
Synthesis of ethanol via a reaction of dimethyl ether with CO2 and H2†
Qingli
Qian
 *a,
Meng
Cui
ab,
Jingjing
Zhang
ab,
Junfeng
Xiang
a,
Jinliang
Song
a,
Guanying
Yang
a and
Buxing
Han
*ab
*a,
Meng
Cui
ab,
Jingjing
Zhang
ab,
Junfeng
Xiang
a,
Jinliang
Song
a,
Guanying
Yang
a and
Buxing
Han
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qianql@iccas.ac.cn; hanbx@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 24th October 2017
Abstract
Ethanol is currently produced via the catalytic hydration of ethylene or fermentation of foods. The synthesis of ethanol from cheap and renewable CO2 is of great importance, but the state of the art routes encounter difficulties, especially in reaction selectivity and activity. Here we show a strategy of ethanol synthesis from CO2, dimethyl ether (DME) and H2. The reaction can be effectively promoted with a Ru–Co bimetallic catalyst using LiI as a promoter in 1,3-dimethyl-2-imidazolidinone (DMI) solvent. The predominant product of this reaction was ethanol and the selectivity of ethanol in total products could reach 71.7 C-mol%. The selectivity of ethanol in the liquid product could reach 94.1%, which was higher than the reported routes using CO2/CO. To the best of our knowledge, this is the first work on ethanol synthesis from DME, CO2 and H2. The reaction mechanism is discussed based on a series of control experiments.
Introduction
Ethanol as an alternative fuel has been widely utilized in the current energy infrastructure.1 It is also a very important bulk chemical. Currently, ethanol is mainly produced via the catalytic hydration of ethylene from fossil raw materials or fermentation of foods such as corn and sugar.2 CO2 is a greenhouse gas and its fixation into value-added products is highly desirable for the sustainable development of our society.3 To date, CO2 has been used as a building block to synthesize various chemicals, such as urea, polymers, carboxylic acids, carbonates, amides and alcohols.4,5 As for the synthesis of alcohols using CO2 as a feedstock, the major research progress has been focused on methanol in the past few decades.6 Efficient synthesis of ethanol is certainly of great importance, but is more difficult due to selective C–C coupling.The previous reports of ethanol production from CO2 were mostly confined to direct CO2 hydrogenation at high temperatures (>250 °C).7–13 In such reports, CO2 usually reacted with H2 to generate reactive C1 intermediates, say CO, CH3 and/or CH3OH, then the C–C bond formation steps took place to generate C2+ products, such as ethanol and higher alcohols.10,11,14–17 Because the in situ formation of C1 intermediates and the C–C bond construction occurred simultaneously, the reaction products usually consisted of various alcohols and hydrocarbons. In addition, the ethanol selectivity in the total products was generally low (<20 C-mol%). To increase the ethanol selectivity, introducing a certain substrate to react with CO2 and H2 is a feasible way. When methanol reacted with CO2 and H2, ethanol was the only alcohol product and 34.2 C-mol% of ethanol selectivity in total products (CO 46.5 C-mol%, methane 19.3 C-mol%) was obtained.18a In addition, the space time yield (STY) of the reaction reached 124.9 C-mmol L−1 h−1. Paraformaldehyde could also be reacted with CO2 and H2 to produce ethanol, during which paraformaldehyde was firstly converted into methanol.18b Currently, CO2 hydrogenation with simultaneous C–C bond formation is still a great challenge in CO2 chemistry.19 Recently, an elegantly designed and highly ordered Pd–Cu nanocatalyst was prepared, and it was discovered that the selectivity of ethanol could reach 92.0%.20 Although significant progress has been achieved in this interesting area, the exploration of a new strategy to produce ethanol efficiently from CO2 under milder conditions using easily prepared catalysts is still highly desirable.
Dimethyl ether (DME) is a cheap and bulk chemical, which can be produced in a single-step process from CO2/CO and H2. DME is also a key intermediate to bulk chemicals in industry (e.g., acetic acid, olefins, hydrocarbons).2 Here we show a protocol to produce ethanol from DME, CO2 and H2 (Scheme 1). The reaction can proceed efficiently over a Ru(PPh3)3Cl2/CoI2 bimetallic homogeneous catalyst under mild conditions. Very interestingly, this reaction has a very high ethanol selectivity. It is confirmed that the ethanol synthesis is through the direct participation of DME (not via methanol or methyl iodide) and/or the synergy of catalyst components, accounting for the distinguished catalytic results. This strategy represents important progress in CO2 chemistry and opens a promising way to fix CO2 into fuels and bulk chemicals.
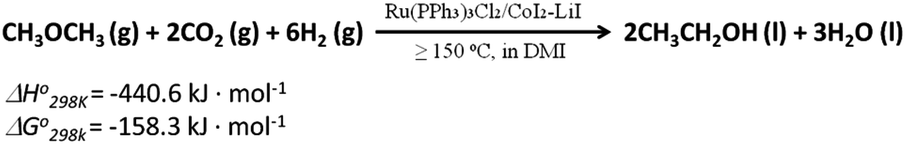 | ||
| Scheme 1 Synthesis of ethanol via the reaction of DME with CO2 and H2. The thermodynamic data were from Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill Book Company, 13th edn, 1985. | ||
Results and discussion
Catalytic system for ethanol synthesis
The reaction could be efficiently catalyzed by Ru(PPh3)3Cl2 and CoI2 bimetallic catalysts using LiI as a promoter in 1,3-dimethyl-2-imidazolidinone (DMI) under milder conditions. In this work, ethanol was the product, and methanol, CO, and methane were the only by-products. The selectivity of the product and all the by-products are given in Table 1. Ethanol was the predominant product in the reaction solution with little methanol as a byproduct (Fig. S1a†). Very interestingly, only minor CO and methane were detected in the gaseous sample (Fig. S1b†). The STY of the reaction was as high as 132.5 C-mmol L−1 h−1 and the selectivity of ethanol was 71.7 C-mol% (entry 1). The selectivity of ethanol in the liquid product could reach 94.1%, which was higher than the reported routes using CO2/CO. Some representative reports of ethanol synthesis from CO2 are listed in Table S1.†| Entry | Catalyst precursor | Promoter | Solvent | STYc | Selectivity [C-mol%] | |||
|---|---|---|---|---|---|---|---|---|
| Ethanol | Methanol | CO | CH4 | |||||
| a Reaction conditions: 30 μmol Ru catalyst, 70 μmol Co catalyst (based on the metal), 2.3 mmol promoter, 2 mL solvent, 0.5 MPa DME (4 mmol), 4 MPa CO2 (30 mmol) and 4 MPa H2 (32 mmol) (at room temperature), 180 °C and 12 h. b A precipitate was observed after the reaction. c STY stands for space time yield (C-mmol L−1 h−1), which is one of the commonly used units, especially when multi-metals are utilized. d The conversion of DME in entry 1 was 20.6%, and the conversions of DME under other conditions were lower than that of entry 1. | ||||||||
| 1d | Ru(PPh3)3Cl2, CoI2 | LiI | DMI | 132.5 | 71.7 | 4.5 | 13.8 | 10.0 |
| 2b | Ru(PPh3)3Cl2, CoI2 | — | DMI | 7.1 | 0.0 | 0.0 | 70.6 | 29.4 |
| 3 | Ru(PPh3)3Cl2, CoI2 | KI | DMI | 80.4 | 39.9 | 7.3 | 40.4 | 12.4 |
| 4b | Ru(PPh3)3Cl2, CoI2 | ZnI2 | DMI | 15.0 | 36.1 | 19.4 | 41.7 | 2.8 |
| 5 | Ru(PPh3)3Cl2, CoI2 | LiCl | DMI | 56.3 | 8.9 | 10.6 | 71.7 | 8.8 |
| 6b | Ru(PPh3)3Cl2, CoI2 | LiBF4 | DMI | 38.3 | 14.1 | 20.6 | 37.0 | 28.3 |
| 7 | Ru(PPh3)3Cl2 | LiI | DMI | 42.5 | 0.4 | 5.5 | 83.3 | 10.8 |
| 8 | CoI2 | LiI | DMI | 9.2 | 2.5 | 20.2 | 40.9 | 36.4 |
| 9b | Ru(acac)3, CoI2 | LiI | DMI | 80.4 | 36.1 | 6.7 | 23.8 | 33.4 |
| 10 | Ru3(CO)12, CoI2 | LiI | DMI | 93.8 | 41.6 | 4.0 | 25.3 | 29.1 |
| 11 | Ru(PPh3)3Cl2, CoCl2 | LiI | DMI | 34.6 | 0.0 | 13.3 | 56.6 | 30.1 |
| 12 | Ru(PPh3)3Cl2, Co4(CO)12 | LiI | DMI | 20.8 | 0.0 | 28.0 | 60.0 | 12.0 |
| 13 | Ru(PPh3)3Cl2, Rh2(CO)4Cl2 | LiI | DMI | 27.5 | 27.3 | 19.7 | 42.4 | 10.6 |
| 14 | Ru(PPh3)3Cl2, Ir4(CO)12 | LiI | DMI | 47.5 | 6.2 | 9.6 | 75.4 | 8.8 |
| 15 | Ru(PPh3)3Cl2, ferrocene | LiI | DMI | 44.2 | 11.3 | 11.3 | 65.1 | 12.3 |
| 16b | NiCl2, CoI2 | LiI | DMI | 45.4 | 23.0 | 22.0 | 12.8 | 42.2 |
| 17b | Mn2(CO)10, CoI2 | LiI | DMI | 15.0 | 5.6 | 63.9 | 19.4 | 11.1 |
| 18b | CuSO4, CoI2 | LiI | DMI | 12.9 | 0.0 | 45.1 | 19.4 | 35.5 |
| 19b | Ru(PPh3)3Cl2, CoI2 | LiI | N(C3H7)3 | 15.4 | 0.0 | 0.0 | 97.3 | 2.7 |
| 20 | Ru(PPh3)3Cl2, CoI2 | LiI | N-Methylpyrrolidine | 9.6 | 0.0 | 0.0 | 91.3 | 8.7 |
| 21b | Ru(PPh3)3Cl2, CoI2 | LiI | Cyclohexanone | 87.9 | 0.0 | 13.3 | 0.0 | 86.7 |
| 22 | Ru(PPh3)3Cl2, CoI2 | LiI | 2-Pyrrolidinone | 19.2 | 0.0 | 0.0 | 93.5 | 6.5 |
| 23 | Ru(PPh3)3Cl2, CoI2 | LiI | NMP | 84.2 | 39.1 | 5.4 | 32.7 | 22.8 |
| 24b | Ru(PPh3)3Cl2, CoI2 | LiI | Cyclohexane | 44.6 | 0.0 | 0.0 | 0.0 | 100.0 |
| 25b | Ru(PPh3)3Cl2, CoI2 | LiI | Benzene | 45.4 | 0.0 | 15.6 | 0.0 | 84.4 |
| 26b | Ru(PPh3)3Cl2, CoI2 | LiI | DMF | 42.9 | 0.0 | 0.0 | 96.1 | 3.9 |
| 27b | Ru(PPh3)3Cl2, CoI2 | LiI | Water | 29.6 | 0.0 | 76.1 | 14.0 | 9.9 |
The promoter was indispensable in this reaction. No alcohol was generated and the catalytic system was not stable without the promoter (entry 2). When the promoters with other cations (K+, Zn2+) or anions (Cl−, BF4−) were used, the results were poor (entries 3–6). Hence LiI was the best promoter in accelerating the target reaction. The strong Lewis acidity and small size of Li+ may be beneficial in coordinating with and/or activating the DME molecule.21 It is well known that the iodide anion is an eminent ligand for transition metal catalysts, which may effectively tailor the stability, selectivity and activity.22 I− helped to maintain the catalyst stability (entries 1 and 6). In addition, the larger size of iodide compared to other halides has a more remarkable steric effect, resulting in better catalytic selectivity (entries 1 and 5).
We tried Ru(PPh3)3Cl2 or CoI2 as the single catalyst respectively, but the results were poor (entries 7 and 8). Obviously, the synergic effect existed between Ru and Co catalysts during the catalytic reaction. The precursors of the Ru–Co bimetallic catalyst were crucial for the catalytic performance. When we utilized Ru(acac)3 or Ru3(CO)12 instead of Ru(PPh3)3Cl2 to conduct the reaction, the catalytic activity was much lower (entries 9 and 10). We also used CoCl2 or Co4(CO)12 instead of CoI2, but the catalytic activity was very low and no ethanol was generated (entries 11 and 12). When we combined Ru(PPh3)3Cl2 with other metal (Rh, Ir, Fe) complexes, the results were not satisfactory (entries 13–15). We also combined CoI2 with other metal (Ni, Mn, Cu) compounds and the catalytic performance was also not satisfactory (entries 16–18). Thus, the Ru(PPh3)3Cl2 and CoI2 cooperated very well for the target reaction.
The solvent also played an important role in the target reaction. It is known that DMI is a cyclic diamine with a ketone group. Using Ru(PPh3)3Cl2/CoI2 as a catalyst and LiI as a promoter, we studied the solvent effect of the reaction. To study the role of the amine group on the solvent molecule, we firstly used tripropylamine as a solvent, but no ethanol was detected and the catalyst decomposed significantly (entry 19). Then we tried a cyclic amine, N-methylpyrrolidine, no ethanol was generated either, but the catalyst was stable (entry 20). So the cyclic amine is beneficial to the stability of the catalyst. To investigate the effect of the ketone group, we utilized cyclohexanone as the solvent, but no ethanol was detected and the catalyst was unstable (entry 21). When 2-pyrrolidinone, which combines a cyclic amine and ketone group, was applied as the solvent, the catalyst was stable, but no ethanol was formed either (entry 22). Ethanol was produced and the catalyst was stable when N-methyl-2-pyrrolidone (NMP) was used as the solvent (entry 23). Thus, it can be deduced that a cyclic amine with the N-methyl group and ketone group in the solvent benefits the reaction. DMI not only has a similar molecular structure to NMP, but also has one more amine group with N-methyl, and it was a better solvent for the reaction than NMP (entries 1 and 23), suggesting that both the amine groups with N-methyl in DMI could effectively improve the reaction activity and selectivity. We also tested other solvents, such as cyclohexane, benzene, DMF and water, but the results were poor (entries 24–27). In brief, the catalytic system composed of Ru(PPh3)3Cl2/CoI2, LiI and DMI showed a better performance than other catalytic systems tested in the work.
Effect of reaction parameters
Based on the optimized catalytic system, we investigated the impact of reaction temperature, pressure, and dosage of each catalyst component on the catalytic reaction. The yields of the products at different temperatures are depicted in Fig. 1. No product was detectable when the reaction was conducted at 140 °C, and obviously ethanol emerged when the temperature was increased to 150 °C. The reaction rate increased rapidly with the increasing temperature until 180 °C. The STY of the reaction at 180 °C was 132.5 C-mmol L−1 h−1 and its growth became slower when the temperature was further raised. The selectivity of ethanol increased steadily with the elevation of temperature until 180 °C. The ethanol selectivity was 71.7 C-mol% at 180 °C, while it decreased evidently on further elevating the temperature. This may be attributed to the evident yield of methane at higher temperatures.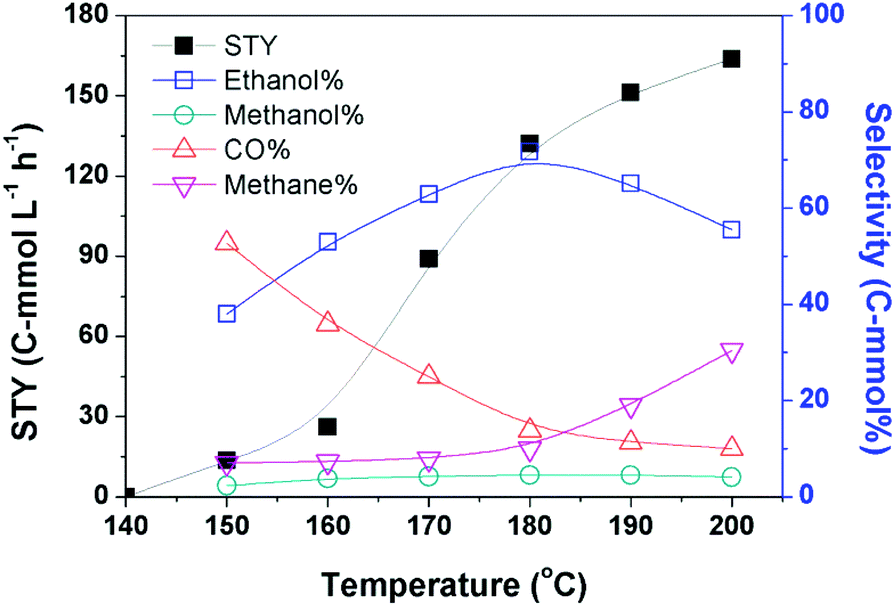 | ||
| Fig. 1 The yield of products at different temperatures. Reaction conditions: 30 μmol Ru(PPh3)3Cl2, 70 μmol CoI2, 2.3 mmol LiI, 2 mL DMI, 0.5 MPa DME, 4 MPa CO2 and 4 MPa H2 (at room temperature), 12 h. | ||
The data in Fig. 1 demonstrate that 180 °C is an appropriate temperature. We further investigated the impact of other parameters on the reaction at this temperature, and the results are given in Table 2. The pressure of CO2 and H2 influenced the reaction significantly. At a fixed ratio of CO2 and H2 (1/1), both the reaction rate and ethanol selectivity increased remarkably when the total pressure was enhanced from 2 MPa to 8 MPa (entries 1–4). They were not sensitive to the total pressure at higher pressure (entry 5). At a fixed total pressure of 8 MPa, the ratio of CO2 and H2 also affected the reaction and the best result was obtained at the ratio of 1/1 (entries 4, 6 and 7). When we tried the experiments without CO2 and/or H2, no alcohol was produced (entries 8–10). Thus, both CO2 and H2 are indispensable for the reaction. The dosage of LiI also evidently impacted the reaction activity and selectivity (entries 4, 11 and 12), and the best performance was obtained when the dosage of LiI was 2.3 mmol. The result revealed that an excess amount of LiI was not favorable to the reaction, which may be due to the occupation of the active sites by the excess iodide anions.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Ru/Co [μmol] | LiI [mmol] | CO2/H2 [MPa] | STY | Selectivity [C-mol%] | |||
|---|---|---|---|---|---|---|---|---|
| Ethanol | Methanol | CO | CH4 | |||||
| a Reaction conditions: Ru(PPh3)3Cl2/CoI2 was used as the catalyst and its dosage was based on the metal, LiI was used as the promoter, 2 mL DMI, DME 0.5 MPa (at room temperature), 180 °C, and 12 h. b No DME was added in the reaction. c Water (2 mmol) was added before the reaction. d CO was used instead of CO2. | ||||||||
| 1 | 30/70 | 2.3 | 1/1 | 15.8 | 31.6 | 36.8 | 23.7 | 7.9 |
| 2 | 30/70 | 2.3 | 2/2 | 32.9 | 46.8 | 15.2 | 29.1 | 8.9 |
| 3 | 30/70 | 2.3 | 3/3 | 83.8 | 67.2 | 6.5 | 18.3 | 8.0 |
| 4 | 30/70 | 2.3 | 4/4 | 132.5 | 71.7 | 4.5 | 13.8 | 10.0 |
| 5 | 30/70 | 2.3 | 5/5 | 144.6 | 73.3 | 4.4 | 12.6 | 9.7 |
| 6 | 30/70 | 2.3 | 2/6 | 103.3 | 68.2 | 1.5 | 19.8 | 10.5 |
| 7 | 30/70 | 2.3 | 6/2 | 23.8 | 35.1 | 5.3 | 49.1 | 10.5 |
| 8 | 30/70 | 2.3 | 0/4 | 29.2 | 0.0 | 0.0 | 0.0 | 100.0 |
| 9 | 30/70 | 2.3 | 4/0 | 0.0 | — | — | — | — |
| 10 | 30/70 | 2.3 | 0/0 | 0.0 | — | — | — | — |
| 11 | 30/70 | 1.15 | 4/4 | 85.0 | 63.2 | 7.8 | 22.1 | 6.9 |
| 12 | 30/70 | 3.45 | 4/4 | 66.7 | 40.6 | 5.6 | 40.0 | 13.8 |
| 13 | 15/85 | 2.3 | 4/4 | 110.0 | 65.9 | 6.1 | 24.2 | 3.8 |
| 14 | 45/55 | 2.3 | 4/4 | 121.3 | 68.5 | 7.9 | 15.4 | 8.2 |
| 15 | 15/35 | 2.3 | 4/4 | 92.5 | 71.6 | 7.2 | 16.7 | 4.5 |
| 16 | 45/105 | 2.3 | 4/4 | 85.0 | 63.7 | 6.8 | 22.1 | 7.4 |
| 17b | 30/70 | 2.3 | 4/4 | 21.3 | 0.0 | 0.0 | 98.7 | 1.3 |
| 18c | 30/70 | 2.3 | 4/4 | 34.2 | 54.9 | 6.1 | 31.7 | 7.3 |
| 19 | 0/0 | 2.3 | 4/4 | 14.6 | 0.0 | 28.6 | 48.5 | 22.9 |
| 20 | 30/0 | 0 | 4/4 | 22.5 | 0.0 | 0.0 | 100.0 | 0.0 |
| 21 | 0/70 | 0 | 4/4 | 5.4 | 0.0 | 69.2 | 0.0 | 30.8 |
| 22b,d | 30/70 | 2.3 | 4/4 | 0.5 | 0.0 | 0.0 | 0.0 | 100.0 |
The atomic ratio of Ru and Co also affected the reaction remarkably. At the same total amount of metals (100 μmol), 30 μmol Ru and 70 μmol Co yielded the best catalytic result (entries 4, 13 and 14). The total catalyst dosage also influenced the reaction. When we fixed the ratio of Ru and Co at 3/7 and changed the total amount of the catalysts, the best dosage of the catalysts was 30 μmol Ru and 70 μmol Co (entries 4, 15 and 16). Therefore, the dosage of the catalysts (30 μmol Ru and 70 μmol Co) was fixed for further investigation.
Recyclability
To investigate the reusability of the catalytic system, the alcohols generated in the reactor were removed at 80 °C for 3 h in a vacuum oven, then GC analysis confirmed that the alcohols in the catalytic system were negligible, and the catalytic system was used directly for the next run. Fig. 2 reveals that the yield of alcohols did not change obviously after five cycles.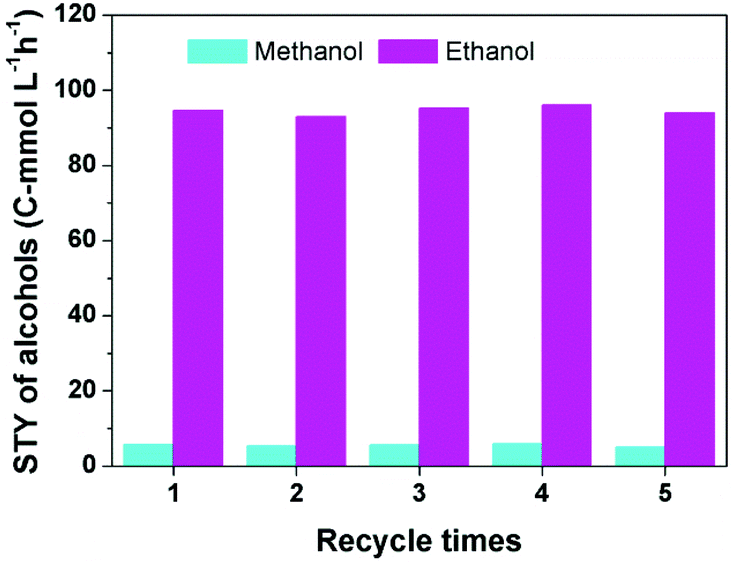 | ||
| Fig. 2 The results of the recycling test. Reaction conditions: 30 μmol Ru(PPh3)3Cl2, 70 μmol CoI2, 2.3 mmol LiI, 2 mL DMI, 0.5 MPa DME, 4 MPa CO2 and 4 MPa H2 (at room temperature), 180 °C and 12 h. | ||
Time course of the reaction
Fig. 3 illustrates the time course of the reaction. The amount of ethanol increased slowly at the beginning (0–3 h), then grew rapidly until 12 h, and the increase of the ethanol production became slower after 12 h. In the whole course, the amount of methane increased slowly and its amount was smaller compared to that of ethanol. In addition, the contents of CO and methanol, which are usually known as intermediates of the ethanol synthesis from CO2, were nearly kept constant after the initial period. While in the reported routes, CO and/or methanol tend to increase at the beginning of the reaction and decrease with time.14–16 This indicates that the reaction would follow a different reaction pathway.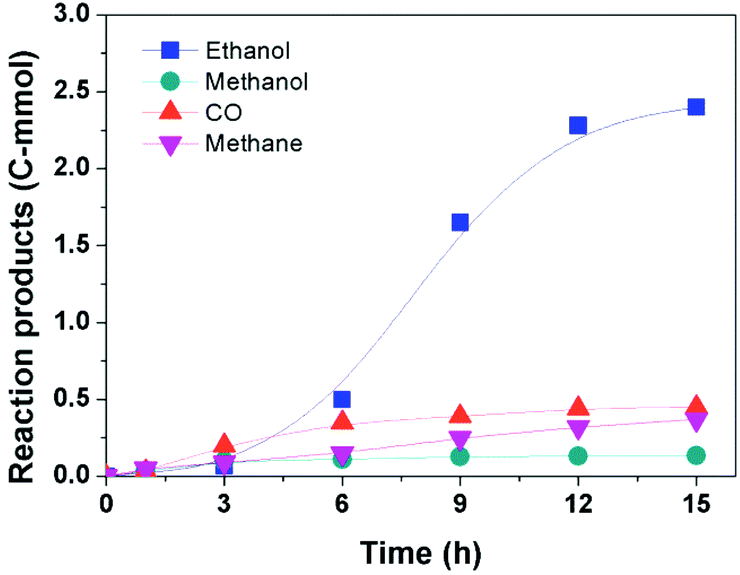 | ||
| Fig. 3 The time course of the reaction of DME with CO2 and H2. Reaction conditions: 30 μmol Ru(PPh3)3Cl2, 70 μmol CoI2, 2.3 mmol LiI, 2 mL DMI, 0.5 MPa DME, 4 MPa CO2 and 4 MPa H2 (at room temperature), 180 °C. | ||
Reaction pathway
In this work DME is necessary for the formation of alcohols. Without DME, no alcohol was formed (entry 17 of Table 2 and Fig. S2a†). In the reported routes, methanol is a common intermediate for the synthesis of ethanol.14–16 It is well known that DME may transform into methanol via hydration over acids.23 If methanol was the intermediate of the target reaction, water in the reactor would promote the reaction. To clarify this possibility, we added water before the reaction but the reaction was significantly inhibited (entry 18 of Table 2). Moreover, in the presence of H2, DME could not effectively transform into methanol, while methanol converted into DME evidently (entry 8 of Table 2 and Fig. S3, S4†). This may be because the DMI solvent rendered a basic condition and suppressed the conversion of DME into methanol. We also found that methanol generated by individual components (Ru catalyst, Co catalyst or LiI) or their combinations (Ru/Co, Ru/LiI or Co/LiI) was little or undetectable (entries 2, 7 and 8 of Table 1 and entries 19–21 of Table 2), suggesting that DME (not via methanol) directly participated in the formation of ethanol. When we used methanol to react with CO2 and H2, considerable methane was produced and the selectivity of ethanol was much lower (Fig. S5†). All the results ruled out the possibility that methanol was the major intermediate for the formation of ethanol in the reaction.Usually, CH3I is a key species for the C–C bond formation from methanol.5a,14,18 CH3I could form spontaneously in the presence of methanol and iodide/iodine at elevated temperatures.24 To make clear whether DME transformed into CH3I during the reaction, we conducted the reaction of DME and LiI at the reaction temperature, but no CH3I was observed (Fig. S6†). We further used CH3I as a substrate to react with CO2 and H2, but it mostly turned into methane and no ethanol was observed (Fig. S7†). It can be deduced that CH3I is not a reactive intermediate of the target reaction in this work, which is in accordance with the previous report on DME homologation with syngas (CO/H2).25 The lack of methanol and/or CH3I intermediates in the reaction could effectively reduce the generation of methane.
As a byproduct, CO was detected after CO2 hydrogenation catalyzed by our reaction system (Fig. S2b†). CO is also regarded as an usual intermediate in ethanol synthesis from CO2, which is generated via the reverse water gas shift reaction (RWGS).14–18 To make this possibility clear, we firstly used CO and H2 to conduct the reaction, but no alcohol was generated (entry 22 of Table 2 and Fig. S8a†), while ethanol was produced when DME reacted with CO and H2, in the presence of water (Fig. S9†). To study whether other C1 species formed by CO2 and H2 participated in the ethanol synthesis, we used formaldehyde and formic acid to react with DME, respectively. The results revealed that no ethanol was detected, but significant methane, H2 and CO2 were observed due to the decomposition of formaldehyde or formic acid (Fig. S10 and S11†). These facts affirmed that CO was the intermediate to react with DME and H2 in the target reaction. In the reported ethanol synthesis from DME and syngas (CO/H2), methanol is usually the major product although dual bed composite catalysts were utilized at high temperatures (≥220 °C).26 In addition, evident methyl acetate and ethyl acetate were observed, while in this work ethanol was the predominant product and no acetate was detected (Fig. S1a†).
Isotopic tracer experiments
To further understand the target reaction, we conducted the tracer test using 13CO2 and D2, respectively. The tracer test using 13CO2 showed that the C atom in the methyl group (CH3-) of ethanol was from DME, while the other C atom (–CH2OH) was from CO2 (Fig. S12†). The NMR spectra of the reaction solution using 13CO2 also supported the above conclusion (Fig. S13†). In addition, the C atoms in methanol and CO originated from DME and CO2 respectively (Fig. S12†). In CO2 hydrogenation or CO hydrogenation, methane was hardly detectable (entries 17 and 22 of Table 2, Fig. S2b and S8b†). So the major origin of methane should be from DME (entry 8 of Table 2, Fig. S3b†).The results using D2 as a tracer showed that D2 entered into the molecules of methanol and ethanol during the reaction (Fig. S14 and S15†). Obviously, evident H–D exchange between D2 and the H atoms in original DME molecules took place. The number of D atoms in the product increased with the elevated D2 pressure. The mass spectra affirmed that the methanol and ethanol molecules produced in the reaction could totally consist of C and D elements, especially at higher D2 pressure. The H atoms in the unreacted DME molecules were intact. It can be deduced that the hydrogen isotope (H–D) exchange reactions proceeded during the activation and/or conversion steps. The detailed study of such exchange reactions has been reported elsewhere.27 Based on the above discussion, we proposed the origin of C and H atoms in the reaction products (Fig. 4).
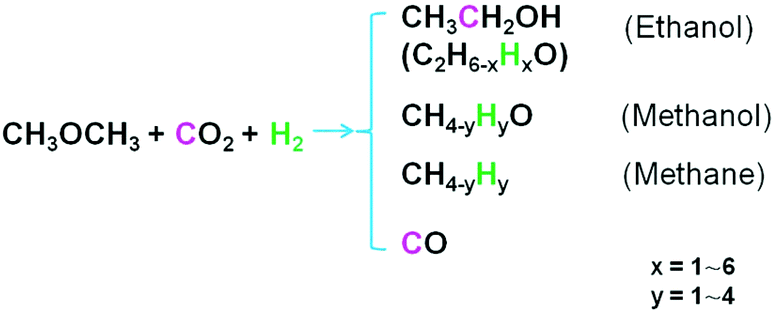 | ||
| Fig. 4 The origin of C and H atoms in the reaction products. | ||
Mechanism
Based on all the results above, we proposed the possible mechanism of the reaction, as is shown in Scheme 2. Firstly, DME coordinated with Li+ (Step 1), during which the CH3–O bond became weaker.21 The activated DME could form CH3Co* with the active Co species (Co*), as shown in Step 2. The Lewis acid promoted methyl Co complex formation has been reported elsewhere.28 CO was generated in situ via a RWGS reaction, promoted by a Ru catalyst (Step 3, Fig. S16†). The Ru catalyzed RWGS reaction has been studied elsewhere.29 Then CO is inserted into the CH3–Co* bond, resulting in the CH3COCo* complex (Step 4, Fig. S9†). The insertion of CO into the metal–carbon bond is a basic step in organic chemistry.25 In the presence of H2, acetaldehyde was formed via the reductive elimination of the acetyl group (Step 5, Fig. S17†). The acetaldehyde was readily reduced into the ethanol product by H2, promoted by the Ru catalyst (Step 6, Fig. S18†). The other moiety of DME generated in Step 2 was transformed into methanol (Step 7), which could be supported by the existence of the methanol byproduct (Fig. S1†). The methanol formed was converted in situ into DME by Ru and/or Co catalysts and started the new reaction cycles (Step 8, Fig. S19†).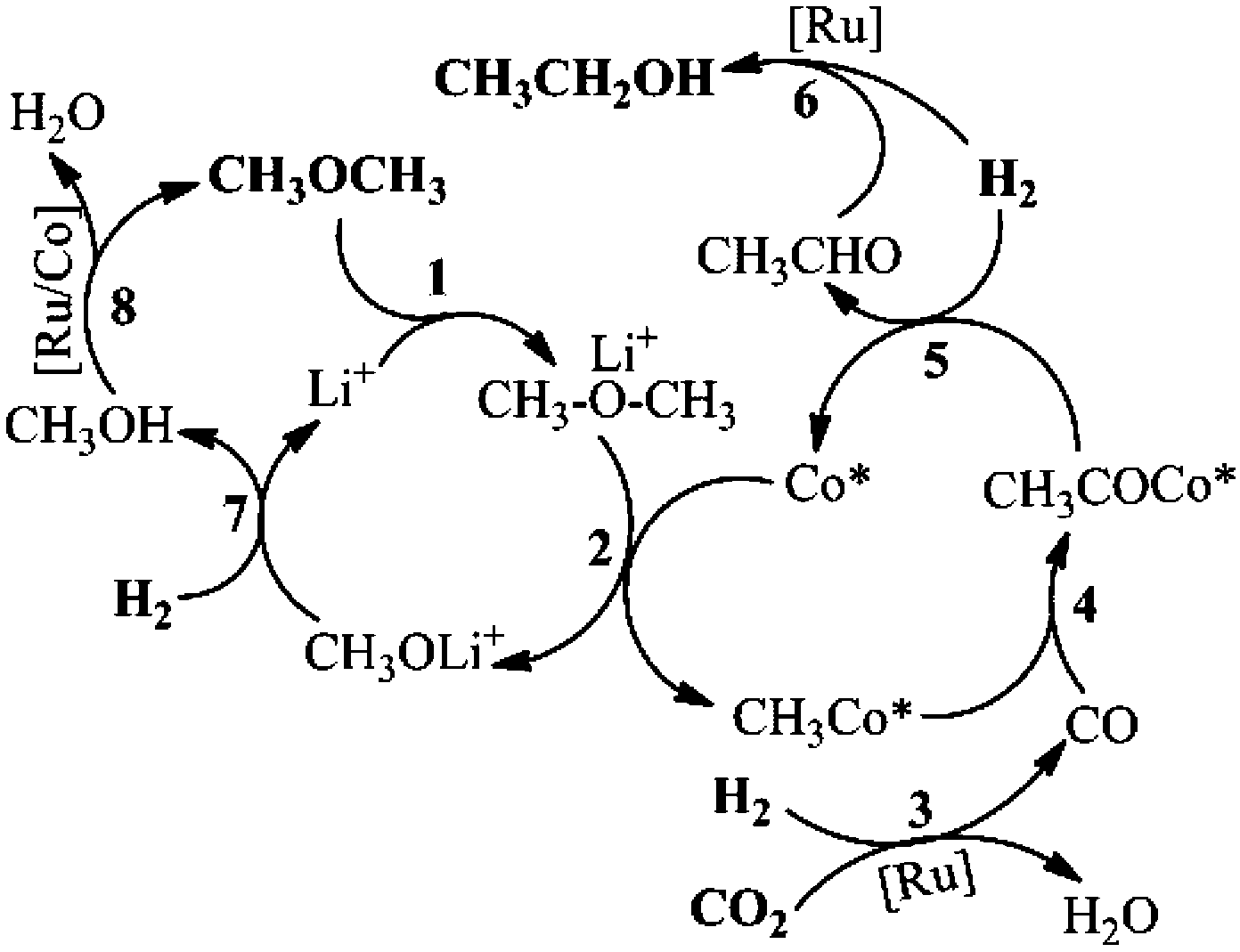 | ||
| Scheme 2 The proposed mechanism of the reaction. | ||
The excellent catalytic results may be attributed to the synergy of the above catalytic cycles. Among these cycles, the rate of the RWGS reaction to form CO was critical to the reaction result. If the catalytic activity of the RWGS reaction was too low, the ethanol formation rate would certainly be low. But when the CO concentration in the reaction was high enough, little ethanol was detected and considerable acetate esters and/or acetic acid emerged (Fig. S20 and S21†). The methyl acetate can be produced via DME carbonylation, catalyzed by a Co catalyst (Fig. S22†). The acetyl group, formed in Step 4, increased with the elevating CO content. If the CO concentration was high, the acetyl group could be converted into methyl acetate with the CH3O generated in Step 2, before its hydrogenation took place. The detailed mechanism of DME carbonylation has been reported in the literature.30 Ethyl acetate and/or acetic acid were produced via further transformation of methyl acetate with CO and/or H2.25 In this work, ethanol was the predominant product, and no acetate or acetic acid was observed. This is mainly because the rate of the RWGS reaction was appropriate. Interestingly, in this work the Co catalyzed DME carbonylation to synthesize methyl acetate could proceed reversely under reaction conditions (Fig. S23†), inhibiting the methyl acetate formation and its further conversion. In the reported DME hydrocarbonylation using syngas (CO/H2), whether by heterogeneous or homogeneous catalysts, methanol is usually the major product.26,28 While in this work, the methanol generated in situ was directly recycled to DME, which accounts for the very low level of methanol during the reaction. It is known that Ru complexes can promote the hydrogenation of CO2/CO into methanol and/or methane,6c whereas in this work the amount of methanol and methane produced from the CO2/CO hydrogenation was hardly detectable (Fig. S2b and S8b†).
Conclusions
In summary, we have developed a route for ethanol synthesis from DME, CO2 and H2. The reaction can be effectively catalyzed by a Ru–Co bimetallic catalyst. Ethanol can be produced at above 150 °C. The catalytic activity can be as high as 132.5 C-mmol L−1 h−1 at 180 °C. The selectivity of ethanol in total products can reach 71.7 C-mol%, and the selectivity of ethanol in the liquid product could reach 94.1%. In addition, the catalyst can be reused at least 5 times without any obvious change in the catalytic performance. The very high efficiency of the reaction resulted from several reasons. Firstly, DME (not via methanol or methyl iodide) directly took part in the formation of ethanol, reducing the methane byproduct. Secondly, the methanol produced can be in situ recycled to DME feedstock, and thus the selectivity to ethanol is enhanced. Finally, Ru and Co cooperate very well to accelerate the desired reaction. The strategy opens a new way of ethanol synthesis and CO2 transformation. We believe that some other value-added chemicals can also be synthesized using DME and CO2 as the starting materials.Experimental
Chemicals
Ruthenium carbonyl (Ru3(CO)12, >98%), tris(triphenylphosphine)ruthenium(II) dichloride (Ru(PPh3)3Cl2, 99%), and ruthenium acetylacetonate (Ru(acac)3, 98+%) were purchased from Adamas Reagent, Ltd. Tetracarbonyl di-u-chlorodirhodium(I) (Rh2(CO)4Cl2, Rh 50.1–52.9%), anhydrous lithium iodide (LiI, 99.95%), potassium iodide (KI, 99.9%), lithium tetrafluoroborate (LiBF4, 98%), cobalt(II) iodide (CoI2, 99.5%), ferrocene (99%), decacarbonyl dimanganese (Mn2(CO)10, C 30.6%) and 2-pyrrolidinone (99%) were obtained from Alfa Aesar China Co, Ltd. Zinc iodide (ZnI2, 98%), nickel(II) chloride (NiCl2, 98%), formaldehyde (HCHO, analytical grade, 40% solution in H2O) and N-methylpyrrolidine (98%) were provided by J&K Chemical Ltd (Shanghai). 1,3-Dimethyl-2-imidazolidinone (DMI, 99%), dicobalt octacarbonyl (Co2(CO)8), lithium chloride (LiCl, 98%) and benzene (99.5%) were purchased from TCI Shanghai Co., Ltd. Formic acid (HCOOH, 88% solution in H2O), N-methyl-2-pyrrolidone (NMP, 99.5%), N,N-dimethylformamide (DMF, 99.5%) and cyclohexane (99.5%) were provided by Sinopharm Chemical Reagent Co., Ltd. Methanol (99.5%), acetic acid (99.5%) and cyclohexanone (99.5%) were obtained from Beijing Chemical Company. Toluene (99.8%, HPLC) was obtained from Xilong Chemical Co., Ltd. Iridium carbonyl (Ir4(CO)12, 98+%), copper(II) sulfate (CuSO4, 98%) and acetaldehyde (CH3CHO, 99.5%) were purchased from Acros Organics. Methyl iodide (CH3I, 99%) was bought from Shandong Xiya Chemical Industry Co., Ltd. Dimethyl ether (DME, 99.9%) was obtained from Zhao Qing Gao Neng Da Chemical Industry Co., Ltd. Deuterium gas (D2, 99.999%) was offered by Zhengzhou Xingdao Chemical Technology Co., Ltd. CO2 (99.99%), H2 (99.99%) and CO (99.99%) were provided by Beijing Analytical Instrument Company.Catalytic reaction
The apparatus and procedures used were similar to our previous work.5a All the reactions were conducted in a 16 mL Teflon-lined stainless steel batch reactor equipped with a magnetic stirrer. The inner diameter of the reactor was 18 mm. In a typical experiment, known amounts of Ru and/or Co catalysts, LiI or another promoter, and 2 mL DMI or another solvent were loaded sequentially into the reactor. The reactor was filled with DME of saturated vapor pressure (0.5 MPa) at room temperature after the reactor was purged three times with the same gas. Then CO2 in the cylinder was charged into the reactor to the desired pressure, and the inlet valve of CO2 was closed. Then H2 was charged into the reactor until a suitable total pressure was reached. The reactor was placed in an air bath of constant temperature, and magnetic stirring was started at 800 rpm. After the reaction, the reactor was cooled in an ice-water bath for 1 h, and the residual gas was released slowly and collected in a gasbag. The liquid mixture was analyzed by GC (Agilent 7890B) equipped with a flame ionization detector and an HP-5 capillary column (0.32 mm in diameter, 30 m in length) using toluene as the internal standard. The identification of the liquid products was done using a GC-MS (Agilent-7890B-5977A) as well as by comparing the retention times of the standards in the GC traces. The yields of the products were calculated from the GC data. NMR spectra were recorded on a Bruker Avance III HD 400 MHz NMR spectrometer (1H NMR, 400 MHz; 13C NMR, 100 MHz). The gaseous samples were analyzed using a GC (Agilent 4890D) equipped with a TCD detector and a packed column (carbon molecular sieve TDX-01, 3 mm in diameter and 1 m in length) using argon as the carrier gas.Recycling test
After the reaction, the reactor was cooled down in an ice bath and the residual gas was released. The amount of the product was determined as discussed above. Then the alcohols formed and unreacted DME in the reactor were removed in a vacuum oven at 80 °C for 3 h, which was confirmed by GC analysis. The catalytic system was used directly for the next run.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21373234, 21533011), the National Key Research and Development Program of China (2017YFA0403102), and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).References
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- X. G. San, Y. Zhang, W. J. Shen and N. Tsubaki, Energy Fuels, 2009, 23, 2843–2844 CrossRef CAS.
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley–VCH, Weinheim, 2010 Search PubMed.
- (a) M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed; (b) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed; (c) W. Wang, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC; (d) P. G. Jessop, F. Joóand and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS; (e) Q. Liu, L. P. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- (a) Q. L. Qian, J. J. Zhang, M. Cui and B. X. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed; (b) J. Wei, Q. J. Ge, R. W. Yao, Z. Y. Wen, C. Y. Fang, L. S. Guo, H. Y. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed; (c) Y. Liu, W. M. Ren, J. Liu and X. B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed; (d) M. Cui, Q. L. Qian, J. J. Zhang, C. J. Chen and B. X. Han, Green Chem., 2017, 19, 3558–3565 RSC; (e) C. Liu, J. H. Xie, G. L. Tian, W. Li and Q. L. Zhou, Chem. Sci., 2015, 6, 2928–2931 RSC; (f) Q. W. Song, Z. H. Zhou and L. N. He, Green Chem., 2017, 19, 3707–3728 RSC; (g) F. Ferretti, M. Sharif, S. Dastgir, F. Ragaini, R. Jackstell and M. Beller, Green Chem., 2017, 19, 3542–3548 RSC; (h) J. Wang, Z. Hao and S. Wohlrab, Green Chem., 2017, 19, 3595–3600 RSC; (i) T. V. Q. Nguyen, J. A. Rodríguez-Santamaría, W. Yoo and S. Kobayashi, Green Chem., 2017, 19, 2501–2505 RSC.
- (a) S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed; (b) J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed; (c) S. Wesselbaum, T. V. Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed; (d) Z. B. Han, L. C. Rong, J. Wu, L. Zhang, Z. Wang and K. L. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS PubMed; (e) H. Yang, S. Qin, H. Wang and J. Lu, Green Chem., 2015, 17, 5144–5148 RSC; (f) J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC; (g) F. Liao, X. Wu, J. Zheng, M. M. Li, A. Kroner, Z. Zeng, X. Hong, Y. Yuan, X. Gong and S. C. E. Tsang, Green Chem., 2017, 19, 270–280 RSC.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Energy, 1997, 22, 343–348 CrossRef CAS.
- K. K. Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67–81 CrossRef.
- H. Kurakata, Y. Izumi and K. Aika, Chem. Commun., 1996, 389–390 RSC.
- T. Inui and T. Yamamoto, Catal. Today, 1998, 45, 209–214 CrossRef CAS.
- T. Inui, T. Yamamoto, M. Inoue, H. Hara, T. Takeguchi and J. B. Kim, Appl. Catal., A, 1999, 186, 395–406 CrossRef CAS.
- L. S. Davy, D. Nieskens, Y. Ferrari, R. Liu and J. Kolonko, Catal. Commun., 2011, 14, 111–113 CrossRef.
- S. G. Li, H. J. Guo, C. R. Luo, H. R. Zhang, L. Xiong, X. D. Chen and L. L. Ma, Chem. Lett., 2013, 143, 345–355 CAS.
- M. Cui, Q. L. Qian, Z. H. He, Z. F. Zhang, J. Ma, T. B. Wu, G. Y. Yang and B. X. Han, Chem. Sci., 2016, 7, 5200–5205 RSC.
- Q. L. Qian, M. Cui, Z. H. He, C. Y. Wu, Q. G. Zhu, Z. F. Zhang, J. Ma, G. Y. Yang, J. J. Zhang and B. X. Han, Chem. Sci., 2015, 6, 5685–5689 RSC.
- K. Tominaga, Y. SaSaki, M. Saito, K. Hagihara and T. Watanabe, J. Mol. Catal., 1994, 89, 51–56 CrossRef CAS.
- Z. H. He, Q. L. Qian, J. Ma, Q. L. Meng, H. C. Zhou, J. L. Song, Z. M. Liu and B. X. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS PubMed.
- (a) K. Tominaga, Y. Sasaki, T. Watanabe and M. Saito, Stud. Surf. Sci. Catal., 1998, 114, 495–498 CrossRef CAS; (b) J. J. Zhang, Q. L. Qian, M. Cui, C. J. Chen, S. S. Liu and B. X. Han, Green Chem., 2017, 19, 4396–4401 RSC.
- (a) M. Beller and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 4527–4528 CrossRef CAS PubMed; (b) G. Prieto, ChemSusChem, 2017, 10, 1056–1070 CrossRef CAS PubMed.
- S. X. Bai, Q. Shao, P. T. Wang, Q. G. Dai, X. Y. Wang and X. Q. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- (a) B. R. Sohnlein, S. G. Li, J. F. Fuller and D. S. Yang, J. Chem. Phys., 2005, 123, 014318 CrossRef PubMed; (b) S. E. Hill, E. D. Glendening and D. Feller, J. Phys. Chem. A, 1997, 101, 6125–6131 CrossRef CAS.
- P. M. Maitlis, A. Haynes, B. R. James, M. Catellani and G. P. Chiusoli, Dalton Trans., 2004, 3409–3419 RSC.
- S. D. Badmaev, V. D. Belyaev, G. G. Volkova, E. A. Paukshtis and V. A. Sobyanin, React. Kinet. Catal. Lett., 2007, 90, 197–204 CrossRef CAS.
- M. Roper and H. Loevenich, J. Organomet. Chem., 1983, 255, 95–102 CrossRef.
- (a) G. Braca, G. Sbrana, G. Valentini, G. Andrich and G. Gregorio, J. Am. Chem. Soc., 1978, 100, 6238–6240 CrossRef CAS; (b) G. Braca, L. Paladini, G. Sbrana, G. Valentini, G. Andrich and G. Gregorio, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 115–122 CrossRef CAS.
- (a) P. Lu, G. H. Yang, Y. Tanaka and N. Tsubaki, Catal. Today, 2014, 232, 22–26 CrossRef CAS; (b) D. Wang, G. H. Yang, Q. X. Ma, Y. Yoneyama, Y. S. Tan, Y. Z. Han and N. Tsubaki, Fuel, 2013, 109, 54–60 CrossRef CAS.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
- R. Bartek, M. M. Habib and W. R. Pretzer, J. Mol. Catal., 1985, 33, 245–248 CrossRef CAS.
- K. Tsuchiya, J. Huang and K. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- (a) D. B. Rasmussen, J. M. Christensen, B. Temel, F. Studt, P. G. Moses, J. Rossmeisl, A. Riisager and A. D. Jensen, Angew. Chem., Int. Ed., 2015, 54, 7261–7264 CrossRef CAS PubMed; (b) P. Cheung, A. Bhan, G. J. Sunley and E. Iglesia, Angew. Chem., Int. Ed., 2006, 45, 1617–1620 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/c7gc02807e |
| This journal is © The Royal Society of Chemistry 2018 |