
An iridium–SPO complex as bifunctional catalyst for the highly selective hydrogenation of aldehydes†
Israel
Cano
 *ab,
Luis M.
Martínez-Prieto
a,
Laure
Vendier
cd and
Piet W. N. M.
van Leeuwen
*a
*ab,
Luis M.
Martínez-Prieto
a,
Laure
Vendier
cd and
Piet W. N. M.
van Leeuwen
*a
aLaboratoire de Physique et Chimie des Nano Objets, LPCNO, UMR5215 INSA-UPS-CNRS, Institut National des Sciences Appliquées, 135 Avenue de Rangueil, 31077 Toulouse, France. E-mail: vanleeuw@insa-toulouse.fr
bGSK Carbon Neutral Laboratory for Sustainable Chemistry, University of Nottingham, NG7 2GA, Nottingham, UK. E-mail: israel.canorico@nottingham.ac.uk
cCNRS, LCC (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, BP44099, F-31077 Toulouse Cedex 04, France
dUniversité de Toulouse, UPS, INPT, F-31077 Toulouse Cedex 04, France
First published on 20th November 2017
Abstract
A secondary phosphine oxide (SPO) ligand (tert-butyl(phenyl)phosphine oxide) was employed to generate an Ir–SPO complex which shows a particular ability to activate dihydrogen under mild conditions without the help of an external base or additive. Such an iridium(I) complex serves as a precursor for homogeneous catalysis since under H2 it is converted to a mixture of several iridium(III) hydride species that are the active catalysts. This system was found to be a highly active catalyst for the hydrogenation of substituted aldehydes, giving very high conversions and chemoselectivities for a wide range of substrates. The SPO ligand presumably plays a key role in the catalytic process through heterolytic cleavage of H2 by metal–ligand cooperation. In addition, an exhaustive characterization of the different iridium hydride species was performed by 1D and 2D NMR spectroscopy. The oxidative addition of H2 to the Ir(I)–SPO complex is highly stereoselective, as all generated Ir(III) hydrides are homochiral. Finally, the crystal structure, as determined by X-ray diffraction, of a dinuclear iridium(III) hydride complex is described.
Introduction
The selective hydrogenation of an aldehyde function in the presence of other reducible groups is an important step in synthetic chemistry. As an example, the chemoselective reduction of α,β-unsaturated aldehydes to their corresponding allylic alcohols has tremendous industrial importance, since these compounds are relevant intermediates and end products in the preparation of fine chemicals, fragrances and pharmaceutical compounds.1 An interesting pathway to reduce polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds is the heterolytic cleavage of dihydrogen into H+ and H−, and the subsequent transfer of hydrogen atoms to substrates such as CO bonds.2 In this approach H2 is frequently cleaved by metal–ligand cooperation; that is, a ligand containing basic sites and a coordinated metal center operate in tandem to activate the hydrogen molecule.3
X bonds is the heterolytic cleavage of dihydrogen into H+ and H−, and the subsequent transfer of hydrogen atoms to substrates such as CO bonds.2 In this approach H2 is frequently cleaved by metal–ligand cooperation; that is, a ligand containing basic sites and a coordinated metal center operate in tandem to activate the hydrogen molecule.3
In the field of homogeneous catalysis, this type of process is accomplished by numerous transition metal complexes, leading to the hydrogenation of aldehydes, ketones, and imines.4 However, iridium-based complexes have shown a limited efficiency in the chemoselective hydrogenation of substituted aldehydes and only a few systems performing this transformation have been reported in the literature.5,6 Additionally, some of these complexes are not able to act as bifunctional catalysts and require the help of an external base to promote the heterolytic splitting of H2.5h,i On the other hand, in the area of metallic nanoparticle (MNP) catalysis, this process is rarely achieved by a heterolytic cleavage mechanism involving the ligand or stabilizing agent. Indeed, the described systems based on iridium produce the H2 activation and its later transfer to aldehydes by the use of heterogeneous catalysts consisting of iridium nanoparticles (IrNPs) immobilized on oxide supports or oxygenated surfaces,7 for which the process takes place by a strong metal–support interaction.8
Along this line, secondary phosphine oxides (SPOs) form an interesting group of phosphorus ligands.9 Once coordinated (via P) as the phosphinous(III) tautomer10 to a suitable transition metal, the resulting complexes display an ability to cleave H2 heterolytically across M and O, as long as there is a vacancy on the metal. Then, the complex can transfer the hydrogen atoms to an appropriate substrate.11 This SPO–metal cooperative effect has been widely utilized in hydrogenation catalysis, in which such reactivity is particularly notorious.12 In that regard, our group has a longstanding experience in the use of this type of ligands, both in homogeneous11 and MNP13 catalysis.
Inspired by these works, we recently reported the synthesis and characterization of an Ir–SPO complex, for which two coordinated phosphine oxide ligands self-assemble after loss of one proton into a monoanionic bidentate ligand held together by an intramolecular hydrogen bond.14 In a preliminary catalytic study, the system showed a very high activity and selectivity in the chemoselective hydrogenation of cinnamaldehyde and p-nitrobenzaldehyde. The complex acts as precursor for homogeneous catalysis, since under H2 it is converted to a mixture of several hydrides.
