 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge transport at a molecular GaAs nanoscale junction
Andrea
Vezzoli
 *a,
Richard J.
Brooke
b,
Nicolò
Ferri
a,
Carly
Brooke
a,
Simon J.
Higgins
a,
Walther
Schwarzacher
*b and
Richard J.
Nichols
*a
*a,
Richard J.
Brooke
b,
Nicolò
Ferri
a,
Carly
Brooke
a,
Simon J.
Higgins
a,
Walther
Schwarzacher
*b and
Richard J.
Nichols
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: andrea.vezzoli@liverpool.ac.uk; nichols@liv.ac.uk
bH. H. Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol BS8 1TL, UK. E-mail: w.schwarzacher@bristol.ac.uk
First published on 15th February 2018
Abstract
In recent years, the use of non-metallic electrodes for the fabrication of single-molecule junctions has developed into an elegant way to impart new properties to nanodevices. Integration of molecular junctions in a semiconducting platform would also speed technological deployment, as it would take advantage of established industrial infrastructures. In a previous proof-of-concept paper, we used simple α,ω-dithiol self-assembled monolayers on a gallium arsenide (GaAs) substrate to fabricate molecular Schottky diodes with a STM. In the devices, we were also able to detect the contribution of a single-molecule to the overall charge transport. The prepared devices can also be used as photodiodes, as GaAs is a III–V direct bandgap (1.42 eV at room temperature) semiconductor, and it efficiently absorbs visible light to generate a photocurrent. In this contribution, we demonstrate that fine control can be exerted on the electrical behaviour of a metal-molecule–GaAs junction by systematically altering the nature of the molecular bridge, the type and doping density of the semiconductor and the light intensity and wavelength. Molecular orbital energy alignment dominates the charge transport properties, resulting in strongly rectifying junctions prepared with saturated bridges (e.g. alkanedithiols), with increasingly ohmic characteristics as the degree of saturation is reduced through the introduction of conjugated moieties. The effects we observed are local, and may be observed with electrodes of only a few tens of nanometres in size, hence paving the way to the use of semiconducting nanoelectrodes to probe molecular properties. Perspectives of these new developments for single molecule semiconductor electrochemistry are also discussed.
Introduction
Since the introduction of techniques to reliably measure the charge transport properties of single molecules trapped between two electrodes,2–5 molecular junctions have been extensively used to study charge transport through organic and organometallic backbones. In the vast majority of such single molecule electrical studies, noble metals have been employed as electrodes, mainly due to their behaviour as simple resistors, their relative insensitivity to oxidation, and the availability of a wide range of chemical groups that can be employed to make chemical and electrical contact to them, through metal-molecule covalent or coordinative bonds. Asymmetric junctions, where one of the electrodes is non-metallic,6 have been recently investigated as a way to impart new functionalities to a molecular device. For instance, metal-molecule–semiconductor junctions show rectifying behaviour, as in a metal-insulator–semiconductor Schottky diode. The charge carrier depletion at the semiconductor interface results in a larger charge flow when the junction is biased in one direction (forward bias) than the other (reverse bias), resulting in asymmetric I–V characteristics. We recently reported this behaviour using gallium arsenide (GaAs) as electrode,1 and it was also demonstrated on silicon,7 in both cases using a molecular wire with appropriate contacting ends and a Au metallic electrode. Using a semiconducting electrode allows for a finer tuning of the junction properties, as the type of its doping (n- or p-type), can be used to control the nature of the majority charge carrier, and the doping density will affect the concentration of charge carriers (and therefore the semiconductor conductivity), and the size of the space charge layer. The rectifying behaviour is not the only new property that the use of a semiconducting electrode imparts to the molecular junction. Illumination with electromagnetic radiation of energy higher than the bandgap of the semiconductor promotes the majority charge carrier between bands, generating an equal number of minority carriers in the valence band. Band bending at the junction separates the photo-generated carriers, giving rise to a spontaneous photocurrent. The photoelectric response is at the basis of Schottky photodiode behaviour, and we recently reported on this effect in single-molecule junctions, by measuring the reverse bias photocurrent through a molecular bridge.8We focussed our efforts on GaAs mainly because of its direct bandgap, which allows for efficient light absorption properties, and higher electron velocity than silicon. Furthermore, extensive literature shows that high-quality self-assembled monolayers (SAMs) can be prepared at a GaAs surface,9–12 mainly through formation of As–S bonds.13 The organic monolayer provides strong and efficient molecule–semiconductor electrical contacts and passivates the surface against oxidation to Ga2O3 and As2O3, with samples stable for days with minimal oxidation as inferred from XPS spectra.14 Multi-molecule, large area (>1 μm) metal-molecules–GaAs devices have been extensively studied in the literature,15–17 using the determination of I–V characteristics to study the charge transport mechanism and overall device behaviour. In these studies, a metallic electrode is deployed on top of a pre-formed monolayer, by high vacuum evaporation or alternative adsorption techniques. In these methods, however, the presence of pinhole defects in the monolayer can lead to short-circuiting of the device, and harsh fabrication methods can damage the organic layer or unpredictably alter the semiconductor surface properties.11,12,17,18 In the method we developed,1 we used a scanning tunnelling microscope (STM) Au tip to make contact to the thiolated termini of an α,ω-dithiol SAM on a 〈100〉 GaAs surface and form metal-molecule–semiconductor junctions. The piezo-transducer controlling the STM tip position on the three axes allows for sub-Å precision, and the mild conditions (room temperature, ambient pressure) ensure the quality of the monolayer is not affected by the measurements. In this contribution, we greatly expand on the two studies we have already performed on GaAs-molecule–Au junctions,1,8 with a systematic study of the effect of the molecular wire nature and the semiconductor type and doping concentration on the final optoelectronic properties.
