
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering oxygen-containing and amino groups into two-dimensional atomically-thin porous polymeric carbon nitrogen for enhanced photocatalytic hydrogen production†
Nannan
Meng‡
a,
Jian
Ren‡
 bc,
Yang
Liu
d,
Yi
Huang
a,
Tristan
Petit
*b and
Bin
Zhang
*a
bc,
Yang
Liu
d,
Yi
Huang
a,
Tristan
Petit
*b and
Bin
Zhang
*a
aDepartment of Chemistry, School of Science, and Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, and Collaborative Innovation Centre of Chemical Science and Engineering (Tianjin), Tianjin, 300072, China. E-mail: bzhang@tju.edu.cn
bMethods for Material Development, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Albert-Einstein-Strasse 15, Berlin, 12489, Germany. E-mail: tristan.petit@helmholtz-berlin.de
cDepartment of Physics, Freie Universität Berlin, Arnimallee 14, Berlin, 14195, Germany
dAnalysis and testing center, Tianjin University, Tianjin, 300072, China
First published on 22nd January 2018
Abstract
Polymeric carbon nitride (PCN) is a promising earth-abundant photocatalyst for solar energy conversion. However, the photocatalytic activities of PCN-based materials remain moderate because of their poor dispersion in water and their fast electron–hole recombination. Here, a facile two-step continuous thermal treatment strategy is presented to endow the bulk PCN nanosheets with an atomically-thin structure, strong hydrophilicity and Lewis basicity to dramatically enhance the photocatalytic hydrogen (H2) generation performance. The formation of the oxygen-containing and amino groups in the atomically-thin PCN sheets improves the charge separation and provides rich active sites for the surface reaction. Such synergistic effects lead to a superior visible-light-driven photocatalytic activity and its H2 evolution rate (1233.5 μmol h−1 g−1) is more than 11 times higher than the bulk PCN using Ni as a cocatalyst. Additionally, the H2 evolution rate can reach 20948.6 μmol h−1 g−1 using Pt as a cocatalyst under AM1.5G solar irradiation.
Broader contextWith the current high energy consumption, the development of efficient cheap materials for energy conversion and storage is a pertinent challenge. Photocatalytic hydrogen evolution is a promising technology that paves the way for a clean and renewable source for hydrogen fuel. Recently, polymeric carbon nitride (PCN) photocatalysts have attracted substantial attention due to their abundant cheap precursors, chemical stability and modification flexibility. However, the photocatalytic performances of pristine PCN are relatively low because of its poor dispersion in water, low electric conductivity and rapid rate of charge-carrier recombination. Thus, the development of various synthetic methods and modification techniques is highly desirable to improve the activity of PCN photocatalysts. Herein, a facile one-pot two-step thermal treatment to synthesize porous ultrathin PCN nanosheets with oxygen-containing and amino groups is reported. In-depth analyses using various characterization methods, especially synchrotron-based X-ray absorption spectroscopy, reveal that the tailored modifications on the structure and chemistry endow the PCN nanosheets with an atomically-thin structure, hydrophilic surfaces and strong Lewis basicity, thus synergistically leading to highly efficient H2 evolution. This research could open new perspectives to design PCN with enhanced photocatalytic properties. |
Polymeric carbon nitride (PCN) is a promising metal-free photocatalyst which has been applied in many photocatalytic reactions such as H2 or O2 evolution from water splitting,1–3 CO2 reduction4–6 or organic waste degradation.7–9 At present, various PCN samples with different morphologies, including nanotube,10 nanosheet,11 nanosphere12 and quantum dot13 have been successfully developed. Among them, two dimensional (2D) few-layer nanosheets show promising performance in photocatalytic applications because of their large surface areas, exposed active sites and short transport distances from the photoexcited center to the active site.14–16 Although some advanced methods have been developed to weaken the interlayer bonding and exfoliate PCN bulk materials, including the ultrasonication-assisted liquid exfoliation approach,17 the H2SO4-intercalated exfoliation method,18 and the thermal shock exfoliation process,19 the fabrication of a 2D atomically-thin nanostructure with a rich porous feature to enhance photocatalytic performance of PCN is still rarely reported.20 On the other hand, grafting heteroatoms or functional groups is another accessible strategy to modulate the performance of PCN. For instance, the introduction of oxygen-containing groups is shown to be profitable for the photocatalytic hydrogen evolution reaction (HER) over PCN, which is mainly due to the increased additional active sites and good dispersion of PCN in water.21,22 Furthermore, the –NH2 group can act as hole-stabilizers and thus prolong the lifetime of the excited states in photocatalysts.23,24 The strong Lewis basicity of the –NH2 group is also favorable for the photoreduction of water to H2. However, PCN with both oxygen-containing and amino groups, especially with atomically-thin thickness, has not yet been reported to our knowledge.
Based on the above discussion, it is conceivable that the PCN endowed with the following two features may show significantly enhanced photocatalytic HER activity: (i) a porous atomically-thin structure with a larger specific surface area and much more catalytic active sites, and (ii) oxygen-containing and amino co-functional groups with strong hydrophilicity and long excited state lifetime. However, it is still a great challenge to develop a facile method to engineer co-functional groups into atomically-thin PCN porous sheets with an enhanced photocatalytic HER.
Herein, we demonstrate a facile one-pot two-step thermal treatment strategy to synthesize atomically-thin porous sheets with oxygen-containing and amino groups from bulk PCN. The microscopic and spectroscopic characterization demonstrates that the consecutive thermal treatment can induce the conversion of bulk PCN into 2D porous sheets with atomically-thin thickness (∼0.4 nm), and oxygen-containing and amino co-functional groups, which can improve the charge separation and provide rich active sites for the surface reaction. Such a synergistic modulation strategy leads to a superior photocatalytic HER activity with a H2 evolution rate of 20948.6 μmol h−1 g−1 using Pt as a cocatalyst under simulated sunlight (AM 1.5G) irradiation, which is a record value, to our knowledge. In addition, by replacing Pt with a non-precious metal Ni cocatalyst, CNPS-NH2 still exhibits over 11 times enhancement of HER performance under visible light irradiation compared to bulk PCN.
As shown in Fig. 1, the two-step thermal treatment process involves the direct oxidation-etching of bulk PCN in air and the subsequent NH3-assisted exfoliation under an NH3 atmosphere. Melamine-derived PCN (CNB) was first oxidized and etched under air at 520 °C for 8 h to form porous oxygen-rich PCN nanosheets (CNPS-O). An additional calcination under an NH3 atmosphere at the same temperature for 1 h was then performed to exfoliate and to form –NH2 groups on the resulting PCN nanosheets (CNPS-NH2) (see the ESI† for details). The volume changes of the as-converted samples with the same mass follow the order: CNB < CNPS-O < CNPS-NH2 (Fig. S1, ESI†), suggesting the stepwise O- and NH3-mediated thermal exfoliation of CNB. Scanning electron microscopy (SEM) images (Fig. 2a–c) show that the two-step thermal treatment can convert the starting macrosized particles to soft and loose tremella-like aggregates. Typical transmission electron microscopy (TEM) images (Fig. 2d, e and Fig. S2, ESI†) suggest the transformation from thick CNB aggregates into apparent porous nanosheets with some wrinkles. The atomic force microscopy (AFM) images and the associated height profiles (Fig. 2f and Fig. S3, ESI†) demonstrate that the thickness of CNPS-O is about 10 nm, and the thickness of CNPS-NH2 is around 0.4 nm, implying that the atomically-thin sheet can be obtained by this facile two-step process. The uniform distributions of C, N and O elements in CNPS-NH2 are visualized by the energy-dispersive X-ray spectroscopy (EDS) mapping images (Fig. 2g). The pore size distributions of the samples (Fig. 2h) manifest that CNPS-NH2 has micropores and mesopores. Furthermore, the Brunauer–Emmett–Teller (BET) surface area is determined to be 195.3 m2 g−1 for CNPS-NH2, which is larger than that of CNPS-O (48.2 m2 g−1) (Fig. S4, ESI†). The high surface area of CNPS-NH2 can be mainly due to its unique ultrathin morphology and rich pores.