Herein we describe the characterization and catalytic applications of such an Ir–SPO hydride system. This catalyst is very active for the chemoselective hydrogenation of substituted aldehydes, providing exceptionally high conversions and selectivities. The SPO ligand presumably plays a crucial double role, as modifying ligand, and as functional ligand acting as heterolytic activator for dihydrogen, since its oxygen atom operates as a basic site and takes a H+ from H2, leaving a H− bound to the metal center.11,13
Results and discussion
Synthesis and characterization of catalytic system
Treatment of (1,5-cyclooctadiene)(methoxy)iridium(I) dimer ([Ir(OMe)(COD)]2) with 4 equivalents of tert-butyl(phenyl)phosphine oxide (1) and 2 equivalents of H2O affords complex 2 (Scheme 1, for further details see ESI†).14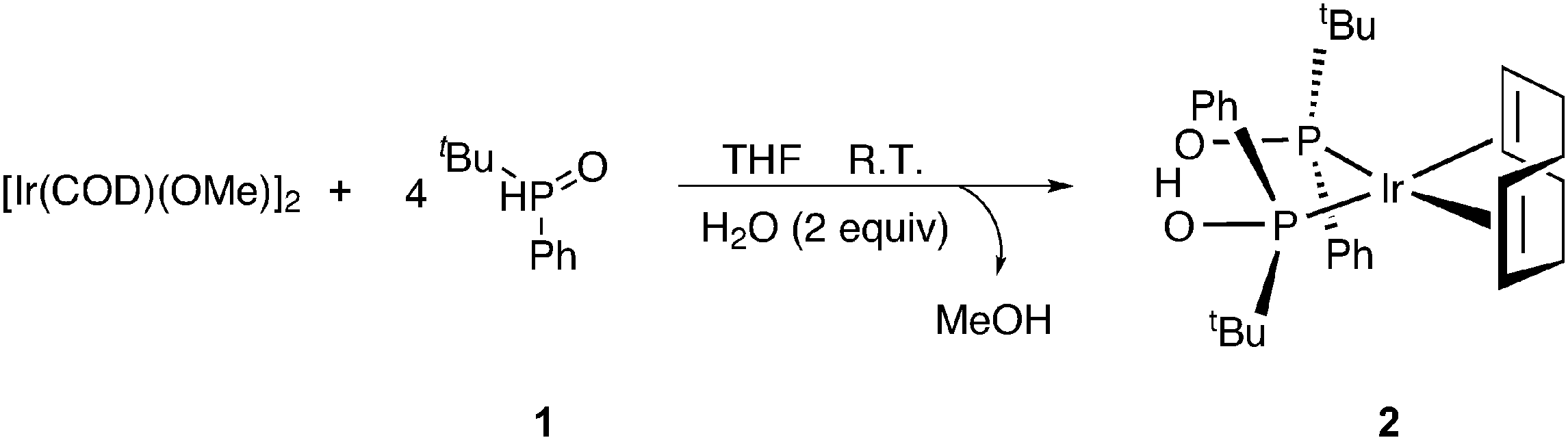 | ||
| Scheme 1 Formation of iridium SPO complex 2. | ||
The structure of 2 was elucidated unambiguously by single crystal X-ray structure analysis and solid state fast magic angle spinning (MAS) 1H NMR.14 Complex 2 adopts a square planar molecular geometry around the metal center with double coordination to cyclooctadiene (COD) and the SPO ligands coordinated to the iridium center as a hydrogen bonded pair of the two, for which SPOs have a strong preference in metal complexes thus obtaining a monoanionic bidentate ligand.15
This complex is a precursor instead of an active catalyst for aldehyde hydrogenation. To investigate the active catalytic species, we treated iridium complex 2 with 5 bar of H2 pressure in acetonitrile (AN) at room temperature (R.T.) for 30 min (Scheme 2, for further details see ESI†).
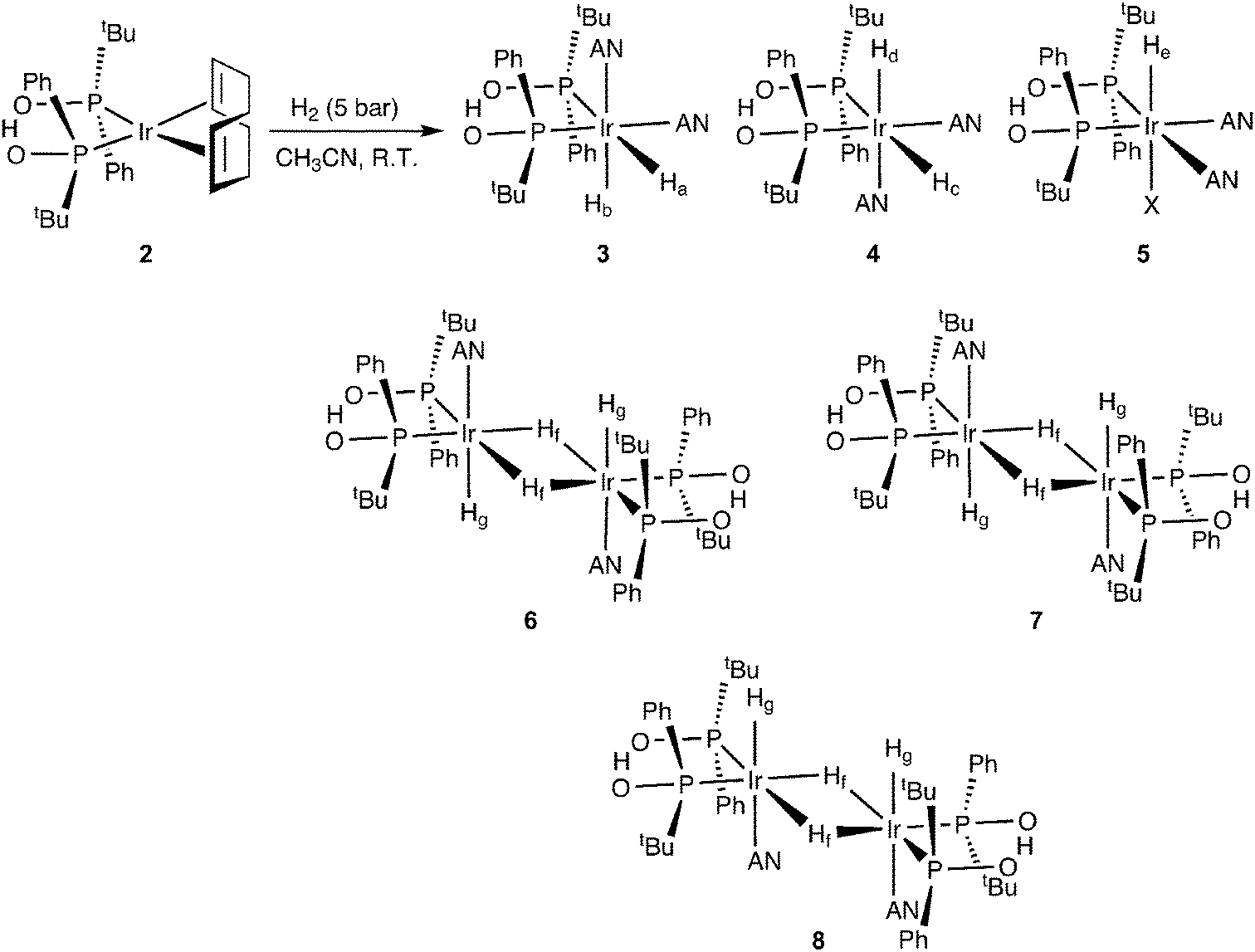 | ||
| Scheme 2 Formation of iridium(III) hydride species under substrate-free hydrogenation conditions. | ||
Under H2, the Ir(I) complex is converted, via H2 oxidative addition, to a mixture of one monohydride Ir((SPO)2H)H(solvent)2X (5), two diastereomeric dihydrides Ir((SPO)2H)H2(solvent)2 (3, 4) and three bridging dihydride dimers (6–8) thereof after loss of a solvent molecule (Scheme 2). The reaction is very fast with an instantaneous change of colour from red-orange to light yellow. The NMR analyses in tetrahydrofuran (THF-d8) led to rapid decomposition and therefore we conducted the experiments in acetonitrile (CD3CN), which has a stabilizing effect on the hydrides.16
The oxidative addition of H2 to Ir(I) complex 2 was studied by 1H and 31P NMR spectroscopy. The hydride region of the 1H NMR spectrum acquired 30 min after introducing H2 into the solution shows several iridium dihydride species (Fig. 1). The hydride resonances with highest intensity correspond to diastereomer 3 and appear as a double doublet of doublets at −10.38 ppm (Ha) due to cis and trans phosphorus couplings (19.1 and 143.3 Hz, respectively) and one hydrogen coupling (4.8 Hz), and as a double doublet of doublets at −21.40 ppm (Hb) attributable to the coupling with two cis phosphorus (14.5 and 20.7 Hz) and one hydrogen (4.8 Hz). Similarly, the hydride resonances for the minor diastereomer 4 appear as a couple of double doublet of doublets at −9.77 ppm (Hc, JP–H = 140.9, 19.8 Hz and JH–H = 4.8 Hz) and −21.52 ppm (Hd, JP–H = 22.7, 12.6 Hz and JH–H = 4.8 Hz), since both diastereomers exhibit the same coordination geometries.17 The ratio between the diastereomers is 2.23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