Results and discussion
In a typical experiment, a pre-formed SAM of the target molecule on a 〈100〉 GaAs substrate is mounted on the STM sample stage and the tip is initially engaged to a low setpoint current (0.1 nA). By further approaching the tip to the monolayer (increasing the setpoint current in a step-wise manner) we bring the tip into shallow contact with a small number of molecules, which results in telegraphic noise in the current versus time profile (Fig. 1a–d). Sudden current jumps have been observed for molecular layers on Au substrates,19,20 and have been attributed to the spontaneous formation of Au–S bonds at the STM tip, which results in a change of charge transport from tunnelling through the bare gap to tunnelling through the molecule. After finding these ideal conditions, where the STM tip is in direct contact with the SAM on GaAs, the feedback loop which controls the STM tip position is disabled, and the charge transport properties are assessed through bias-dependent and time-dependent measurements. Such a procedure ensures the fabrication of Au-molecule(s)–GaAs junctions, with molecular contacts at both electrodes. Details of the sample preparation can be found in the Methods section of the manuscript (vide infra).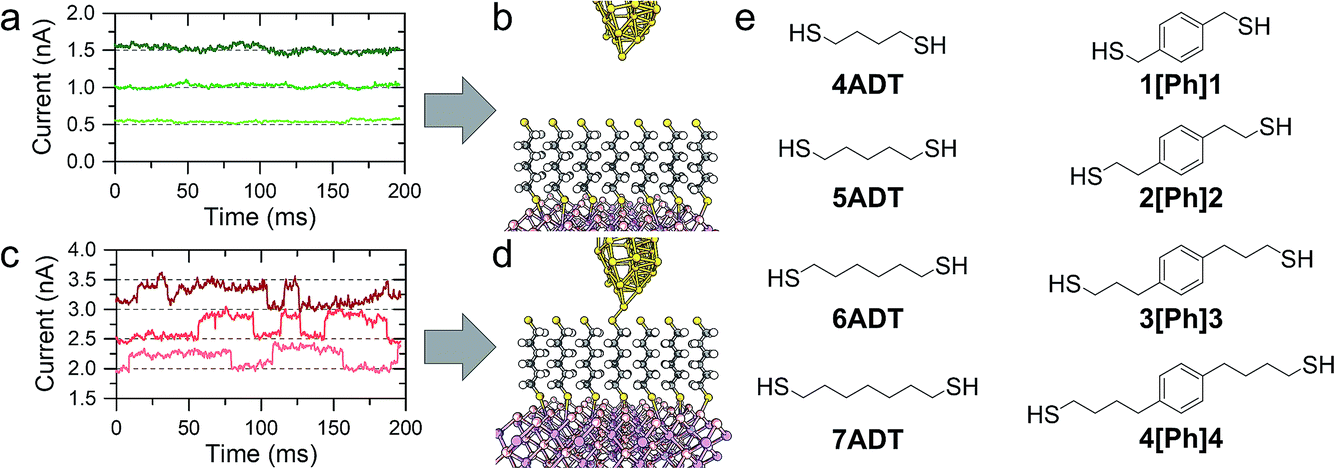 | ||
| Fig. 1 (a) Tunnelling readout at low setpoint bias, where the tip is not in contact with the monolayer, as depicted in (b). (c) Tunnelling readout at higher setpoint bias, where the sudden jumps are related to the formation of Au–S bonds between the tip and the monolayer thiolated termini (d). (e) Structure of molecular wires employed in this study. | ||
The prepared metal-molecule(s)–semiconductor devices behave like Schottky diodes as discussed in the Introduction, and we reported on the effect of the molecular wire (the insulator of the Schottky diode) in our previous publication.1 We found that on heavily doped nGaAsHD the rectification ratio RR, which is defined as the ratio of the current flowing in forward bias to the current flowing in reverse bias at a fixed magnitude of bias potential, was remarkably dependent on the nature of the molecular wire employed. The saturated α,ω-alkanedithiols (ADT) 4ADT, 5ADT, 6ADT and 7ADT showed an almost constant RR at ±1 V of approximately 12 (Fig. 2a), which is reduced to approximately 3.7 for 1[Ph]1, and a fully-conjugated biphenyl-4,4′-dithiol showed near-ohmic behaviour. In this present work, we have now synthesised molecular wires retaining the central conjugated unit of 1[Ph]1, but incorporating alkyl spacers of increasing length (2[Ph]2, 3[Ph]3, and 4[Ph]4). In this nomenclature, the number refers to the length of the alkyl chain of the thiolated arm at each side of the phenyl group. Using this series of molecules, we are now able to demonstrate here the fine control which can be exerted on the RR of the junction by small chemical alterations of the molecular backbone linking the metal and the semiconductor. Devices prepared with this X[Ph]X series of molecular wires showed increasing RR with increasing alkyl chain length, reaching the RR of ADTs in 4[Ph]4 as can be observed by comparing Fig. 2a and b. In the model we propose to rationalise our results (Fig. 2e), the charge transport in reverse bias is assisted by the molecular LUMO, and its alignment with the metal Fermi level and the semiconductor conduction band edge is key to the RR value. In the X[Ph]X series, the LUMO energy (and the HOMO–LUMO gap) increases with increasing alkyl chain length21 as the conjugation across the system is broken, and this trend further validates the proposed mechanism. The further the LUMO is from the metal Fermi level, the lower the net current transported under reverse bias conditions, and therefore the higher the RR (Fig. 3).
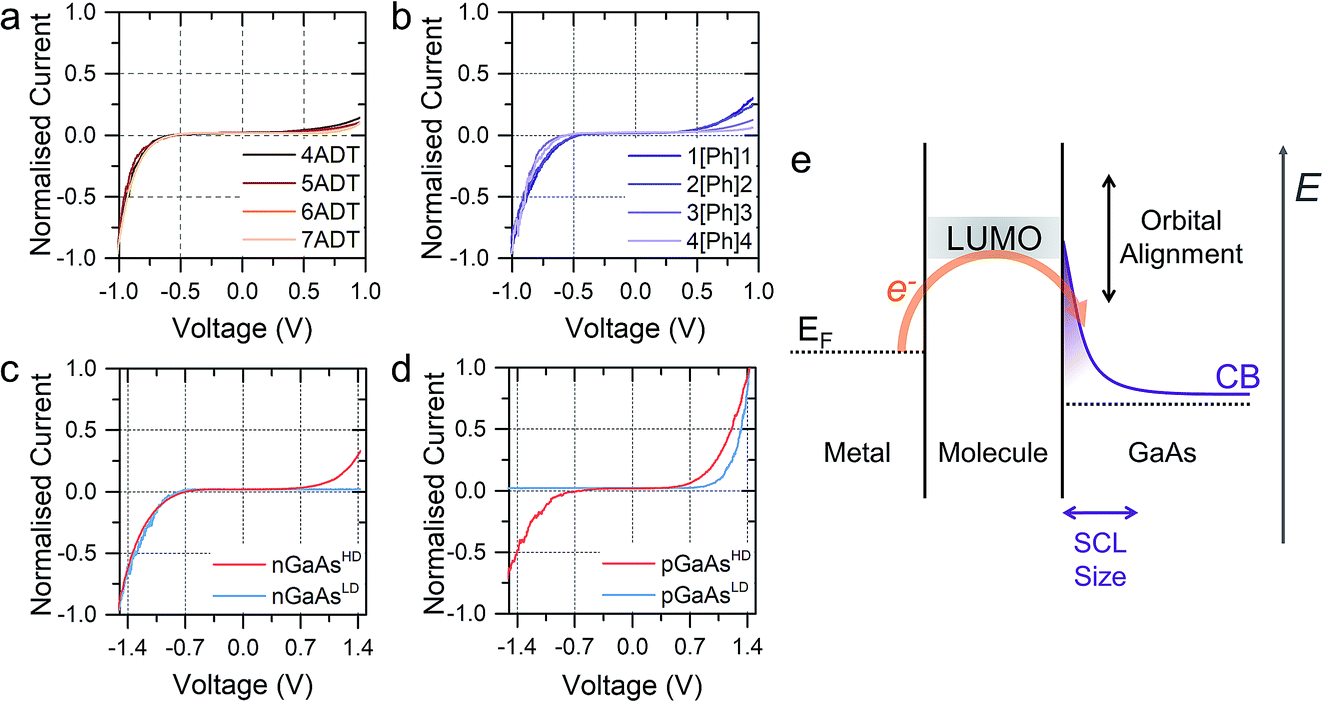 | ||
| Fig. 2 (a) I–V characteristics of α,ω-alkanedithiols of increasing length on nGaAsHD. (b) I–V characteristics of the X[Ph]X series on nGaAsHD. (c) I–V characteristics of 5ADT on nGaAs of different doping concentration. (d) I–V characteristics of 5ADT on pGaAs of different doping concentration. (e) Band diagram for the reverse biased junction with nGaAs, where E is the electron energy. All data shown in this Figure is obtained in the dark. Note that the sense corresponding to forward bias is opposite between panel c and d. Junctions made at n-type GaAs have forward bias at negative potential, and junctions made at p-type GaAs have forward bias at positive potential. | ||
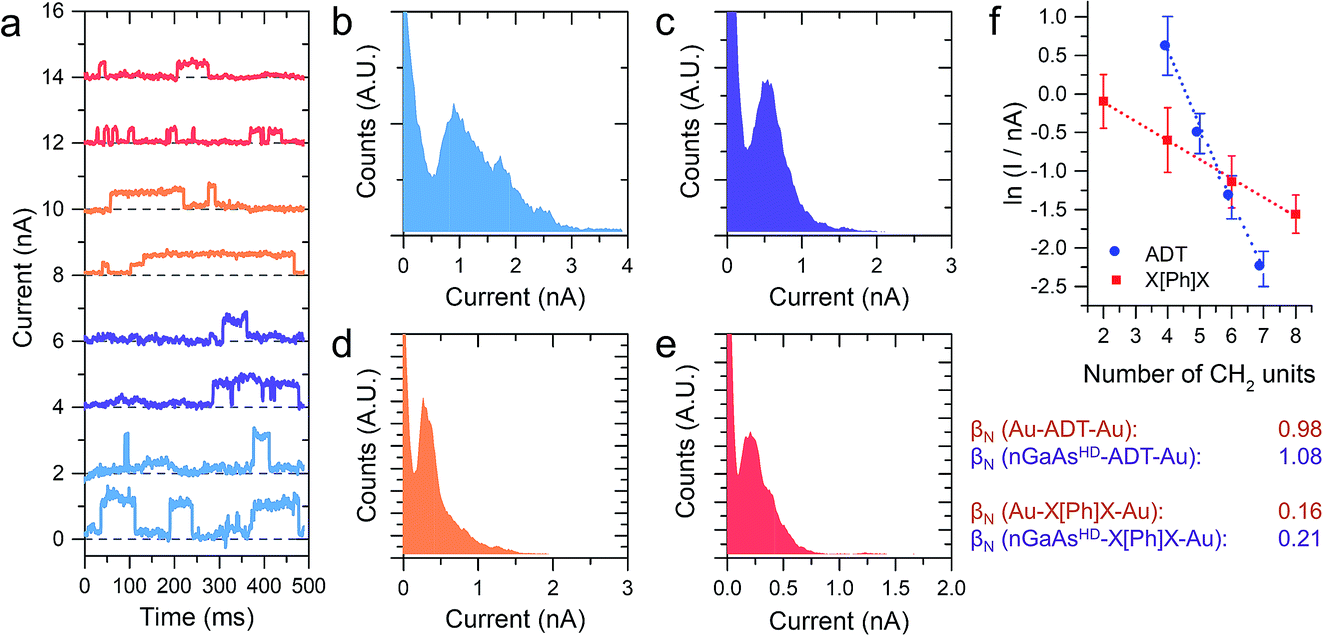 | ||
| Fig. 3 (a) Example I(t) traces for nGaAsHD–X[Ph]X–Au junctions. Traces are offset on the y axis for clarity. (b) Histogram constructed from 1154 individual current jumps for nGaAsHD–1[Ph]1–Au junctions. (c) Histogram constructed from 532 individual current jumps for nGaAsHD–2[Ph]2–Au junctions. (d) Histogram constructed from 1196 individual current jumps for nGaAsHD–3[Ph]3–Au junctions. (e) Histogram constructed from 970 individual current jumps for nGaAsHD–4[Ph]4–Au junctions. (f) Plot of current jump magnitude versus number of CH2 units plot used to determine the value of βN. All data shown in this Figure is obtained in the dark. | ||
The molecular wire bridging the metal–semiconductor gap is not the only variable that can be used to adjust the RR of the junction, but the type and doping density of the semiconductor was found to have a strong influence too. Using 5ADT as an example, the RR of the final device was found to be extremely high, >103, when using poorly doped semiconductors (GaAsLD: 1015–1017 cm−3 carrier concentration), and less pronounced in heavily doped (GaAsHD: 1018–1019 cm−3 carrier concentration) n- or p-type GaAs. This can be observed in the I–V characteristics presented in Fig. 2c (n-type) and 2d (p-type). In this case, the reason for the observed behaviour lies in the size of the space charge layer (SCL) at the semiconductor–molecule interface. Heavily doped semiconductors will have a smaller SCL, which will allow for a more efficient tunnelling than the much larger SCL found in lightly doped GaAs.8 It is interesting to note, however, that there is a key difference between p- and n-type GaAs, as the SCL size is also influenced by the zero-bias Schottky barrier, which is lower for p-type (approx. 0.6 V) than for n-type (approx. 0.8 V), resulting in less band bending and therefore a smaller SCL. This phenomenon accounts for the poor rectification found for 5ADT on pGaAsHD (RR ≈ 2 at ±1.5 V).
The I–V characteristics presented in the preceding text have been taken with the tip making contact to an undefined (albeit small) number of molecules, through which the charge is transported. With the tip in shallow contact with the monolayer the current as a function of time readout showed a series of sudden jumps, that have been ascribed to the formation and rupture of Au–S bonds at the tip–molecule interface. The bond formation/rupture process results in a change in the number of molecules bridging the tip–semiconductor junction, and the magnitude of the jump is therefore representative of the current flowing through an integer number of molecular wires.1,8,19 We collected for each sample current versus time traces containing hundreds of jumps in forward bias conditions, and analysed them statistically to quantify the single-molecule contribution to the overall current. Forward bias conditions were chosen simply because current is naturally larger than in reverse bias, allowing precise determination of single molecule events. We reported on the current jumps for the α,ω-alkanedithiol series in our previous publication,1 and we found that the decay constant β, as in the tunnelling relationship correlating molecular conductance and length G ∝ e−βL (where G is conductance and L is molecular length), was in excellent accordance with the value found for junctions made with the same molecules sandwiched between metallic electrodes. This strong molecular signature was taken as a further validation of the hypothesis that the current jumps were related to single-molecule charge transport, and prompted us to further study the current decay in GaAs-molecule–Au junctions.
The X[Ph]X series has already been characterised in Au-molecule–Au junctions, and an unusually low value of β was experimentally determined.21,22 This phenomenon was later ascribed to the presence of two orbitals located at the metal–sulfur interface of the molecular junction, which act as charge transport “gateways” that reduce the effect of molecular length on the overall conductance.22 The effect was only observed in covalently-bonded molecular wires, where the thiol proton is lost upon chemisorption at the Au electrodes, with the formation of strong Au–S bonds. In nGaAs–X[Ph]X–metal junctions, β was found again to be in good accordance with the value obtained employing the same molecular wires in Au-molecule–Au junctions, thus suggesting that the same “gateway” states are present at the semiconductor–molecule interface. Strongly polarised interfacial bonds are required for the generation of the charge transport “gateway”, so that this result is consistent with thiol chemisorption at the GaAs surface with formation of covalent bonds,23 and the presence of S–GaAs localised electronic states, as postulated by a previous combined DFT and internal photoemission study.24
We discussed in the introduction the possibility of generating a photocurrent upon illumination of the GaAs wafer with light of appropriate wavelength, and we reported initial results for the photocurrent transport across a molecular bridge in our previous publication.8 We found that highly rectifying junctions prepared at the nGaAsLD 〈100〉 surface show high values of reverse-bias photocurrent, while poorly rectifying junctions prepared with nGaAsHD electrodes provide only limited photocurrent of the order of a few pA. This effect was attributed to the larger SCL found in the poorly doped GaAs, which in addition to providing a sizeable tunnelling barrier that reduces reverse-bias dark current, also yields a larger volume for the generation of the charge carriers contributing to the photocurrent. This inference was made for nGaAs (Fig. 4a) and now, to further validate this model, we perform the same comparison between p-type GaAs of different doping density. The same behaviour is now observed, with pGaAsHD showing poor rectification in the dark, as can be observed in Fig. 2d, and failing to generate an appreciable photocurrent upon illumination with a HeNe laser as in Fig. 4b (blue = in the dark; red = under illumination). On the other hand, pGaAsLD was found to be highly rectifying in the dark (Fig. 4b, green curve, >103 RR) and a small photocurrent is generated upon illumination (Fig. 4b, orange curve). The absolute photocurrent values recorded for the two GaAsLD substrates is different, with the n-type semiconductor showing a much stronger response upon illumination. This can be attributed to the molecular LUMO states now being energetically far away from the semiconductor bands and unable to facilitate the minority carrier tunnelling (electrons for p-type GaAs) and to the difference in the zero-bias Schottky barrier, which is lower for p-type (approx. 0.6 V) than for n-type (approx. 0.8 V), resulting in less band bending and therefore a smaller SCL.
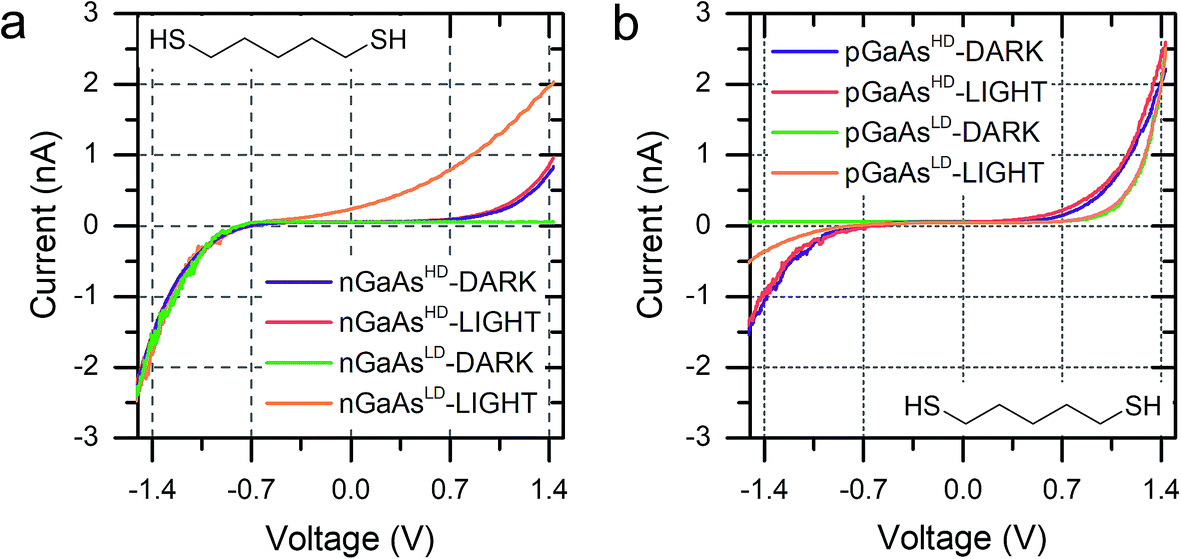 | ||
| Fig. 4 (a) I–V characteristics of nGaAs–5ADT–Au junctions in the dark and under laser illumination for the two doping density semiconductors used in this study. (b) I–V characteristics of pGaAs–5ADT–Au junctions in the dark and under laser illumination for the two doping density semiconductors used in this study. As in Fig. 2, junctions made at n-type GaAs have forward bias at negative potential, and junctions made at p-type GaAs have forward bias at positive potential. | ||
Interestingly, the photocurrent does not saturate as happens in metal–semiconductor planar junctions (Fig. 5a), but instead it increases with increasing reverse bias until breakdown, at bias >5 V. We attributed this behaviour to the strong anisotropy of the junction, where the STM tip generates a SCL only of a few nanometres in radius, illuminated by a laser spot several orders of magnitude bigger. As the bias is increased, the SCL increases in size by changes in band bending, and the fraction of illuminated area that contributes charge carriers to the overall photocurrent also increases (Fig. 5b).8 The geometry therefore makes the device sensitive to small changes of bias, and it ensures excellent performances as photodiode even when illuminated with low-intensity light, as can be observed in Fig. 5c.
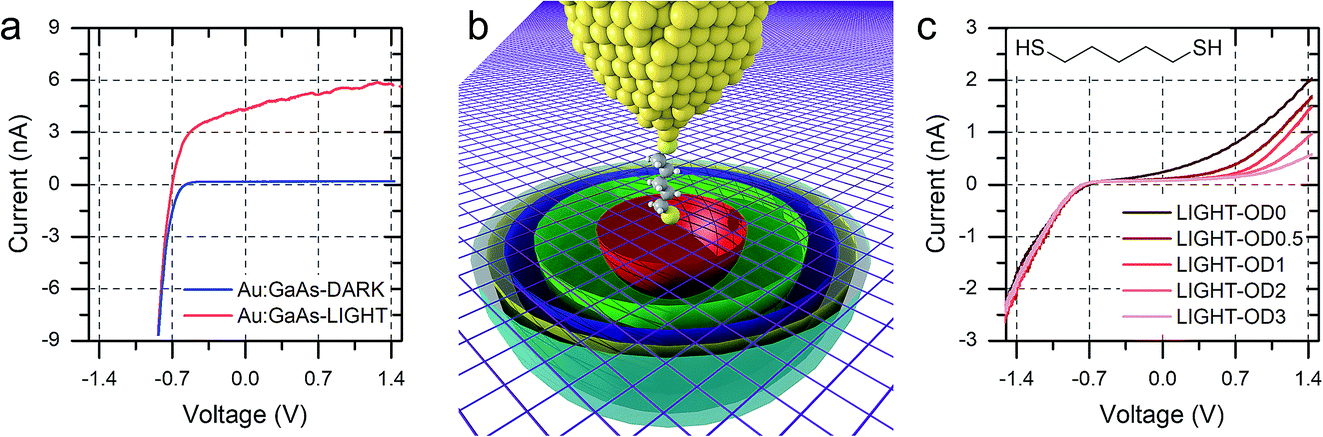 | ||
| Fig. 5 (a) I–V characteristics of a “hard contact” between Au and nGaAsLD obtained by crashing the tip several μm into a freshly etched GaAs surface. (b) Simplified representation of the hemispherical space charge layer at different bias values. Ideally, the SCL is the small red hemisphere at low bias, and it size increases with increasing bias due to increasing band bending, shown here as green, blue, yellow and cyan hemispheres. The semiconductor surface is depicted as a purple mesh for clarity. (c) I–V characteristics of nGaAsLD–5ADT–Au junctions under illuminations with light attenuated with optical filters of variable neutral density. | ||
Conclusions
Molecular electronics at semiconducting electrodes is currently a poorly explored area, with only a few reports on the fabrication and characterisation of hybrid metal-molecule–semiconductor devices. In this contribution, we demonstrated that rectifying properties and optoelectronic response are direct consequences of the use of GaAs as a junction component, and several parameters can be adjusted to tune the final properties of the fabricated device. Type and doping density of the semiconductor have a direct effect on the SCL size, which in turn offers a convenient way to tune the rectification ratio and the amount of photocurrent generated. The molecular bridge linking GaAs and Au was also found to be an important variable, and the energy alignment of the molecular orbital to the semiconductor band edges was found to be key to the imparted properties. Additionally, further control can be exerted through electrochemical gating, which alters charge transport by modifying the orbital alignment to the semiconductor band edges, and this is a direction we are currently exploring. In our previous contribution,8 we demonstrated that trapped carriers at GaAs surface state have an exquisite effect on charge transport, and here we discussed the presence of additional electronic “gateway” states at the GaAs–S interface, which promote charge transport over large distances and reduce its attenuation with length.In summary, semiconducting electrodes offer not only a straightforward way to impart additional properties to a molecular junction, but their use also provide insights on phenomena occurring at the molecule/semiconductor interface, valuable to researchers who might employ these hybrid materials in technologically relevant fields such as memory storage25 and flexible electronics.26
Experimental
Chemicals and synthesis
4ADT, 5ADT, 6ADT and reagents used throughout the syntheses and monolayer preparation were purchased from Sigma-Aldrich. 1[Ph]1 was purchased from TCI UK. Solvents and HCl 37% were purchased from Thermo Fisher Scientific. All chemicals were used without further purification except where noted. 7DT27 and BPDT28 were prepared as bis(thioacetate) following published procedures. The preparation of 3[Ph]3 and 4[Ph]4 is described elsewhere.212[Ph]2 was prepared as bis(thioacetate) by reduction of 2,2′-(1,4-phenylene)diacetic acid to the corresponding diol using sodium borohydride, followed by tosylation of the alcoholic functions and nucleophilic displacement with potassium thioacetate to obtain the target compound.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 8:2, followed by ethyl acetate:dichloromethane 1:1) to give the title compound as white powder (0.5 g, 38%). 1H NMR (400 MHz, CDCl3) δ: 7.70 (d, 4H, J = 8 Hz, Ph.), 7.30 (d, 4H, J = 8.4 Hz, Ph.), 7.02 (s, 4H, Ph.) 4.18 (t, 4H, J = 6.8 Hz, CH2), 2.92 (t, 4H, J = 6.8 Hz, CH2), 2.44 (s, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ: 144.78, 134.86, 132.92, 129.83, 129.14, 127.86, 70.45, 34.94, 21.66. m/z (HRMS, CI, CH4): 497.1058 [M + Na]+. C24H26O6S2Na calc. 497.1068.
:ethyl acetate 8:2, followed by ethyl acetate:dichloromethane 1:1) to give the title compound as white powder (0.5 g, 38%). 1H NMR (400 MHz, CDCl3) δ: 7.70 (d, 4H, J = 8 Hz, Ph.), 7.30 (d, 4H, J = 8.4 Hz, Ph.), 7.02 (s, 4H, Ph.) 4.18 (t, 4H, J = 6.8 Hz, CH2), 2.92 (t, 4H, J = 6.8 Hz, CH2), 2.44 (s, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ: 144.78, 134.86, 132.92, 129.83, 129.14, 127.86, 70.45, 34.94, 21.66. m/z (HRMS, CI, CH4): 497.1058 [M + Na]+. C24H26O6S2Na calc. 497.1068.
Sample preparation
An ohmic contact (GaIn eutectic) was painted with a small brush on the back of the GaAs slide (nGaAsHD: Si-doped, n-type, 〈100〉 ±0.05°, carrier concentration 3 × 1018 cm−3, Wafer Technology Ltd.; nGaAsLD: Si-doped, n-type, 〈100〉 ±0.03°, carrier concentration 1.5–1.7 × 1017 cm−3, Wafer Technology Ltd.; pGaAsHD p-type, Zn-doped 〈100〉 ±0.05°, carrier concentration 5 × 1018 to 5 × 1019 cm−3 Wafer Technology Ltd.; pGaAsLD p-type, Zn-doped 〈100〉 ±0.05°, carrier concentration 9 × 1015 to 2.9 × 1016 cm−3, El-Cat Inc.) and then annealed for 90 minutes in vacuum (∼10−2 mbar) at 400 °C. The wafer was chemically etched (n-type: concentrated ammonia, 5 minutes; p-type: concentrated HCl, 1 minute, ultrasonic bath) to remove the native oxides, rinsed with ultrapure Milli-Q water and absolute ethanol, and immediately immersed in a degassed ethanol solution containing 1 mM of the desired molecular wire and 5% concentrated ammonia solution (to deprotect the thioacetate function29 and avoid oxide layer regrowth). Samples were incubated under Ar atmosphere for 24 h, removed from solution, copiously rinsed with ethanol, dried under a stream of inert gas, and placed on a Au substrate (gold-on-glass, Arrandee), with an additional layer of fresh GaIn eutectic painted to provide optimal contact.STM measurements
An STM (Keysight Technology 5500 SPM) equipped with an electrochemically etched Au tip (ethanol:HCl 37%, 1:1, 2.5 V) is used to fabricate and characterize the molecular junctions presented in this study. The sample was mounted on the STM stage, and the gold tip was advanced towards the substrate in forward bias conditions (n-type: −1.5 V; p-type: +1.5 V; bias applied to the sample) by increasing the setpoint current until sudden jumps in the current profile are observed. These jumps have been related to a change in transport from tunnelling through air to tunnelling through the molecular backbone.19,30,31 Once the tip was engaged to the monolayer, we recorded I–V characteristics by sweeping the bias between 1.5 V in forward bias to 1.5 in reverse bias, at 3 V s−1. The measurements were performed in the dark and under laser illumination (Toshiba LHG-3220, 3 mW HeNe tube 632.8 nm). A filter wheel (Thorlabs FW1AND, with Thorlabs NE05B, NE10B, NE20B, NE30B and NE40B ND filters) was used to reduce the laser intensity, and a periscope (Thorlabs RS99, equipped with two Thorlabs BB1-E02 broadband mirrors) was used to raise the laser beam to the STM sample stage level, and to precisely align it to the tip–substrate position. Data presented in the manuscript is the average of 25 individual I–V characteristics, obtained from different regions of the substrate. I(t) traces were recorded by increasing the setpoint current of 100–200 pA respective to the value used for the I–V measurements, in order to have the tip slightly embedded in the monolayer. The feedback loop was then disabled to “freeze” the tip z position, and the tunnelling current readout was recorded for a few seconds. After recording each trace, the feedback loop was re-engaged to minimise the effect of z drift. Data was processed using software written in Python, which has been described previously1 and is available upon request. The obtained slices were compiled in statistical histograms for further analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC for funding under Grant No. EP/M005046/1 (Liverpool) and EP/M00497X/1 (Bristol).References
- A. Vezzoli, R. J. Brooke, N. Ferri, S. J. Higgins, W. Schwarzacher and R. J. Nichols, Nano Lett., 2017, 17, 1109–1115 CrossRef PubMed.