 | ||
| Fig. 1 Schematic illustration of the preparation process of CNPS-NH2. | ||
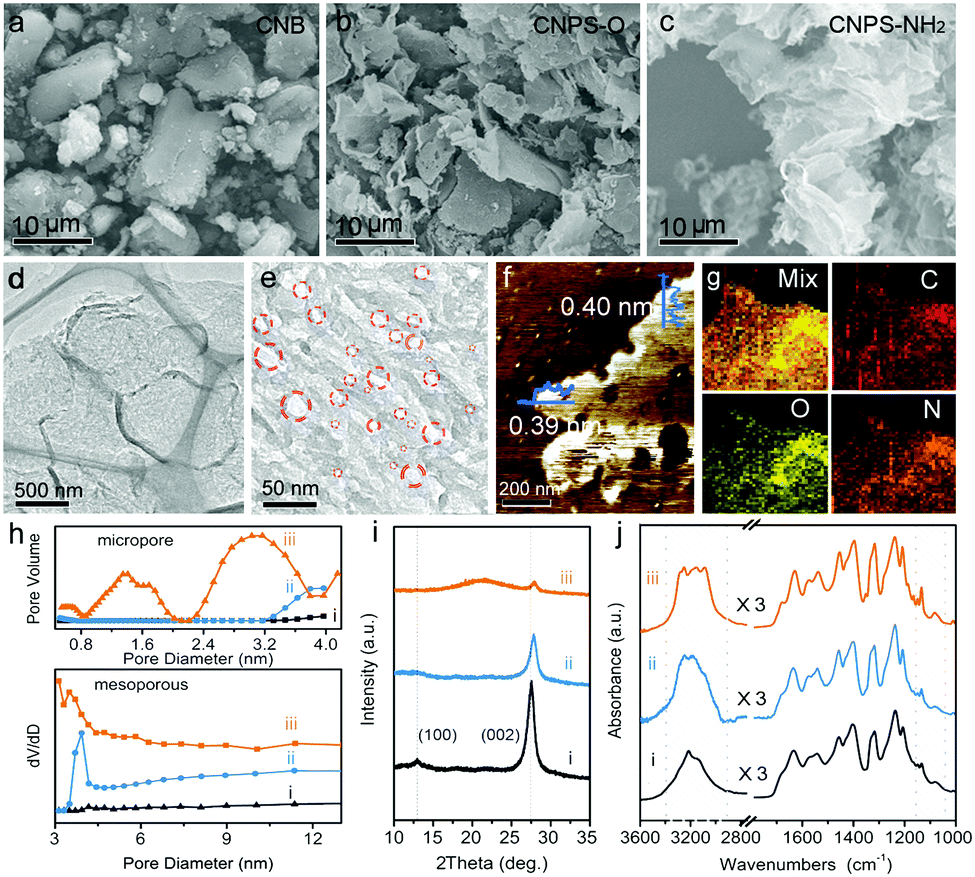 | ||
| Fig. 2 SEM images of (a) CNB, (b) CNPS-O and (c) CNPS-NH2. (d and e) TEM images, (f) AFM image and (g) EDS elemental mapping images of CNPS-NH2. (h) Micropore and mesopore size distributions, (i) XRD patterns and (j) ATR-FTIR spectra of the different samples: (i) CNB, (ii) CNPS-O and (iii) CNPS-NH2. | ||
The X-ray diffraction (XRD) pattern was further applied to characterize the structures of the three samples. As shown in Fig. 2i, the (100) and (002) diffraction peaks in the XRD pattern of CNB at about 13.1° and 27.5° can be associated with the repeated tri-s-heterocycle packing in the conjugated PCN planes and the interlayer stacking of graphite-like materials, respectively.25,26 Compared with the (100) peak of CNB, the (100) peaks of CNPS-O and CNPS-NH2 become much weaker, and almost disappear. This is mainly attributed to the damage in the inplane repeated packing of PCN, which is a typical feature of porous PCN.27,28 Since the electronegativity of oxygen (3.44) is higher than nitrogen (3.04), the interaction between the adjacent layers can be strengthened by powerful attraction. This result can be reflected by the notable shift of (002) diffraction from 27.5° (CNB) to 27.8° (CNPS-O). After NH3-assisted exfoliation, the diffraction peak position of (002) remains unchanged when compared with CNPS-O, implying the presence of the oxygen component in the CNPS-NH2 ultrathin porous nanosheet.29 In addition, a broad peak at around 22° can be attributed to the amorphous phase, suggesting the low crystalline nature of CNPS-NH2, as confirmed by the high resolution TEM image and the selected area electron diffraction (SAED) pattern (Fig. S5, ESI†). The attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra (Fig. 2j and Fig. S6, ESI†) present the typical bands in the as-prepared PCN samples. The increased intensity of the C–O stretching at 1062 and 1134 cm−1 from CNB to CNPS-NH2 indicates the richer oxygen-containing groups in CNPS-NH2. The peaks between 1200 and 1700 cm−1 are mostly due to the stretching and bending modes of the heptazine heterocyclic ring, and the broad feature at 3000–3350 cm−1 is assigned to the stretching modes of the amine group, alcohol hydroxyl group and sp/sp2 C–H.30 There are distinct shapes for the amine group, which is in agreement with the bending mode in the range of 1550–1640 cm−1. Primary amino groups with two N–H stretch absorptions (3252 and 3285 cm−1) are observed in CNPS-NH2, while the two other samples mostly present secondary amino groups (only one peak around 3250 cm−1).31 The distinct change of the zeta potential from −14.0 to +19.1 mV before and after NH3 thermal treatment of CNPS-O further proves the grafting of -NH2 into CNPS-NH2 (Fig. S6, ESI†).32 These results imply that the facile oxidation etching and subsequent NH3-assisted exfoliation treatment can lead to the formation of atomically-thin PCN sheets co-modified by oxygen-containing and primary amino groups. These bifunctional groups in CNPS-NH2 can not only enhance its hydrophilicity (Fig. S7, ESI†) leading to an improved solvent accessible surface area, but can also facilitate hybridization with metal cocatalysts,21,22 which are both beneficial to the photocatalytic HER process.
 | ||
| Fig. 3 (a) C K-edge, N K-edge and O K-edge XAS spectra. (b) C 1s, N 1s and O 1s high-resolution XPS spectra of the as-prepared samples: (i) CNB, (ii) CNPS-O and (iii) CNPS-NH2. | ||
To investigate the local electronic configuration and the chemical structure of PCN-based photocatalysts, X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS, Fig. S8, ESI†) were adopted. The C, N, and O K-edge XAS spectra of these samples are presented in Fig. 3a. XAS probes unoccupied electronic states and is particularly sensitive to the local electronic configuration. The C K-edge XAS spectra consist of contributions from transitions from 1s to π* at 285.7, 287.5, 288.6, and 289.9 eV and σ* at 293–298 eV. The π* feature at 285.7 eV is typical of the out-of-plane C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, related to interlayer bonding.33 The intensity of this feature decreases from CNB to CNPS-NH2 because the delocalized π bonds existing in the interlayer of the stacked CNB are weakened after the sequence of calcination treatments. Together with the decrease in the percentage of the aromatic carbon at 288.6 eV, it confirms that CNB has been exfoliated and converted to ultrathin sheets, as determined by the above-mentioned results. This phenomenon can be more clearly seen from the C 1s XPS spectra (Fig. 3b). Moreover, the features at 287.5 and 289.9 eV in the C K-edge XAS spectrum, which are attributed to the carbon species in different carbon–oxygen functional groups,34 increase from CNB to CNPS-NH2. At the nitrogen K-edge region (Fig. 3a), four main characteristic resonances are observed at 399.6, 401.7, 402.5, and 404–412 eV, which correspond to the 1s → π* transition in aromatic nitrogen atoms of heterocyclic rings (πCN–C*), graphitic three-fold nitrogen atoms (πN–3C*), sp3 N–C bridging among tri-s-triazine moieties (πN–C*), and 1s → σN–C* transition, respectively.35 These results reveal the preservation of the tri-s-triazine moieties in the molecular structure of the CNPS-NH2, which is favorable for channeling photogenerated charges in CNPS-NH2 to boost its photocatalytic activity. It is notable that XAS gives the information about the local structure, which is difficult to be obtained from the XRD pattern. The O K-edge XAS and O 1s XPS spectra further reveal the changes of surface chemistry during successive thermal treatments. Significantly, new features appear in the pre-edge region of O K-edge XAS for CNPS-O and CNPS-NH2. The feature at 533.6 eV on CNPS-O is assigned to the 1s → π* transition in the epoxide structure,36 which is weakened after the treatment under an NH3 atmosphere, probably due to the replacement of related oxygen atoms by nitrogen atoms. The feature at around 529 eV could refer to the electronic transition from O 1s to O 2p holes.37 During the successive thermal treatments, some bonds involving oxygen atoms are broken due to defect formation, thus leading to the appearance of O 2p holes. This increase of O 2p hole concentration at oxygen defects are likely to prolong the lifetime of photogenerated electrons as displayed subsequently. Moreover, the characters around 536 and 538 eV for 1s → σC–O–H* transition and 1s → σO–C* gradually disappear from CNB to CNPS-NH2.29
C bond, related to interlayer bonding.33 The intensity of this feature decreases from CNB to CNPS-NH2 because the delocalized π bonds existing in the interlayer of the stacked CNB are weakened after the sequence of calcination treatments. Together with the decrease in the percentage of the aromatic carbon at 288.6 eV, it confirms that CNB has been exfoliated and converted to ultrathin sheets, as determined by the above-mentioned results. This phenomenon can be more clearly seen from the C 1s XPS spectra (Fig. 3b). Moreover, the features at 287.5 and 289.9 eV in the C K-edge XAS spectrum, which are attributed to the carbon species in different carbon–oxygen functional groups,34 increase from CNB to CNPS-NH2. At the nitrogen K-edge region (Fig. 3a), four main characteristic resonances are observed at 399.6, 401.7, 402.5, and 404–412 eV, which correspond to the 1s → π* transition in aromatic nitrogen atoms of heterocyclic rings (πCN–C*), graphitic three-fold nitrogen atoms (πN–3C*), sp3 N–C bridging among tri-s-triazine moieties (πN–C*), and 1s → σN–C* transition, respectively.35 These results reveal the preservation of the tri-s-triazine moieties in the molecular structure of the CNPS-NH2, which is favorable for channeling photogenerated charges in CNPS-NH2 to boost its photocatalytic activity. It is notable that XAS gives the information about the local structure, which is difficult to be obtained from the XRD pattern. The O K-edge XAS and O 1s XPS spectra further reveal the changes of surface chemistry during successive thermal treatments. Significantly, new features appear in the pre-edge region of O K-edge XAS for CNPS-O and CNPS-NH2. The feature at 533.6 eV on CNPS-O is assigned to the 1s → π* transition in the epoxide structure,36 which is weakened after the treatment under an NH3 atmosphere, probably due to the replacement of related oxygen atoms by nitrogen atoms. The feature at around 529 eV could refer to the electronic transition from O 1s to O 2p holes.37 During the successive thermal treatments, some bonds involving oxygen atoms are broken due to defect formation, thus leading to the appearance of O 2p holes. This increase of O 2p hole concentration at oxygen defects are likely to prolong the lifetime of photogenerated electrons as displayed subsequently. Moreover, the characters around 536 and 538 eV for 1s → σC–O–H* transition and 1s → σO–C* gradually disappear from CNB to CNPS-NH2.29
The optical absorption properties of the samples were studied by UV-Vis absorption spectra (Fig. S9, ESI†). The light absorption edge of CNPS-O is red-shifted relative to that of CNB, mainly owning to the presence of oxygen-containing group.38 The blue-shift in the absorption edge of CNPS-NH2, compatible with its white color, is attributed to the quantum confinement effect in small PCN domains in atomically-thin products.39 From Kubelka–Munk function versus the energy of exciting wavelength derived from UV-Vis spectra,40,41 the band gaps of these samples are calculated to be 2.75, 2.70, and 2.96 eV for CNB, CNPS-O, and CNPS-NH2, respectively. The result is in agreement with the variation of the steady state photoluminescence emission peak (Fig. S10, ESI†). Thus, the band edges of the as-prepared catalysts could be consequently deduced based on bandgaps and valence-band XPS spectra (Fig. S11, ESI†).42 It should be noted that the potential of conduction-band edge of CNPS-NH2 is much higher than those of other samples, which will provide a huge thermodynamic driving force for the HER (Fig. 4a and Fig. S12, ESI†). Next, the charge transfer properties, which are the key parameters in determining the photocatalytic performance, were also studied. Time-resolved photoluminescence (PL) spectra (Fig. 4b) indicate that the emission lifetime of CNPS-NH2 is much longer than those of the other two samples. Such photogenerated electrons with long lifetime can boost surface reactions and thus give rise to the enhanced photocatalytic activity.43 In fact, if charges are efficiently trapped at these defective sites (probably involving oxygen as determined by XAS), the probability that they react with adsorbed species largely increases, with a parallel decrease in the undesired electron–hole recombination. In the electrochemical impedance spectroscopy (EIS) Nyquist plot (Fig. S13, ESI†), a remarkable decrease in the arc radius for CNPS-NH2 is observed, which reflects that the charge transfer resistance of CNPS-NH2 is smaller compared to the other samples.44,45 Additionally, consistent conclusion is also supported from the greatly increased transient photocurrent of CNPS-NH2 over CNPS-O and CNB (inset in Fig. 4b). These results reveal that the mobility of the photo-generated charge carriers is promoted in CNPS-NH2, and thus a better photocatalytic performance can be anticipated.46,47
 | ||
| Fig. 4 (a) Schematic band structures of the as-prepared samples, (b) time-resolved PL spectra and transient photocurrent response (inset) of CNB, CNPS-O and CNPS-NH2. (c) The photocatalytic HER over the as-prepared samples under visible light irradiation and simulated sunlight irradiation. (d) Cycling tests of CNPS-NH2/Ni under visible light irradiation. | ||
The photocatalytic HER performances of the samples were first measured under visible light (λ > 420 nm) illumination, wherein Ni with an optimized content (Fig. S14, ESI†) and triethanolamine (TEOA) were used as a cocatalyst and a sacrificial agent, respectively. As shown in Fig. 4c, CNB and CNPS-O exhibit H2 evolution rates of 110.1 and 472.1 μmol h−1 g−1, respectively. As expected, CNPS-NH2 shows the highest performance of H2 evolution, a remarkable evolution rate of 1233.5 μmol h−1 g−1, which is 11 times higher than that of CNB. It is noteworthy that the H2 evolution rate achieved by CNPS-NH2 represents an outstanding photocatalytic activity for the noble-metal-free cocatalyst based PCN system reported so far (Table S1, ESI†). The excellent activity of CNPS-NH2 verifies that the rationally precise structure control and surface design are the promising approach to yield the efficient solar-to-fuel photocatalyst. The HER driven by the simulated sunlight (AM 1.5 G) irradiation is further carried out with other conditions remaining unchanged. CNPS-NH2 presents the highest hydrogen production rate of 8134.1 μmol h−1 g−1, which is much higher than CNPS-O (4375.8 μmol h−1 g−1) and CNB (1403.5 μmol h−1 g−1). In addition, the cycling stability of CNPS-NH2 was testified. As displayed in Fig. 4d, the activity could last for over 12 h without any obvious decay. The TEM image and XRD pattern of the CNPS-NH2 showed no obvious change after long-term cycling test (Fig. S15, ESI†), indicating its high stability during the photocatalytic test.48,49
At present, Pt is wildly used as a cocatalyst in the PCN-based photocatalytic system. For comparison, CNPS-NH2/Pt was further detected (Fig. S16, ESI†). It is impressive that the H2 production rate of CNPS-NH2/Pt is up to 20948.6 μmol h−1 g−1 under AM 1.5G solar irradiation. To the best of our knowledge, this performance is one of the most robust self-modified PCN/Pt systems (Table S1, ESI†). Compared with visible-light-driven H2 production activity between CNB/Pt (582.7 μmol h−1 g−1) and CNPS-NH2/Ni (1233.5 μmol h−1 g−1), the superior performance of CNPS-NH2/Ni proves that rational structure designs are significant in the solar-to-fuel energy community for replacing noble metals with low cost co-catalysts.
Conclusions
In summary, we demonstrate an effective approach to construct atomically-thin PCN porous sheets with oxygen-containing and –NH2 co-functional groups for the enhanced photocatalytic HER. With strong Lewis basic sites, abundant in-plane holes, a good solvent accessible surface and a long photogenerated carrier lifetime, the efficient photoexcited charge utilization can be significantly improved with respect to its generation, diffusion and reaction, and thus the CNPS-NH2 is found to be a highly efficient photocatalyst for H2 production. These findings suggest that introducing oxygen-containing and amino groups into ultrathin PCN porous nanosheets through tailoring modification on their structure and chemistry may provide a possible avenue to develop low-cost highly efficient photocatalysts for various applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21422104), the Natural Science Foundation of Tianjin City (No. 17JCJQJC44700 and 16JCZDJC30600). J. R. and T. P. acknowledge the Volkswagen Foundation (Freigeist Fellowship No. 89592) for financial support. We acknowledge the kind support of Ms S Choudhury, Dr L. Puskar and staff members of the BESSY II Synchrotron Facility.Notes and references
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- W. Adam, Nat. Nanotechnol., 2017, 12, 1019 Search PubMed.
- M. Marszewski, S. W. Cao, J. G. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- Y. Xu, M. Kraft and R. Xu, Chem. Soc. Rev., 2016, 45, 3039–3052 RSC.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. C. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- J. S. Zhang, Y. Chen and X. C. Wang, Energy Environ. Sci., 2015, 8, 3092–3180 CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. W. Fu, J. G. Yu, C. J. Jiang and B. Cheng, Adv. Energy Mater., 2017, 1701503 Search PubMed.
- S. E. Guo, Z. P. Deng, M. X. Li, B. J. Jiang, C. G. Tian, Q. J. Pan and H. G. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- J. S. Zhang, M. W. Zhang, C. Yang and X. C. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- Q. H. Liang, Z. Li, Y. Bai, Z. H. Huang, F. Y. Kang and Q. H. Yang, Sci. China Mater., 2017, 60, 109–118 CrossRef.
- Y. F. Sun, S. Gao, F. C. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- J. Di, J. X. Xia, H. M. Li and Z. Liu, Nano Energy, 2017, 35, 79–91 CrossRef CAS.
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- H. H. Ou, L. H. Lin, Y. Zheng, P. J. Yang, Y. X. Fang and X. C. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed.
- J. Xu, L. W. Zhang, R. Shi and Y. F. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 CAS.
- P. J. Yang, H. H. Ou, Y. X. Fang and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992–3996 CrossRef CAS PubMed.
- H. Han., B. Wang, J. Gao, Z. H. Cheng, Y. Zhao, Z. P. Zhang and L. T. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef PubMed.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- V. W. Lau, V. W. Yu, F. Ehrat, T. Botari, I. Moudrakovski, T. Simon, V. Duppel, E. Medina, J. K. Stolarczyk, J. Feldmann, V. Blum and B. V. Lotsch, Adv. Energy Mater., 2017, 7, 1602251 CrossRef.
- J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. M. Spoor, K. Rock, L. D. A. Siebbeles, F. Kapteijn, F. Grozema, A. Houtepen, J. Gascon, J. Hunger and M. A. van der Veen, ChemSusChem, 2016, 9, 388–395 CrossRef CAS PubMed.
- M. B. Chambers, X. Wang, L. Ellezam, O. Ersen, M. Fontecave, C. Sanchez, L. Rozes and C. Mellot-Draznieks, J. Am. Chem. Soc., 2017, 139, 8222–8228 CrossRef CAS PubMed.
- Y. Wang, H. R. Li, J. Yao, X. C. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- J. S. Zhang, M. W. Zhang, L. H. Lin and X. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 6297–6301 CrossRef CAS PubMed.
- Y. Y. Kang, Y. Q. Yang, L. C. Yin, X. D. Kang, L. Z. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- X. J. She, J. J. Wu, J. Zhong, H. Xu, Y. C. Yang, R. Vajtai, J. Lou, Y. Liu, D. L. Du, H. M. Li and P. M. Ajayan, Nano Energy, 2016, 27, 138–146 CrossRef CAS.
- D. J. Martin, K. P. Qiu, S. A. Shevlin, A. D. Handoko, X. W. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- J. J. Chen, Z. Y. Mao, L. X. Zhang, D. J. Wang, R. Xu, L. J. Bie and B. D. Fahlman, ACS Nano, 2017, 11, 12650–12657 CrossRef CAS PubMed.
- Y. Huang, Y. Liu, D. Y. Zhu, Y. N. Xin and B. Zhang, J. Mater. Chem. A, 2016, 4, 13626–13635 CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CAS.
- W. Che, W. R. Cheng, T. Yao, F. M. Tang, W. Liu, H. Su, Y. Y. Hang, Q. H. Liu, J. K. Liu, F. C. Hu, Z. Y. Pan, Z. H. Sun and S. Q. Wei, J. Am. Chem. Soc., 2017, 139, 3021–3026 CrossRef CAS PubMed.
- V. Lee, L. Whittaker, C. Jaye, K. M. Baroudi, D. A. Fischer and S. Banerjee, Chem. Mater., 2009, 21, 3905–3916 CrossRef CAS.
- T. Mizokawa, Y. Wakisaka, T. Sudayama, C. Iwai, K. Miyoshi, J. Takeuchi, H. Wadati, D. G. Hawthorn, T. Z. Regier and G. A. Sawatzky, Phys. Rev. Lett., 2013, 111, 056404 CrossRef CAS PubMed.
- S. N. Guo, Y. Zhu, Y. Y. Yan, Y. L. Min, J. C. Fan and Q. J. Xu, Appl. Catal., B, 2016, 185, 315–321 CrossRef CAS.
- X. D. Zhang, X. Xie, H. Wang, J. J. Zhang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- W. G. Ma, D. X. Han, M. Zhou, H. Sun, L. N. Wang, X. D. Dong and L. Niu, Chem. Sci., 2014, 5, 3946–3951 RSC.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Haung, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Y. O. Wang, M. K. Bayazit, S. J. A. Moniz, Q. S. Ruan, C. C. Lau, N. Martsinovich and J. W. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 CAS.
- Y. S. Jun, E. Z. Lee, X. C. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- G. G. Liu, G. X. Zhao, W. Zhou, Y. Y. Liu, H. Pang, H. B. Zhang, D. Han, X. G. Meng, P. Li, T. Kako and J. H. Ye, Adv. Funct. Mater., 2016, 26, 6822–6829 CrossRef CAS.
- J. R. Ran, T. Y. Ma, G. P. Gao, X. W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 CAS.
- F. Razip, L. Q. Sun, Y. Y. Wang, X. L. Zhang, M. Humayun, S. Ali, L. L. Bai, Y. Qu, H. T. Yu and L. Q. Jing, Adv. Energy Mater., 2017, 1701580 Search PubMed.
- S. B. Wang, B. Y. Guan, Y. Lu and X. W. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS PubMed.
- J. X. Feng, H. Xu, S. S. Ye, G. F. Ouyang, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2017, 56, 8120–8124 CrossRef CAS PubMed.
- J. X. Feng, J. Q. Wu, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee03592f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |