
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of a 12-week high-α-linolenic acid intervention on EPA and DHA concentrations in red blood cells and plasma oxylipin pattern in subjects with a low EPA and DHA status†
Theresa
Greupner‡
 a,
Laura
Kutzner‡
b,
Fabian
Nolte
b,
Alena
Strangmann
a,
Heike
Kohrs
a,
Andreas
Hahn
a,
Nils Helge
Schebb
bc and
Jan Philipp
Schuchardt
*a
a,
Laura
Kutzner‡
b,
Fabian
Nolte
b,
Alena
Strangmann
a,
Heike
Kohrs
a,
Andreas
Hahn
a,
Nils Helge
Schebb
bc and
Jan Philipp
Schuchardt
*a
aInstitute of Food Science and Human Nutrition, Leibniz University Hannover, Germany. E-mail: schuchardt@nutrition.uni-hannover.de; Fax: +49 (0)511-7625729; Tel: +49 (0)511-762 2987
bInstitute for Food Toxicology, University of Veterinary Medicine Hannover, Germany
cChair of Food Chemistry, Faculty of Mathematics and Natural Sciences, University of Wuppertal, Germany
First published on 15th February 2018
Abstract
The essential omega-3 fatty acid alpha-linolenic acid (ALA, 18:3n3) can be converted into EPA and DHA. The aim of the present study was to determine the effect of a high-ALA diet on EPA and DHA levels in red blood cells (RBCs) and their oxylipins in the plasma of subjects with a low EPA and DHA status. Fatty acid concentrations [μg mL−1] and relative amounts [% of total fatty acids] in the RBCs of 19 healthy men (mean age 26.4 ± 4.6 years) were analyzed by means of GC-FID. Free plasma oxylipin concentrations were determined by LC-MS based targeted metabolomics. Samples were collected and analyzed at baseline (week 0) and after 1 (week 1), 3 (week 3), 6 (week 6), and 12 (week 12) weeks of high dietary ALA intake (14.0 ± 0.45 g day−1). ALA concentrations significantly (p < 0.001) increased from 1.44 ± 0.10 (week 0) to 4.65 ± 0.22 (week 1), 5.47 ± 0.23 (week 3), 6.25 ± 0.24 (week 6), and 5.80 ± 0.28 (week 12) μg mL−1. EPA concentrations increased from 6.13 ± 0.51 (week 0) to 7.33 ± 0.33 (week 1), 8.38 ± 0.42 (p = 0.021, week 3), 10.9 ± 0.67 (p < 0.001, week 6), and 11.0 ± 0.64 (p < 0.001, week 12) μg mL−1. DHA concentrations unexpectedly decreased from 41.0 ± 1.93 (week 0) to 37.0 ± 1.32 (week 1), 36.1 ± 1.37 (week 3), 35.1 ± 1.06 (p = 0.010, week 6), and 30.4 ± 1.09 (p < 0.001, week 12) μg mL−1. Relative ΣEPA + DHA amounts were unchanged during the intervention (week 0: 4.63 ± 0.19, week 1: 4.67 ± 0.16, week 3: 4.61 ± 0.13, week 6: 4.73 ± 0.15, week 12: 4.52 ± 0.11). ALA- and EPA-derived hydroxy- and dihydroxy-PUFA increased similarly to their PUFA precursors, although in the case of ALA-derived oxylipins, the concentrations increased less rapidly and to a lesser extent compared to the concentrations of their precursor FA. LA-derived oxylipins remained unchanged and arachidonic acid and DHA oxylipin concentrations were not significantly changed. Our results confirm that the intake of ALA is not a sufficient source for the increase of EPA + DHA in subjects on a Western diet. Specifically, a high-ALA diet results in increased EPA and declined DHA concentrations. However, the changes effectively balance each other out so that ΣEPA + DHA in RBCs – which is an established marker for health protective effects of omega-3-PUFA – remains constant. The PUFA levels in RBCs reflect the concentration and its changes in plasma hydroxy- and dihydroxy-PUFA concentrations for ALA and EPA.
Introduction
Health benefits including lower risk of cardiovascular disease and mortality,1–5 reduction of inflammatory conditions,2,6 angiogenesis, tumor growth and metastasis2,7 and a better visual and neurological development8 are associated with long chain (LC) omega-3 (n3) polyunsaturated fatty acids (PUFAs), namely eicosapentaenoic acid (EPA, C20:5n3) and docosahexaenoic acid (DHA, C22:6n3). The positive health effects are believed to be mediated in part by the oxylipins formed from EPA and DHA in the arachidonic acid (AA, C20:4n6) cascade via different enzymatic pathways: cyclooxygenase (COX) action leads to prostanoid formation, lipoxygenases (LOX) give rise to hydroperoxy-PUFA which can be reduced to hydroxy-PUFA, whereas cytochrome P450 (CYP) enzymes act as epoxygenases and ω-hydroxylases.9,10 Epoxy-PUFA can be hydrolyzed to the corresponding diols by soluble epoxide hydrolase (sEH).11 Additionally, EPA and DHA can also be converted to oxidative products by autoxidation.10The primary dietary source of EPA and DHA is oily cold water fish such as salmon, tuna, herring and mackerel. In most Western diets, the intake of EPA and DHA is low due to low fish consumption. In the U.S. the daily mean EPA and DHA intake of adults is 30–40 mg and 70–80 mg, respectively.12 In Germany, the median EPA and DHA intake is 65–78 mg and 107–135 mg, respectively.13 Thus, the intake of EPA and DHA is about 2.5- to 5-fold lower compared to the recommendation of a minimum intake of 500 mg EPA and DHA per day for cardiovascular health.14 Another source of LC n3 PUFAs is the essential fatty acid alpha-linolenic acid (ALA, C18:3n3), which is present in high amounts in plant oils, particularly linseed, chia, perilla and walnut oil. The essential n3 fatty acid ALA can be converted into EPA, docosapentaenoic acid (DPAn3, C22:5n3) and DHA in a multistep elongation and desaturation reaction.15 However, several studies, including studies with stable isotopes, suggest that the conversion rate from ALA to EPA (5–8%), to DPAn3 (5–8%) and especially to DHA (0.5–5%) is low in subjects on a Western diet16–18 as reviewed in ref. 19–21. One reason is the high intake of the essential omega-6 (n6) PUFA linoleic acid (LA, C18:2n6), which competes with ALA for the rate-limiting enzyme Δ6-desaturase transforming LA into gamma-linolenic acid (GLA, C18:3n6) while blocking ALA into stearidonic acid (C18:4 n3).22–25
The mean intake of ALA and LA in the U.S. is 1.5–1.6 g d−1 and 15.1–15.9 g d−1,12 respectively, while the median intake of ALA and LA in Germany is 0.9–1.3 g d−1 and 7.3–10.1 g d−1, respectively.13 The LA intake is particularly high in the U.S. due to the high consumption of LA-rich plant oils such as corn, sunflower, and soybean oil.26 Although the intake recommendations for ALA (1.1–1.6 g d−1) and LA (12–17 g d−1) in the U.S.27 and Germany (ALA: 1.1 g d−1, LA: 5.4 g d−1 calculated on the basis of 0.5% of total energy (en%) and 2.5 en%, respectively, and 2000 kcal d−1 (ref. 28)) are basically met, the blood levels of EPA and DHA in the general population of both countries are low29 probably due to the overall low n3 PUFA intake and the resulting high LA/ALA ratio (about 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the U.S. and 8–10:1 in Germany). The low levels of EPA and DHA in RBCs, i.e. omega-3 index, are associated with an increased risk of death from cardiovascular disease,30 neurodegeneration and cognitive impairment31,32 and total mortality.33,34
:1 in the U.S. and 8–10:1 in Germany). The low levels of EPA and DHA in RBCs, i.e. omega-3 index, are associated with an increased risk of death from cardiovascular disease,30 neurodegeneration and cognitive impairment31,32 and total mortality.33,34
The question whether low LC n3 PUFA blood levels can be increased by supplementing ALA-rich oils remains controversial. Few studies have investigated the effect of supplementing ALA on blood EPA and DHA content in subjects with a low LC n3 PUFA status on a Western diet. Li et al.35 studied the effect of moderate-ALA diet (3.7 ± 1.4 g d−1) and high-ALA diet (15.4 ± 7.5 g d−1) in vegetarians, while Fokkema et al.36 observed the effect of a short-term low-dose ALA-enriched diet (2.01 g d−1) in vegans. The effect of a high-ALA diet on absolute LC PUFA concentrations in red blood cells (RBCs) has not been studied in a long-term study with healthy omnivores.
The aim of the present study was therefore to determine the short (1 and 3 weeks) and long term (6 and 12 weeks) effect of a high-ALA diet providing a daily ALA dose of 14.0 ± 0.45 g on EPA and DHA concentrations and relative EPA and DHA amounts in RBCs and their oxylipins in the plasma of subjects with a low EPA and DHA status consuming a Western (German) diet.
Materials and methods
This investigator initiated study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects were approved by the ethics committee at the medical chamber of Lower Saxony (Hannover, Germany). Written informed consent was obtained from all subjects. The study is registered in the German clinical trial register (no. DRKS00006765).Study design
The study was conducted at the Institute of Food Science and Human Nutrition, Leibniz University Hannover, Germany and consisted of a screening phase, a 12-week lasting intervention period and an 8-week lasting follow-up period. During the intervention period, five examinations were carried out, at the beginning (week 0) and after one (week 1), three (week 3), six (week 6) and twelve (week 12) weeks. Two further examinations were carried out in the follow-up period (week 14 and week 20). During the intervention period, the subjects daily ingested 22.3 g of linseed oil (All Organic Trading GmbH, Kempten, Germany) with an ALA content of 58% of total fatty acids (Table S1†). Hence, the ALA intake from linseed oil was 12.9 g day−1. The peroxide value of the linseed oil was 1.0 meq kg−1 and the acid value was 0.28 mg KOH per g.During each visit, fasting blood was collected, blood pressure was measured and the subjects completed a questionnaire to obtain information about changes in medication, lifestyle habits (e.g. physical activity), and tolerability of linseed oil. The questionnaire additionally included questions about dietary changes during the study. Moreover, the participants were requested to restrict their dietary n3 PUFA intake four weeks before and throughout the study, e.g. abstain from fatty fish (salmon, herring, tuna and mackerel) and ALA-rich vegetable foodstuffs such as linseeds, chia or walnuts (including oils), to minimize the effects on the variability of ALA and LC PUFA intake and blood levels. Prior to visit week 0, week 6, and week 12, the subjects completed a 3-day dietary questionnaire including two working days and one weekend day. The dietary questionnaires were analyzed using PRODI (Nutri-Science GmbH, Freiburg, Germany) to obtain the data on energy and nutrient intake. The total ALA intake was calculated retrospectively by adding the ALA intake from the background diet to the ALA dose from the linseed oil intake.
Blood samples were collected in the morning between 6:30 and 11:00 a.m. after overnight fasting. The examinations were scheduled at the same time for each subject. The samples were obtained by venipuncture of an arm vein using Multiflyneedles (Sarstedt, Nümbrecht, Germany) into serum and EDTA monovettes (Sarstedt). For the analysis of fatty acids in the plasma, EDTA blood monovettes were centrifuged for 10 min at 1500g and 4 °C and the plasma was transferred into 1.5 mL plastic tubes (Sarstedt) and immediately frozen and stored at −80 °C until extraction and analysis. For the analysis of fatty acids in RBCs, the cell sediment after centrifugation for 10 min at 1500g and 4 °C and removal of the plasma was washed twice with PBS (containing 1.5 mg mL−1 EDTA). Finally, the RBCs were reconstituted in PBS to the initial blood volume, transferred into 1.5 mL Eppendorf tubes and immediately frozen and stored at −80 °C until extraction and analysis. All transfer steps were carried out on ice. Other sets of blood samples (serum and EDTA monovettes) were sent to external laboratories for the measurement of clinical parameters. Serum lipid levels, liver enzymes and small blood picture were determined in the LADR laboratory (Laborärztliche Arbeitsgemeinschaft für Diagnostik und Rationalisierung e.V.), Hannover, Germany.
Study population
Participants were recruited from the general population in Hannover, Germany through advertisements. Several selection criteria were defined to assemble a homogeneous study collective. In particular, only men within a narrow age range from 20 to 40 years were included to minimize potential fluctuations in lipid profiles due to age or hormonal influence. Subjects were preselected via screening questionnaires according to the following inclusion criteria: male sex, age between 20 and 40 years, body mass index (BMI) between 20 and 27 kg m−2, and mixed diet with low meat and fish consumption. The exclusion criteria were defined as follows: smoking, serum triglyceride levels ≥150 mg dl−1 (≥1.7 mmol l−1), serum total cholesterol levels ≥200 mg dl−1 (≥5.2 mmol l−1), a relative amount of ΣEPA + DHA in whole blood ≤3 and ≥6%, intake of fish (>2 times per week) as well as addiction to alcohol, drugs and/or medications and diseases: chronic diseases (e.g. malignant tumors, manifest cardiovascular disease, insulin-dependent type 1 and 2 diabetes, and severe renal or liver diseases); chronic gastrointestinal disorders (especially small intestine, pancreas, and liver) as well as prior gastrointestinal surgical procedures (e.g. gastrectomy); hormonal disorders (e.g. Cushing's syndrome and untreated hyperthyroidism); uncontrolled hypertension; blood coagulation disorders and intake of coagulation-inhibiting drugs; periodic intake of laxatives; intake of anti-inflammatory drugs (including acetylsalicylic acid); and intake of lipid lowering drugs or supplements during the last 3 months before baseline examination. The inclusion and exclusion criteria were assessed via questionnaires. The pre-selected subjects were invited for a screening examination to collect fasting blood for the analysis of serum lipid levels, liver enzymes and fatty acid profiles in whole blood.Fatty acid analysis
The concentrations of fatty acids were determined by means of gas chromatography (GC) with flame ionization detection as described37 with slight modifications. In brief, lipids were extracted with MTBE/MeOH and derivatized with methanolic hydrogen chloride, and the resulting fatty acid methyl esters (FAME) were quantified using methyl pentacosanoate (C25:0 FAME) as the internal standard (IS). In addition to the determined concentration, reported as μg fatty acid per mL blood, the relative amount (% of total fatty acids) of each fatty acid was calculated directly based on peak areas as described.37Oxylipin analysis
The concentrations of oxylipins in the plasma were determined by means of an established liquid chromatography-mass spectrometry (LC-MS) based targeted metabolomics platform (Table S2†) as described38–40 with slight modifications. In brief, after the addition of the IS and antioxidant solution, the plasma samples were diluted with 1 M sodium acetate in water:MeOH (95:5 v:v) adjusted to pH 6.0 with acetic acid. Additionally, 10 μL of a solution of the LOX inhibitor 2-(1-thienyl)ethyl 3,4-dihydroxybenzylidenecyanoacetate (2 μM) and the protease inhibitor phenylmethylsulfonyl fluoride (5 mM) was added. The extraction was carried out on a nonpolar (C8)/anion exchange mixed mode material (Bond Elut Certify II, 200 mg, Agilent) utilizing ethyl acetate/n-hexane (75:25, v:v) with 1% acetic acid as the eluent. The LC-MS measurement was carried out in scheduled selected reaction monitoring following negative electrospray ionization and the quantification of oxylipins was performed by external calibration utilizing 13 deuterated IS.38–40
Calculations and statistics
The results of anthropometrical measures, serum lipid levels, dietary energy and fat intake are presented as mean ± standard deviation (SD), while the PUFA levels in RBC membranes and their relative change (%) are presented as mean ± standard error (SE). If the concentration of an analyte was below the lower limit of quantification (LLOQ) in more than 50% of the samples at one time point, the LLOQ is given for this analyte. Relative changes in the variables (v) were calculated individually for each subject at each time point (x) as Δ%, calculated by: Δ% = 100 × (vtx − vweek 0)/vweek 0.The activities of delta-5 desaturase (D5D) and delta-6 desaturase (D6D) were calculated using product-to-precursor ratios: (C20:4n6/C20:3n6) and (C20:3n6/C18:2n6), respectively, as previously described.41 The indices of highly unsaturated fatty acids (HUFA) were calculated as follows: % n3 in HUFA = 100 × (C20:5n3 + C22:5n3 + C22:6n3)/(C20:3n6 + C20:4n6 + C22:4n6 + C20:5n3 + C22:5n3 + C22:6n3); % n6 in HUFA = 100 × (C20:3n6 + C20:4n6 + C22:4n6)/(C20:3n6 + C20:4n6 + C22:4n6 + C20:5n3 + C22:5n3 + C22:6n3), modified from ref. 42.
The distributions of the sample sets were analyzed by means of the Kolmogorov–Smirnov test. The differences between baseline (week 0) levels and different time points after high-ALA diet (week 1, week 3, week 6, week 12) as well as between week 12 and week 14 and week 20 were analyzed by ANOVA for repeated measurements with the acceptance of statistical significance at p ≤ 0.05. To determine the statistical significance between baseline levels and each time point (as well as between week 12 and week 14 and week 20), t-tests for paired samples were carried out. For t-tests, the significance levels were adjusted according to the Holm–Bonferroni method. All statistical analyses were carried out with the SPSS software (Version 24, SPSS Inc., Chicago, IL, USA).
Results
Study population
Twenty male subjects met the criteria and thus were included in the study. All participants (mean age 26.2 ± 4.53 years) were healthy and had a normal BMI (24.9 ± 2.0 kg m−2) and serum lipid profile (Table 1). The eating habits (especially fish consumption) and the physical activity of the probands did not change during the intervention period as investigated by dietary questionnaires. The study collective consumed a normal mixed diet (including meat) with low fish consumption (≤1 fish serving per week) and low fruit and vegetable consumption (1–2 portions per day) and had a medium physical activity status (3–5 hours of sports per week) and a high education level (advanced technical college certificate). One subject withdrew his consent after 1 week of the intervention. All other 19 participants completed the 12-week intervention period and attended at all five intervention time points. At week 14 of the follow-up period, only 17, and at week 20, 13 participants attended the examinations. Linseed oil was well tolerated and no adverse effects were reported during the intervention period.| Week 0 | Week 1 | t-Testa | Week 3 | t-Testa | Week 6 | t-Testa | Week 12 | t-Testa | An reMb | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | p (week 1–week 0) | Mean ± SD | p (week 3–week 0) | Mean ± SD | p (week 6–week 0) | Mean ± SD | p (week 12–week 0) | p | |
| The levels are shown at week 0, 1, 3, 6, and 12 of the high-ALA diet (14.0 ± 0.45 g d−1). BMI: body mass index; dias BP: diastolic blood pressure; HDL: high density lipoprotein; LDL: low density lipoprotein; n.s.: not significant; SD: standard deviation; sys BP: systolic blood pressure; TC: total cholesterol; TG: triglycerides, wk: week.a t-Test for paired samples with Holm–Bonferroni correction; significance level p ≤ 0.05.b ANOVA for repeated measures (An reM); significance level p ≤ 0.05. | ||||||||||
| Age (years) | 26.2 ± 4.53 | |||||||||
| Weight (kg) | 82.7 ± 8.25 | 84.4 ± 7.83 | — | n.s. | ||||||
| BMI (kg m−2) | 24.7 ± 2.08 | 25.3 ± 1.87 | — | n.s. | ||||||
| Sys BP (mmHg) | 128 ± 12.0 | 125 ± 12.1 | — | 128 ± 9.27 | — | 131 ± 10.9 | — | 131 ± 14.9 | — | n.s. |
| Dias BP (mmHg) | 77.8 ± 9.39 | 77.0 ± 6.37 | — | 77.2 ± 5.21 | — | 78.2 ± 5.06 | — | 76.3 ± 5.23 | — | n.s. |
| TC (mg dl−1) | 168 ± 22.9 | 162 ± 23.2 | — | 159 ± 22.9 | — | 167 ± 25.8 | — | 167 ± 26.3 | — | n.s. |
| LDL (mg dl−1) | 108 ± 18.5 | 96.9 ± 16.6 | 0.010 | 102 ± 17.9 | n.s. | 105 ± 19.5 | n.s. | 101 ± 17.0 | n.s. | 0.022 |
| HDL (mg dl−1) | 54.1 ± 7.88 | 52.4 ± 8.28 | n.s. | 50.6 ± 8.47 | n.s. | 55.4 ± 7.8 | n.s. | 54.8 ± 10.9 | n.s. | 0.002 |
| TG (mg dl−1) | 77.1 ± 20.4 | 65.9 ± 18.8 | n.s. | 76.1 ± 27.7 | n.s. | 86.0 ± 35.4 | n.s. | 87.1 ± 36.4 | n.s. | 0.026 |
The evaluation of the 3-day dietary questionnaires showed that the variability of PUFA intake other than ALA was minimal (Table 2). For LA, AA, EPA, DPAn3 and DHA intake, no significant changes occurred during the intervention period. The intake of ALA from the background diet was low with minimal variability. The total ALA intake was 1.39 ± 1.31 g d−1 at week 0, 13.9 ± 0.34 g d−1 at week 6 and 14.0 ± 0.53 g d−1 at week 12. Due to the high-ALA diet, the total PUFA intake significantly (p < 0.001) increased from week 0 (11.2 ± 7.02 g d−1) to week 6 (23.3 ± 3.14 g d−1) and week 12 (22.9 ± 5.87 g d−1). The energy and total fat intake did not change significantly; however, the energy intake was lower at week 12 (2403 ± 524 kcal d−1) compared to week 0 (2910 ± 1181 kcal d−1) and week 6 (2924 ± 820 kcal d−1) (Table 2). In addition, for saturated fatty acid (SFA) and monounsaturated fatty acid (MUFA) intake, no significant changes were detected during the intervention period. The intake of SFA decreased (n.s.) from week 0 (31.1 ± 15.5 g d−1) to week 6 (26.9 ± 11.0 g d−1) and week 12 (24.4 ± 9.83 g d−1). Although not significant, the intake of EPA, DPAn3 and DHA slightly decreased during the intervention period (Table 2).
| Week 0 | Week 6 | t-Testa | Week 12 | t-Testa | An reMb | |
|---|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | p | |||
| The levels are shown at week 0, 6 and 12 of the high-ALA diet (14.0 ± 0.45 g d−1). AA: arachidonic acid; ALA: α-linolenic acid; EPA: eicosapentaenoic acid; DHA: docosahexaenoic acid; DPAn3: docosapentaenoic acid; LA: linoleic acid; MUFA: monounsaturated fatty acids; PUFA: polyunsaturated fatty acids; SFA: saturated fatty acids; wk: week.a t-Test for paired samples with Holm–Bonferroni correction; significance level p ≤ 0.05.b ANOVA for repeated measures (An reM); significance level p ≤ 0.05. | ||||||
| Energy intake (kcal) | 2910 ± 1181 | 2924 ± 820 | 2403 ± 524 | n.s. | ||
| Total fat intake (g) | 113 ± 40.3 | 128 ± 42.0 | 108 ± 24.3 | n.s. | ||
| SFA (g) | 31.1 ± 15.5 | 26.9 ± 11.0 | n.s. | 24.4 ± 9.83 | n.s. | 0.026 |
| MUFA (g) | 14.3 ± 6.88 | 14.6 ± 8.44 | 13.9 ± 6.14 | n.s. | ||
| PUFA (g) | 11.2 ± 7.02 | 23.3 ± 3.14 | 0.002 | 22.9 ± 5.87 | 0.004 | <0.001 |
| LA (g) | 9.25 ± 5.93 | 9.14 ± 2.87 | 9.58 ± 3.34 | n.s. | ||
| ALA (g) | 1.39 ± 1.31 | 13.9 ± 0.34 | <0.001 | 14.0 ± 0.53 | <0.001 | <0.001 |
| AA (g) | 0.09 ± 0.07 | 0.16 ± 0.18 | 0.10 ± 0.07 | n.s. | ||
| EPA (g) | 0.19 ± 0.52 | 0.02 ± 0.03 | 0.01 ± 0.03 | n.s. | ||
| DPAn3 (g) | 0.06 ± 0.06 | 0.04 ± 0.06 | 0.03 ± 0.06 | n.s. | ||
| DHA (g) | 0.15 ± 0.18 | 0.06 ± 0.06 | 0.05 ± 0.06 | n.s. | ||
Body weight, BMI, blood pressure and total cholesterol (TC) levels were unchanged during the intervention (Table 1). Also, low density lipoprotein (LDL), high density lipoprotein (HDL) and triglyceride (TG) levels did not change during the intervention period even though statistically significant lower LDL levels (108 ± 18.5 to 96.9 ± 16.6 mg dl−1) were detected after 1 week (no significant differences after 3, 6 and 12 weeks).
Changes of fatty acid profile in RBCs
At baseline, the relative amount of ΣEPA + DHA in the RBCs of the study collective was 4.63 ± 0.19% of total fatty acids (Table 3). Thus, all subjects had a low ΣEPA + DHA status within a narrow range.| Week 0 | Week 1 | t-Testa | Week 3 | t-Testa | Week 6 | t-Testa | Week 12 | t-Testa | An reMb | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SE | Mean ± SE | p (week 1–week 0) | Mean ± SE | p (week 3–week 0) | Mean ± SE | p (week 6–week 0) | Mean ± SE | p (week 12–week 0) | p | |
| The levels are shown as concentration [μg mL−1] in blood and as relative amount [%] of total fatty acids at week 0, 1, 3, 6, and 12 of the high-ALA diet (14.0 ± 0.45 g d−1). AA: arachidonic acid; D5D/D6D index, delta-5/6 desaturase index: calculated according to ref. 41: D5D = C20:4n6/C20:3n6 and D6D = C20:3n6/C18:2n6; EPA: eicosapentaenoic acid; HUFA: highly unsaturated fatty acids; indices of HUFA calculated as follows, modified from ref. 42: % n3 in HUFA = 100 × (C20:5n3 + C22:5n3 + C22:6n3)/(C20:3n6 + C20:4n6 + C22:4n6 + C20:5n3 + C22:5n3 + C22:6n3); % n6 in HUFA = 100 × (C20:3n6 + C20:4n6 + C22:4n6)/(C20:3n6 + C20:4n6 + C22:4n6 + C20:5n3 + C22:5n3 + C22:6n3); MUFA: monounsaturated fatty acids: C14:1n5, C15:1n5, C16:1n7, C17:1n7, C18:1n9, C18:1n7, C20:1n9, C22:1n9, 24:1n9; n.s.: not significant; SFA: saturated fatty acids: C10:0, C11:0, C12:0, C13:0, C14:0, C15:0, C16:0, C17:0, C18:0, C20:0, C21:0, C22:0, C24:0; PUFA: polyunsaturated fatty acids: C18:2n6, C18:3n6, C18:3n3, C20:2n6, C20:3n6, C20:4n6, C20:5n3, C22:4n6, C22:5n3, C22:6n3; SE: standard error; TFA: total fatty acids; Σ n3 PUFA: C18:3n3, C20:3n3, C20:5n3, C22:5n3, C22:6n3; Σ n6 PUFA: C18:2n6, C18:3n6, C20:2n6, C20:3n6, C20:4n6, C22:2n6, C22:4n6; wk: week.a t-Test for paired samples with Holm–Bonferroni correction; significance level p ≤ 0.05.b ANOVA for repeated measures (An reM); significance level p ≤ 0.05. | ||||||||||
| C12:0 μg mL−1 | <0.25 | <0.25 | — | <0.25 | — | <0.25 | — | <0.25 | — | — |
| % of total FA | — | — | — | — | — | — | — | — | — | — |
| C14:0 μg mL−1 | 3.74 ± 0.10 | 3.02 ± 0.15 | 0.001 | 2.73 ± 0.16 | <0.001 | 3.03 ± 0.15 | 0.026 | 3.09 ± 0.17 | n.s. | <0.001 |
| % of total FA | 0.37 ± 0.01 | 0.32 ± 0.01 | n.s. | 0.28 ± 0.01 | n.s. | 0.31 ± 0.01 | n.s. | 0.34 ± 0.02 | n.s. | <0.001 |
| C14:1n5 μg mL−1 | <0.25 | <0.25 | — | <0.25 | — | <0.25 | — | <0.25 | — | — |
| % of total FA | — | — | — | — | — | — | — | — | — | — |
| C15:0 μg mL−1 | 1.75 ± 0.05 | 1.62 ± 0.06 | — | 1.67 ± 0.09 | — | 1.61 ± 0.07 | — | 1.68 ± 0.19 | — | n.s. |
| % of total FA | 0.17 ± 0.01 | 0.17 ± 0.01 | — | 0.17 ± 0.01 | — | 0.16 ± 0.01 | — | 0.18 ± 0.02 | — | n.s. |
| C16:0 μg mL−1 | 196 ± 4.60 | 188 ± 3.00 | — | 187 ± 3.86 | — | 191 ± 5.60 | — | 184 ± 5.58 | — | n.s. |
| % of total FA | 19.3 ± 0.08 | 19.7 ± 0.15 | — | 19.4 ± 0.16 | — | 19.5 ± 0.19 | — | 20.1 ± 0.15 | 0.002 | 0.029 |
| C16:1n7 μg mL−1 | 2.65 ± 0.15 | 2.15 ± 0.12 | — | 2.30 ± 0.15 | n.s. | 2.44 ± 0.17 | n.s. | 2.35 ± 0.18 | n.s. | 0.032 |
| % of total FA | 0.26 ± 0.01 | 0.22 ± 0.01 | — | 0.24 ± 0.01 | — | 0.25 ± 0.01 | — | 0.25 ± 0.01 | — | n.s. |
| C17:0 μg mL−1 | 3.27 ± 0.07 | 3.03 ± 0.07 | — | 3.01 ± 0.12 | n.s. | 3.02 ± 0.09 | n.s. | 2.86 ± 0.10 | 0.039 | 0.012 |
| % of total FA | 0.32 ± 0.01 | 0.32 ± 0.01 | — | 0.31 ± 0.01 | — | 0.31 ± 0.01 | — | 0.31 ± 0.01 | — | — |
| C18:0 μg mL−1 | 154 ± 2.63 | 147 ± 1.92 | — | 148 ± 2.91 | — | 150 ± 3.41 | — | 143 ± 4.19 | — | n.s. |
| % of total FA | 15.1 ± 0.11 | 15.4 ± 0.13 | — | 15.3 ± 0.08 | — | 15.4 ± 0.10 | 0.004 | 15.6 ± 0.13 | <0.001 | <0.001 |
| C18:1n9 μg mL−1 | 135 ± 3.91 | 120 ± 2.44 | 0.036 | 121 ± 2.56 | n.s. | 122 ± 3.69 | n.s. | 115 ± 4.00 | 0.035 | 0.001 |
| % of total FA | 13.3 ± 0.18 | 12.5 ± 0.15 | 0.013 | 12.5 ± 0.16 | 0.014 | 12.5 ± 0.16 | 0.017 | 12.5 ± 0.16 | 0.023 | <0.001 |
| C18:1n7 μg mL−1 | 14.5 ± 0.46 | 12.9 ± 0.28 | 0.044 | 12.6 ± 0.34 | 0.016 | 12.4 ± 0.34 | 0.008 | 11.9 ± 0.42 | 0.010 | <0.001 |
| % of total FA | 1.43 ± 0.02 | 1.35 ± 0.02 | 0.001 | 1.30 ± 0.02 | <0.001 | 1.27 ± 0.02 | <0.001 | 1.30 ± 0.02 | 0.002 | <0.001 |
| C18:2n6 μg mL−1 | 104 ± 3.60 | 97.9 ± 2.94 | — | 102 ± 3.60 | — | 103 ± 3.10 | — | 97.9 ± 3.69 | — | n.s. |
| % of total FA | 10.3 ± 0.32 | 10.3 ± 0.29 | — | 10.5 ± 0.29 | — | 10.6 ± 0.30 | — | 10.7 ± 0.28 | — | n.s. |
| C18:3n6 μg mL−1 | <0.25 | <0.25 | — | <0.25 | — | <0.25 | — | <0.25 | — | — |
| % of total FA | — | — | — | — | — | — | — | — | — | — |
| C19:0 μg mL−1 | <0.25 | <0.25 | — | <0.25 | — | <0.25 | — | <0.25 | — | — |
| % of total FA | — | — | — | — | — | — | — | — | — | — |
| C18:3n3 μg mL−1 | 1.44 ± 0.10 | 4.65 ± 0.22 | <0.001 | 5.47 ± 0.23 | <0.001 | 6.25 ± 0.24 | <0.001 | 5.80 ± 0.28 | <0.001 | <0.001 |
| % of total FA | 0.14 ± 0.01 | 0.49 ± 0.02 | <0.001 | 0.57 ± 0.03 | <0.001 | 0.64 ± 0.02 | <0.001 | 0.63 ± 0.03 | <0.001 | <0.001 |
| C20:0 μg mL−1 | 5.44 ± 0.12 | 4.41 ± 0.12 | <0.001 | 4.34 ± 0.10 | <0.001 | 4.30 ± 0.12 | <0.001 | 3.91 ± 0.12 | <0.001 | <0.001 |
| % of total FA | 0.54 ± 0.01 | 0.46 ± 0.01 | 0.003 | 0.45 ± 0.01 | <0.001 | 0.44 ± 0.01 | <0.001 | 0.43 ± 0.01 | <0.001 | <0.001 |
| C20:1n9 μg mL−1 | 3.14 ± 0.11 | 2.90 ± 0.07 | n.s. | 2.98 ± 0.09 | n.s. | 2.85 ± 0.11 | 0.003 | 2.67 ± 0.10 | 0.003 | 0.002 |
| % of total FA | 0.31 ± 0.01 | 0.30 ± 0.01 | — | 0.31 ± 0.01 | — | 0.29 ± 0.01 | — | 0.29 ± 0.01 | — | n.s. |
| C20:2n6 μg mL−1 | 2.04 ± 0.09 | 1.92 ± 0.09 | — | 2.00 ± 0.12 | — | 1.87 ± 0.10 | — | 1.83 ± 0.12 | — | n.s. |
| % of total FA | 0.20 ± 0.01 | 0.20 ± 0.01 | — | 0.21 ± 0.01 | — | 0.19 ± 0.01 | — | 0.20 ± 0.01 | — | n.s. |
| C20:3n6 μg mL−1 | 15.5 ± 0.81 | 13.7 ± 0.68 | 0.014 | 13.1 ± 0.71 | 0.024 | 13.3 ± 0.71 | 0.009 | 12.2 ± 0.86 | 0.001 | <0.001 |
| % of total FA | 1.53 ± 0.08 | 1.43 ± 0.07 | n.s. | 1.35 ± 0.06 | n.s. | 1.36 ± 0.06 | n.s. | 1.32 ± 0.07 | 0.035 | <0.001 |
| C20:4n6 μg mL−1 | 150 ± 3.90 | 136 ± 2.33 | n.s. | 138 ± 2.69 | n.s. | 137 ± 3.24 | 0.043 | 124 ± 3.53 | 0.001 | <0.001 |
| % of total FA | 14.8 ± 0.14 | 14.3 ± 0.14 | 0.042 | 14.3 ± 0.12 | 0.035 | 14.0 ± 0.12 | <0.001 | 13.5 ± 0.14 | <0.001 | <0.001 |
| C20:5n3 μg mL−1 | 6.13 ± 0.51 | 7.33 ± 0.33 | n.s. | 8.38 ± 0.42 | 0.021 | 10.9 ± 0.67 | <0.001 | 11.0 ± 0.64 | <0.001 | <0.001 |
| % of total FA | 0.60 ± 0.04 | 0.77 ± 0.03 | 0.006 | 0.87 ± 0.04 | 0.002 | 1.12 ± 0.06 | <0.001 | 1.20 ± 0.06 | <0.001 | <0.001 |
| C22:0 μg mL−1 | 17.7 ± 0.56 | 16.6 ± 0.39 | n.s. | 16.9 ± 0.39 | n.s. | 16.9 ± 0.54 | n.s. | 15.3 ± 0.51 | 0.026 | <0.001 |
| % of total FA | 1.75 ± 0.04 | 1.75 ± 0.04 | n.s. | 1.75 ± 0.04 | n.s. | 1.73 ± 0.04 | n.s. | 1.67 ± 0.03 | n.s. | 0.020 |
| C22:1n9 μg mL−1 | 2.42 ± 0.16 | 2.81 ± 0.30 | n.s. | 2.41 ± 0.16 | n.s. | 2.34 ± 0.23 | n.s. | 1.90 ± 0.20 | n.s. | 0.016 |
| % of total FA | 0.24 ± 0.02 | 0.29 ± 0.03 | n.s. | 0.25 ± 0.02 | n.s. | 0.25 ± 0.03 | n.s. | 0.21 ± 0.02 | n.s. | 0.042 |
| C22:4n6 μg mL−1 | 31.6 ± 1.01 | 31.6 ± 1.14 | n.s. | 30.7 ± 1.02 | n.s. | 29.3 ± 1.15 | n.s. | 25.1 ± 1.07 | <0.001 | <0.001 |
| % of total FA | 3.13 ± 0.11 | 3.20 ± 0.10 | n.s. | 3.18 ± 0.09 | n.s. | 2.99 ± 0.08 | n.s. | 2.73 ± 0.07 | <0.001 | <0.001 |
| C22:5n3 μg mL−1 | 25.9 ± 0.82 | 25.2 ± 0.59 | n.s. | 28.2 ± 0.80 | n.s. | 30.2 ± 1.03 | 0.033 | 32.3 ± 1.35 | 0.014 | <0.001 |
| % of total FA | 2.55 ± 0.05 | 2.64 ± 0.05 | n.s. | 2.92 ± 0.06 | <0.001 | 3.09 ± 0.07 | <0.001 | 3.52 ± 0.10 | <0.001 | <0.001 |
| C24:0 μg mL−1 | 45.6 ± 1.25 | 45.3 ± 0.81 | — | 46.6 ± 1.04 | — | 47.1 ± 1.15 | — | 44.0 ± 1.34 | — | n.s. |
| % of total FA | 4.50 ± 0.08 | 4.75 ± 0.08 | 0.001 | 4.83 ± 0.10 | <0.001 | 4.83 ± 0.08 | 0.007 | 4.81 ± 0.10 | 0.004 | <0.001 |
| C22:6n3 μg mL−1 | 41.0 ± 1.93 | 37.0 ± 1.32 | n.s. | 36.1 ± 1.37 | n.s. | 35.1 ± 1.06 | 0.010 | 30.4 ± 1.09 | <0.001 | <0.001 |
| % of total FA | 4.03 ± 0.16 | 3.90 ± 0.15 | n.s. | 3.74 ± 0.13 | n.s. | 3.62 ± 0.12 | 0.017 | 3.33 ± 0.11 | 0.001 | <0.001 |
| C24:1n9 μg mL−1 | 51.8 ± 1.88 | 50.4 ± 1.13 | n.s. | 50.6 ± 1.03 | n.s. | 50.0 ± 1.65 | n.s. | 44.4 ± 1.52 | 0.019 | <0.001 |
| % of total FA | 5.10 ± 0.11 | 5.28 ± 0.10 | 0.003 | 5.25 ± 0.09 | 0.035 | 5.11 ± 0.11 | n.s. | 4.84 ± 0.09 | n.s. | <0.001 |
| ΣTFA μg mL−1 | 1016 ± 22.0 | 955 ± 12.1 | n.s. | 966 ± 17.3 | n.s. | 977 ± 22.2 | n.s. | 918 ± 26.1 | n.s. | 0.015 |
| ΣSFA μg mL−1 | 427 ± 8.49 | 408 ± 5.39 | — | 411 ± 7.59 | — | 418 ± 10.3 | — | 399 ± 11.3 | — | n.s. |
| % of total FA | 42.1 ± 0.16 | 42.8 ± 0.24 | n.s. | 42.5 ± 0.16 | n.s. | 42.7 ± 0.16 | 0.001 | 43.5 ± 0.23 | <0.001 | <0.001 |
| ΣMUFA μg mL−1 | 210 ± 6.24 | 191 ± 3.68 | n.s. | 192 ± 3.80 | n.s. | 193 ± 5.53 | n.s. | 179 ± 5.89 | 0.019 | <0.001 |
| % of total FA | 20.7 ± 0.28 | 20.0 ± 0.23 | n.s. | 19.9 ± 0.24 | n.s. | 19.7 ± 0.24 | 0.023 | 19.4 ± 0.23 | 0.003 | <0.001 |
| ΣPUFA μg mL−1 | 378 ± 8.35 | 355 ± 4.74 | n.s. | 364 ± 7.20 | n.s. | 367 ± 7.43 | n.s. | 340 ± 9.73 | n.s. | 0.017 |
| % of total FA | 37.3 ± 0.23 | 37.2 ± 0.24 | — | 37.7 ± 0.20 | — | 37.6 ± 0.26 | — | 37.1 ± 0.26 | — | n.s. |
| Σn3 PUFA μg mL−1 | 74.4 ± 2.83 | 74.2 ± 1.43 | n.s. | 78.1 ± 1.80 | n.s. | 82.4 ± 2.01 | n.s. | 79.4 ± 2.24 | n.s. | 0.023 |
| % of total FA | 7.32 ± 0.19 | 7.79 ± 0.15 | <0.001 | 8.10 ± 0.13 | <0.001 | 8.46 ± 0.17 | <0.001 | 8.68 ± 0.13 | <0.001 | <0.001 |
| Σn6 PUFA μg mL−1 | 303 ± 6.55 | 280 ± 4.66 | n.s. | 286 ± 6.14 | n.s. | 285 ± 6.33 | n.s. | 261 ± 8.01 | 0.004 | <0.001 |
| % of total FA | 29.9 ± 0.27 | 29.4 ± 0.29 | n.s. | 29.6 ± 0.25 | n.s. | 29.2 ± 0.26 | n.s. | 28.5 ± 0.25 | 0.001 | <0.001 |
| ΣEPA & DHA μg mL−1 | 47.1 ± 2.36 | 46.4 ± 1.44 | n.s. | 44.5 ± 1.46 | n.s. | 46.0 ± 1.32 | n.s. | 41.3 ± 1.27 | n.s. | 0.023 |
| % of total FA | 4.63 ± 0.19 | 4.67 ± 0.16 | — | 4.61 ± 0.13 | — | 4.73 ± 0.15 | — | 4.52 ± 0.11 | — | n.s. |
| Σn6/Σn3 PUFA | 4.15 ± 0.13 | 3.81 ± 0.10 | <0.001 | 3.68 ± 0.08 | 0.002 | 3.48 ± 0.09 | <0.001 | 3.30 ± 0.07 | <0.001 | <0.001 |
| AA/EPA | 26.5 ± 1.55 | 19.3 ± 0.90 | <0.001 | 17.0 ± 0.69 | <0.001 | 13.3 ± 0.80 | <0.001 | 11.8 ± 0.59 | <0.001 | <0.001 |
| D5D index | 10.1 ± 0.51 | 10.3 ± 0.46 | — | 10.9 ± 0.47 | — | 10.7 ± 0.52 | — | 10.8 ± 0.68 | — | n.s. |
| D6D index | 0.15 ± 0.01 | 0.14 ± 0.01 | n.s. | 0.13 ± 0.01 | <0.001 | 0.13 ± 0.01 | <0.001 | 0.12 ± 0.01 | <0.001 | <0.001 |
| % n3 in HUFA | 27.0 ± 0.64 | 27.9 ± 0.55 | 0.010 | 28.6 ± 0.47 | 0.002 | 29.8 ± 0.49 | <0.001 | 31.4 ± 0.47 | <0.001 | <0.001 |
| % n6 in HUFA | 73.1 ± 0.64 | 72.2 ± 0.55 | 0.010 | 71.5 ± 0.47 | 0.002 | 70.2 ± 0.49 | <0.001 | 68.6 ± 0.47 | <0.001 | <0.001 |
Prior to the intervention, AA was present in the highest concentrations in the RBCs (150 ± 3.90 μg mL−1) among all PUFAs, followed by LA (104 ± 3.60 μg mL−1), DHA (41.0 ± 1.93 μg mL−1), C22:4n6 (31.6 ± 1.01 μg mL−1), DPAn3 (25.9 ± 0.82 μg mL−1) and EPA (6.13 ± 0.51 μg mL−1) (Table 3). The ALA concentrations in the RBCs were low with 1.44 ± 0.10 μg mL−1 at week 0. In the course of the high-ALA diet, the concentrations and relative amount of ALA, EPA and DPAn3 in the RBCs (Fig. 1A–C) increased significantly and decreased again in the follow-up period, whereas the DHA concentrations decreased in response to the high-ALA diet (Fig. 1D). In the following, only the concentrations are discussed unless the relative fatty acid distribution showed a different trend. It is noteworthy that the relative amount of ΣEPA + DHA in the RBCs did not change in response to the high-ALA diet (Table 3).
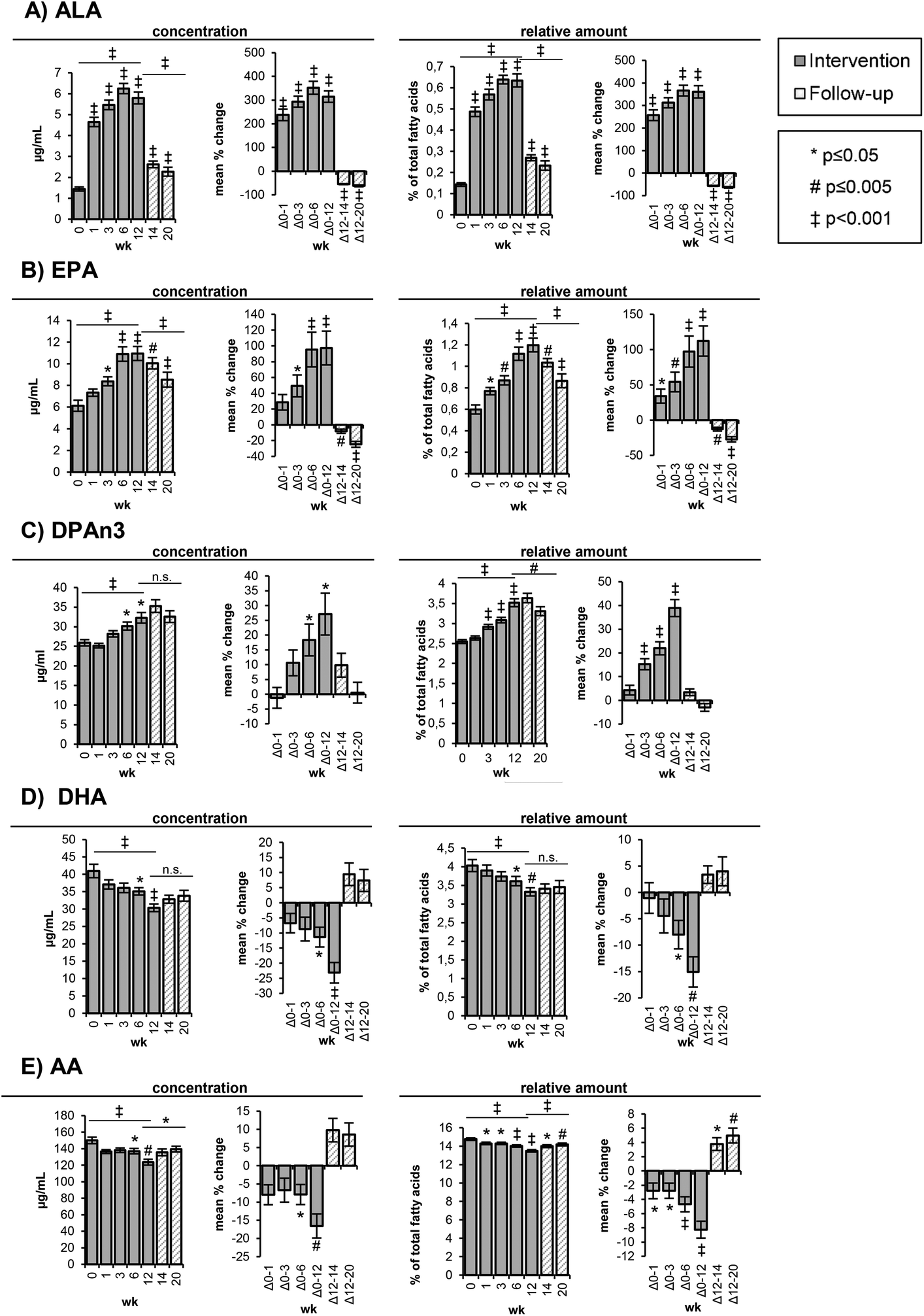 | ||
| Fig. 1 Content of selected polyunsaturated fatty acids in red blood cells. Bars represent mean ± SE. The levels are shown as concentrations [μg mL−1] and as relative amounts [%] of total fatty acids at week 0, 1, 3, 6, and 12 of the high-ALA diet (14.0 ± 0.45 g d−1) and at week 14 and 20 of follow-up. AA: arachidonic acid (C20:4n6); ALA: alpha-linolenic acid (C18:3n3); DHA: docosahexaenoic acid (C22:6n3); DPAn3: docosapentaenoic acid (C22:5n3); EPA: eicosapentaenoic acid (C20:5n3); wk: week. | ||
The ALA concentrations in the RBCs increased time-dependently (p < 0.001) from 1.44 ± 0.10 μg mL−1 (week 0) to 4.65 ± 0.22 μg mL−1 (week 1) and 5.47 ± 0.23 μg mL−1 (week 3) corresponding to a mean change of 238 ± 24% and 294 ± 23%, respectively (Table 3, Fig. 1A). The highest ALA levels (6.25 ± 0.24 μg mL−1) were observed after 6 weeks of the high-ALA diet. In the follow-up period, the ALA concentrations dropped (p < 0.001) rapidly from 5.80 ± 0.28 μg mL−1 at week 12 to 2.62 ± 0.16 μg mL−1 after 2 weeks (week 14) and 2.27 ± 0.21 μg mL−1 after 8 weeks (week 20) (Table S3†).
The EPA levels in the RBCs increased linearly and almost doubled (p < 0.001) in concentration from baseline (6.13 ± 0.51 μg mL−1) to 10.9 ± 0.67 μg mL−1 at week 6 and 11.0 ± 0.64 μg mL−1 at week 12 (Table 3, Fig. 1B). In the follow-up period, the concentrations decreased more slowly compared to ALA with 10.0 ± 0.52 μg mL−1 after 2 weeks (week 14) and 8.53 ± 0.69 μg mL−1 after 8 weeks (week 20) (Table S3†).
The changes in DPAn3 concentrations were smaller and only significant after 6 (p = 0.033) and 12 weeks (p = 0.014) of the high-ALA diet, whereas the changes of the relative amounts were statistically significant (p < 0.001) after 3, 6 and 12 weeks (Table 3, Fig. 1C). From baseline to week 12, the DPAn3 concentrations increased from 25.9 ± 0.51 μg mL−1 to 32.3 ± 1.35 μg mL−1 and remained high over the follow-up period (35.3 ± 1.63 μg mL−1 at week 14 and 32.6 ± 1.49 at week 20) (Table S3†).
A linear and significant (p < 0.001) reduction of DHA concentrations was observed between baseline (41.0 ± 1.93 μg mL−1) and week 12 (30.4 ± 1.09 μg mL−1) (Table 3, Fig. 1D). After 2 and 8 weeks of follow-up, the DHA concentrations slightly increased to 33.8 ± 1.57 μg mL−1 (n.s.) (Table S3†).
The initial AA concentrations in the RBCs (150 ± 3.90 μg mL−1) were only marginally reduced (136–138 μg mL−1) in the first 6 weeks and then significantly (p = 0.001) dropped to 124 ± 3.53 μg mL−1 at week 12 (Table 3, Fig. 2), corresponding to a mean decrease of 16.6 ± 3.31%. The relative AA amount was statistically significantly reduced at all time points of the intervention. In the follow-up period, the AA concentrations increased, although not significantly, whereas the relative increase was significant (p = 0.012 (week 14) and p = 0.001 (week 20)) (Table S3†).
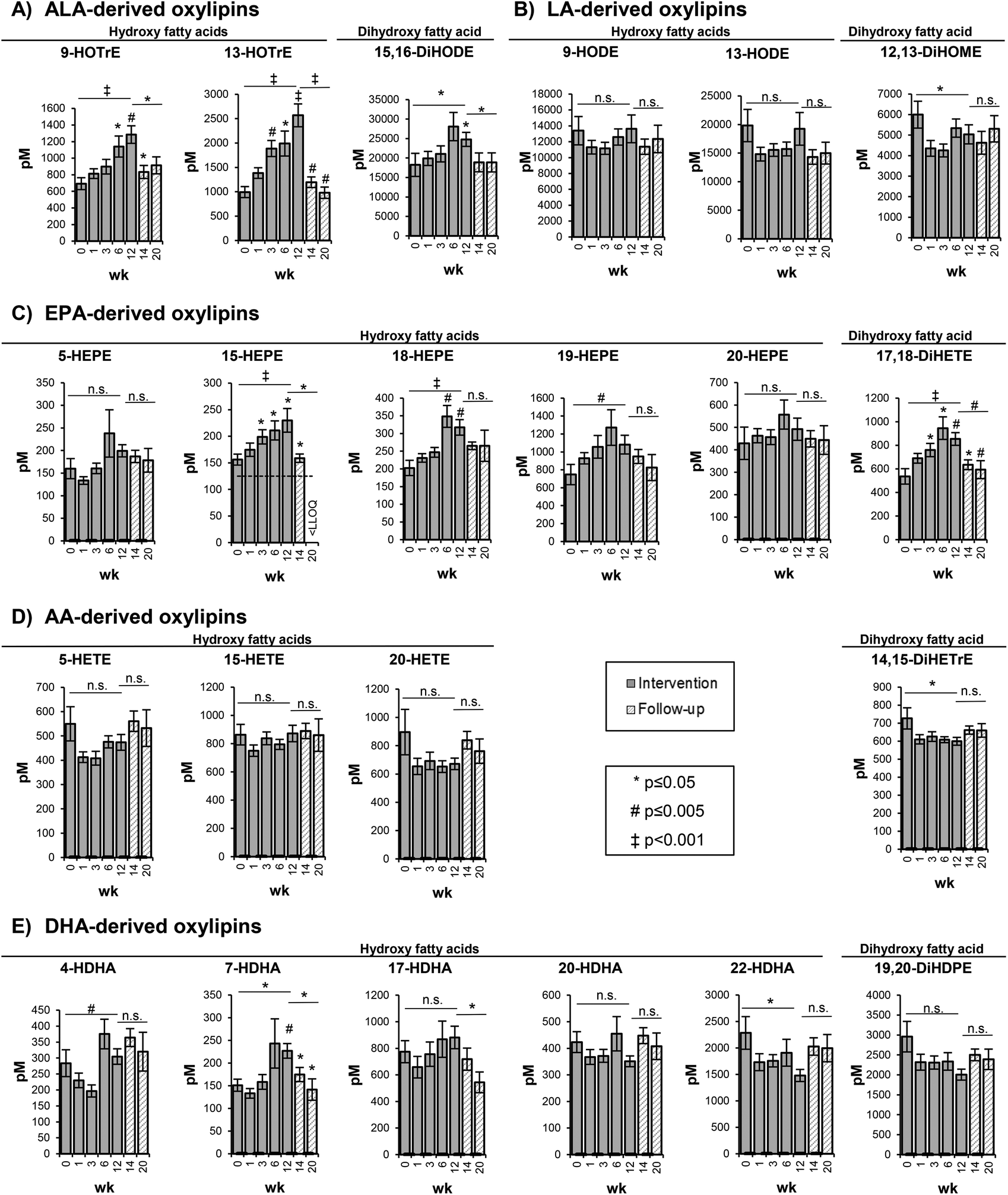 | ||
| Fig. 2 Concentration of selected free oxylipins in the plasma. Bars represent mean ± SE [pM]. The plasma concentrations of free oxylipins are shown at week 0, 1, 3, 6, and 12 of the high-ALA diet (14.0 ± 0.45 g d−1) and at week 14 and 20 of follow-up. AA: arachidonic acid (C20:4n6); ALA: alpha-linolenic acid (C18:3n3); DHA: docosahexaenoic acid (C22:6n3); DiHETE: dihydroxy eicosatetraenoic acid; DiHETrE: dihydroxy eicosatrienoic acid; DiHDPE: dihydroxy docosapentaenoic acid; DiHODE: dihydroxy octadecadienoic acid; DiHOME: dihydroxy octadecenoic acid; EPA: eicosapentaenoic acid (C20:5n3); HDHA: hydroxy docosahexaenoic acid; HEPE: hydroxy eicosapentaenoic acid; HETE: hydroxy eicosatetraenoic acid; HODE: hydroxy octadecadienoic acid; HOTrE: hydroxy octadecatrienoic acid; LA: linoleic acid (C18:2n6); LLOQ: lower limit of quantification; SE: standard error; wk: week. | ||
Both the absolute concentrations and relative amount of LA in the RBCs did not change in the course of the high-ALA diet and follow-up period (Tables 3 and S3†). The concentration of SFA, PUFA and Σn3 PUFA in the RBCs remained constant during the study. However, the concentrations of MUFA and Σn6 PUFA in the RBCs were significantly decreased after 12 weeks of the high-ALA diet (Table 3). The MUFA concentrations decreased (p = 0.019) from 210 ± 6.24 μg mL−1 at week 0 to 179 ± 5.89 μg mL−1 at week 12, while the Σn6 PUFA concentrations decreased (p = 0.004) from 303 ± 6.55 μg mL−1 at week 0 to 261 ± 8.01 μg mL−1 at week 12 (Table 3).
The ratio of Σn6 to Σn3 PUFA significantly declined (p < 0.001) after 1 week of the high-ALA diet from 4.15 ± 0.13 (week 0) to 3.81 ± 0.10 (week 1) and further to 3.30 ± 0.07 after week 12 (Table 3). In the follow-up period, the Σn6/Σn3 PUFA ratio increased (p = 0.002) again to 3.73 ± 0.13 (week 20) (Table S3†).
Consistently, the ratio of AA to EPA dropped (p < 0.001) time-dependently from 26.5 ± 1.55 (week 0) to 11.8 ± 0.59 at week 12 and slowly increased (p < 0.001) in the follow-up period to 17.3 ± 1.06 (Table 3). The activity of D6D decreased (p < 0.001) in response to the high-ALA diet from 0.15 ± 0.01 (week 0) to 0.12 ± 0.01 at week 12 and increased (p < 0.001) again in the follow-up period (week 20: 0.17 ± 0.01) (Table 3). The percentage of n3 in HUFA increased from 27.0 ± 0.64 (week 0) to 31.4 ± 0.47 (week 12), whereas the percentage of n6 in HUFA decreased from 73.1 ± 0.64 (week 0) to 68.6 ± 0.47 (week 12) following the high-ALA diet (Table 3). A reversion of this change could be observed in the follow-up period: n3 in HUFA decreased again from 31.4 ± 0.47% (week 12) to 28.9 ± 0.63% (week 20) and n6 in HUFA increased from 68.6 ± 0.47% (week 12) to 71.1 ± 0.63 (Table S3†).
Changes of oxylipin concentrations in the plasma
The changes in the concentration of free hydroxy- and dihydroxy-PUFA in the plasma were represented by the shift in concentrations of their precursor PUFAs in the RBCs. As shown exemplarily for selected oxylipins, representing the metabolites of 5-LOX, 15-LOX and CYP catalyzed epoxygenation and hydrolysis by sEH, a similar but less pronounced trend compared to the precursor PUFA was observed (Fig. 2).5- and 15-LOX-derived hydroxy-PUFA from ALA, i.e. 9-HOTrE and 13-HOTrE, increased time-dependently from 693 ± 71.7 pM (week 0) to 1285 ± 106 pM (week 12) (p = 0.001) and from 993 ± 113 pM (week 0) to 2569 ± 234 pM (week 12) (p < 0.001) (Fig. 2, Table S4†). The highest concentrations of 9- and 13-HOTrE were observed at week 12. The concentration of the terminal dihydroxy-PUFA 15,16-DiHODE also increased from 18243 ± 2970 pM (week 0) to 24795 ± 1802 pM (week 12) (p = 0.033); however, it was less pronounced compared to ALA-derived hydroxy-PUFA and, though not significant, the highest concentration was observed at week 6 (28086 ± 3631 pM).
LA-derived oxylipins remained unchanged; however, the concentrations varied in the course of the trial (Fig. 2, Table S4†). This is consistent with the LA concentration in the RBCs that did not show significant changes in response to the high-ALA diet.
For EPA-derived oxylipins, as exemplarily shown in Fig. 2 for 5-, 15-, 18- and 20-HEPE and 17,18-DiHETE, an increase from week 0 to week 6 was observed followed by a slight decrease at week 12 (except 15-HEPE).
For most of the AA-derived hydroxy- and dihydroxy-PUFA, such as 5-, 15-, 20-HETE and 14,15-DiHETrE, a slight though statistically not significant decrease in concentration after 1 week of ALA supplementation was observed (Fig. 2). Similarly, no consistent shift of DHA-derived oxylipins was observed (Fig. 2, Table S4†). Overall, the oxylipin levels were reflected by the respective precursor fatty acid concentrations in the RBCs, though changes in the oxylipin levels occurred at later time points and were, due to higher SE, less pronounced compared to the precursor fatty acids.
Discussion
Factors like age,43,44 BMI,44 smoking45 and genotype46 showed an influence on the LC n3 PUFA status, especially on the conversion from ALA to DHA. Recently, it has been shown that the activity of endogenous EPA and DHA synthesis adapted during evolution in the presence of these fatty acids in the diet.47 Several studies observed higher circulating relative DHA amounts in women compared to men independent of dietary intake48–50 and higher conversion rates from ALA and EPA to DHA51 possibly due to the influence of estrogen on the PUFA metabolism.52 Consequently, to minimize the variability, a homogeneous collective of healthy, non-smoking men within a narrow range regarding age (mean age 26.2 ± 4.53 years) and BMI (24.9 ± 2.0 kg m−2) was chosen to investigate the effect of ALA on the LC n3-PUFA levels.In addition, to study the effect of a high daily ALA intake in a Western diet on the EPA and DHA blood levels, it is crucial to choose a study collective with low baseline EPA/DHA levels, since the expected conversion of ALA to EPA and DHA is the highest compared to subjects with a moderate or high EPA/DHA status.
We selected probands basically eating a mixed Western diet with a low meat and fish consumption and explicitly screened for low blood LC n3 PUFA status. The relative ΣEPA + DHA level in RBCs, similar to omega-3 index, was 4.15 ± 0.13% of total fatty acids and comparable to the (low) mean omega-3 index of men in the U.S. and Germany which is associated with the risk of cardiovascular disease.53
As expected, the high-ALA diet – providing an ALA amount of about 4.7 en% – resulted in a strong increase in the ALA levels in the RBCs. The incorporation of ALA (and other PUFAs) into the RBCs is determined by the blood cell turnover (the mean life span of a red blood cell is approximately 120 days in circulation) and thus the ALA and its bioconverted longer chain n3 PUFAs EPA and DHA do not fully reach the RBCs. Nevertheless, a 238 ± 24% increase of ALA concentration in the RBCs was observed after one week.
The ratio of LA to ALA in the RBCs (74.8 ± 3.54) at baseline is much higher than expected from the dietary supply of these C18 PUFAs (LA/ALA intake ratio: 7.70 ± 3.75). Even after a 12-week intake of similarly high amounts of LA (9.38 ± 3.08 g d−1) and ALA (14.0 ± 0.45 g d−1) with a ratio of 0.67 ± 0.21, the ratio of LA to ALA in the RBCs remained high (17.5 ± 0.97). The possible reasons for this could be that ALA significantly differs from LA in absorption, tissue distribution, membrane incorporation, and/or degradation. Most likely, a high percentage of 60–85% of ALA is rapidly degraded by beta-oxidation54 before it becomes available for tissue distribution and membrane integration as well as elongation and desaturation to EPA.
The high-ALA diet also affected the concentrations of other PUFAs with significantly increased EPA and DPAn3 as well as significantly decreased DHA and AA concentrations in the RBCs. Of note, the concentration of EPA was with a change of about 5 μg mL−1 similarly elevated as the ALA concentration. The percentage increase in the concentrations and relative amounts was more pronounced for EPA (week 12: 97 ± 21% and 112 ± 17%, respectively) compared to DPAn3 (week 12: 27 ± 7% and 39 ± 4%, respectively). It is likely that increasing EPA levels are the result of a conversion from ALA; however, it cannot be excluded that the retroconversion of DHA to DPAn3 and EPA occurred, which would also (partly) explain decreasing DHA concentrations. These results are consistent with previous studies, where a dose-dependent and higher increase of EPA compared to DPAn3 was observed in the RBCs55,56 and plasma and platelet phospholipids.35 However, supplementing EPA is more efficacious compared to ALA in raising the EPA blood levels.19 A six-week supplementation of 600 mg EPA per day, which corresponds to a ∼10-fold increase compared to the median intake in Germany, resulted in a 138% increase of the relative serum phospholipid EPA amount.57 A higher dose of 2.0 g EPA per day caused a 325% increase of the relative EPA amount in the RBCs.58
The initial ALA concentrations were also low (1.44 ± 0.10 μg mL−1) compared to the EPA concentrations (6.13 ± 0.51 μg mL−1) and the ratio of ALA to EPA increased in the first three weeks and slightly decreased thereafter. The high-ALA diet led to a strong increase of ALA in the RBCs. However, as discussed before, with increasing ALA concentrations, the rate of ALA catabolism increases24 as observed in the moderate drop of the ALA levels between week 6 and week 12. This may also explain the steady state in the EPA concentrations between week 6 (10.9 ± 0.67 μg mL−1) and week 12 (11.0 ± 0.64 μg mL−1).
The observed reduction of the DHA levels is surprising and in contrast to earlier studies, which determined a conversion (rate) of ALA to DHA of 0.5–5%.16–21 However, in these studies, the ingested ALA amount was low (1.0–3.5 g)17,59 compared to ours. A study with a similar design as our study including a high daily ALA dose (15.4 ± 7.5 g) showed no significant effect on relative DHA amounts in plasma and platelet phospholipids,35 possibly due to low sample size (n = 7), an inhomogeneous initial LC n3 PUFA status and a highly variable ALA intake (high SD). In contrast, we observed significantly decreased DHA concentrations and relative amounts in the RBCs in response to the high-ALA diet. It should be noted that the biological variability of fatty acid concentrations in the RBCs is small compared to plasma and plasma phospholipids.60 Nonetheless, several studies indicated a tendency for declining DHA levels in response to high ALA consumption, which was in most cases marginal and not statistically significant.35,61–69 One possible explanation for the decreasing DHA concentrations in the RBCs may be that DHA accumulates in the nerve cells of the brain.70 While human studies are limited to blood as the medium of investigation, animal studies demonstrated that the conversion of ALA to EPA and DHA is tissue specific. In a rat study with high-ALA chia seed supplementation, the accumulation of DHA in the heart and liver was observed, while the plasma DHA concentrations remained constant.71
In our study, the ALA intake from the background diet was tightly controlled and hence the variability of total daily ALA intake in the intervention period was low (∼1.9 g). Only a few other studies found a significant reduction of the relative DHA levels in mononuclear cell phospholipids66 and platelet phosphatidylcholine69 in response to an ALA enriched diet. However, the LA amount in the diet of the studies by Kew et al. (13.1 and 16.2 g d−1)66 and Weaver et al. (22.5 g d−1)69 was much higher than in our study (<10 g) based on food questionnaires (Table 2).
The conversion efficiency of ALA to DHA appears to be affected by a high LA, ALA and total PUFA intake. A rat study observed the highest conversion of ALA to DHA as a result of feeding a narrow dietary range of 1–2 en% LA and 1–3 en% ALA, while the DHA levels were suppressed to basal levels (∼2% total fatty acids) with the total PUFA levels above 3 en%.72 Excessive LA and ALA compete with LC n3 PUFAs for the rate-limiting enzyme Δ6-desaturase.73 Δ6-desaturase catalyzes the desaturation of LA to GLA, of ALA to stearidonic acid (C18:4n3) and also of tetracosapentaenoic acid (C24:5n3) to tetracosahexaenoic acid (C24:6n3), which is finally shortened to form DHA by peroxisomal β-oxidation.15
The intake of LA, AA, EPA, DPAn3, and DHA did not significantly change during the intervention compared to baseline (3-day dietary questionnaires Table 2). In addition, the ALA intake from the background diet (minus the ALA intake via the daily linseed oil ingestion) did not change between week 0, week 6 and week 12. On the other hand, the intake of EPA, DPAn3, and DHA via the background diet was slightly decreased, possibly due to the advice to avoid oily fish meals during the intervention time. However, it seems unlikely that this statistically insignificant decline caused the observed decrease in the DHA concentrations in the RBCs.
However, it should be noted that estimates of dietary fat intake relied on self-reported data and are potentially biased by food choice, incomplete dietary protocols and methodological limitations associated with accurate fatty acid composition data in food databases.74
The beneficial health effects of LC n3 PUFA are believed to be (partly) mediated by oxidized mediators formed in the AA cascade.10 A correlation between higher levels of precursor n3 PUFAs (e.g. EPA and DHA) and their oxylipins was demonstrated in different intervention studies.75–79 Accordingly, in the present study, the changes of the oxylipin levels in the plasma are generally reflected by the changes of the respective precursor fatty acids in the RBCs. As expected, the levels of ALA-derived oxylipins increased in response to the higher dietary intake of ALA. In contrast to the ALA concentrations in the RBCs, which increased more than 3-fold after only 1 week of ALA supplementation, ALA-derived oxylipins were only slightly but not significantly elevated. Moreover, whilst the ALA concentration in the RBCs seemed to reach a steady state after 6 weeks of the high-ALA diet, ALA-derived oxylipins, such as hydroxy-PUFA 9- and 13-HOTrE, increased steadily until week 12 up to 1.9-fold and 2.6-fold, respectively. The slower and less pronounced rise of ALA-derived oxylipins compared to their precursor fatty acid was not observed for DHA-derived oxylipins compared to the blood cell concentrations of DHA in a similar study.78,79 A possible explanation is the lower baseline concentration of ALA compared to DHA, which might have led to a more rapid increase and the higher supplemented dose.77
With regard to oxylipin formation, ALA is mostly discussed as a precursor of LC n3 PUFA;80 therefore, the biological role of ALA-derived oxylipins is only poorly understood. Some studies demonstrated positive biological effects of ALA-derived oxylipins;81,82 however, further investigation of these mediators needs to be carried out as ALA-derived oxylipins are present in relevant concentrations in humans on a Western diet.10
Consistent with the elevated levels of EPA in the RBCs (1.8-fold increase at week 6), a high-ALA diet leads to increasing concentrations of EPA-derived hydroxy- and dihydroxy-PUFA in the plasma (∼1.3- to 1.8-fold at week 6). The higher levels of EPA-derived oxylipins upon a high-ALA diet may have beneficial health effects, e.g. 18-HEPE – a precursor of pro-resolving and anti-inflammatory E-series resolvins83 – concentrations increased 1.7-fold at week 6. However, it has to be noted that direct supplementation with EPA raises the EPA-derived oxylipin levels more efficiently.84
Although the DHA concentrations in the RBCs were significantly lowered in response to the high-ALA diet, the DHA-derived oxylipin levels showed no consistent shift towards lower levels.
The conversion rates of ALA to EPA and DHA as well as the formation of oxylipins from n3 PUFA are influenced by the presence of n6 PUFA competing for the same enzymes.15,85 Several studies demonstrated that n3 PUFA supplementation leads to declining AA and AA-derived oxylipin concentration; however, the results were heterogeneous between different intervention studies.84 Despite the significant decrease of AA in the RBCs in the present study, only a slight but not significant decline of AA-derived oxylipins, e.g. hydroxy-PUFA 5- and 20-HETE, was observed, while no effect was observed for, e.g., the 15-LOX product 15-HETE.
Similar results were obtained for LA-derived oxylipins and no relevant reduction in response to high-ALA intake was observed. Most likely, the excess of LA in the diet (and consequently in the RBC membrane concentration) was too high to be modified by a high-ALA diet. A decrease of the LA/ALA ratio (from 74.8 ± 3.54 (week 0) to 17.5 ± 0.97 (week 12)) and the ratio of LOX-derived hydroxy-PUFA 9-HODE/9-HOTrE (19.4 ± 1.04 (week 0) to 10.6 ± 0.68 (week 12)) and 13-HODE/13-HOTrE (from 20.5 ± 1.39 (week 0) to 7.73 ± 0.78 (week 12)) results from elevated ALA and ALA-derived oxylipin concentrations with constant LA and LA-derived oxylipin concentrations. A reduction of LA and its oxylipins is assumed to be beneficial, as negative health effects were observed for LA metabolites such as sEH products of the CYP-derived epoxy-PUFA.86–88 A reduction of the LA metabolite 9,10-DiHOME by supplementation with a lower (6 g d−1) dose of ALA compared to our study was demonstrated by Caligiuri et al. in young individuals (19–28 years),89 after only 4 weeks of the intervention. However, the participants had to abstain from dietary oils, which might have altered their normal eating habits, thus, leading to shifts in the fatty acid and oxylipin pattern.
Conclusions
Our results demonstrate that a high-ALA diet of 14.0 ± 0.45 g day−1 – which is 8–12 times higher than the common intake recommendation for this essential fatty acid – results in a significant increase in ALA, EPA and DPAn3 concentrations in RBCs and a significant decline in DHA concentrations. However, the ΣEPA + DHA concentration in RBCs – which is associated with cardiac, cerebral, and general health status – was not affected in response to a high-ALA diet. The changes in the plasma oxylipin levels were generally reflected by their precursor fatty acids in RBCs. The high-ALA diet failed to modulate LA and LA-derived oxylipins. Our results demonstrate on both the fatty acid as well as the oxylipin level that on a Western diet (with high LA intake), ALA is not a significant source for endogenous EPA and DHA levels.Abbreviations
| AA | Arachidonic acid |
| ALA | Alpha-linolenic acid |
| BMI | Body mass index |
| COX | Cyclooxygenase |
| CYP | Cytochrome P450 |
| DHA | Docosahexaenoic acid |
| DiHETE | Dihydroxy eicosatetraenoic acid |
| DiHETrE | Dihydroxy eicosatrienoic acid |
| DiHODE | Dihydroxy octadecadienoic acid |
| DiHOME | Dihydroxy octadecenoic acid |
| DPAn3 | n3 docosapentaenoic acid |
| D5D | Delta-5 desaturase |
| D6D | Delta-6 desaturase |
| en% | Percent of total energy |
| EPA | Eicosapentaenoic acid |
| FAME | Fatty acid methyl ester |
| HDHA | Hydroxy docosahexaenoic acid |
| HDL | High density lipoprotein |
| HEPE | Hydroxy eicosapentaenoic acid |
| HETE | Hydroxy eicosatetraenoic acid |
| HODE | Hydroxy octadecadienoic acid |
| HOTrE | Hydroxy octadecatrienoic acid |
| HUFA | Highly unsaturated fatty acids |
| IS | Internal standard |
| LA | Linoleic acid |
| LC | Long chain |
| LC-MS | Liquid chromatography-mass spectrometry |
| LDL | Low density lipoprotein |
| LLOQ | Lower limit of quantification |
| LOX | Lipoxygenases |
| MUFA(s) | Monounsaturated fatty acid(s) |
| n.s. | Not significant |
| n3 | Omega-3 |
| n6 | Omega-6 |
| PUFA(s) | Polyunsaturated fatty acid(s) |
| RBCs | Red blood cells |
| SD | Standard deviation |
| SE | Standard error |
| sEH | Soluble epoxide hydrolase |
| SFA(s) | Saturated fatty acid(s) |
| TC | Total cholesterol |
| TG | Triglycerides |
| wk(s) | Week(s) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the German Research Foundation (Grant SCHE 1801 and SCHU 2516) to NHS and JPS.We would like to thank the participants who contributed their time to this project.
References
- P. C. Calder, Clin. Sci., 2004, 107, 1–11 CrossRef CAS PubMed.
- P. C. Calder, Eur. J. Lipid Sci. Technol., 2014, 116, 1280–1300 CrossRef CAS.
- W. S. Harris, Curr. Atheroscler. Rep., 2009, 11, 411–417 CrossRef CAS PubMed.
- P. E. Marik and J. Varon, Clin. Cardiol., 2009, 32, 365–372 CrossRef PubMed.
- D. Mozaffarian and J. H. Y. Wu, J. Am. Coll. Cardiol., 2011, 58, 2047–2067 CrossRef CAS PubMed.
- P. C. Calder, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2015, 1851, 469–484 CrossRef CAS PubMed.
- G. Zhang, D. Panigrahy, L. M. Mahakian, J. Yang, J.-Y. Liu, K. S. Stephen Lee, H. I. Wettersten, A. Ulu, X. Hu, S. Tam, S. H. Hwang, E. S. Ingham, M. W. Kieran, R. H. Weiss, K. W. Ferrara and B. D. Hammock, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6530–6535 CrossRef CAS PubMed.
- J. P. SanGiovanni, S. Parra-Cabrera, G. A. Colditz, C. S. Berkey and J. T. Dwyer, Pediatrics, 2000, 105, 1292–1298 CrossRef CAS PubMed.
- C. Arnold, M. Markovic, K. Blossey, G. Wallukat, R. Fischer, R. Dechend, A. Konkel, C. von Schacky, F. C. Luft, D. N. Muller, M. Rothe and W.-H. Schunck, J. Biol. Chem., 2010, 285, 32720–32733 CrossRef CAS PubMed.
- M. Gabbs, S. Leng, J. G. Devassy, M. Monirujjaman and H. M. Aukema, Adv. Nutr., 2015, 6, 513–540 CrossRef CAS PubMed.
- C. Morisseau and B. D. Hammock, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 37–58 CrossRef CAS PubMed.
- S. Raatz, Z. Conrad, L. Johnson, M. Picklo and L. Jahns, Nutrients, 2017, 9, 438 CrossRef PubMed.
- P. Stehle, Eur. J. Food Res. Rev., 2014, 4, 14 Search PubMed.
- International Society for the Studies of Fatty Acids and Lipids.
- B. Lands, Nutrients, 2012, 4, 1338–1357 CrossRef CAS PubMed.
- E. A. Emken, R. O. Adlof and R. M. Gulley, Biochim. Biophys. Acta, 1994, 1213, 277–288 CrossRef CAS.
- N. Salem, R. Pawlosky, B. Wegher and J. Hibbeln, Prostaglandins Leukot. Essent. Fatty Acids, 1999, 60, 407–410 CrossRef CAS.
- S. H. Vermunt, R. P. Mensink, M. M. Simonis and G. Hornstra, Lipids, 2000, 35, 137–142 CrossRef CAS PubMed.
- J. T. Brenna, N. Salem, A. J. Sinclair and S. C. Cunnane, Prostaglandins Leukot. Essent. Fatty Acids, 2009, 80, 85–91 CrossRef CAS PubMed.
- G. Burdge, Curr. Opin. Clin. Nutr. Metab. Care, 2004, 7, 137–144 CrossRef CAS PubMed.
- M. Plourde and S. C. Cunnane, Appl. Physiol., Nutr., Metab., 2007, 32, 619–634 CrossRef CAS PubMed.
- L. M. Arterburn, E. B. Hall and H. Oken, Am. J. Clin. Nutr., 2006, 83, S1467–1476S CrossRef.
- J. T. Brenna, Curr. Opin. Clin. Nutr. Metab. Care, 2002, 5, 127–132 CrossRef CAS PubMed.
- G. C. Burdge and P. C. Calder, Reprod., Nutr., Dev., 2005, 45, 581–597 CrossRef CAS PubMed.
- K. E. Wood, E. Mantzioris, R. A. Gibson, C. E. Ramsden and B. S. Muhlhausler, Prostaglandins Leukot. Essent. Fatty Acids, 2015, 95, 47–55 CrossRef CAS PubMed.
- T. L. Blasbalg, J. R. Hibbeln, C. E. Ramsden, S. F. Majchrzak and R. R. Rawlings, Am. J. Clin. Nutr., 2011, 93, 950–962 CrossRef CAS PubMed.
- Institute of Medicine, Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids, National Academy Press, Washington, DC, 2005 Search PubMed.
- Deutsche Gesellschaft für Ernährung, Referenzwerte für die Nährstoffzufuhr, Bonn, 2. Aufl., 2016.
- K. D. Stark, M. E. Van Elswyk, M. R. Higgins, C. A. Weatherford and N. Salem, Prog. Lipid Res., 2016, 63, 132–152 CrossRef CAS PubMed.
- W. S. Harris and C. von Schacky, Prev. Med., 2004, 39, 212–220 CrossRef CAS PubMed.
- K. Lukaschek, C. von Schacky, J. Kruse and K.-H. Ladwig, Dementia Geriatr. Cognit. Disord., 2016, 42, 236–245 CrossRef CAS PubMed.
- A. V. Witte, L. Kerti, H. M. Hermannstädter, J. B. Fiebach, S. J. Schreiber, J. P. Schuchardt, A. Hahn and A. Flöel, Cereb. Cortex, 2014, 24, 3059–3068 CrossRef PubMed.
- W. S. Harris, K. F. Kennedy, J. H. O'Keefe and J. A. Spertus, Int. J. Cardiol., 2013, 168, 53–59 CrossRef PubMed.
- M. E. Kleber, G. E. Delgado, S. Lorkowski, W. März and C. von Schacky, Atherosclerosis, 2016, 252, 175–181 CrossRef CAS PubMed.
- D. Li, A. Sinclair, A. Wilson, S. Nakkote, F. Kelly, L. Abedin, N. Mann and A. Turner, Am. J. Clin. Nutr., 1999, 69, 872–882 CAS.
- M. R. Fokkema, D. A. J. Brouwer, M. B. Hasperhoven, I. A. Martini and F. A. J. Muskiet, Prostaglandins Leukot. Essent. Fatty Acids, 2000, 63, 287–292 CrossRef CAS PubMed.
- A. I. Ostermann, M. Müller, I. Willenberg and N. H. Schebb, Prostaglandins Leukot. Essent. Fatty Acids, 2014, 91, 235–241 CrossRef CAS PubMed.
- A. I. Ostermann, I. Willenberg and N. H. Schebb, Anal. Bioanal. Chem., 2015, 407, 1403–1414 CrossRef CAS PubMed.
- I. Willenberg, K. Rund, S. Rong, N. Shushakova, F. Gueler and N. H. Schebb, Inflammation Res., 2016, 65, 133–142 CrossRef CAS PubMed.
- K. M. Rund, A. I. Ostermann, L. Kutzner, J.-M. Galano, C. Oger, C. Vigor, S. Wecklein, N. Seiwert, T. Durand and N. H. Schebb, Anal. Chim. Acta, 2017 DOI:10.1016/J.aca.2017.11.002 , in press.
- S. Bokor, J. Dumont, A. Spinneker, M. Gonzalez-Gross, E. Nova, K. Widhalm, G. Moschonis, P. Stehle, P. Amouyel and S. De Henauw, et al. , J. Lipid Res., 2010, 51, 2325–2333 CrossRef CAS PubMed.
- B. Lands, Prog. Lipid Res., 2008, 47, 77–106 CrossRef CAS PubMed.
- A. Patenaude, D. Rodriguez-Leyva, A. L. Edel, E. Dibrov, C. M. C. Dupasquier, J. A. Austria, M. N. Richard, M. N. Chahine, L. J. Malcolmson and G. N. Pierce, Eur. J. Clin. Nutr., 2009, 63, 1123–1129 CrossRef CAS PubMed.
- S. A. Sands, K. J. Reid, S. L. Windsor and W. S. Harris, Lipids, 2005, 40, 343 CrossRef CAS PubMed.
- R. J. Pawlosky, J. R. Hibbeln and N. Salem, J. Lipid Res., 2007, 48, 935–943 CrossRef CAS PubMed.
- R. N. Lemaitre, T. Tanaka, W. Tang, A. Manichaikul, M. Foy, E. K. Kabagambe, J. A. Nettleton, I. B. King, L.-C. Weng, S. Bhattacharya, S. Bandinelli, J. C. Bis, S. S. Rich, D. R. Jacobs, A. Cherubini, B. McKnight, S. Liang, X. Gu, K. Rice, C. C. Laurie, T. Lumley, B. L. Browning, B. M. Psaty, Y.-D. I. Chen, Y. Friedlander, L. Djousse, J. H. Y. Wu, D. S. Siscovick, A. G. Uitterlinden, D. K. Arnett, L. Ferrucci, M. Fornage, M. Y. Tsai, D. Mozaffarian and L. M. Steffen, PLoS Genet., 2011, 7, e1002193 CAS.
- M. T. Buckley, F. Racimo, M. E. Allentoft, M. K. Jensen, A. Jonsson, H. Huang, F. Hormozdiari, M. Sikora, D. Marnetto, E. Eskin, M. E. Jørgensen, N. Grarup, O. Pedersen, T. Hansen, P. Kraft, E. Willerslev and R. Nielsen, Mol. Biol. Evol., 2017, 34, 1307–1318 CrossRef PubMed.
- L. Bakewell, G. C. Burdge and P. C. Calder, Br. J. Nutr., 2006, 96, 93 CrossRef CAS PubMed.
- F. L. Crowe, C. Murray Skeaff, T. J. Green and A. R. Gray, Br. J. Nutr., 2008, 99, 168–174 CrossRef CAS PubMed.
- E. J. Giltay, L. J. Gooren, A. W. Toorians, M. B. Katan and P. L. Zock, Am. J. Clin. Nutr., 2004, 80, 1167–1174 CrossRef CAS PubMed.
- G. C. Burdge, A. E. Jones and S. A. Wootton, Br. J. Nutr., 2002, 88, 355 CrossRef CAS PubMed.
- G. C. Burdge and S. A. Wootton, Br. J. Nutr., 2002, 88, 411 CrossRef CAS PubMed.
- S. Thuppal, C. von Schacky, W. Harris, K. Sherif, N. Denby, S. Steinbaum, B. Haycock and R. Bailey, Nutrients, 2017, 9, 930 CrossRef PubMed.
- G. Barceló-Coblijn and E. J. Murphy, Prog. Lipid Res., 2009, 48, 355–374 CrossRef PubMed.
- G. Barceló-Coblijn, E. J. Murphy, R. Othman, M. H. Moghadasian, T. Kashour and J. K. Friel, Am. J. Clin. Nutr., 2008, 88, 801–809 CrossRef.
- K. Kuhnt, S. Weiß, M. Kiehntopf and G. Jahreis, Lipids Health Dis., 2016, 15, 32 CrossRef PubMed.
- I. B. Asztalos, J. A. Gleason, S. Sever, R. Gedik, B. F. Asztalos, K. V. Horvath, M. L. Dansinger, S. Lamon-Fava and E. J. Schaefer, Metabolism, 2016, 65, 1636–1645 CrossRef CAS PubMed.
- M. Peet, J. Brind, C. N. Ramchand, S. Shah and G. K. Vankar, Schizophr. Res., 2001, 49, 243–251 CrossRef CAS PubMed.
- E. A. Emken, R. O. Adlof, H. Rakoff, W. K. Rohwedder and R. M. Gulley, et al. , Biochem. Soc. Trans., 1990, 18, 766–769 CrossRef CAS PubMed.
- W. S. Harris and R. M. Thomas, Clin. Biochem., 2010, 43, 338–340 CrossRef CAS PubMed.
- M. A. Allman, M. M. Pena and D. Pang, Eur. J. Clin. Nutr., 1995, 49, 169–178 CAS.
- W. J. Bemelmans, J. Broer, E. J. Feskens, A. J. Smit, F. A. Muskiet, J. D. Lefrandt, V. J. Bom, J. F. May and B. Meyboom-de Jong, Am. J. Clin. Nutr., 2002, 75, 221–227 CrossRef CAS PubMed.
- G. E. Caughey, E. Mantzioris, R. A. Gibson, L. G. Cleland and M. J. James, Am. J. Clin. Nutr., 1996, 63, 116–122 CrossRef CAS PubMed.
- J. K. Chan, B. E. McDonald, J. M. Gerrard, V. M. Bruce, B. J. Weaver and B. J. Holub, Lipids, 1993, 28, 811–817 CrossRef CAS PubMed.
- D. S. Kelley, G. J. Nelson, J. E. Love, L. B. Branch, P. C. Taylor, P. C. Schmidt, B. E. Mackey and J. M. Iacono, Lipids, 1993, 28, 533–537 CrossRef CAS PubMed.
- S. Kew, T. Banerjee, A. M. Minihane, Y. E. Finnegan, R. Muggli, R. Albers, C. M. Williams and P. C. Calder, Am. J. Clin. Nutr., 2003, 77, 1287–1295 CrossRef CAS PubMed.
- T. A. B. Sanders and K. M. Younger, Br. J. Nutr., 1981, 45, 613 CrossRef CAS PubMed.
- F. A. Wallace, E. A. Miles and P. C. Calder, Br. J. Nutr., 2003, 89, 679–689 CrossRef CAS PubMed.
- B. J. Weaver, E. J. Corner, V. M. Bruce, B. E. McDonald and B. J. Holub, Am. J. Clin. Nutr., 1990, 51, 594–598 CrossRef CAS PubMed.
- G. Barceló-Coblijn, L. W. Collison, C. A. Jolly and E. J. Murphy, Lipids, 2005, 40, 787–798 CrossRef.
- H. Poudyal, S. K. Panchal, J. Waanders, L. Ward and L. Brown, J. Nutr. Biochem., 2012, 23, 153–162 CrossRef CAS PubMed.
- R. A. Gibson, M. A. Neumann, E. L. Lien, K. A. Boyd and W. C. Tu, Prostaglandins Leukot. Essent. Fatty Acids, 2013, 88, 139–146 CrossRef CAS PubMed.
- R. Portolesi, B. C. Powell and R. A. Gibson, J. Lipid Res., 2007, 48, 1592–1598 CrossRef CAS PubMed.
- E. Archer, G. A. Hand and S. N. Blair, PLoS One, 2013, 8, e76632 CAS.
- R. Fischer, A. Konkel, H. Mehling, K. Blossey, A. Gapelyuk, N. Wessel, C. von Schacky, R. Dechend, D. N. Muller, M. Rothe, F. C. Luft, K. Weylandt and W.-H. Schunck, J. Lipid Res., 2014, 55, 1150–1164 CrossRef CAS PubMed.
- M. L. Nording, J. Yang, K. Georgi, C. Hegedus Karbowski, J. B. German, R. H. Weiss, R. J. Hogg, J. Trygg, B. D. Hammock and A. M. Zivkovic, PLoS One, 2013, 8, e76575 CAS.
- J. P. Schuchardt, S. Schmidt, G. Kressel, I. Willenberg, B. D. Hammock, A. Hahn and N. H. Schebb, Prostaglandins Leukot. Essent. Fatty Acids, 2014, 90, 27–37 CrossRef CAS PubMed.
- J. P. Schuchardt, A. I. Ostermann, L. Stork, L. Kutzner, H. Kohrs, T. Greupner, A. Hahn and N. H. Schebb, Prostaglandins Leukot. Essent. Fatty Acids, 2016, 115, 12–23 CrossRef CAS PubMed.
- J. P. Schuchardt, A. I. Ostermann, L. Stork, S. Fritzsch, H. Kohrs, T. Greupner, A. Hahn and N. H. Schebb, Prostaglandins Leukot. Essent. Fatty Acids, 2017, 121, 76–87 CrossRef CAS PubMed.
- G. Burdge, Curr. Opin. Clin. Nutr. Metab. Care, 2004, 7, 137–144 CrossRef CAS PubMed.
- S. P. B. Caligiuri, K. Love, T. Winter, J. Gauthier, C. G. Taylor, T. Blydt-Hansen, P. Zahradka and H. M. Aukema, J. Nutr., 2013, 143, 1421–1431 CrossRef CAS PubMed.
- N. Kumar, G. Gupta, K. Anilkumar, N. Fatima, R. Karnati, G. V. Reddy, P. V. Giri and P. Reddanna, Sci. Rep., 2016, 6, 31649 CrossRef CAS PubMed.
- N. Tejera, W. E. Boeglin, T. Suzuki and C. Schneider, J. Lipid Res., 2012, 53, 87–94 CrossRef CAS PubMed.
- A. I. Ostermann and N. H. Schebb, Food Funct., 2017, 8, 2355–2367 CAS.
- B. Lands, BioMed Res. Int., 2015, 2015, 1–8 CrossRef PubMed.
- J. F. Greene and B. D. Hammock, in Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury, ed. K. V. Honn, L. J. Marnett, S. Nigam and E. A. Dennis, Springer US, Boston, MA, 1999, vol. 4, pp. 471–477 Search PubMed.
- J. H. Moran, R. Weise, R. G. Schnellmann, J. P. Freeman and D. F. Grant, Toxicol. Appl. Pharmacol., 1997, 146, 53–59 CrossRef CAS PubMed.
- J. Zheng, C. G. Plopper, J. Lakritz, D. H. Storms and B. D. Hammock, Am. J. Respir. Cell Mol. Biol., 2001, 25, 434–438 CrossRef CAS PubMed.
- S. P. B. Caligiuri, H. M. Aukema, A. Ravandi and G. N. Pierce, Exp. Gerontol., 2014, 59, 51–57 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fo01809f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |