
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origin of enhanced Brønsted acidity of NiF-modified synthetic mica–montmorillonite clay†
Chong
Liu‡
 a,
Evgeny A.
Pidko‡
*ab and
Emiel J. M.
Hensen
*a
a,
Evgeny A.
Pidko‡
*ab and
Emiel J. M.
Hensen
*a
aInorganic Materials Chemistry Group, Schuit Institute of Catalysis, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.a.pidko@tudelft.nl; e.j.m.hensen@tue.nl
bITMO University, Lomonosova 9, St. Petersburg, 191002, Russia
First published on 28th November 2017
Abstract
The Brønsted acidity of synthetic mica–montmorillonite (SMM) clay was studied by periodic DFT calculations. Different structural models were compared to determine the Brønsted acidity of protons of the SMM clay based on (i) isomorphous substitution of Si4+ by Al3+ in the tetrahedral silicate layer and additional NiF-doping (ii) in the platelets and (iii) at the edge terminations of the clay platelets. The acid strength was judged from the computed adsorption energies of ammonia and pyridine. The SMM acidity is mainly determined by the composition of the clay platelets. The strongest acidity is found in structures in which octahedral [AlO]+ is replaced by [NiF]+ adjacent to tetrahedral [Si–(OH)–Al] moieties in the tetrahedral layer. For the Brønsted acid sites in the interlayer of SMM, modification with either Ni2+ or F− in the octahedral layers has only a minor influence on the acidity. Our data indicate that Brønsted acid sites, properly modified in the second coordination shell by electron-withdrawing F, in the interlayer and at defect sites at the edges of clay platelets (intralayer sites) can contribute to the enhanced acidity in NiF-modified SMM. Although the predicted acidity of SMM by ammonia adsorption is higher than that of faujasite zeolite, the reactivity judged from propene protonation demonstrates that zeolites are more reactive than clays. This difference seems to be the result of the curved nature of the micropores of zeolites, which stabilizes the transition states for an acid-catalyzed reaction more than flat surfaces of clays do.
Introduction
Solid acids are widely used as heterogeneous catalysts in industrial chemical processes.1,2 Porous aluminosilicates like zeolites, clays, and amorphous silica–alumina phases are applied as acid catalysts in processes such as fluid catalytic cracking, hydrocracking, and hydroisomerization of petroleum feedstock. The acidity of these aluminosilicate catalysts is due to the substitution of Al3+ for Si4+ in the silica network and the compensation of the resulting negative lattice charge by protons. It has been experimentally shown that the strength of Brønsted acid sites (BAS) is similar in highly crystalline zeolites, clays and amorphous silica–aluminas, while activity differences in hydrocarbon conversion can be mainly correlated with different acid site densities.3 Acid-leached natural clays were the first solid acid catalysts used in commercial catalytic cracking units for the upgrading of crude petroleum to gasoline.4 These clays were later replaced by amorphous silica–alumina and zeolite catalysts.Zeolites and amorphous silica–alumina have three-dimensional silica networks in which diluted tetrahedral Al sites give rise to BAS. Clays have a two-dimensional layered structure, which can be classified as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 depending on the stacked structure of tetrahedral silicate sheets and octahedral hydroxide sheets.5Fig. 1 shows the layered structure of a 2:1 clay, which contains two tetrahedral (T) sheets sandwiching the central octahedral (O) sheet. There are two ways to generate negative charge in the clay lattice, one is by substitution of Al3+ for Si4+ in the T layer and the other involves a variation of the composition of the O layer.6 The compensation of the resulting negative charges by protons will generate Brønsted acid sites within the interlayer space. Doping of the parent aluminosilicate lattice with either univalent anionic species or bivalent metal ions provides a practical means for adjusting the acidity of the BAS in clay materials.
:1 or 2:1 depending on the stacked structure of tetrahedral silicate sheets and octahedral hydroxide sheets.5Fig. 1 shows the layered structure of a 2:1 clay, which contains two tetrahedral (T) sheets sandwiching the central octahedral (O) sheet. There are two ways to generate negative charge in the clay lattice, one is by substitution of Al3+ for Si4+ in the T layer and the other involves a variation of the composition of the O layer.6 The compensation of the resulting negative charges by protons will generate Brønsted acid sites within the interlayer space. Doping of the parent aluminosilicate lattice with either univalent anionic species or bivalent metal ions provides a practical means for adjusting the acidity of the BAS in clay materials.
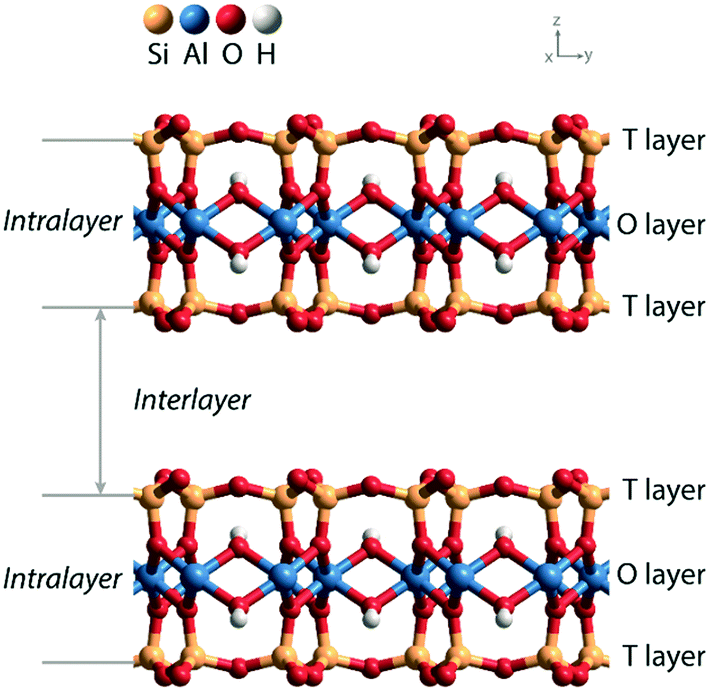 | ||
| Fig. 1 The layered structure of a 2:1 clay (T–O–T layers) composed of one octahedral (O) sheet with Al in the coordination center between two surface tetrahedral (T) sheets with Si in the coordination center. The layers are typically negatively charged due to substitutions in the T and O layers. This framework charge is compensated by cations in the interlayer. | ||
Synthetic mica–montmorillonite (SMM) clays have been used as acid catalysts for hydrocarbon transformations such as oligomerization, hydrocracking, and hydroisomerization.7–11 In particular, metal-substituted SMM such as Ni–SMM displays high Brønsted acidity.9 The name SMM is derived from the understanding that it is a 2:1 layer aluminosilicate composed of interstratified expandable (montmorillonite-like) and non-expandable (mica-like) layers.12 It was later realized that these materials have the beidellite structure, in which the induced charge mainly originates from isomorphous substitution of Al3+ for Si4+ in the silica sheet.10 Experimental studies demonstrated that the Ni–SMM containing F− anions exhibits very high Brønsted acidity, which even exceeds that of the zeolites as evidenced by a very high activity in alkane hydroisomerization.13 However, the reason for the enhanced acidity of the NiF-modified SMM is not well understood yet. An alternative interpretation is that Lewis acid sites on exposed defect sites of the clay structures play a role in hydrocarbon conversion.14
In this work, we employed periodic DFT calculations to investigate the acidity and reactivity of the SMM clay. Two structural models were considered, one involving BAS in the intralayer of SMM generated by exposing the edge defects of the clay platelets and the other one involving zeolite-like BAS due to isomorphous substitution of Si4+ by Al3+ within the tetrahedral silicate layer of SMM. The impact of Ni2+ and F− substitutions in the clay interior on acidity was then explored in a systematic manner. The acid strength of the BAS was determined by computing the adsorption energies of ammonia and pyridine. Propene protonation was used as a model reaction for hydrocarbon activation. The acidic and catalytic properties of SMM were further compared with those of faujasites to unravel the activity differences between clay and zeolite materials.
Computational methods
Density functional theory (DFT) calculations with periodic boundary conditions were performed using the Vienna ab initio simulation package (VASP).15–17 The generalized gradient approximation Perdew–Burke–Ernzerhof (PBE) exchange and correlation functional was used.18 The electron–ion interactions were described with the projected augmented wave (PAW) method.19,20 The valence electrons were treated using a plane-wave basis set with a kinetic energy cutoff of 500 eV. Convergence was assumed when the forces on each atom were below 0.05 eV Å−1. A modest Gaussian smearing of 0.05 eV was applied to band occupations around the Fermi level, and the total energies were extrapolated to σ → 0. Van der Waals interactions were described by the DFT-D2 dispersion correction method of Grimme.21Two types of computational models (SMM-intra and SMM-inter) were constructed to simulate the BAS in the intra- and interlayer of SMM (Fig. 2) on the basis of the crystal structure reported by Viani et al.22 For the SMM-intra model, a 2 × 3 × 1 supercell was constructed. From this, a nanorod structure was built by exposing the (010) surface edge (Fig. 2a). The dangling bonds were saturated by hydrogen atoms. This model has a composition of Al8Si16O38(OH)12, in which a BAS with an octahedrally coordinated Al3+ is formed at the defect edge. The second type of clay model (SMM-inter, Fig. 2b) was constructed using a 2 × 1 × 1 supercell, in which a Si atom was substituted by an Al in the T layer of SMM. The SMM-inter model has a composition of Al9Si15O39(OH)9, with a Brønsted proton located in the interlayer of SMM. The experimental parameters of the unit cell (a = 5.17, b = 8.98, c = 15.00 Å, and α = β = γ = 90°) were used in the simulations and kept fixed during the optimizations, except that the cell parameter c was adjusted to 20 Å to minimize the interaction between the slabs. Full geometry optimizations with guest molecules were performed with fixed cell parameters. For the SMM-intra and SMM-inter models, Monkhorst–Pack grids of 2 × 1 × 1 and 2 × 2 × 1, respectively, were used to sample the Brillouin zone.23 The climbing image nudged elastic band (CI-NEB) method was employed to determine the minimum-energy reaction paths and the corresponding transition states.24 The nature of the transition states was confirmed by determining the vibrational frequencies using the finite difference method. Small displacements of 0.02 Å were used to determine the numerical Hessian matrix. Gibbs free energies were computed within the ideal gas approximation at a pressure of 1 atm and a temperature of 473 K using the results of frequency analysis. Only vibrational degrees of freedom were considered for the intermediates and transition states in the models.
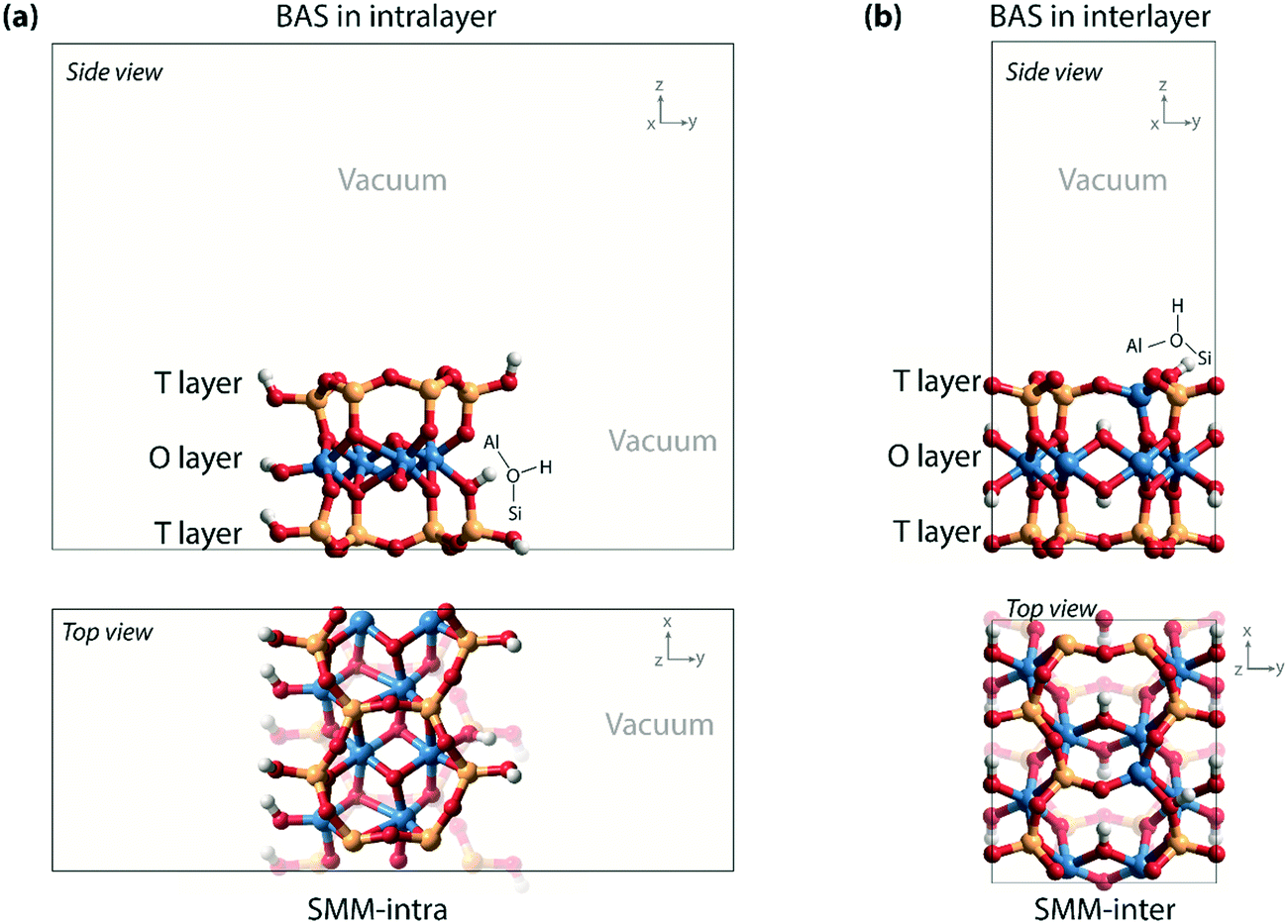 | ||
| Fig. 2 (a) Nanorod model with BAS in the intralayer of SMM (SMM-intra); (b) clay model with BAS in the interlayer of SMM (SMM-inter). | ||
To compare the acidity and reactivity of SMM clays with zeolites, four faujasite (FAU) models with varying chemical composition and acidity were also employed. The calculations were carried out at the same level as that for SMM clays, except that the Brillouin zone sampling was restricted to the Γ point. The effects of both the framework Al (AlF) density and the presence of extraframework Al (EFAl) species were considered. A rhombohedral unit cell (Si48O96) was used,25 and three defect-free models with a Si/AlF ratio of 2.4, 7, and 47 (FAU-2.4, FAU-7, and FAU-47, Fig. S1†) were constructed by introducing, respectively, 14, 6, and 1 AlF substitutions per unit cell. All the charge-compensating protons were introduced at the O1 positions, which were identified as the preferred proton-acceptor sites. For the three defect-free FAU models, the cell parameters were firstly optimized (FAU-2.4: a = b = c = 17.65 Å, α = β = γ = 60°; FAU-7: a = b = c = 17.44 Å, α = β = γ = 60°; FAU-47: a = b = c = 17.29 Å, α = β = γ = 60°). The fourth model containing EFAl species (FAU-EFAl, Fig. S1†) was made from a unit cell with Si/AlF = 7, which contains a trinuclear [Al3O4H3]4+ EFAl cluster, which was shown to be highly stable in previous works.26,27 This cationic EFAl complex is preferentially located inside faujasite's sodalite cage, close to a vicinal supercage BAS. The induced positive charge by EFAl is compensated by removing the protons to keep the model system neutral. Further details of the structural parameters of the FAU models can be found in ref. 27.
Results and discussion
As a starting point of this study, we evaluated the structural and acidic properties of the BAS present in the intralayer and interlayer space of the parent aluminosilicate SMM clay by periodic DFT calculations (Fig. 2). The acid strength of the BAS was judged from a comparison of computed adsorption energies (ΔEads) of ammonia and pyridine, which are commonly used as probe molecules for experimental acidity characterization on solid acids.28 The optimized structures of the adsorption complexes for the SMM-intra and SMM-inter Al-containing models are shown in Fig. 3. The adsorption of ammonia or pyridine results in the complete deprotonation of the BAS resulting in contact ion-pair complexes. For ammonia, the resulting NH4+ ion interacts with the framework O atoms of SMM via multiple H-bonding interactions. The structural parameters of the optimized adsorption complex reveal the presence of one particularly strong H-bonding contact between NH4+ and the basic lattice O atom corresponding to the original deprotonated BAS (dOH = 1.50 Å in SMM-intra and dOH = 1.58 Å in SMM-inter). In addition, there are two weaker H-bonds formed with dOH = 2.14 Å/2.31 Å for SMM-intra and dOH = 1.86 Å/1.88 Å for SMM-inter. In the adsorption complexes of pyridine, the transferred proton interacts with the original O in the deprotonated BAS at very short distances of 1.53 Å and 1.55 Å for the SMM-intra and SMM-inter models, respectively.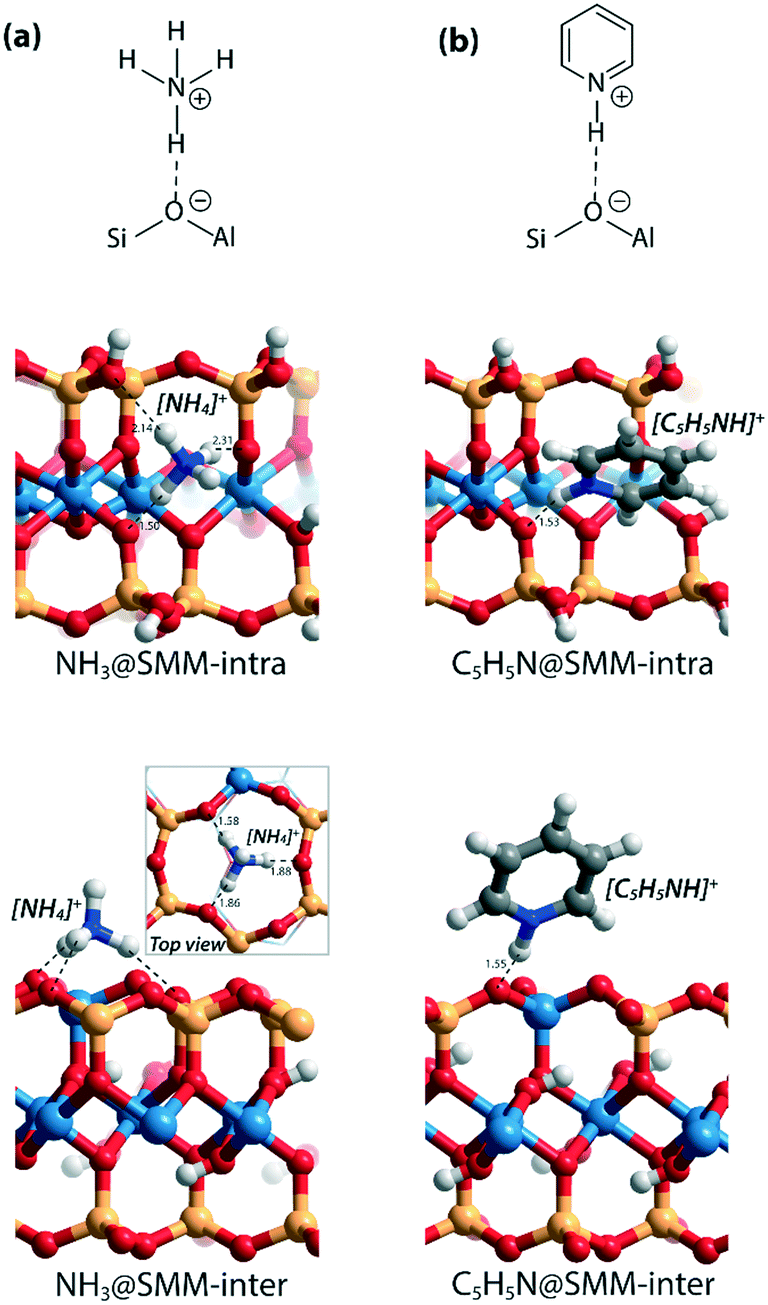 | ||
| Fig. 3 Optimized structures of the adsorption complexes of (a) ammonia (NH3) and (b) pyridine (C5H5N) in SMM-intra and SMM-inter models. Bond distances (d) are in Å. | ||
The DFT-computed adsorption energies (ΔEads) are listed in Table 1. The acid strength of structural defects in SMM-intra is substantially lower than the acid strength of the SMM-inter layers (Table 1, SMM-intra vs. SMM-inter, ΔEads[NH3] = −134 kJ mol−1vs. −168 kJ mol−1 and ΔEads[C5H5N] = −138 kJ mol−1vs. −167 kJ mol−1). The BAS in the SMM-intra model are due to tetrahedrally coordinated Si4+ adjacent to an octahedrally coordinated Al3+, while the BAS in the SMM-inter model feature a conventional bridging hydroxyl group as in zeolites. The difference in acidity of the two sites can be readily understood by using Pauling's valence rules.29 The bridging O atom of the BAS [Si–(OH)–Al] in the SMM-intra model has a more negative excess charge, resulting in a stronger OH bond and, therefore, a weaker Brønsted acidity than the BAS in the SMM-inter model.
| Model | Substitution | ΔEads(NH3) | ΔEads(C5H5N) | ΔE≠(C3H6) | |
|---|---|---|---|---|---|
| T layer | O layer | ||||
| SMM-intra | — | — | −134 | −138 | 68 |
| SMM-intra-1NiF | — | 1 × [NiF]+ for [AlO]+ | −155 | −146 | 62 |
| SMM-intra-2NiF | — | 2 × [NiF]+ for [AlO]+ | −167 | −157 | 55 |
| SMM-intra-3NiF | — | 3 × [NiF]+ for [AlO]+ | −181 | −161 | 48 |
| SMM-inter | 1 × Al3+ for Si4+ | — | −168 | −167 | 58 |
| SMM-inter-NiF | 1 × Al3+ for Si4+ | 1 × [NiF]+ for [AlO]+ | −179 | −177 | 52 |
| SMM-inter-AlF | 1 × Al3+ for Si4+, 1 × [AlF]2+ for [SiO]2+ | — | −197 | −185 | 49 |
| SMM-inter-NiF-AlF | 1 × Al3+ for Si4+, 1 × [AlF]2+ for [SiO]2+ | 1 × [NiF]+ for [AlO]+ | −209 | −196 | 44 |
| SMM-inter-8F | 1 × Al3+ for Si4+ | 8 × F− for [OH]− | −169 | −167 | 54 |
| SMM-inter-12Ni | 1 × Al3+ for Si4+ | 12 × Ni2+ for 8 × Al3+ | −171 | −175 | 62 |
| FAU-2.4 | Si/AlF = 2.4 | — | −104 | −146 | 83 |
| FAU-7 | Si/AlF = 7 | — | −125 | −163 | 60 |
| FAU-47 | Si/AlF = 47 | — | −142 | −199 | 51 |
| FAU-EFAl | Si/AlF = 7 | EFAl = [Al3O4H3]4+ | −182 | −274 | 22 |
We next evaluated the effect of Ni2+ and F− substitutions in the SMM-intra and SMM-inter models on the acidic properties. Experimental findings have shown that the acidity of aluminosilicate clays is substantially enhanced by replacement of [AlO]+ with [NiF]+.13 NiF-modified SMM-intra models were constructed by replacing [AlO]+ in the octahedral layer with [NiF]+ (Fig. 4). To study the influence of increasing [NiF]+ loading, three distinct models with 1, 2, and 3 lattice substitutions (SMM-intra-1NiF, SMM-intra-2NiF and SMM-intra-3NiF, respectively) were constructed. DFT calculations confirmed the strong acidity enhancement upon introduction of [NiF]+ units in the structure. Table 1 shows that replacement of one unit already results in an increase of the ammonia adsorption energy from −134 kJ mol−1 to −155 kJ mol−1 and the pyridine adsorption energy from −138 kJ mol−1 to −146 kJ mol−1. Introducing more [NiF]+ in the lattice results in a further increase of the acidity. The acidity of the BAS in the interlayer space in the SMM-inter model is also increased by introduction of [NiF]+ units. The substitution of a single [AlO]+ pair with a [NiF]+ unit in SMM-inter (Fig. 5, SMM-inter-NiF) results in increased adsorption energies of ammonia from −168 kJ mol−1 to −179 kJ mol−1 and pyridine from −167 kJ mol−1 to −177 kJ mol−1. This is consistent with the earlier noted increased OH shift upon CO adsorption for a similar SMM-inter model.13
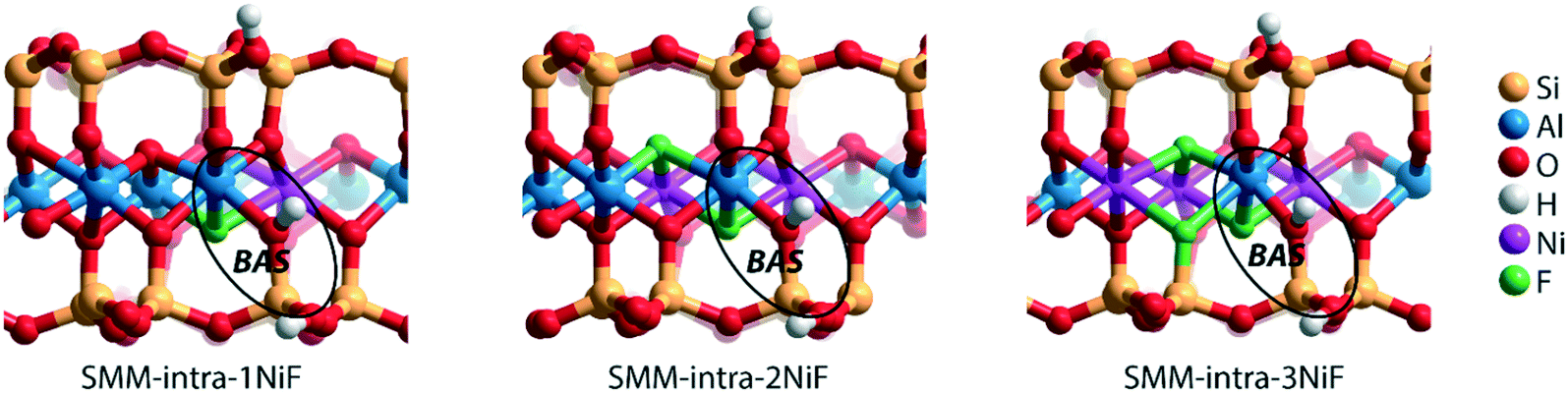 | ||
| Fig. 4 Clay models with BAS in the intralayer of SMM with substitutions of [NiF]+ for [AlO]+ in the octahedral layer. | ||
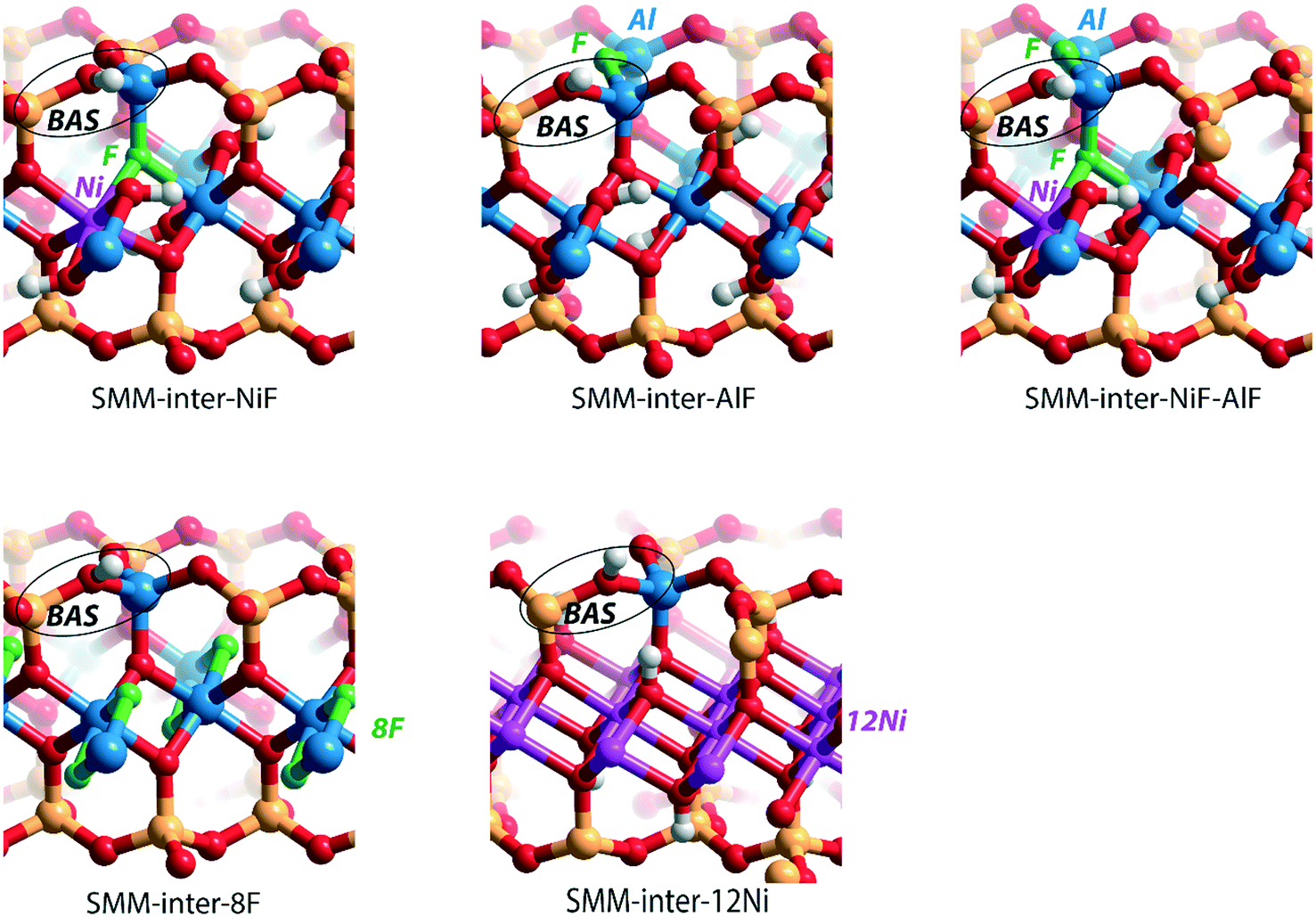 | ||
| Fig. 5 Clay models with BAS in the interlayer of SMM with different substitutions in the SMM-inter model: 1 × [NiF]+ for [AlO]+ in the octahedral layer (SMM-inter-NiF), 1 × [AlF]2+ for [SiO]2+ in the tetrahedral layer (SMM-inter-AlF), both 1 × [NiF]+ for [AlO]+ in the octahedral layer and 1 × [AlF]2+ for [SiO]2+ in the tetrahedral layer (SMM-inter-NiF-AlF), 8 × F− for [OH]− in the octahedral layer (SMM-inter-8F), and 12 × Ni2+ for 8 × Al3+ in the octahedral layer (SMM-inter-12Ni). | ||
To gain a better insight into the origin of the acidity enhancement, we constructed additional SMM-inter models featuring a single F− substitution in a Ni-free clay and [NiF]+ and F−-substituted models. The Ni-free model with a BAS featuring direct Al–F coordination was obtained by substituting [SiO]2+ in the tetrahedral layer with an [AlF]2+ moiety (Fig. 5, SMM-inter-AlF). The modification of this structure with a [NiF]+ unit in the octahedral layer is denoted as SMM-inter-NiF-AlF (Fig. 5). Both these structural modifications lead to strong acidity enhancement compared to the parent model, exceeding the acidity of the initial NiF-modified structures (Table 1). The acidity enhancement is related to the high electronegativity of F− ions in the first coordination sphere of the Al center, resulting in polarization and weakening of the vicinal OH group.
To separately study the effect of F− and Ni2+ substitutions in the octahedral layer, fully exchanged SMM-inter models with either all Al3+ substituted with Ni2+ or all [OH]− substituted with F− were compared. The SMM-inter-8F (Fig. 5) model shows very similar ΔEads(NH3) and ΔEads(C5H5N) (−169 kJ mol−1 and −167 kJ mol−1) to the Ni and F-free SMM-inter model (Table 1, −168 kJ mol−1 and −167 kJ mol−1). This indicates that F− substitution only slightly impacts clay acidity, unless it is placed in the direct coordination environment of the BAS. The Ni-rich model was obtained by replacing the Al3+ cations in the octahedral layer with Ni2+, based on the crystal structure of the trioctahedral sheet silicate.30 The Ni-only substituted SMM model (Fig. 5, SMM-inter-12Ni) did not lead to a significant acidity enhancement (Table 1). Similar to the case of full substitution, the partially exchanged SMM-inter clays involving only F− or Ni2+ modifications display similar acidities to the original model (Table S1†).
It has been experimentally found that only a small fraction of BAS in F-containing Ni–SMM clay displays enhanced acidity and also that synthesis of Ni–SMM without F leads to poor acidity.13 Accordingly, we infer that the enhanced acidity of NiF-containing SMM clays is due to the replacement of [AlO]+ by [NiF]+ in close proximity to the BAS. The introduction of highly electronegative F− ions in the first coordination sphere of the Al center decreases the electron density of the O atom of the bridging hydroxyl group by the electron-withdrawing effect. This leads to weakening of the OH bond resulting in an increased acidity of the BAS. The use of Ni2+ facilitates the isomorphous substitution of [AlO]+ by [NiF]+. The higher acidity computed when more [NiF]+ is introduced in the structure is consistent with this assertion because of the stronger total electronegativity of the framework F anions close to the acid site. We expect that additional substitutions in the model will further increase the acidity. It is worthwhile mentioning that restructuring at the edges of the clay platelets may take place,31 which should be included in future studies.
We also explored the activity of the model BAS in SMM clays using propene protonation as a model reaction. Propene protonation leads to primary or secondary propoxy species.32 Here, we only considered the formation of a sec-propoxy intermediate, which is the kinetically favored pathway (Fig. 6a). DFT-calculated activation barriers [ΔE≠(C3H6)] of propene protonation are in line with the acidity trends derived from the base adsorption energies (Table 1). The ΔE≠(C3H6) for the SMM-intra model is 68 kJ mol−1. This barrier decreases to 62 kJ mol−1, 55 kJ mol−1, and 48 kJ mol−1 for SMM-intra-1NiF, SMM-intra-2NiF, and SMM-intra-3NiF, respectively. For the BAS in the interlayer of SMM clays, the introduction of F− directly attached to Al3+ also strongly decreases the ΔE≠(C3H6) from 58 kJ mol−1 for SMM-inter to 52 kJ mol−1, 49 kJ mol−1, and 44 kJ mol−1 for SMM-inter-NiF, SMM-inter-AlF, and SMM-inter-NiF-AlF, respectively. No activity enhancement was observed for SMM-inter-8F and SMM-inter-12Ni. These data indicate that the acid strength of clay is the main factor affecting catalytic activity. Earlier, we concluded on the basis of the computed red shift of the bridging hydroxyl group OH for an interlayer SMM model that the BAS with enhanced acidity are located on the basal plane of the clay platelets.13 The present data show that also intralayer BAS, whose electronic properties are properly modified by F in the second coordination shell, can be strongly acidic. Given that these acid materials operate in the dehydrated state during hydrocarbon hydroconversion, we speculate that the interlayer sites at defect sites at the edges of the clay platelets are the more likely active sites.
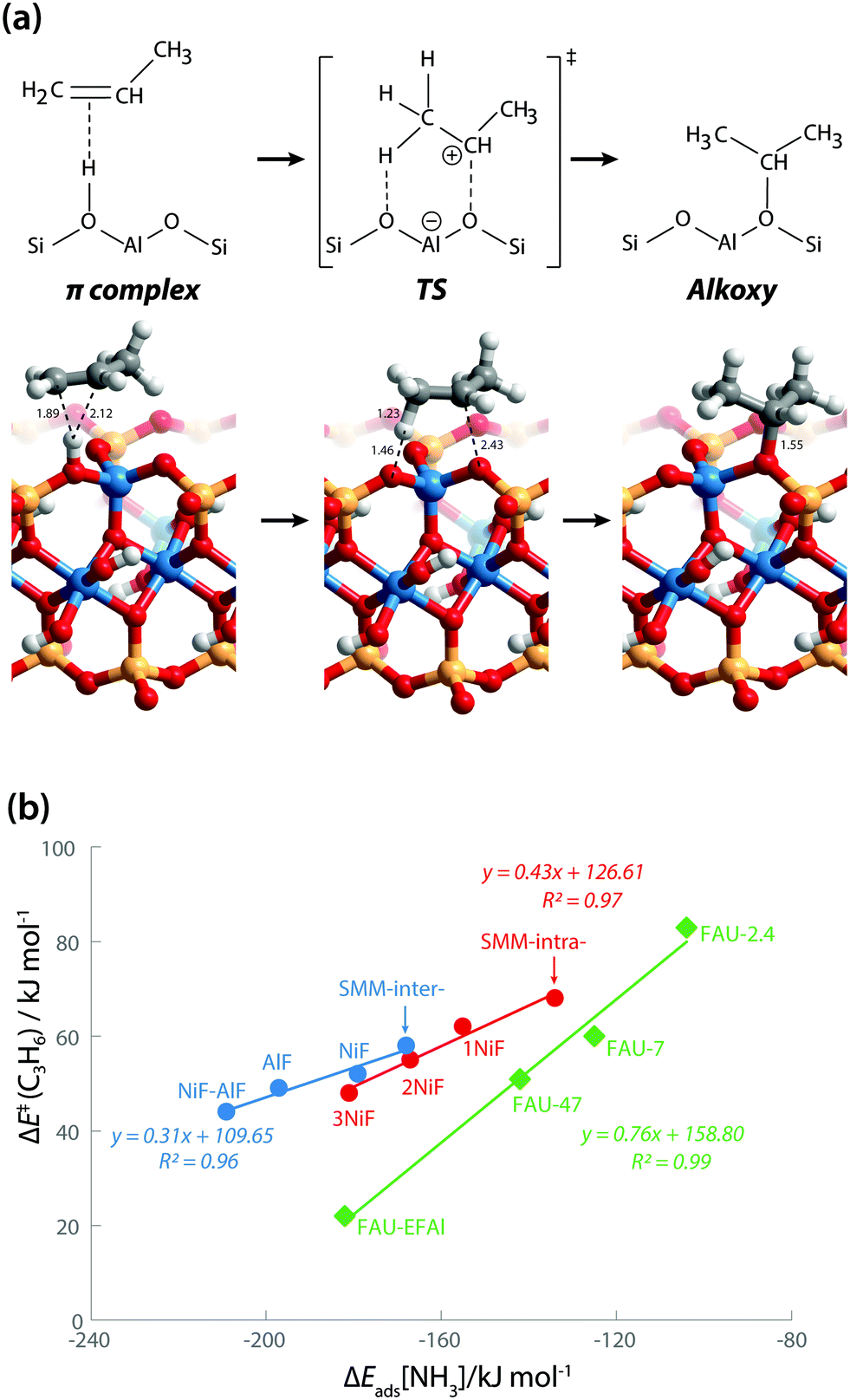 | ||
| Fig. 6 (a) Protonation of propene into sec-propoxy on the BAS of clay (SMM-inter). Bond distances are in Å. (b) Reactivity of clays in comparison with faujasite zeolites. | ||
We also compared the acid activity of clays and zeolites by including four faujasite zeolites with varying Si/AlF ratios and models containing EFAl species (Fig. S1†). The acidity was determined by comparing ΔEads(NH3) and ΔEads(C5H5N) (Table 1 and Fig. S2†). Expectedly, the intrinsic acidity of protons in faujasite increases with decreasing framework Al density (i.e., increasing Si/AlF ratio).27 The introduction of a multinuclear EFAl–oxide cluster in the sodalite cage enhances the acidity of the vicinal supercage BAS.26,27 Different from SMM for which the values of ΔEads(NH3) and ΔEads(C5H5N) are similar for a particular type of BAS, ΔEads(C5H5N) is much higher than ΔEads(NH3) for the FAU models. This may be due to several factors, including the differences in the interaction of the protonated base with the conjugate anionic zeolite framework as well as the dispersion interactions. For FAU and SMM, pyridine is adsorbed in the form of contact ion-pair complexes with one hydrogen bond between [C5H5NH]+ and the deprotonated BAS, while in the adsorption complexes of ammonia, the resulting [NH4]+ cations are bidentate-coordinated in FAU models but tridentate-coordinated in SMM models (Fig. 3 and S2†). The occurrence of additional hydrogen bonding in SMM models provides additional structural stabilization and brings ΔEads(NH3) much closer to ΔEads(C5H5N). In addition, the difference between the dispersion contributions of ΔEads(C5H5N) and ΔEads(NH3) is larger for FAU than for SMM (Table S2†), which also contributes to the observed difference between the adsorption energies of the two bases.
The larger molecular size of pyridine results in stronger interaction with the clay and zeolite surfaces due to the dispersion forces (Table S2†). We postulate that ammonia is a more sensitive probe to intrinsic acid strength than pyridine. Therefore, we used ΔEads(NH3) as the acidity descriptor to correlate acidity and reactivity. As discussed above, other factors also strongly affect the adsorption energy and, accordingly, the acid strength, which includes the dispersion interaction. For each model (SMM-inter, SMM-intra, FAU), the activation barrier for propene protonation [ΔE≠(C3H6)] correlates well with the acid strength determined using ΔEads[NH3] (Fig. 6b). When comparing the SMM models with FAU containing BAS of similar acid strength as judged from ΔEads[NH3], a higher ΔE≠(C3H6) and lower reactivity of SMM are predicted (Fig. 6b). We speculate that this is due to the specific curvature of the zeolite channels, which stabilizes the transition state in the zeolite pore. Fig. 7 shows the transition state geometries in propene protonation on the BAS of three models with similar acid strength as determined using ΔEads[NH3], namely SMM-inter-NiF, SMM-intra-3NiF, and FAU-EFAl. The bond distances in the transition state indicate that the extent of proton transfer follows the trend FAU-EFAl > SMM-intra-3NiF > SMM-inter-NiF, which also leads to a higher formal charge (+0.82) of the [C3H7]+ fragment in FAU-EFAl than those in SMM-intra-3NiF (+0.76) and SMM-inter-NiF (+0.74). In the transition state, the cationic [C3H7]+ fragments are mainly stabilized by the electronic interactions with negative framework oxygen atoms. For the clay model SMM-inter-NiF, the negative framework oxygen atoms are distributed on a flat surface, and the electronic stabilization of [C3H7]+ is weaker than that for FAU-EFAl with its curved surface around the BAS. This stabilization is due to the interaction of the cationic transition state with the negatively charged oxygen atoms. This explains the lower activation barrier for FAU-EFAl. For SMM-intra-3NiF, the surface is also curved in a nearly similar way to that for FAU zeolite, but the terminal silanol group and the associated basic O atoms have a lower negative charge, resulting in decreased stabilization of the transition state in SMM-intra-3NiF. The influence of dispersion interactions on the activation barriers was further explored by separately analyzing the dispersion contributions. By excluding these interactions, the activation barriers are raised by 4, 6, and 1 kJ mol−1 for SMM-inter-NiF, SMM-intra-3NiF, and FAU-EFAl models, respectively (Table S3†). This indicates a minor influence of the dispersion interactions on the adsorbed and transition states. Recently, entropic effects on the reaction kinetics in zeolite-catalyzed hydrocarbon conversion have been emphasized.33,34 To separate the enthalpic and entropic contributions, the free energy contributions at 473 K were computed for SMM-inter-NiF, SMM-intra-3NiF, and FAU-EFAl (Table S4†). For all the three models, the entropic contributions (−TΔS≠) to the free-energy barriers (ΔG≠) are similar, implying that the enthalpic contributions (ΔH≠) dominate the reaction kinetics. These results suggest that although clays possess BAS with a higher acid strength than zeolites, this does not result in higher reactivity because of the presence of these acid sites on an open surface that lacks the stabilization of small molecules induced by electrostatic interactions due to the curvature of the zeolite surface.35,36 Experimentally, it has been observed that NiF-modified SMM clay is substantially more active than FAU-based catalysts in the bifunctional hydroisomerization of long hydrocarbons.13 We speculate that the stronger dispersion stabilization of hydrocarbon reactants heavier (longer) than propene with the clay and zeolite negates the difference in electrostatic stabilization of the cationic transition state.
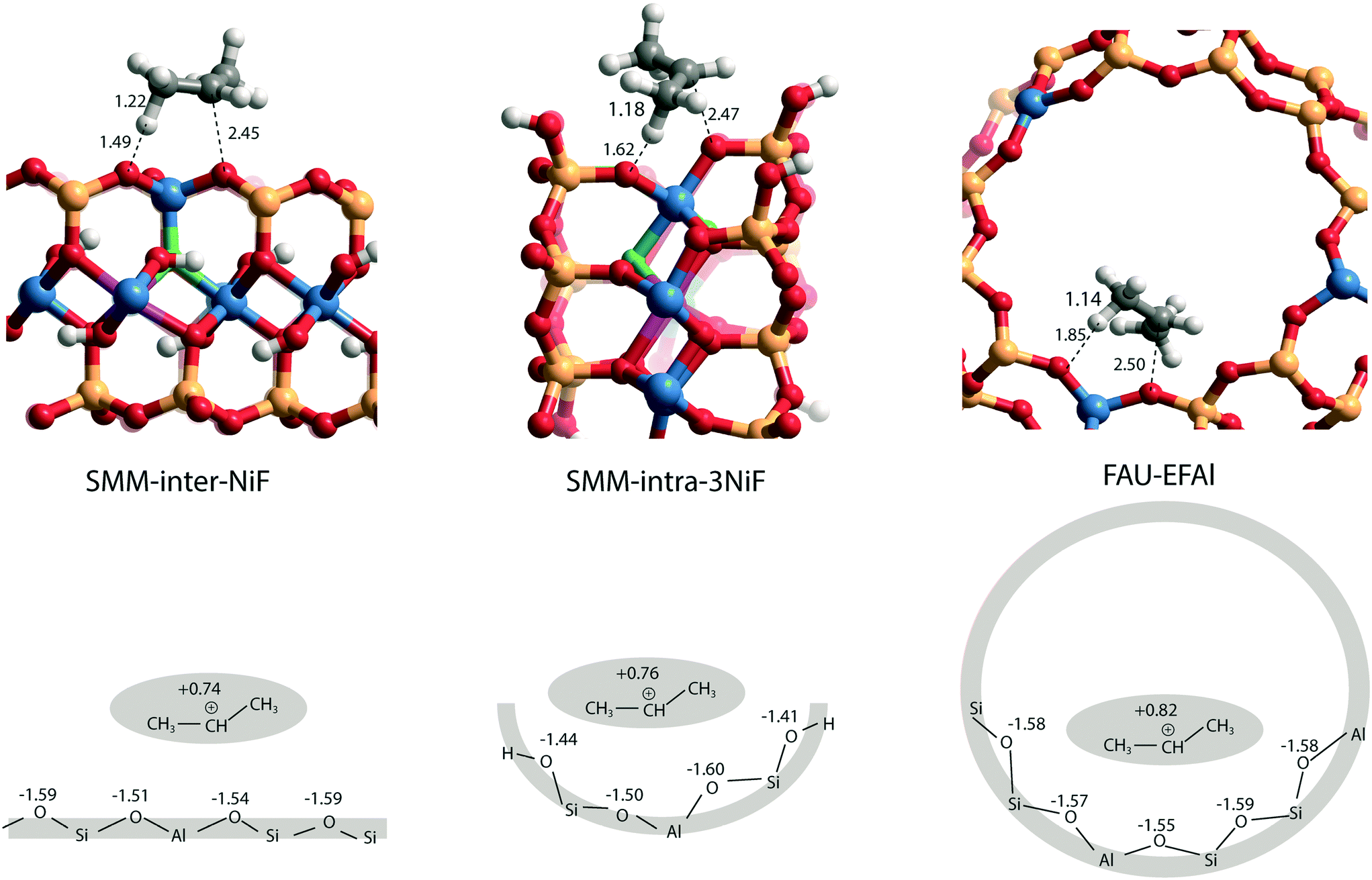 | ||
| Fig. 7 Transition state geometries in protonation of propene into sec-propoxy on the BAS of SMM-inter-NiF, SMM-intra-3NiF, and FAU-EFAl (bond distances are in Å), with respective illustrations of the local structures below (values are shown next to the [C3H7]+ fragments and O atoms indicate Bader charges). | ||
Conclusions
The Brønsted acidity of SMM clay was studied by periodic DFT calculations. Two structural models were considered, one involving BAS at the edge defects located in the intralayer of SMM and one bridging hydroxyl groups in the interlayer resulting from isomorphous substitution of Si4+ by Al3+ within the tetrahedral silicate layer of SMM. The presence of F− and Ni2+ lattice substitutions vicinal to the [Si–(OH)–Al] moiety of both types of structures strongly enhances the acid strength as probed using the adsorption energies of ammonia and pyridine. For the BAS in the interlayer of SMM, it is found that introduction of Ni2+ or F− in the octahedral layers has only a minor influence on the acidity. This difference implies that the experimentally observed acidity enhancement is mainly due to simultaneous Ni2+ and F− substitution ([NiF]+ for [AlO]+) in close proximity to the BAS. The role of F− is to withdraw electrons from the OH bond, and the role of Ni2+ is to facilitate the isomorphous substitution of O by F. Increasing F for O substitution in the first coordination shell of the Al atom to which the acidic OH group coordinates increases the acidity. The adsorption energies of ammonia indicate that clays contain acid sites stronger than those in zeolites, but their catalytic activity in the activation of a small olefin model compound is lower than that of zeolites. We speculate that the lower barrier for the zeolite was due to the curvature of its surface, resulting in more effective electrostatic stabilization of the cationic transition state by negatively charged surface oxygen atoms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. L. thanks the China Scholarship Council (CSC) for financial support. The Netherlands Organization for Scientific Research (NWO) is acknowledged for providing access to the supercomputer facilities. The authors acknowledge the support from the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC). E. A. P. thanks the Government of the Russian Federation (Grant 074-U01) and the Ministry of Education and Science of the Russian Federation (Project 11.1706.2017/4.6) for support.References
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- G. Busca, Chem. Rev., 2007, 107, 5366–5410 CrossRef CAS PubMed.
- E. J. M. Hensen, D. G. Poduval, D. A. J. M. Ligthart, J. A. R. van Veen and M. S. Rigutto, J. Phys. Chem. C, 2010, 114, 8363–8374 CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- J. T. Kloprogge, S. Komarneni and J. E. Amonette, Clays Clay Miner., 1999, 47, 529–554 CAS.
- P. Komadel, in Natural Microporous Materials in Environmental Technology, ed. P. Misaelides, F. Macášek, T. J. Pinnavaia and C. Colella, Springer Netherlands, Dordrecht, 1999, pp. 3–18 Search PubMed.
- J. C. Q. Fletcher, M. Kojima and C. T. O'Connor, Appl. Catal., 1986, 28, 181–191 CrossRef CAS.
- J. P. Giannetti and D. C. Fisher, in Hydrocracking and Hydrotreating, American Chemical Society, 1975, vol. 20, pp. 52–64 Search PubMed.
- H. E. Swift and E. R. Black, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 106–110 CAS.
- K. H. W. Robschlager, C. A. Emeis and R. A. van Santen, J. Catal., 1984, 86, 1–8 CrossRef CAS.
- J. J. L. Heinerman, I. L. C. Freriks, J. Gaaf, G. T. Pott and J. G. F. Coolegem, J. Catal., 1983, 80, 145–153 CrossRef CAS.
- A. C. Wright, W. T. Granquist and J. V. Kennedy, J. Catal., 1972, 25, 65–80 CrossRef CAS.
- C. Yue, C. Liu, B. Mezari, A. Brückner, E. A. Pidko, M. S. Rigutto and E. J. M. Hensen, Appl. Catal., B, 2016, 191, 62–75 CrossRef CAS.
- P. Vogel, C. T. O'Conner and M. Kolma, Clay Miner., 1990, 25, 355–362 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Viani, A. F. Gualtieri and G. Artioli, Am. Mineral., 2002, 87, 966–975 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- M. Czjzek, H. Jobic, A. N. Fitch and T. Vogt, J. Phys. Chem., 1992, 96, 1535–1540 CrossRef CAS.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 7024–7033 CrossRef CAS.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 344, 570–577 CrossRef CAS.
- L.-E. Sandoval-Díaz, J.-A. González-Amaya and C.-A. Trujillo, Microporous Mesoporous Mater., 2015, 215, 229–243 CrossRef.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, 1960 Search PubMed.
- B. Perdikatsis and H. Burzlaff, Kristallografiya, 1981, 156, 177–186 CAS.
- A. G. Newton, K. D. Kwon and D.-K. Cheong, Minerals, 2016, 6, 1–15 CrossRef.
- X. Rozanska, T. Demuth, F. Hutschka, J. Hafner and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 3248–3254 CrossRef CAS.
- R. Gounder and E. Iglesia, Acc. Chem. Res., 2012, 45, 229–238 CrossRef CAS PubMed.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2014, 316, 93–102 CrossRef CAS.
- R. Gounder and E. Iglesia, Chem. Commun., 2013, 49, 3491–3509 RSC.
- D. Lesthaeghe, V. Van Speybroeck and M. Waroquier, Phys. Chem. Chem. Phys., 2009, 11, 5222–5226 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy02053h |
| ‡ Current address: Inorganic Systems Engineering Group, Department of Chemical Engineering, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, the Netherlands. |
| This journal is © The Royal Society of Chemistry 2018 |