference catalyst.
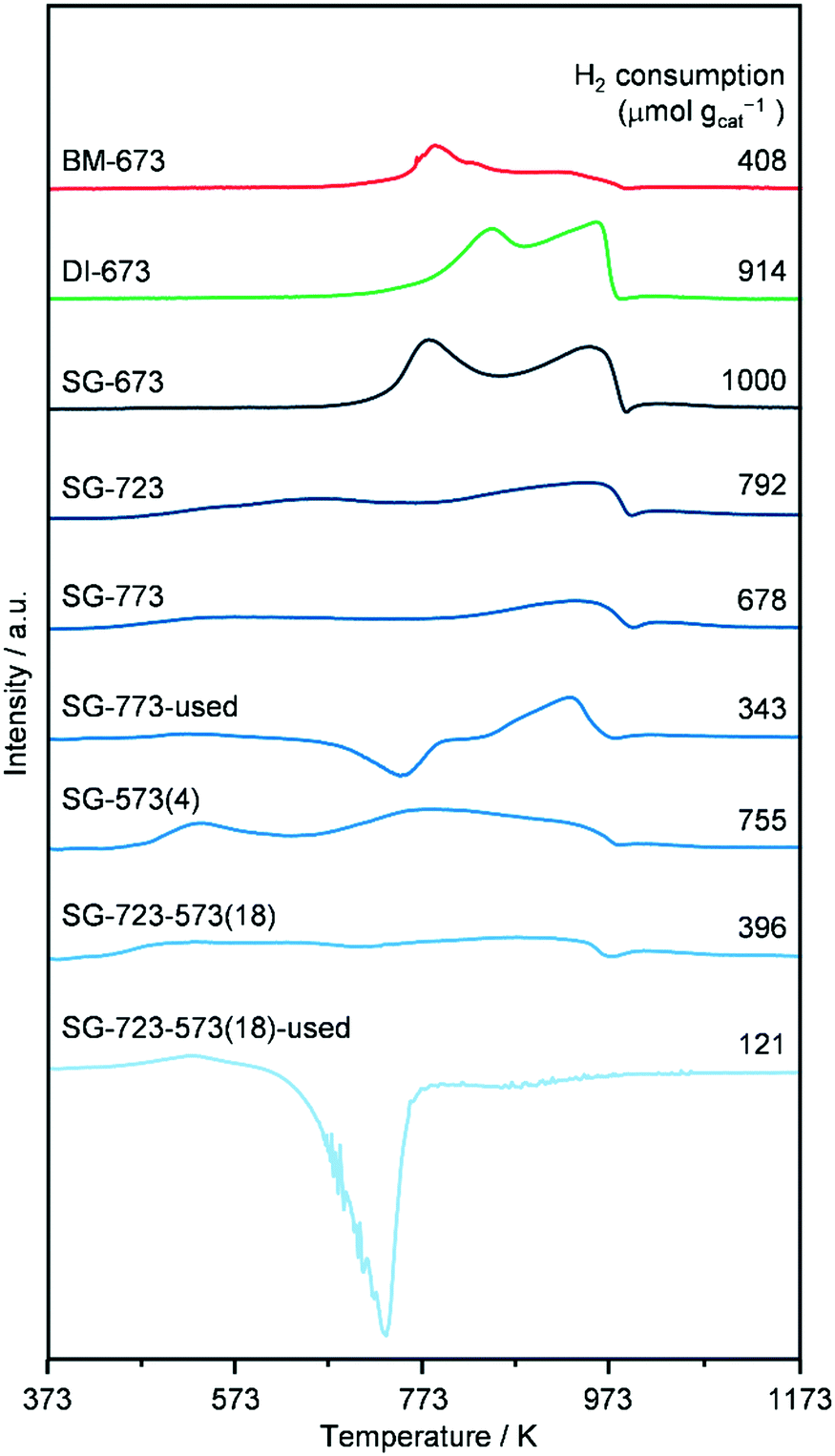
XPS indicated a Co/Mo ratio of 1.8, 1.2, 1.4 and 0.9 for as-reduced SG-673, SG-773 and SG-723-573(18) and used SG-723-573(18), respectively. This implies that the surface of the as-reduced catalysts is moderately to largely enriched in Co and that a ratio closer to unity is approached if harsher conditions are applied upon activation, i.e., higher temperature and/or pressure. This matches the greater abundance of crystalline CoMo mixed oxides detected in SG-773. The use in the reaction provoked a significant alteration of the composition leading to a minor enrichment in Mo. Accordingly, the environment experienced by the catalysts upon HA synthesis caused substantial restructuring, as suggested by the XRD analysis, leading to a relative exposure of Co and Mo analogous to their bulk ratio. It has to be noted that K remained undetected due to its very low loading and the overlapping of possible weak K 2p signals with features of the C 1s spectra. Compared to the as-reduced materials (SG-723-573(18)), the used catalyst contained more carbon (99 versus 96–97 at%) and less active metals (1.1–1.3 at% of Co + Mo), indicating that C-containing species remained adsorbed on the surface. Even if the catalysts were exposed to air prior to the XPS analysis, which has been shown to lead to partial oxidation of the active metals,19 the Co2p, and, especially, the Mo3d core level spectra were quite different for the various catalysts (Fig. S9†). Accordingly, the spectra were further analysed to determine the oxidation state of the two metals and, thus, attain indications about their relative reduction degree in the samples (Fig. 7). The analysis is simply qualitative for Co and semi-quantitative for Mo, since screening effects were not taken into account in the fitting of its peaks. Regarding Co, all as-reduced catalysts only featured Co2+ species, while an additional signal related to Co0 was detected in the case of the used material.38 Concerning Mo, SG-673 mainly contained Mo6+ species, SG-773 comprised Mo6+ and Mo5+ species in about equal amounts and SG-723-573(18) featured half of the metal in oxidation state 6 and half as a combination of 4 and 2 states. The latter two species became predominant after use in the reaction, which also determined the full reduction of oxidised Mo species to metallic Mo to a little extent. This is in line with the H2-TPR measurements and might be the origin of the simultaneous enhancement of HA synthesis and water gas shift over SG-723-573(18).39 Indeed, it has been reported that the molecular adsorption of CO on Mo is favoured when species with low oxidation states are present.17 A higher availability of CO is advantageous for alkyl insertion and the generation of HA but is also favourable for reaction with adsorbed O atoms and the formation of CO2. Thus, under these conditions, the production of HA could be independently maximised only if O atoms more easily reacted with H atoms to form water. Overall, a more reduced state for the catalyst was achieved by increasing the temperature of reduction, adding a high-pressure reduction step, and, even more, exposing the material to the reaction conditions. It should be noted that the O content of the used catalyst was 5-times lower than that of the other samples, possibly due to the fact that the adsorbates prevented re-oxidation of Mo and Co. Thus, the XPS data for this catalyst are considered highly indicative of its actual state upon HA synthesis.
 | ||
| Fig. 7 Distribution of surface Mo species for KCoMo/CNF catalysts reduced in one or two steps at variable temperatures and pressures for a distinct time in as-reduced and used forms. | ||
To shed further light on the electronic properties of the metals, a catalytic test was conducted over SG-723-573(18) in a diffuse-reflectance reaction chamber located in an infrared spectrometer exploiting the usefulness of CO as a probe for the oxidation state of metals. The SG-derived material activated at 723 K was loaded in the cell and further treated at 0.5 MPa in diluted H2 and then at 573 K in pure H2 for 18 h, as done prior to the catalytic testing. Thereafter, a reaction mixture comprising diluted CO and H2 was fed at 5 MPa and 543 K and spectra were collected regularly for 14 h on stream. The absorption bands of gaseous CO as well as C–H stretching signals were visualised, indicating that the reaction took place at least to a minimal extent, in spite of the low concentration of the reactive gases required to avoid saturation of the CO absorption. Trace amounts of methane, ethanol and CO2, i.e., the most abundant products, were also detected by the online GC analysis. Since the catalyst had to be diluted for the measurement, signals arising from CO bound to surface Co and Mo species could be detected only by subtracting the contribution of gaseous CO from the spectra. This data processing unveiled a pattern comprising two main multicomponent bands between 2250–2020 cm−1, which remained almost unaltered throughout the whole reaction time (Fig. 8). In view of the quite low signal to noise ratio, an unequivocal attribution of the absorptions to specific surface species is unfeasible. Nevertheless, previous literature indicates that signals would be generated in this spectral region by CO linearly bound to Co0 (2035 cm−1), Co+ (2116 cm−1), Co2+ (2160–2180 cm−1), Moδ+ (2103 cm−1), Mo2+ (2105–2110 cm−1), Mo4+ (2180–2190 cm−1) and Mo–OH (2138–2175 cm−1).40–43 Interestingly, a weak absorption at low frequencies (2020–1995 cm−1) appeared at the end of the run, suggesting the reduction of Mo to even the metallic state.41 Thus, overall the operando DRIFTS analysis suggests the presence of Co and Mo in various but low oxidation states, corroborating the XRD, H2-TPR and XPS spectroscopy data.
 | ||
| Fig. 8 DRIFT spectra collected upon reaction at 543 K and 5 MPa over SG-723-573(18). | ||
Finally, electron microscopy was utilised to visualise the morphology and size of the supported metal phases and the distribution of Co, Mo and K in the samples (Fig. 9). STEM analysis of SG-673 indicated the formation of nanoparticles as small as ca. 5 nm, mostly forming larger aggregates located in the interior and at the exterior of the CNF. Based on EDX, all of the structures comprise Co and Mo, in line with the detection of CoMoO4 as a principal phase in this catalyst by XRD. Analysis of the lattice fringes detected in the HRTEM micrographs of the same specimen (Fig. S10a†) revealed d spacing values of 6.7 and 3.4 Å, which are consistent with the (001) and (002) planes of this mixed oxide. Imaging of SG-773 showed particles of ca. 10–20 nm mostly placed in the channels of the CNF in an inhomogeneous manner, exhibiting a more defined morphology compared to those in SG-673. The coexistence of Co and Mo in these particles, based on EDX, hinted to a high intermixing of the metals. The d lattice spacing values of 4.9, 3.5 and 2.5 Å measured for the fringes visible in the HRTEM images of this sample (Fig. S10b†) correspond to the (002) and (102) and (200) planes of CoMoO3. Thus, this evidence is in line with the XRD analysis in terms of the higher crystallinity of the mixed oxide and the lower oxidation state of Mo in SG-773 with respect to SG-673. STEM of SG-723-573(18) uncovered the existence of much smaller particles along with clusters, here detected at the outer surface of a coil of CNF. The mapping of the metals suggested again a high proximity of Co and Mo. When inspecting the micrographs and EDX mapping of the same catalyst after use in the reaction, a moderate degree of sintering and metal segregation was evident. Still, the particle size remained inferior to that observed in SG-673. Thus, we concluded that the vicinity of Co and Mo, which was a common denominator for the samples, was associated with the HA selectivity, and was enhanced if Mo is in a more reduced state (SG-773 and SG-723-573(18)). In contrast, the metals dispersion, which increased in the order SG-773 < SG-673 < SG-723-573(18), had a clear correlation with the activity. It is worth noting that very weak signals of K were identified by EDX in the same areas analysed for the four samples, suggesting its very fine distribution.
 | ||
| Fig. 9 STEM images and EDX mappings of C (blue), Co (red), Mo (green) and K (white) of (a) SG-673, (b) SG-773 and SG-723-573(18) (c) prior to and (d) after reaction. | ||
Conclusions
In this study, descriptors for the activity and selectivity of higher alcohols synthesis catalysts based on Co and Mo and promoted by K have been identified. The first step in this direction comprised the selection of a suitable support. In this respect, CNF were introduced as a carrier enabling superior HA selectivity and stability compared to conventional oxides and amorphous C-based solids in spite of the low surface area, likely due to electronic effects. The impact of synthesis and activation parameters on the performance of CNF-supported catalysts was explored combining testing with in-depth ex situ and operando characterisation of the structural and electronic properties of the metals. A sol–gel route using citric acid emerged as a superior preparation method with respect to traditional and ball milling-assisted dry impregnation to enhance the HA yield, due to the better intermixing and dispersion of the metals in the presence of a complexant. A unitary Co/Mo ratio was shown as more adequate for the same purpose. Activation of the as-prepared catalyst in hydrogen at a higher temperature promoted the formation of mixed oxides of Co and Mo and a higher reduction degree for Mo, leading to a higher selectivity to C2+ alcohols, but produced moderately larger and more defined particles, which diminished the activity. A positive effect on both HA selectivity and CO conversion was attained when conducting the reduction at a lower temperature but at the same pressure as that of the reaction and for a longer time. A maximal HA yield was attained when combining higher temperature and higher pressure treatments due to the generation of very small nanoparticles or clusters of atomically-mixed Co and Mo, which majorly exhibited low oxidation states in the fresh catalyst and further reduced to even the metallic state upon reaction. Hence high metal proximity, dispersion and reduction degree stand as crucial properties for the conversion of syngas to HA over CoMo catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Total Research & Technology Feluy. Dr. S. Mitchell is thanked for the electron microscopy measurements and the Microscopy Centre of ETH Zurich (ScopeM) for granting access to its facilities. Dr. S. Siol acknowledges funding from the COST project IZCNZ0-174856 C16.0075, in the COST Action MP1407 (e-MINDS).References
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- M. Gupta, M. L. Smith and J. J. Spivey, ACS Catal., 2011, 1, 641–656 CrossRef CAS.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- H. T. Luk, C. Mondelli, J. A. Stewart, D. Curulla-Ferré and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- T. Lidderdale, Motor Gasoline Outlook and State MTBE Bans, 2003 Search PubMed.
- S. P. S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- G. Quarderer and G. Cochran, EP0119609A1, 1984.
- M. Dunster and J. Korchnak, EP0303438A2, 1988.
- R. Stevens, US Pat., 4752622, 1986 Search PubMed.
- B. E. Concha, G. L. Bartholomew and C. H. Bartholomew, J. Catal., 1984, 89, 536–541 CrossRef CAS.
- K. Fujimoto and T. Oba, Appl. Catal., 1985, 13, 289–293 CrossRef CAS.
- A. Juan and D. E. Damiani, Catal. Today, 1992, 15, 469–480 CrossRef CAS.
- X. Li, L. Feng, Z. Liu, B. Zhong, D. B. Dadyburjor and E. L. Kugler, Ind. Eng. Chem. Res., 1998, 37, 3853–3863 CrossRef CAS.
- X. Li, L. Feng, L. Zhang, D. B. Dadyburjor and E. L. Kugler, Molecules, 2003, 13–30 CrossRef.
- M. Lv, W. Xie, S. Sun, G. Wu, L. Zheng, S. Chu, C. Gao and J. Bao, Catal. Sci. Technol., 2015, 5, 2925–2934 CAS.
- X. Wu, Y. Guo, J. Zhou, G. Lin, X. Dong and H. Zhang, Appl. Catal., A, 2008, 340, 87–97 CrossRef CAS.
- M. Zhang, W. Zhang, W. Xie, Z. Qi, G. Wu, M. Lv, S. Sun and J. Bao, J. Mol. Catal. A: Chem., 2014, 395, 269–275 CrossRef CAS.
- G. Wu, J. Zhou, M. Lv, W. Xie, S. Sun, C. Gao, W. Wang and J. Bao, Chin. J. Chem. Phys., 2015, 28, 604–610 CrossRef CAS.
- W. Xie, J. Zhou, L. Ji, S. Sun, H. Pan, J. Zhu and J. Bao, RSC Adv., 2016, 6, 38741–38745 RSC.
- R. Khalid Karim and R. Asad Khan, US Pat., 2014/0142206A1, 2014 Search PubMed.
- Y. Zhang, Y. Sun and B. Zhong, Catal. Lett., 2001, 76, 249–253 CrossRef CAS.
- J. Bao, Y. Fu, Z. Sun and C. Gao, Chem. Commun., 2003, 746–747 RSC.
- J. Baltrusaitis, B. Mendoza-sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- G. M. Lari, O. G. Groeninger, Q. Li, C. Mondelli, N. López and J. Pérez-Ramírez, ChemSusChem, 2016, 9, 3407–3418 CrossRef CAS PubMed.
- G. M. Lari, R. García-Muelas, C. Mondelli, N. López and J. Pérez-Ramírez, Green Chem., 2016, 18, 4682–4692 RSC.
- T. C. Keller, S. Isabettini, D. Verboekend, E. G. Rodrigues and J. Pérez-Ramírez, Chem. Sci., 2014, 5, 677–684 RSC.
- S. J. Ardakani, M. Alyani and K. J. Smith, Can. J. Chem. Eng., 2016, 94, 655–661 CrossRef CAS.
- J. Heremans, Carbon, 1985, 23, 431–436 CrossRef CAS.
- A. Lekawa-raus, J. Patmore, L. Kurzepa, J. Bulmer and K. Koziol, Adv. Funct. Mater., 2014, 24, 3661–3682 CrossRef CAS.
- M. Davari, S. Karimi, A. Tavasoli and A. Karimi, Appl. Catal., A, 2014, 485, 133–142 CrossRef CAS.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed.
- X. Chen, Y. Zhang, X. P. Gao, G. L. Pan, X. Y. Jiang, J. Q. Qu, F. Wu, J. Yan and D. Y. Song, Int. J. Hydrogen Energy, 2004, 29, 743–748 CrossRef CAS.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- J. Bao, Z. Sun, Y. Fu, G. Bian, Y. Zhang and N. Tsubaki, Top. Catal., 2009, 52, 789–794 CrossRef CAS.
- T. Tatsumi, A. Muramatsu and H. Tominaga, Appl. Catal., 1987, 34, 77–88 CrossRef CAS.
- Y. Xiang, R. Barbosa and N. Kruse, ACS Catal., 2014, 4, 2792–2800 CrossRef CAS.
- J. E. Herrera, L. Balzano, A. Borgna, W. E. Alvarez and D. E. Resasco, J. Catal., 2001, 145, 129–145 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. R. de la Osa, A. de Lucas, A. Romero, P. Casero, J. L. Valverde and P. Sánchez, Fuel, 2012, 97, 428–434 CrossRef CAS.
- D. Song, J. Li and Q. Cai, J. Phys. Chem. C, 2007, 111, 18970–18979 CAS.
- N. D. Subramanian, C. S. S. R. Kumar, K. Watanabe, P. Fischer, R. Tanoanka and J. J. Spivey, Catal. Sci. Technol., 2012, 2, 621–631 CAS.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2010, 385, 153–162 CrossRef CAS.
- B. Müller, A. D. van Langeveld, J. A. Moulijn and H. Knözinger, J. Phys. Chem., 1993, 97, 9028–9033 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Picture of the reactor setup; explanation of the catalyst codes; and XRD, XPS, TGA, HRTEM and catalytic data. See DOI: 10.1039/c7cy01908d |
| This journal is © The Royal Society of Chemistry 2018 |