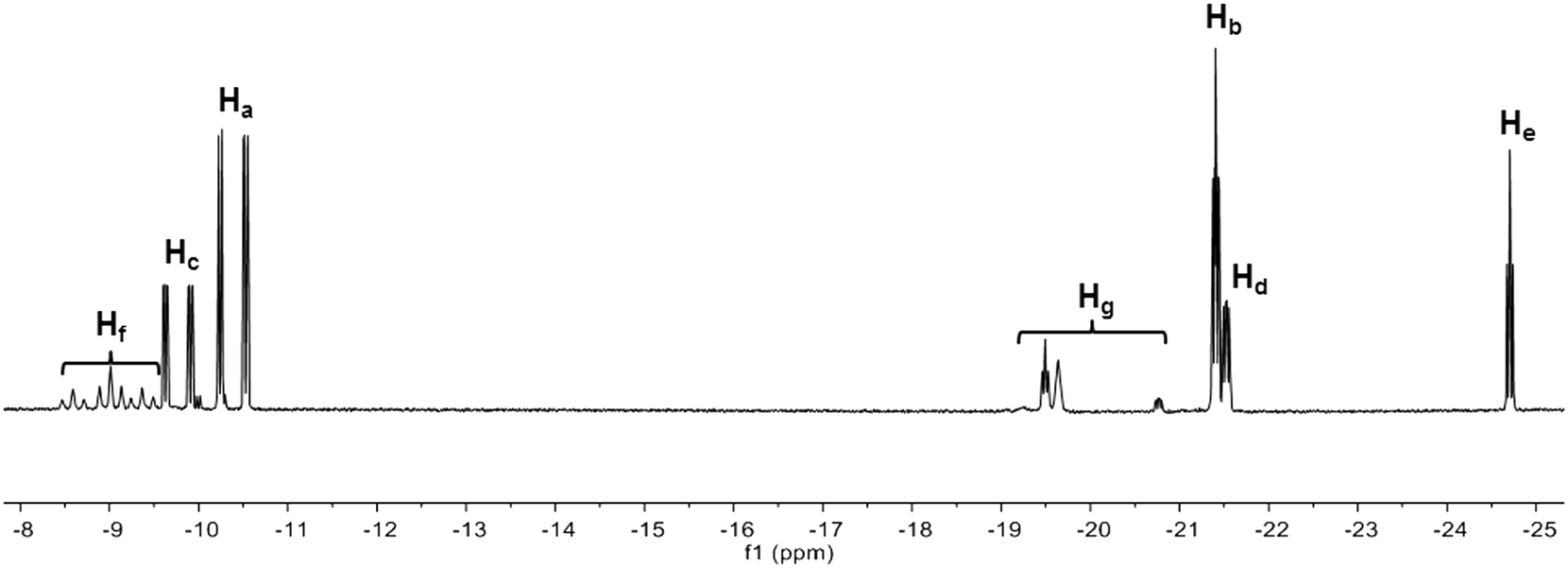 | ||
| Fig. 1 1H NMR spectrum (500 MHz) in the hydride region after reaction of 2 with H2 (5 bar) in CD3CN. | ||
The identity of the major diastereomer 3 was established by 2D NOESY NMR experiments (Fig. S3†). As the tert-butyl groups are non-equivalent, both diastereomers display 2 signals for these substituents in the 1H NMR spectrum (Fig. S2†), being the most intense peaks those that belong to 3 (0.71 and 0.94 ppm). Only the tert-butyl group at 0.71 ppm shows NOESY correlations with both hydrides Ha and Hb (Fig. S3†) in this species, whereas the other tert-butyl substituent does not give signal with the axial hydride Hb. This suggests that the tert-butyl groups of 3 are placed in trans position. Indeed, no interaction between the tert-butyl groups of 3 was detected in the NOESY experiment (Fig. S4†). In addition, the tert-butyl resonances for 4 appear at 0.80 and 0.82 ppm (Fig. S2†). The 31P{H} NMR spectrum (Fig. S5†) exhibits two doublet signals for each diastereomer (70.2 and 77.2 ppm with JP–P = 14.6 Hz for 3, and 62.5 and 84.2 ppm with JP–P = 14.9 Hz for 4). The assignation of signals was performed through a 1H–31P HMBC 2D experiment (Fig. S6 and S7†), which also enabled us to confirm the identity of the tert-butyl resonances corresponding to 4. Both iridium dihydride complexes 3 and 4 are homochiral (Scheme 2 shows the RR isomers), which is the most stable configuration, and there is no meso isomer.
The 1H NMR spectrum in the hydride region also shows a signal attributable to a species 5 with one hydride (He) and one non-identified anion (X) generated by decomposition, both located in axial position (Scheme 2). The hydride resonance appears at −24.70 ppm as a well-resolved triplet with JP–H of 13.6 Hz (Fig. 1) due to the coupling with two cis phosphorus nuclei (the signal would be located at −5–(−10) ppm in case of coupling with one trans phosphorus nucleus), whereas the phosphorus signals arise as two doublets (AB pattern) at 77.6 (JP–P = 12.7 Hz) and 78.1 ppm (JP–P = 13.2 Hz) in the 31P{H} NMR spectrum (Fig. S5 and S6†). This AB system indicates that the molecule is homochiral (RR/SS), since a meso compound would give a singlet signal for the two phosphorus in the 31P{1H} NMR spectrum. The integration of hydride signals in the 1H NMR spectrum reveals that the iridium monohydride complex is the third most abundant species. Consequently, the peaks for the tert-butyl substituents are those observable at 0.64 and 0.97 ppm in the 1H NMR spectrum, which was corroborated by analysis of the 1H–31P HMBC 2D experiment in the tert-butyl zone (Fig. S7†).
On the other hand, we observed the formation of three dimers 6, 7 and 8 with bridging and terminal hydrides as minor species (Scheme 2 and Fig. 1). The hydride region of the 1H NMR spectrum displays three triplets at δ −8.59 (JP–H = 60.5 Hz), −9.01 (JP–H = 61.5 Hz) and −9.37 ppm (JP–H = 62.3 Hz) corresponding to the bridging hydrides (Hf) of each dimer, and small signals around −19.5 and −20.8 ppm attributable to the terminal hydrides (Hg).18 Each dimeric species exhibits two different phosphorus environments, giving a pair of signals in the 31P{H} NMR spectrum (Fig. S5†). As was previously described for the mononuclear iridium complexes 3–5, all the dimers are homochiral and there are no meso isomers because all the dimeric species generate two peaks with cis phosphorus coupling in the 31P{H} NMR spectrum (Fig. S5†). The homochiral nature of all species as caused by the bulky tert-butyl groups reduces the number of possible diastereoisomers and allows the present interpretation.
Evaporation of solvent favors a displacement of the equilibrium between the hydride species toward the formation of the dimers. As a result, crystals of the dinuclear iridium(III) hydride complex 7 suitable for X-ray diffraction were obtained. The crystal structure of 7 is shown in Fig. 2. The Ir–Ir distance is 2.77 Å, in good agreement with the values reported in the literature for dinuclear Ir(III)/Ir(III) complexes containing an (IrH)2(μ-H2) unit.16,18,19 The Ir2 core possesses a 36-electron configuration with no M–M bond. The two acetonitrile molecules are orientated mutually trans. However, the two terminal hydrides were not located by Fourier differences, but one vacant site in the coordination sphere of each iridium atom trans to the acetonitrile molecule were assumed to be their positions. Fourier differences revealed a Ir1–Hy2 distance of 1.75 Å and an Ir1–Ir1i–Hy2 angle of 42.9°, the latter very close to the 45° required for the bridging hydrides being equidistant to both iridium atoms. Interestingly, Fourier differences also showed that the intramolecular hydrogen bond O–H–O contained in the monoanionic bidentate ligand is essentially linear (O⋯O is 2.398 Å) as was also found by DFT calculations for related Rh complexes,11e and as was already proposed by Palenik et al. on the basis of the short O⋯O distance of 2.5 Å in Pd–SPO complexes,20 although routinely the structures are drawn showing the O–H–O atoms as part of a regular 6-membered ring.11a–d
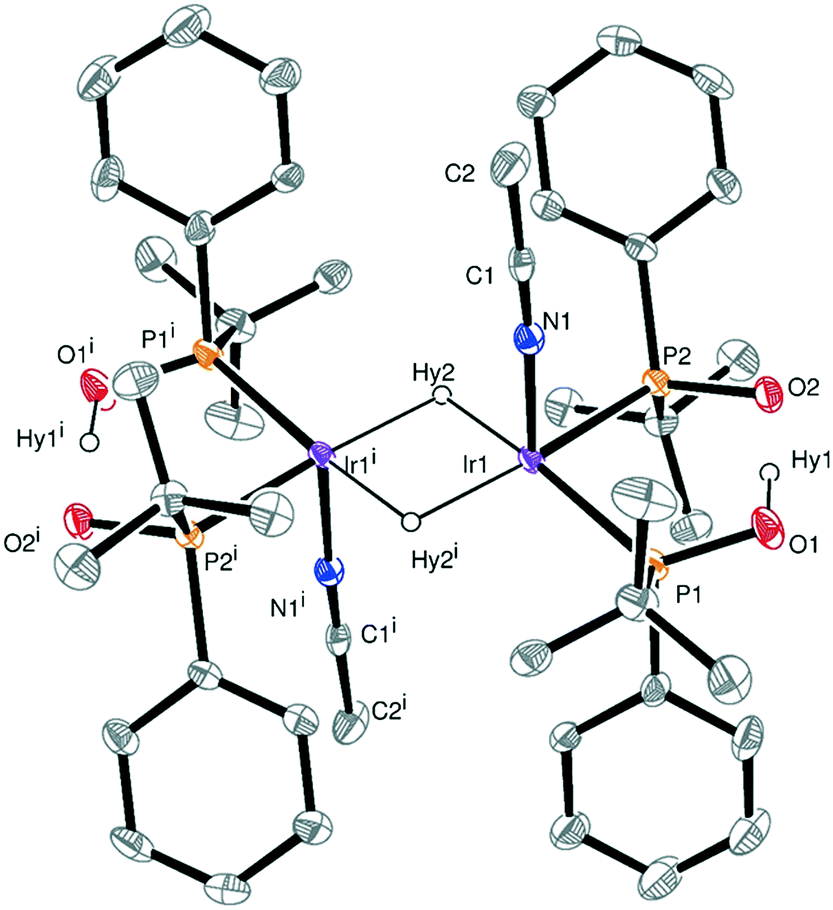 | ||
| Fig. 2 X-ray structure of 7 showing thermal ellipsoids set at 40% probability level. Selected bond lengths (Å): Ir1–P1, 2.2930(13); Ir1–P2, 2.2794(12); Ir1–N1, 2.120(4); Ir1–Hy2, 1.75(6); P1–O1, 1.555(4); P2–O2, 1.558(4); O1–Hy1, 0.97(6); O2–Hy1, 1.43(6). Selected angles (deg): N1–Ir1–P2, 91.61(11); N1–Ir1–Hy2, 89(2); P2–Ir1–P1, 89.01(5); P2–Ir1–Hy2, 90.3(19). | ||
As concerns the symmetry, the crystal structure of 7 contains an inversion center but has neither a plane of symmetry (due to the chiral ligands) nor a 2-fold axis due to the acetonitrile molecules and hydrides in trans axial positions. The tert-butyl groups are placed in trans position and the pairs of SPOs exhibit an R configuration at the phosphorus atoms in one monoanionic bidentate ligand and S in the other one. Thus, one side of the dimer is RR and the other side is SS, giving two doublets at approximately 72 (JP–P = ∼22 Hz) and ∼82 ppm (JP–P = ∼22 Hz) in the 31P{1H} NMR spectrum (Fig. S5†).
Catalytic hydrogenation of substituted aldehydes
This encouraging demonstration of hydrogen activation prompted us to evaluate the ability of the Ir–SPO system as hydrogenation catalyst. The hydrogenation of a range of substituted aldehydes was investigated using catalytic quantities of 2, which was found to be the precursor for a highly active and almost exclusively selective catalyst. Firstly, we decided to evaluate the activity of the catalytic system in a screening set of experiments for the hydrogenation reaction of cinnamaldehyde (Table 1). At R.T. and 5 bar H2 pressure, complete selectivity toward the unsaturated alcohol was observed in 1 h, albeit with low conversion (entry 1, 25%). In 2.5 h, 97% conversion and 99% selectivity to the expected allylic alcohol were obtained (entry 2). Increasing the time to 4.5 h, quantitative conversion of the substrate with >99% selectivity was achieved (entry 4). With the pressure maintained at 5 bar but increasing the temperature to 60 °C, a decrease in the conversion was observed (entry 11, 45%), which points to some decomposition and/or deactivation process of the catalyst. Furthermore, a loss of selectivity to the α,β-unsaturated alcohol was produced. We observed hydrogenation of the CC bond and both 3-phenylpropanal (6%) and 3-phenylpropanol (4%) were generated in addition to the expected product (cinnamyl alcohol, 90%). The solvent was found to be relatively important in that THF consistently provided good results (entry 2–5), while other solvents gave much lower conversions (entry 6, toluene; entry 7, CH2Cl2). Moreover, the use of methanol led to a decrease in the selectivity, since the acetal derivative was generated as by-product (entry 8).
| Entry | Solvent | T (K) | Time (h) | P (bar) | Conversionb (%) | Selectivityc (%) |
|---|---|---|---|---|---|---|
| a Reagents and conditions: 2 (0.00125 mmol), cinnamaldehyde (3.75 mmol), solvent (0.75 mL). b Conversions determined by 1H NMR spectroscopy and refer to the selective conversion of cinnamaldehyde (average of two runs). c UA = unsaturated alcohol, A = saturated alcohol, Al = saturated aldehyde. d The formation of the acetal product was observed. | ||||||
| 1 | THF | 295 | 1 | 5 | 25 | >99 |
| 2 | THF | 295 | 2.5 | 5 | 97 |
99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 (UA:A) :1 (UA:A)
|
| 3 | THF | 295 | 3 | 5 | 99 |
99:1 (UA:Al + A)
|
| 4 | THF | 295 | 4.5 | 5 | >99 | >99 |
| 5 | THF | 295 | 5 | 5 | >99 | 99:1 (UA:A) |
| 6 | Toluene | 295 | 4.5 | 5 | 2 | >99 |
| 7 | CH2Cl2 | 295 | 4.5 | 5 | 4 | >99 |
| 8 | CH3OH | 295 | 4.5 | 5 | 50 | 56d |
| 9 | THF | 295 | 18 | 5 | >99 | 96:4 (UA:A) |
| 10 | THF | 295 | 18 | 10 | >99 | 98:2 (UA:A) |
| 11 | THF | 333 | 4.5 | 5 | 45 | 90:6:4 (UA:Al:A) |
It is worth noting that the use of higher pressures or longer reaction times involved only a slight reduction in the chemoselectivity (entries 5, 9 and 10), which highlights the preference of the catalytic system toward the aldehyde functionality. With these optimized reaction parameters based on 2.5 h as reaction time, R.T., 5 bar of hydrogen pressure and THF as solvent, a TON of 2910 and a TOF of 1164 h−1 (entry 2) were achieved.
We studied the rate dependence on the reaction time (Fig. 3). The profile clearly shows an incubation time of ca. 1 h, during which 2 generates the catalytically active hydrides species. Since the hydrides are formed in acetonitrile on the timescale of the NMR sample preparation, we cannot say what this incubation time involves. Nanoparticle formation can be excluded on the basis of rate – IrNP being >50 times slower catalysts for entry 1 – and selectivity, vide infra.14 Indeed, the formation of nanoparticles requires more than 12 h under 5 bar of H2 pressure, whereas no nanoparticles generation was observed under these conditions in the NMR tube employed for the characterization of hydrides.14 From the profile we deduced a maximum TOF of 2040 h−1 at 1.5–2 h of reaction for the hydrogenation of cinnamaldehyde. To the best of our knowledge, the catalytic system described herein performs as one of the best catalysts in terms of rate and selectivity compared to iridium-based systems reported to date.5,7 The oxidative addition of H2 observed is the same as that described for diphosphine complexes17 and thus there is no indication that in this instance we are dealing with a heterolytic cleavage.21 Since the present Ir–SPO catalyst shows poor activity for alkenes compared to iridium catalysts containing neutral ligands (monophosphines, bisphosphines, Phox ligands), mechanistically the SPO function might be involved in the hydrogenation, but firm evidence is lacking.
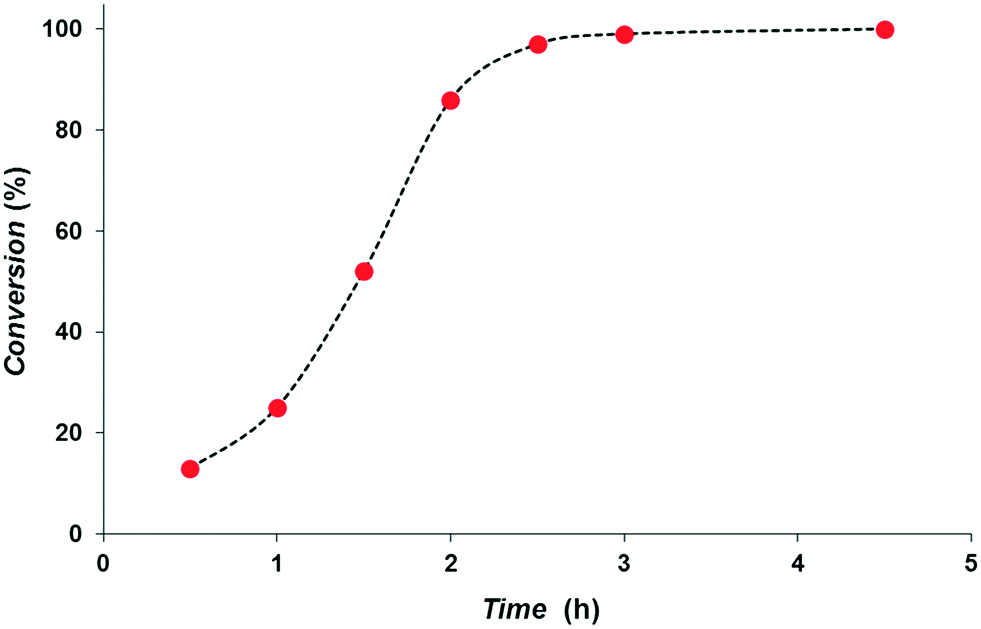 | ||
| Fig. 3 Profile of the evolution of the reaction with the time for the hydrogenation of cinnamaldehyde. Reagents and conditions: 2 (0.00125 mmol), cinnamaldehyde (3.75 mmol), THF (0.75 mL), 5 bar, 295 K. | ||
With optimized conditions in hand, we were keen to study the substrate scope and functional groups tolerated by 2 (Table 2). The catalyst showed a very high activity and selectivity in the hydrogenation of aldehydes over other functional groups. In terms of activity, the reaction appears to be very general and, in nearly all cases, very high conversions were obtained. Nevertheless, we observed differences in the TOF depending on the reactant, since some substrates required longer reaction times to complete the catalytic process (Table 2 and section 4†).
| Entry | Substrate | Product | t (h) | Conv.b (%) | Selectivityc (%) | TOF (h−1) |
|---|---|---|---|---|---|---|
| a Reagents and conditions: 2 (0.00125 mmol), substrate (3.75 mmol), THF (0.75 mL), 295 K, 5 bar of H2. b Conversions and product identities were determined by 1H NMR spectroscopy (average of two runs). c UA = unsaturated alcohol, A = saturated alcohol, Al = saturated aldehyde. d Maximum TOF. | ||||||
| 1 |

|

|
2.5 | 97 | 99:1 (UA:A) |
2040d |
| 2 |
|
|
5 | 38 | 99:1 (UA:A) |
228 |
| 3 |
|

|
5 | 94 | 97:3 (UA:A) |
564 |
| 4 |
|
|
5 | >99 | >99 | 600 |
| 5 |
|
|
5 | >99 | >99 | 600 |
| 6 |
|
|
20 | >99 | >99 | 150 |
| 7 |
|
|
5 | 96 | >99 | 720 |
| 8 |
|
|
5 | 98 | >99 | 588 |
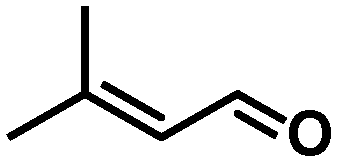
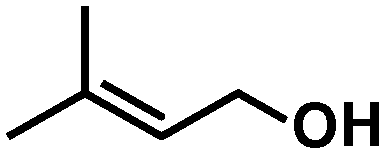
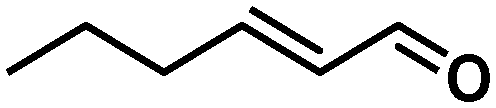
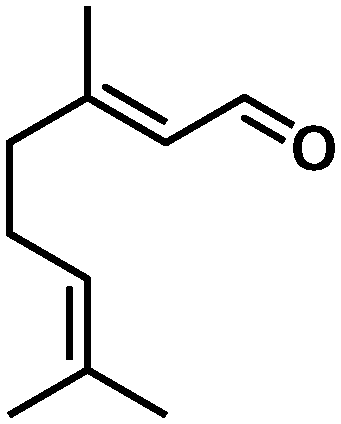
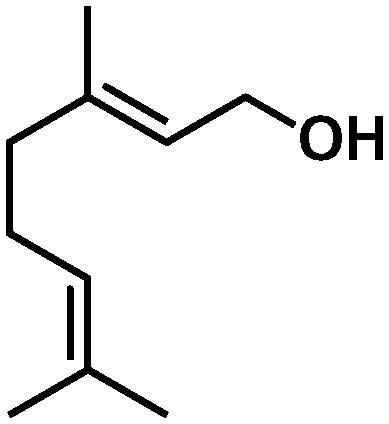
In all cases, high selectivities were observed for several α,β-unsaturated aldehydes (entries 1–4), including some that are of particular interest in the production of perfumes and fragrances.22 The complex was very selective to the carbonyl functionality in cinnamaldehyde and prenal (entries 1–2). However, a slight reduction in the selectivity was produced in the hydrogenation of trans-2-hexen-1-al in comparison with the previous substrates (entry 3). The steric impediment in the former probably avoids a higher reduction of the CC bond. Of particular importance is the selective hydrogenation of citral, which proceeded with complete chemoselectivity (entry 4). Interestingly, in contrast to other systems based on Ru,23 no reaction was observed in the reduction of 2-octynal. Indeed, this substrate poisoned the catalyst, as we reported in a preliminary communication.14
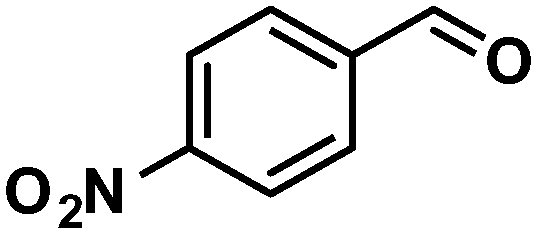
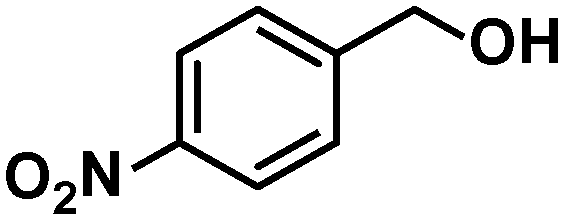
In addition to this selectivity to CO over alkenes, the catalyst is highly tolerant to several other functional groups. For example, the hydrogenation of p-nitrobenzaldehyde yielded the corresponding nitrobenzyl alcohol with perfect retention of the nitro group (entry 5). This is the second indication that nanoparticles are not responsible for the catalytic activity, because IrNPs gave formation of aminoaldehyde and aminoalcohol when used as the catalyst.14 In addition, we found poisoning of the catalytic system for the reactions with p-cyanobenzaldehyde and 2-octynal, while IrNPs on the contrary showed high conversions and chemoselectivities in the hydrogenation of these substrates.14 Esters were also tolerated excellently and the aldehyde group was selectively reduced to alcohol (entry 6). Finally, complete chemoselectivity was observed in compounds containing reducible heteroaromatic substituents (entries 7–8), such as furfural (compound derived from biomass) and 2-thiophenecarboxaldehyde.
Conclusions
In summary, we have successfully used an Ir–SPO complex to activate dihydrogen under mild conditions. The treatment of the iridium(I) complex with H2 led, via H2 oxidative addition, to the formation of a mixture of one monohydride Ir((SPO)2H)H(solvent)2X, two diastereomeric dihydrides Ir((SPO)2H)H2(solvent)2, and three bridging dihydride dimers as minor species. Such a process undergoes a high degree of diastereoselectivity, since a thorough characterization of the different iridium(III) hydride complexes by 1D and 2D NMR spectroscopy showed that all monomeric species are homochiral. In addition, the crystal structure of a dinuclear iridium(III) hydride complex was determined by X-ray diffraction and discussed. This dimer shows the preferred homochiral coordination for each half of the molecule, with opposite chirality resulting in an inversion center for the dimer.This catalytic system was highly efficient for the chemoselective hydrogenation of substituted aldehydes, displaying exceptionally high activities and selectivities for a wide variety of substrates. For instance, the hydrogenation of cinnamaldehyde proceeded with a TOF of 2040 h−1 and a selectivity of 99%. The catalyst works without the help of an external base or additive presumably through a ligand–metal cooperative mechanism in which the SPO ligand might play a crucial double role, as modifying ligand, and as functional ligand acting as heterolytic activator for dihydrogen. Finally, it is worth noting that with the study reported herein, SPO ligands have shown their merit in homogeneous hydrogenation catalysis, which may inspire the design of new homogeneous SPO-based catalysts that incorporate earth-abundant metals and exhibit similar catalytic properties.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank UPS-Toulouse and INSA for financial support. Dr. Israel Cano acknowledges financial support from the European Community through a Marie Skłodowska-Curie Individual Fellowships (IF-EF; Programme/Call: H2020-MSCA-IF-2015; Proposal No: 704710–Sdchirnanocat). Prof. P. W. N. M. van Leeuwen thanks the Université Fédérale Toulouse Midi-Pyrénées for an IDEX Chaire d'Attractivité.Notes and references
- (a) P. Gallezot and D. Richard, Catal. Rev.: Sci. Eng., 1998, 40, 81–126 CrossRef CAS; (b) P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- H. Berke, ChemPhysChem, 2010, 11, 1837–1849 CAS.
- R. H. Crabtree, in The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, Hoboken, NJ, 5th edn, 2009 Search PubMed.
- (a) E. T. Papish, M. P. Magee and J. R. Norton, in Recent Advances in Hydride Chemistry, ed. M. Peruzzini and R. Poli, Elsevier, Amsterdam, 2001, ch. 2 Search PubMed; (b) G. J. Kubas, in Metal Dihydrogen and σ-Bond Complexes, Kluwer Academic/Plenum Publishers, New York, 2001 Search PubMed; (c) Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For chemoselective hydrogenation of substituted aldehydes catalyzed by iridium-based complexes, see: (a) W. Strohmeier and H. Steigerwald, J. Organomet. Chem., 1977, 129, 43–46 CrossRef; (b) E. Farnetti, M. Pesce, J. Kaspar, R. Spogliarich and M. Graziani, J. Chem. Soc., Chem. Commun., 1986, 746–747 RSC; (c) C. S. Chin, B. Lee and S. C. Park, J. Organomet. Chem., 1990, 393, 131–135 CrossRef CAS; (d) C. S. Chin and B. Lee, J. Chem. Soc., Dalton Trans., 1991, 1323–1327 RSC; (e) L. Dahlenburg, K. Herbst and A. Zahl, J. Organomet. Chem., 2000, 616, 19–28 CrossRef CAS; (f) R.-X. Li, X.-J. Li, N.-B. Wong, K.-C. Tin, Z.-Y. Zhou and T. C. W. Mak, J. Mol. Catal. A: Chem., 2002, 178, 181–190 CrossRef CAS; (g) F. López-Linares, G. Agrifoglio, A. Labrador and A. Karam, J. Mol. Catal. A: Chem., 2004, 207, 117–122 CrossRef; (h) X. Wu, C. Corcoran, S. Yang and J. Xiao, ChemSusChem, 2008, 1, 71–74 CrossRef CAS PubMed; (i) M. G. Manas, J. Graeupner, L. J. Allen, G. E. Dobereiner, K. C. Rippy, N. Hazari and R. H. Crabtree, Organometallics, 2013, 32, 4501–4506 CrossRef CAS.
- For chemoselective transfer hydrogenation of substituted aldehydes catalyzed by iridium-based complexes, see: X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718–6722 CrossRef CAS PubMed.
- (a) B. F. Machado, H. T. Gomes, P. Serp, P. Kalck and J. L. Faria, ChemCatChem, 2010, 2, 190–197 CrossRef CAS; (b) P. Chen, J.-Q. Lu, G.-Q. Xie, G.-S. Hu, L. Zhu, L.-F. Luo, W.-X. Huang and M.-F. Luo, Appl. Catal., A, 2012, 433–434, 236–242 CrossRef CAS; (c) H. Rojas, J. L. Martinez, S. Mancipe, G. Borda and P. Reyes, React. Kinet., Mech. Catal., 2012, 106, 445–455 CrossRef CAS; (d) H. Rojas, G. Diaz, J. J. Martinez, C. Castaneda, A. Gomez-Cortes and J. Arenas-Alatorre, J. Mol. Catal. A: Chem., 2012, 363–364, 122–128 CrossRef CAS; (e) M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Chem. Commun., 2013, 49, 7034–7036 RSC; (f) J. Zhao, J. Ni, J. Xu, J. Xu, J. Cen and X. Li, Catal. Commun., 2014, 54, 72–76 CrossRef CAS.
- (a) M. Consonni, D. Jokic, D. Y. Murzin and R. Touroude, J. Catal., 1999, 188, 165–175 CrossRef CAS; (b) P. Concepcion, A. Corma, J. Silvestre-Albero, V. Franco and J. Y. Chane-Ching, J. Am. Chem. Soc., 2004, 126, 5523 CrossRef CAS PubMed; (c) M. Boronat, F. Illas and A. Corma, J. Phys. Chem. A, 2009, 113, 3750–3757 CrossRef CAS PubMed; (d) M. Boronat, P. Concepción and A. Corma, J. Phys. Chem. C, 2009, 113, 16772–16784 CrossRef CAS; (e) D. A. Panayotov, S. P. Burrows, J. T. Yates and J. R. Morris, J. Phys. Chem. C, 2011, 115, 22400–22408 CrossRef CAS.
- (a) D. Martin, D. Moraleda, T. Achard, L. Giordano and G. Buono, Chem. – Eur. J., 2011, 17, 12729–12740 CrossRef CAS PubMed; (b) P. Sutra and A. Igau, Coord. Chem. Rev., 2016, 308, 97–116 CrossRef CAS.
- B. Hoge, S. Neufeind, S. Hettel, W. Wiebe and C. Thösen, J. Organomet. Chem., 2005, 690, 2382–2387 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen and C. F. Roobeek, Eur. Pat. Appl., EP 82576, 1983, Chem. Abstr., 1983, 99, 121813 Search PubMed; (b) P. W. N. M. van Leeuwen, C. F. Roobeek, R. L. Wife and J. H. G. Frijns, J. Chem. Soc., Chem. Commun., 1986, 31–33 RSC; (c) P. W. N. M. van Leeuwen, C. F. Roobeek, J. H. G. Frijns and A. G. Orpen, Organometallics, 1990, 9, 1211–1222 CrossRef CAS; (d) P. W. N. M. van Leeuwen and C. F. Roobeek, New J. Chem., 1990, 14, 487–493 CAS; (e) P. M. Castro, H. Gulyas, J. Benet-Buchholz, C. Bo, Z. Freixa and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2011, 1, 401–407 RSC.
- See the following for a variety of examples related to hydrogenation via ligand–metal cooperation performed by SPO-based catalysts: (a) X.-B. Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. F. Boogers and J. G. de Vries, Org. Lett., 2003, 5, 1503–1506 CrossRef CAS PubMed; (b) X.-b. Jiang, M. van den Berg, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Tetrahedron: Asymmetry, 2004, 15, 2223–2229 CrossRef CAS; (c) N. V. Dubrovina and A. Börner, Angew. Chem., Int. Ed., 2004, 43, 5883–5886 CrossRef CAS PubMed; (d) L. Ackermann, Synthesis, 2006, 1557–1571 CrossRef CAS; (e) H. Landert, F. Spindler, A. Wyss, H.-U. Blaser, B. Pugin, Y. Ribourduoille, B. Gschwend, B. Ramalingam and A. Pfaltz, Angew. Chem., Int. Ed., 2010, 49, 6873–6876 CrossRef CAS PubMed; (f) T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771–803 CrossRef CAS; (g) K. Dong, Z. Wang and K. Ding, J. Am. Chem. Soc., 2012, 134, 12474–12477 CrossRef CAS PubMed.
- (a) E. Rafter, T. Gutmann, F. Low, G. Buntkowsky, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 595–599 RSC; (b) I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2014, 136, 2520–2528 CrossRef CAS PubMed; (c) I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS PubMed; (d) I. Cano, M. J.-L. Tschan, L. M. Martínez-Prieto, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2016, 6, 3758–3766 RSC; (e) N. Almora-Barrios, I. Cano, P. W. N. M. van Leeuwen and N. Lopez, ACS Catal., 2017, 7, 3949–3954 CrossRef CAS.
- I. Cano, L. M. Martínez-Prieto, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 1444–1450 CrossRef CAS PubMed.
- (a) D. M. Roundhill, R. P. Sperline and W. B. Beaulieu, Coord. Chem. Rev., 1978, 26, 263–279 CrossRef CAS; (b) J. A. S. Duncan, D. Hedden, D. M. Roundhill, T. A. Stephenson and M. D. Walkinshaw, Angew. Chem., Int. Ed. Engl., 1982, 21, 452–453 CrossRef; (c) J. A. S. Duncan, T. A. Stephenson, W. B. Beaulieu and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1983, 1755–1761 RSC; (d) J. A. S. Duncan, T. A. Stephenson, M. D. Walkinshaw, D. Heden and D. M. Roundhill, J. Chem. Soc., Dalton Trans., 1984, 801–807 RSC; (e) B. Patel, S. J. A. Pope and G. Reid, Polyhedron, 1998, 17, 2345–2351 CrossRef CAS; (f) C. Waloch, J. Wieland, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2007, 46, 3037–3039 CrossRef CAS PubMed; (g) B. Breit, in Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen, Wiley-VCH Verlag, GmbH, Weinheim, Germany, 2008, pp. 29–55 Search PubMed; (h) J. Meeuwissen, R. J. Detz, A. J. Sandee, B. de Bruin and J. N. H. Reek, Dalton Trans., 2010, 39, 1929–1931 RSC.
- S. Gruber, M. Neuburger and A. Pfaltz, Organometallics, 2013, 32, 4702–4711 CrossRef CAS.
- (a) A. Ç. Atesin, S. B. Duckett, C. Flaschenriem, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2007, 46, 1196–1204 CrossRef CAS PubMed; (b) E. P. K. Olsen, T. Singh, P. Harris, P. G. Andersson and R. Madsen, J. Am. Chem. Soc., 2015, 137, 834–842 CrossRef CAS PubMed.
- F. M. Chadwick, N. Olliff and A. S. Weller, J. Organomet. Chem., 2016, 812, 268–271 CrossRef CAS.
- (a) G. Robertson and P. Tucker, Aust. J. Chem., 1984, 37, 257–263 CrossRef CAS; (b) C. Bianchini, E. Farnetti, M. Graziani, J. Kaspar and F. Vizza, J. Am. Chem. Soc., 1993, 115, 1753–1759 CrossRef CAS; (c) R. C. Schnabel and D. M. Roddick, Organometallics, 1993, 12, 704–711 CrossRef CAS; (d) R. C. Schnabel, P. S. Carroll and D. M. Roddick, Organometallics, 1996, 15, 655–662 CrossRef CAS; (e) J. D. Feldman, J. C. Peters and T. D. Tilley, Organometallics, 2002, 21, 4050–4064 CrossRef CAS; (f) A. Choualeb, A. J. Lough and D. G. Gusev, Organometallics, 2007, 26, 5224–5229 CrossRef CAS; (g) R. Peloso, R. Pattacini, C. S. J. Cazin and P. Braunstein, Inorg. Chem., 2009, 48, 11415–11424 CrossRef CAS PubMed; (h) A. Franzke, F. Voss and A. Pfaltz, Tetrahedron, 2011, 67, 4358–4363 CrossRef CAS; (i) A. J. Pontiggia, A. B. Chaplin and A. S. Weller, J. Organomet. Chem., 2011, 696, 2870–2876 CrossRef CAS.
- D. V. Naik, G. J. Palenik, S. Jacobson and A. J. Carty, J. Am. Chem. Soc., 1974, 96, 2286–2288 CrossRef CAS.
- A heterolytic cleavage in which the H2 molecule is cleaved by a metal-ligand cooperative mechanism could occur. The monoanionic bidentate ligand acts as heterolytic activator for dihydrogen and takes a H+ from H2, leaving a H− bound to the metal center. In this scenario, a referee suggested that the monoanionic ligand would be converted in two neutral hydroxyphosphine ligands and a monohydride Ir(I) complex would be formed. Subsequently, fast migration of one H+ regenerates the monoanionic bidentate ligand and leads to the formation of an iridium(III) dihydride complex.
- N. Herron and D. L. Thorn, Adv. Mater., 1998, 10, 1173–1184 CrossRef CAS.
- See the following for a variety of examples related to the selective hydrogenation and transfer hydrogenation of alkynyl ketones catalyzed by Ru–based complexes: (a) T. Ohkuma, H. Ooka, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 10417–10418 CrossRef CAS; (b) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, experimental procedure and supporting data. CCDC 1574554. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy01953j |
| This journal is © The Royal Society of Chemistry 2018 |