- M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252–254 CrossRef.
- B. Xu and N. Tao, Science, 2003, 301, 1221–1223 CrossRef PubMed.
- X. D. Cui, A. Primak, X. Zarate, J. Tomfohr, O. F. Sankey, A. L. Moore, T. A. Moore, D. Gust, G. Harris and S. M. Lindsay, Science, 2001, 294, 571–574 CrossRef PubMed.
- W. Haiss, H. van Zalinge, S. J. Higgins, D. Bethell, H. Höbenreich, D. J. Schiffrin and R. J. Nichols, J. Am. Chem. Soc., 2003, 125, 15294–15295 CrossRef PubMed.
- T. Kim, Z.-F. Liu, C. Lee, J. B. Neaton and L. Venkataraman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10928–10932 CrossRef PubMed.
- A. C. Aragonès, N. Darwish, S. Ciampi, F. Sanz, J. J. Gooding and I. Díez-Pérez, Nat. Commun., 2017, 8, 15056 CrossRef PubMed.
- A. Vezzoli, R. J. Brooke, S. J. Higgins, W. Schwarzacher and R. J. Nichols, Nano Lett., 2017, 17, 6702–6707 CrossRef PubMed.
- S. Lodha, P. Carpenter and D. B. Janes, J. Appl. Phys., 2006, 99, 24510 CrossRef.
- C. L. McGuiness, A. Shaporenko, C. K. Mars, S. Uppili, M. Zharnikov and D. L. Allara, J. Am. Chem. Soc., 2006, 128, 5231–5243 CrossRef PubMed.
- J. W. P. Hsu, Y. L. Loo, D. V. Lang and J. A. Rogers, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2003, 21, 1928 CrossRef.
- Y. L. Loo, D. V. Lang, J. A. Rogers and J. W. P. Hsu, Nano Lett., 2003, 3, 913–917 CrossRef.
- H. Lu and M. Zharnikov, J. Phys. Chem. C, 2015, 119, 27401–27409 CrossRef.
- C. L. McGuiness, D. Blasini, J. P. Masejewski, S. Uppili, O. M. Cabarcos, D. Smilgies and D. L. Allara, ACS Nano, 2007, 1, 30–49 CrossRef PubMed.
- E. Moons, M. Bruening, A. Shanzer, J. Beier and D. Cahen, Synth. Met., 1996, 76, 245–248 CrossRef.
- S. Lodha and D. B. Janes, J. Appl. Phys., 2006, 100, 24503 CrossRef.
- A. Vilan, A. Shanzer and D. Cahen, Nature, 2000, 404, 166–168 CrossRef PubMed.
- A. S. Martin, J. R. Sambles and G. J. Ashwell, Phys. Rev. Lett., 1993, 70, 218–221 CrossRef PubMed.
- W. Haiss, R. J. Nichols, H. van Zalinge, S. J. Higgins, D. Bethell and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2004, 6, 4330–4337 RSC.
- S. Huang, J. He, S. Chang, P. Zhang, F. Liang, S. Li, M. Tuchband, A. Fuhrmann, R. Ros and S. Lindsay, Nat. Nanotechnol., 2010, 5, 868–873 CrossRef PubMed.
- C. Brooke, A. Vezzoli, S. J. Higgins, L. A. Zotti, J. J. Palacios and R. J. Nichols, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 195438 CrossRef.
- S. Sangtarash, A. Vezzoli, H. Sadeghi, N. Ferri, H. M. O’Brien, I. Grace, L. Bouffier, S. J. Higgins, R. J. Nichols and C. J. Lambert, Nanoscale, 2018, 10, 3060 RSC.
- O. Voznyy and J. J. Dubowski, J. Phys. Chem. B, 2006, 110, 23619–23622 CrossRef PubMed.
- J. W. P. Hsu, D. V Lang, K. W. West, Y.-L. Loo, M. D. Halls and K. Raghavachari, J. Phys. Chem. B, 2005, 109, 5719–5723 CrossRef PubMed.
- Z. Liu, A. A. Yasseri, J. S. Lindsey and D. F. Bocian, Science, 2003, 302, 1543–1545 CrossRef PubMed.
- R. J. Hamers, Nature, 2001, 412, 489–490 CrossRef PubMed.
- W. Haiss, S. Martin, L. E. Scullion, L. Bouffier, S. J. Higgins and R. J. Nichols, Phys. Chem. Chem. Phys., 2009, 11, 10831–10838 RSC.
- B. de Boer, H. Meng, D. F. Perepichka, J. Zheng, M. M. Frank, Y. J. Chabal and Z. Bao, Langmuir, 2003, 19, 4272–4284 CrossRef.
- A. Singh, D. H. Dahanayaka, A. Biswas, L. A. Bumm and R. L. Halterman, Langmuir, 2010, 26, 13221–13226 CrossRef PubMed.
- S. Chang, J. He, A. Kibel, M. Lee, O. Sankey, P. Zhang and S. Lindsay, Nat. Nanotechnol., 2009, 4, 297–301 CrossRef PubMed.
- S. Chang, J. He, L. Lin, P. Zhang, F. Liang, M. Young, S. Huang and S. Lindsay, Nanotechnology, 2009, 20, 185102 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |