
An artificial synthetic pathway for acetoin, 2,3-butanediol, and 2-butanol production from ethanol using cell free multi-enzyme catalysis†
Liaoyuan
Zhang‡
 *ab,
Raushan
Singh‡
b,
Sivakumar
D
b,
Zewang
Guo
a,
Jiahuan
Li
a,
Fanbing
Chen
a,
Yuanzhi
He
a,
Xiong
Guan
a,
Yun Chan
Kang
*c and
Jung-Kul
Lee
*b
*ab,
Raushan
Singh‡
b,
Sivakumar
D
b,
Zewang
Guo
a,
Jiahuan
Li
a,
Fanbing
Chen
a,
Yuanzhi
He
a,
Xiong
Guan
a,
Yun Chan
Kang
*c and
Jung-Kul
Lee
*b
aKey Laboratory of Biopesticide and Chemical Biology, Ministry of Education, College of Life Sciences, Gutian Edible Fungi Research Institute, Fujian Agriculture and Forestry University, Fuzhou, Fujian Province 350002, PR China. E-mail: zliaoyuan@126.com; Fax: +86-591-83789121; Tel: +86-591-83789492
bDepartment of Chemical Engineering, Konkuk University, Seoul 05029, Republic of Korea. E-mail: jkrhee@konkuk.ac.kr; Fax: +82-2-4583504; Tel: +82-2-4503505
cDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: yckang@korea.ac.kr; Fax: +82-2-4583504; Tel: +82-2-4503505
First published on 23rd November 2017
Abstract
Upgrading ethanol to higher order alcohols is desired but difficult using current biotechnological methods. In this study, we designed a completely artificial reaction pathway for upgrading ethanol to acetoin, 2,3-butanediol, and 2-butanol in a cell-free bio-system composed of ethanol dehydrogenase, formolase, 2,3-butanediol dehydrogenase, diol dehydratase, and NADH oxidase. Under optimized conditions, acetoin, 2,3-butanediol, and 2-butanol were produced at 88.78%, 88.28%, and 27.25% of the theoretical yield from 100 mM ethanol, respectively. These results demonstrate that this artificial synthetic pathway is an environmentally-friendly novel approach for upgrading bio-ethanol to acetoin, 2,3-butanediol, and 2-butanol.
Introduction
The production of biofuels from renewable biomass has been given increased attention because of the finite supply of fossil fuels.1–3 It is estimated that the global biofuel market consists of approximately 15% biodiesel and 85% bioethanol.4,5 Bioethanol, a direct product of biomass fermentation, is considered a renewable energy source that can replace or blend with gasoline for transportation use.6,7 Gasoline blended with an appropriate bioethanol as a fuel additive may increase the octane level and reduce toxic emission of pollutants.8 However, several disadvantages including the low energy density, high hygroscopicity, and corrosivity to engine technology and fuel pipelines limit the broad implementation of bioethanol in global transportation.7,9,10 This has shifted research interest to higher-order alcohols such as 2,3-butanediol and butanol.7,11,122,3-Butanediol is an important platform chemical and potential aviation fuel with a heating value of 27.2 kJ g−1 and can be used to produce butadiene (a monomer of synthetic rubber), acetoin (a volatile compound used in foods, plant growth promoters, and biological pest control), diacetyl (a flavor enhancer), and methyl ethyl ketone (2-butanone, an excellent organic solvent).13–15 Furthermore, 2-butanone can be hydrogenated to produce 2-butanol, which shows the highest octane number and the lowest boiling point among the four stereoisomers of butanol.1,16 Compared to bioethanol, butanol has a higher energy density and lower hygroscopicity and can be directly blended with gasoline without the need for modifying the current vehicle system.17,18 In recent years, many studies have been conducted to engineer microbes for 1-butanol and isobutanol production through genetic modification.19,20 However, bio-production of 2-butanol has not been widely explored. A few studies on 2-butanol production have been conducted by extending the terminal product of 2,3-butanediol or acetolactate using metabolic engineering methods.1,16,21 The final 2-butanol concentration was still quite low because of metabolic flux limitations and its inevitable toxicity to microbial cells.
One of the potential solutions for overcoming these limitations is to construct a simplified pathway in vitro using a limited number of enzymes, a process known as cell-free metabolic engineering (CFME). CFME is a cell-free bio-system that uses in vitro ensembles of catalytic proteins prepared from purified enzymes or crude lysates of cells to produce target products.22,23 It can efficiently eliminate cell-associated process barriers, such as substrate or product toxicity, intracellular flux balance that results in low target product yield and unwanted by-products, and product excretion constraints by intracellular transport barriers.4,22,24 Compared with traditional metabolic engineering, CFME has many unique advantages including no requirement for cell growth, shorter synthetic pathway, faster reaction rate, higher theoretical yield and productivity, and easier manipulation of reaction conditions, although some challenges such as activity, stability, and cost of enzymes must be resolved.25,26
The costs of acetoin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 $ per ton), 2,3-butanediol (10000–50000 $ per ton), and 2-butanol (10000–78000 $ per ton) are over 10-fold that of ethanol (800–950 $ per ton), according to the global trade data. To upgrade ethanol to higher-order alcohols, we designed a completely artificial CFME-based reaction pathway composed of ethanol dehydrogenase, formolase, 2,3-butanediol dehydrogenase, diol dehydratase, and NADH oxidase for the conversion of ethanol into C4 alcohols. Furthermore, protein engineering was conducted to improve the catalytic efficiencies of formolase and diol dehydratase to increase the conversion rate of this artificial pathway. In addition, a novel NAD(P)H purge valve regulatory node was developed to prevent NADH buildup in this artificial reaction. The results showed that C4 compounds including acetoin, 2,3-butanediol, and 2-butanol can be efficiently produced from ethanol using the CFME-based artificial reaction pathway.
000–30000 $ per ton), 2,3-butanediol (10000–50000 $ per ton), and 2-butanol (10000–78000 $ per ton) are over 10-fold that of ethanol (800–950 $ per ton), according to the global trade data. To upgrade ethanol to higher-order alcohols, we designed a completely artificial CFME-based reaction pathway composed of ethanol dehydrogenase, formolase, 2,3-butanediol dehydrogenase, diol dehydratase, and NADH oxidase for the conversion of ethanol into C4 alcohols. Furthermore, protein engineering was conducted to improve the catalytic efficiencies of formolase and diol dehydratase to increase the conversion rate of this artificial pathway. In addition, a novel NAD(P)H purge valve regulatory node was developed to prevent NADH buildup in this artificial reaction. The results showed that C4 compounds including acetoin, 2,3-butanediol, and 2-butanol can be efficiently produced from ethanol using the CFME-based artificial reaction pathway.
Experimental
Enzymes and chemicals
Restriction enzymes, DNA Polymerase High Fidelity and T4 DNA ligase were purchased from TaKaRa Biotech (Shiga, Japan) and New England Biolabs (Ipswich, MA, USA), respectively. DNA and protein markers were obtained from Tiangen Biotech (Shanghai, China). Isopropyl-beta-D-thiogalactopyranoside (IPTG), dithiothreitol (DTT), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St Louis, MO, USA) and Sinopharm (Shanghai, China), respectively. The standards, including (3S/3R)-acetoin, (2S,3S)-2,3-butanediol, (2R,3R)-2,3-butanediol, meso-2,3-butanediol, butanone, and 2-butanol, were obtained from Sigma-Aldrich. All other chemicals, unless otherwise indicated, were of analytical grade and commercially available.Bacterial strains, plasmids, and bacterial growth conditions
The strains and plasmids used in this study are presented in Table 1. Escherichia coli DH5α and BL21(DE3) as the cloning and expression hosts were cultured at 37 °C. The plasmid pET28a was used to construct the expression vector. Luria-Bertani (LB) medium was used for strain cultivation and recombinant protein expression. Kanamycin was added to the LB medium for cultivation of recombinant strains at a final concentration of 50 μg mL−1.| Strain or plasmid | Relevant genotype and description | Source |
|---|---|---|
| Strains | ||
| E. coli DH5α | Host of plasmid for cloning | Lab stock |
| E. coli BL21(DE3) | Host of plasmid for expression, F-, ompT, hsdSB(rB-mB-), gal(λ c I 857, ind1, Sam7, nin5, lacUV-T7 gene1), dcm(DE3) | Lab stock |
| Plasmids | ||
| pET28a | Expression vector, KmR | Novagen |
| pET-EtDH | pET28a carries EtDH gene | This study |
| pET-EtDH:D46G | pET28a carries EtDH mutant gene | This study |
| pET-FLS | pET28a carries FLS gene | This study |
| pET-FLS:L482S | pET28a carries FLS mutant gene | This study |
| pET-FLS:L482R | pET28a carries FLS mutant gene | This study |
| pET-FLS:L482E | pET28a carries FLS mutant gene | This study |
| pET-BDH | pET28a carries BDH gene | This study |
| pET-BDH:S199A | pET28a carries BDH mutant gene | This study |
| pET-DDH | pET28a carries DDH gene | This study |
| pET-DDH:S302A | pET28a carries DDH mutant gene | This study |
| pET-DDH:Q337A | pET28a carries DDH mutant gene | This study |
| pET-DDH:F375I | pET28a carries DDH mutant gene | This study |
| pET-DDH:S302A/Q337A | pET28a carries DDH mutant gene | This study |
| pET-DDH:S302A/F375I | pET28a carries DDH mutant gene | This study |
| pET-DDH:Q337A/F375I | pET28a carries DDH mutant gene | This study |
| pET-DDH:S302A/Q337A/F375I | pET28a carries DDH mutant gene | This study |
| pET-dhaR | pET28a carries dhaR gene | This study |
| pET-DDH-dhaR | pET28a carries DDH and dhaR genes | This study |
| pET-NOX | pET28a carries NOX gene | This study |
Recombinant protein expression and purification
The genes for ethanol dehydrogenase (EtDH), formolase (FLS), 2,3-butanediol dehydrogenase (BDH), diol dehydratase (DDH), and NADH oxidase (NOX) from Cupriavidus necator,27Pseudomonas fluorescens,28Clostridium autoethanogenum,29Lactobacillus brevis,1,30 and Lactobacillus rhamnosus31 were synthesized by General Biosystems, Inc. (Anhui, China) and cloned into the expression plasmid pET28a (the details of enzyme sequences are presented in the ESI†). The protein expression plasmids were introduced into E. coli BL21(DE3). Recombinant E. coli BL21(DE3) harboring pET-EtDH, pET-FLS, pET-BDH, pET-DDH, pET-dhaR, pET-DDH-dhaR, and pET-NOX were cultured at 37 °C in LB medium and induced by adding 0.5 mM IPTG when the optical density was 0.6 at 600 nm. After induction for 24 h at 18 °C, the cells were harvested by centrifugation and disrupted by sonication in an ice bath. The cell lysate was centrifuged at 8000g for 10 min to remove the cell debris. For the EtDH, FLS, BDH, dhaR, and NOX enzymes, the soluble fraction was subjected to purification with a HisTrap HP column according to the purification protocol (GE Healthcare, Little Chalfont, UK). DDH purification was carried out as described previously.32 The purified enzymes were concentrated and desalted by ultrafiltration, and then detected by SDS-PAGE.Development of the enzyme variants
EtDH:D46G and BDH:S199A variants were generated by site-directed mutagenesis, which was performed using the primers EtDH1/EtDH2 and BDH1/BDH2 as shown in Table S1.† The recombinant plasmids pET-EtDH and pET-BDH containing the wild-type EtDH and BDH genes were used as DNA templates for PCR-amplification, respectively. Recombinant plasmids harboring the correct mutant genes were transformed into E. coli BL21(DE3) and colonies selected via kanamycin resistance were used for protein expression. After purification, the activities and kinetic parameters of the EtDH and BDH variants were determined.To improve the catalytic efficiency of the FLS enzyme, the HotSpot Wizard 2.0 server was used to analyze hot spots by inputting the FLS structure (PDB no.: 4QPZ).28,33 Six residuals as hot spots (T396, T446, M473, S477, L482, and L499) were identified and subjected to site-saturated mutagenesis using the primers FLS1–FLS12 (Table S1†). The recombinant plasmid pET-FLS containing the wild-type FLS gene was used as a DNA template. Recombinant plasmids containing the mutant genes were transformed into E. coli BL21(DE3). FLS variants were screened using the whole-cell biocatalytic method with acetaldehyde as a substrate. Briefly, the colonies were inoculated in LB medium and protein expression was induced by adding 0.5 mM IPTG for 24 h at 18 °C when the optical density at 600 nm reached 0.6. The cells were harvested by centrifugation and used to carry out whole-cell biocatalysis in a reaction mixture containing 50 mM phosphate buffer (pH 8.0), 100 mM acetaldehyde, and 40 g L−1 wet cell weight (WCW) at 30 °C for 6 h. The product acetoin was quantified using the Voges–Proskauer (VP) reaction (see the ESI†). Moreover, positive variants were verified by enzyme activity and kinetic parameter assays after purification. Variants were also analyzed by commercial sequencing.
DDH variants including S302A, Q337A, F375I, S302A/Q337A, S302A/F375I, Q337A/F375I, and S302A/Q337A/F375I were developed to assess their catalytic efficiencies compared with the wild-type DDH enzyme. Site-directed mutagenesis was performed using the primers DDH1–DDH6 as shown in Table S1.† The recombinant plasmid pET-DDH-dhaR harboring the wild-type DDH and its reactivating factor dhaR genes were used as DNA templates for PCR amplification. The PCR products were transformed into E. coli BL21(DE3) for protein expression, which was conducted in LB medium containing 0.5 mM IPTG at 18 °C for 24 h. These variants were used to evaluate the catalytic activities using whole-cell biocatalysis with meso-2,3-butanediol as a substrate. The reaction mixture consisted of 50 mM HEPES buffer (pH 7.0), 50 mM meso-2,3-butanediol, 20 μM coenzyme B12, and 40 g L−1 wet cell weight, and whole-cell biocatalysis was carried out at 30 °C for 6 h in the dark. The product butanone was analyzed and quantified by gas chromatography.
Enzyme activity assays
Determination of kinetic parameters
The kinetic parameters of EtDH and EtDH:D46G were determined in a reaction mixture containing 100 mM glycine–NaOH buffer (pH 9.5), 5 mM Mg2+, 3 mM NAD+/NADP+, and 0.5–100 mM ethanol at room temperature. The Km and kcat values were obtained by a nonlinear regression fitting of the Michaelis–Menten equation. All assays were carried out in triplicate.The kinetic parameters of FLS and its variants were assayed in a reaction mixture containing 100 mM phosphate buffer (pH 8.0), 1 mM Mg2+, 0.1 mM TPP, and 0.5–20 mM acetaldehyde at room temperature. The Km and kcat values were obtained by a nonlinear regression fitting of the Michaelis–Menten equation. All assays were carried out in triplicate.
The kinetic parameters of BDH and BDH:S199A were measured in a reaction mixture containing 50 mM Tris-HCl buffer (pH 7.5), 0.2 mM NADPH, 1 mM DTT, and 0.5–100 mM acetoin or 0.5–10 mM butanone at room temperature. The Km and kcat values were obtained by a nonlinear regression fitting of the Michaelis–Menten equation. All assays were carried out in triplicate.
Cell-free multi-enzyme catalysis system
The synthesis of acetoin, 2,3-butanediol, and 2-butanol from ethanol by cell-free multi-enzyme catalysis was conducted in a 0.5 mL reaction mixture containing the substrate, coenzymes, metal ions, and the corresponding enzymes. The reaction conditions including temperature, pH, coenzyme, and metal ions were optimized to improve the flux of the artificial reaction pathway (see the ESI†). The optimal reaction conditions for acetoin, 2,3-butanediol, and 2-butanol from ethanol were as follows.Acetoin production was carried out in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 0.1 mg mL−1 EtDH, 0.2 mg mL−1 FLS:L482S, 0.1 mg mL−1 NOX, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. The reaction was conducted at 30 °C.
2,3-Butanediol was produced in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1 mM NADP+, 0.1 mg mL−1 EtDH:D46G, 0.2 mg mL−1 FLS:L482S, 0.1 mg mL−1 NOX, 0.1 mg mL−1 BDH:S199A, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. The reaction was conducted at 30 °C.
2-Butanol was produced in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1 mM NADP+, 0.1 mg mL−1 EtDH:D46G, 0.2 mg mL−1 FLS:L482S, 0.1 mg mL−1 NOX, 0.1 mg mL−1 BDH:S199A, 0.2 mg mL−1 DDH:Q337A/F375I, 0.2 mg mL−1 dhaR, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, 1 mM coenzyme B12, 100 mM ATP, and 100 mM ethanol. The reaction was carried out at 30 °C.
All reactions were carried out for 6 h and the products were identified by gas chromatography (GC). The product percentage yields were calculated using the following equation: percentage yield (%) = product yield (mM)/theoretical yield (mM). Theoretically, two moles of ethanol can generate one mole of acetoin or 2,3-butanediol or 2-butanol.
Recyclability of cascade reactions
Purified enzymes were mixed with activated silicon oxide nanoparticles and incubated for 12 h at 4 °C. Before immobilization, the silicon oxide nanoparticles (4830HT; Nanostructured & Amorphous Materials, Houston, TX, USA) were activated by treating the nanoparticles with glutaraldehyde (Sigma). Immobilization yield (%) and immobilization efficiency (%) were calculated for the immobilized enzymes as follows: immobilization efficiency = (αi/αf) × 100, immobilization yield = [{Pi − (Pw + Ps)}/Pi] × 100, where αi is the total activity of the immobilized enzyme and αf is the total activity of the free enzyme. Pi is the total protein content of the crude enzyme preparation and Pw and Ps are the protein concentrations in the wash solution and supernatant after immobilization, respectively.For acetoin production, the reaction was performed in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1.06 U mL−1 EtDH, 0.05 U mL−1 FLS:L482S, 0.98 U mL−1 NOX, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. Similarly, 2,3-butanediol production was performed in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1 mM NADP+, 0.1 mg mL−1 EtDH:D46G, 0.2 mg mL−1 FLS:L482S, 0.1 mg mL−1 NOX, 0.1 mg mL−1 BDH:S199A, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. The reaction was conducted at 30 °C for 6 h. The reusability of immobilized enzymes was examined under the same reaction conditions. After each reaction cycle, the immobilized enzyme was removed by centrifugation at 4000g for 30 min. The immobilized enzyme was collected and washed with deionized water and buffer. For the second reaction cycle, the immobilized enzyme was dissolved in fresh buffer, added to the substrate, and processed as in the first cycle.
Analytical methods
Cell growth was determined by measuring the optical density at 600 nm with a spectrophotometer (UV-1800, MAPADA). Protein concentration was determined using the Bradford method, and bovine serum albumin served as the standard protein. The concentrations of ethanol, acetaldehyde, acetoin, 2,3-butanediol, butanone, and 2-butanol were determined with the addition of isoamylol as an internal standard and quantified using a gas chromatograph system (Agilent GC9860, Santa Clara, CA, USA) equipped with a chiral column (Supelco β-DEX™ 120, 30 m length, 0.25 mm inner diameter). The operating conditions were as follows: N2 was used as the carrier gas at a flow rate of 1.2 mL min−1; the injector temperature and detector temperature were 215 and 245 °C, respectively; and the column temperature was maintained at 50 °C for 1.5 min, and then increased to 180 °C at a rate of 15 °C min−1.Results and discussion
Construction of an artificial pathway for C4 compound production from ethanol
We designed a novel artificial reaction pathway for the conversion of ethanol into the C4 compounds acetoin, 2,3-butanediol, and 2-butanol, which required cascade enzymes (Fig. 1). Ethanol is first dehydrogenated by the NAD(P)H-dependent EtDH to afford acetaldehyde, which undergoes a condensation reaction to yield acetoin by FLS, a computationally designed enzyme. Subsequently, acetoin is reduced by the NADPH-dependent BDH to produce 2,3-butanediol. Finally, 2-butanol is obtained via a dehydration and hydrogenation reaction by DDH and BDH, respectively. NOX is used to regenerate NAD+ throughout the reaction process. Considering that oxygen is used as a substrate for NOX, aeration may be required for a large-scale reaction. By stoichiometry, 1 M ethanol is converted completely into acetoin or 2,3-butanediol and generates 1 or 0.5 M NADH, respectively, which requires 0.5 M oxygen for acetoin production or 0.25 M oxygen for 2,3-butanediol production (1 M NADH oxidized by NOX requires 0.5 M oxygen as a substrate). Thus, 1 L of the reaction mixture containing 1 M ethanol as a substrate requires 11.2 or 5.6 L oxygen for acetoin or 2,3-butanediol production throughout the reaction process. For 2-butanol production, the oxygen demand in the reaction was lower than that of 2,3-butanediol production. Compared to traditional fermentation, the aeration demand in the current reaction system is much lower supporting the scale-up potential of the artificial reaction pathway. In this artificial reaction pathway, we chose EtDH from C. necator,27 BDH from C. autoethanogenum,29 DDH from L. brevis1,30 with its reactivating factor dhaR, and NOX enzymes from L. rhamnosus31 as candidate enzymes because of their relatively high catalytic efficiencies for their corresponding substrates as demonstrated in previous studies. However, the FLS enzyme was only reported to catalyze three molecules of formaldehyde into one molecule of dihydroxyacetone.28 To verify the catalytic ability of acetaldehyde conversion to acetoin by the FLS enzyme, the catalytic reaction was carried out using whole-cell biocatalysts over-expressing FLS with acetaldehyde as a substrate. The VP test and GC/GC-MS analysis indicated that racemic acetoin ((3S)-acetoin and (3R)-acetoin) was produced from acetaldehyde, demonstrating that the FLS enzyme can catalyze the conversion of acetaldehyde into acetoin (Fig. S1, S14 and S15†). These results show that C4 compounds including acetoin, 2,3-butanediol, and 2-butanol can be produced from ethanol using the artificial reaction pathway.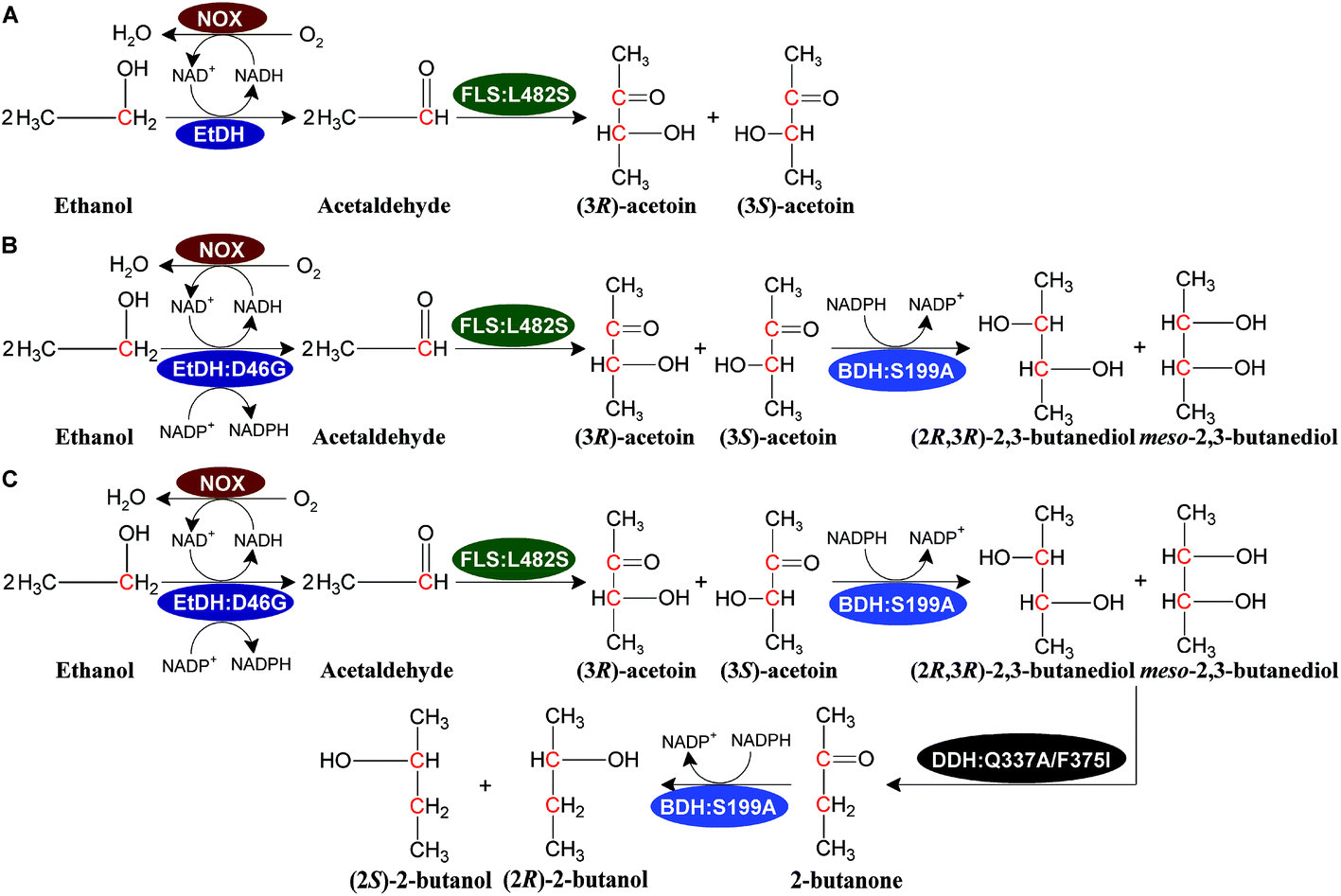 | ||
| Fig. 1 Bio-system based on the artificial reaction cascade for the conversion of ethanol to acetoin (A), 2,3-butanediol (B), and 2-butanol (C). The enzymes are ethanol dehydrogenase (EtDH and its variant EtDH:D46G from C. necator), formolase (FLS:L482S, a newly designed enzyme originate from P. fluorescens), 2,3-butanediol dehydrogenase (BDH:S199A from C. autoethanogenum), diol dehydratase (DDH:Q337/F375I from L. brevis), and NADH oxidase (NOX from L. rhamnosus). | ||
Characterization of enzymes in the artificial pathway
Furthermore, to determine the catalytic efficiencies of these enzymes in the artificial pathway, we systematically determined the kinetic parameters of EtDH, FLS, BDH, and DDH after purification (the results of enzyme expression and purification are presented in ESI, Fig. S2 and S3†). As shown in Table 2, the kcat/Km values of EtDH for ethanol with NAD+, FLS for acetaldehyde with TPP, and BDH for acetoin/butanone with NADPH were 17.09, 7.69 × 10−3, 18.50, and 19.31 s−1 mM−1, respectively. However, the purified DDH enzyme with its reactivating factor dhaR showed a low activity (0.02 U mg−1) towards meso-2,3-butanediol as a substrate, partly because of its low stability.32 A whole-cell catalytic method was used to determine the conversion of three 2,3-butanediol isomers (meso-2,3-butanediol, (2R,3R)-2,3-butanediol, and (2S,3S)-2,3-butanediol) to butanone by E. coli/pET-DDH-dhaR cells. The results showed that 20.56 mM butanone was produced from 50 mM meso-2,3-butanediol after 6 h (Fig. S4†), indicating that the DDH enzyme with dhaR had a high catalytic activity towards meso-2,3-butanediol in vivo. In contrast, no butanone was detected during whole-cell catalysis in the presence of (2R,3R)-2,3-butanediol and (2S,3S)-2,3-butanediol as substrates (Fig. S4†). These results are consistent with those of a previous study showing that only meso-2,3-butanediol was dehydrated by the DDH enzyme.34 The enzyme thermostability assay showed that the three enzymes had relatively higher stabilities and retained 87.91% (EtDH), 70.43% (FLS), and 91.30% (NOX) of their initial activities after incubation at 30 °C for 6 h, while all enzymes except NOX showed a decreased stability at 37 and 45 °C (Fig. S5†).| Enzyme | Substrate/coenzyme | K m (mM) | k cat (s−1) | k cat/Km (s−1 mM−1) | Ref. |
|---|---|---|---|---|---|
| EtDH | Ethanol/NAD+ | 0.37 ± 0.05 | 6.28 ± 0.11 | 17.09 | This study |
| Ethanol/NADP+ | 0 | 0 | 0 | This study | |
| EtDH:D46G | Ethanol/NAD+ | 0.57 ± 0.03 | 5.65 ± 0.09 | 9.97 | This study |
| Ethanol/NADP+ | 0.60 ± 0.02 | 0.99 ± 0.03 | 1.65 | This study | |
| FLS | Acetaldehyde/TPP | 58.46 ± 2.32 | 0.45 ± 0.03 | 7.69 × 10−3 | This study |
| FLS:L482S | Acetaldehyde/TPP | 47.45 ± 1.20 | 0.63 ± 0.01 | 1.33 × 10−2 | This study |
| FLS:L482R | Acetaldehyde/TPP | 50.27 ± 1.44 | 0.53 ± 0.02 | 1.06 × 10−2 | This study |
| FLS:L482E | Acetaldehyde/TPP | 50.95 ± 1.51 | 0.49 ± 0.03 | 9.66 × 10−3 | This study |
| BDH | Acetoin/NADPH | 84.86 ± 7.98 | 157.0 ± 9.0 | 18.50 | This study |
| Acetoin/NADH | 0 | 0 | 0 | This study | |
| Butanone/NADPH | 1.94 ± 0.05 | 29.90 ± 1.02 | 15.41 | This study | |
| Butanone/NADH | 0 | 0 | 0 | This study | |
| BDH:S199A | Acetoin/NADPH | 116.1 ± 8.7 | 224.3 ± 9.7 | 19.31 | This study |
| Acetoin/NADH | 0 | 0 | 0 | This study | |
| Butanone/NADPH | 1.08 ± 0.03 | 40.78 ± 1.42 | 37.76 | This study | |
| Butanone/NADH | 0 | 0 | 0 | This study | |
| DDH | 2,3-Butanediol/B12 | 10.4 | 35 | 3.4 | 36 |
| NOX | O2/NADH | 5.8 × 10−3 | 218.7 | 3.77 × 104 | 31 |
Acetoin production from ethanol via the artificial pathway
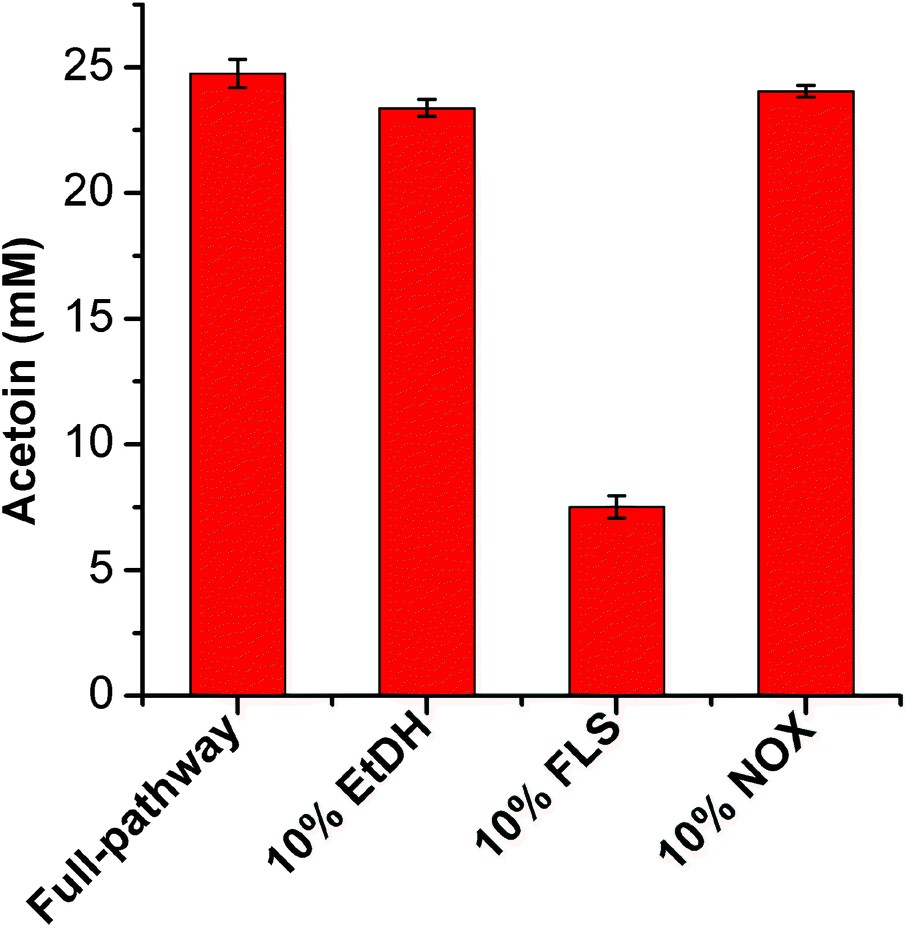 | ||
| Fig. 2 Analysis of the rate-limiting step in the conversion of ethanol to acetoin. Results are the means ± SD of three parallel replicates. | ||
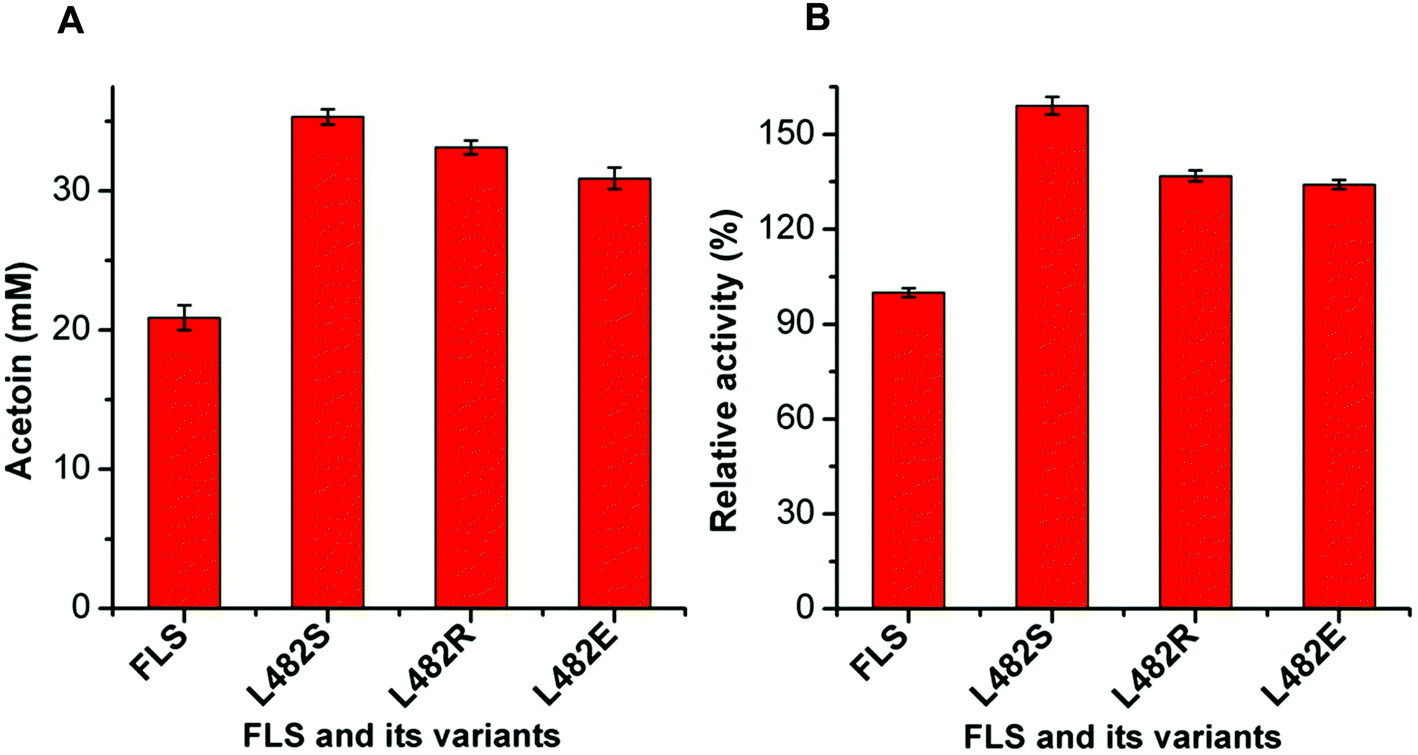 | ||
| Fig. 3 Screening of positive FLS variants by whole-cell biocatalysis using acetaldehyde (100 mM) as a substrate (A) and activity assays of FLS and its variants after purification (B). (A) The product acetoin from acetaldehyde through whole-cell biocatalysis was determined using the VP test. (B) Percentage ratios were calculated based on specific activities (specific activity of FLS was 0.16 U mg−1). Results are the means ± SD of three parallel replicates. | ||
The mutant FLS:L482S showed a higher activity than the wild-type FLS. Computational studies for FLS variants at atomic resolution using molecular dynamics simulation for 100 ns provided insight into the structural changes for the mutation L482S and its correlation with enzymatic activity. Computational analysis revealed that W480 in the mutant FLS:L482S made stronger contacts (2.1 Å) with the substrate acetaldehyde than the wild-type (2.8 Å) (Fig. S9A and B†). Siegel et al. (2015) reported W480 as an active site residue in FLS (4QPZ), supporting the higher activity of FLS:L482S.28 The 100 ns molecular dynamics analysis also shows that the FLS:L482S retained more hydrogen bond contacts than the wild-type enzyme (Fig. S9C and D†).
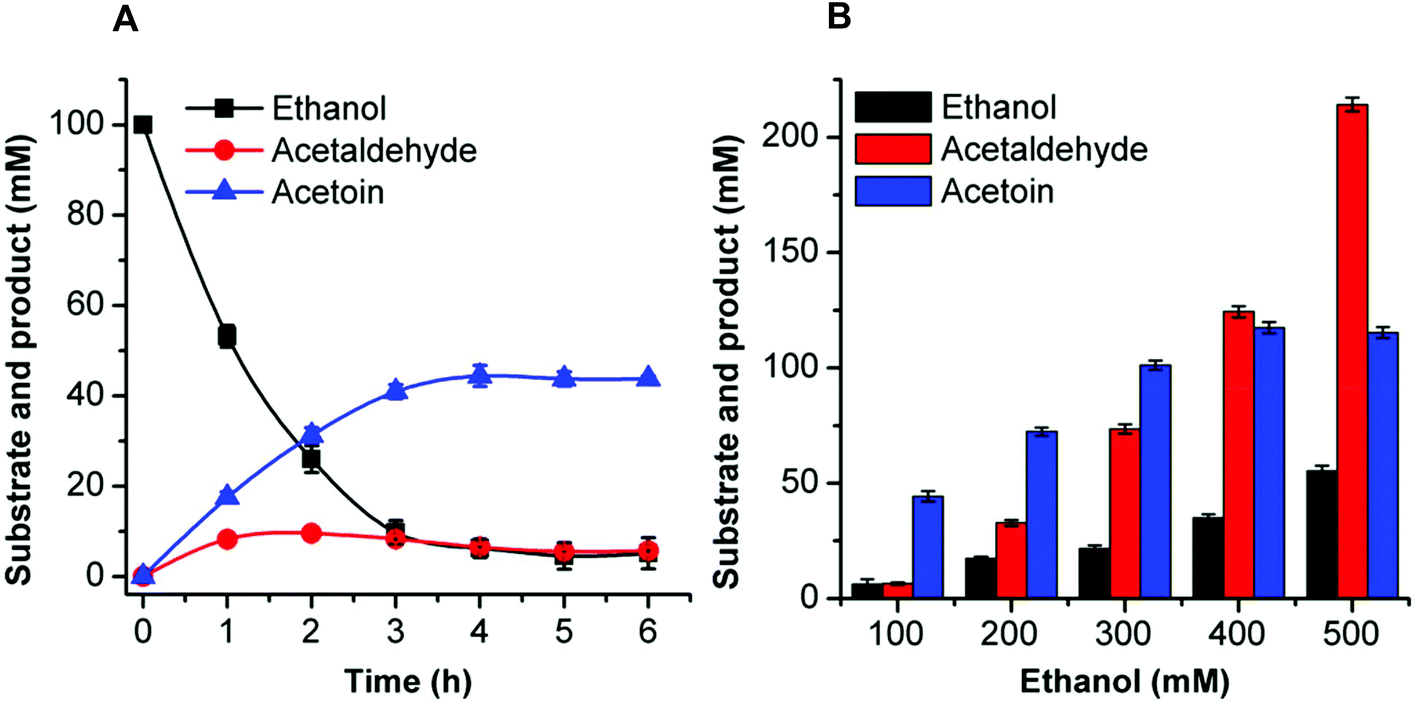 | ||
| Fig. 4 Time-course of acetoin production from 100 mM ethanol (A) and effects of different substrate concentrations on acetoin production (B) by the artificial reaction cascade. (A) The reaction was performed in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1.06 U ml−1 EtDH, 0.05 U mL−1 FLS:L482S, 0.98 U mL−1 NOX, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. (B) The reactions were carried out under the same conditions, except that the substrate concentration was 200–500 mM ethanol. These reactions were conducted at 30 °C for 6 h. Results are the means ± SD of three parallel replicates. | ||
2,3-Butanediol production from ethanol via the artificial pathway
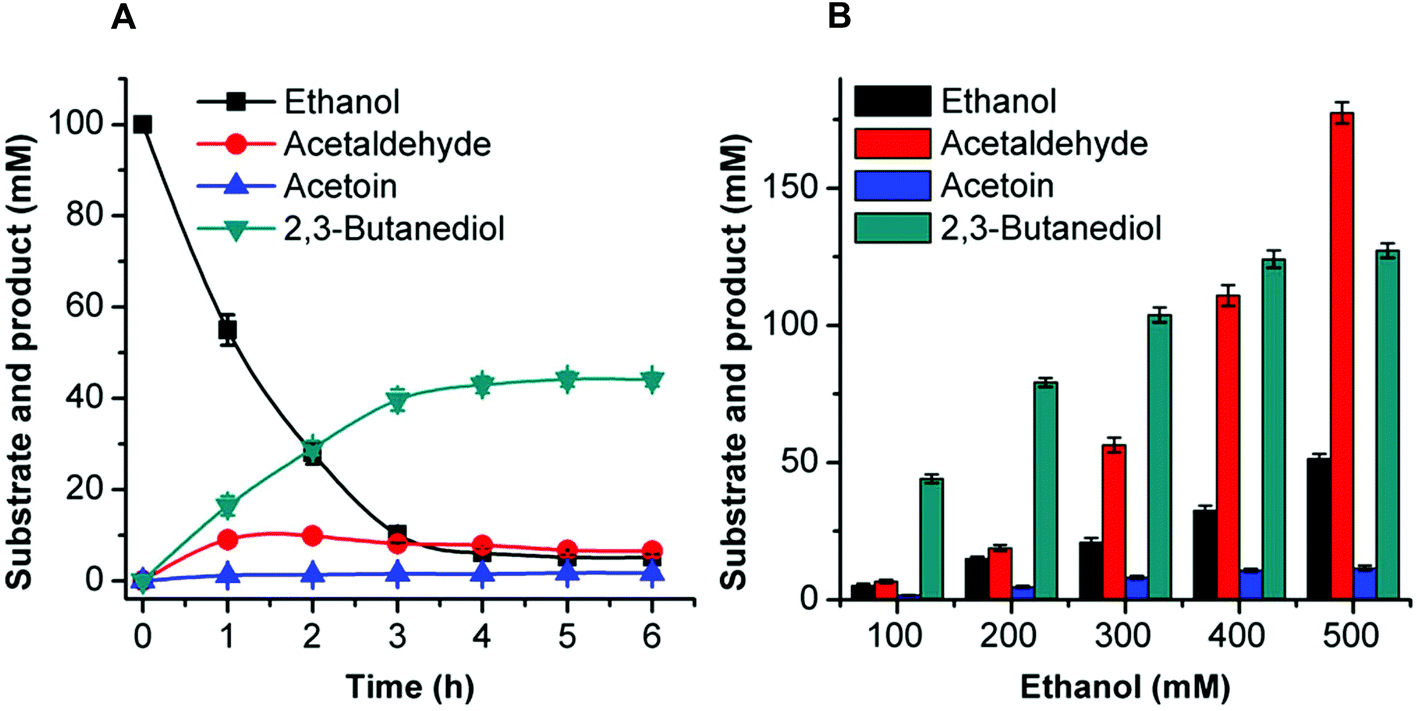 | ||
| Fig. 5 Time-course of 2,3-butanediol production from 100 mM ethanol (A) and effects of different substrate concentrations on 2,3-butanediol production (B) by the artificial reaction cascade. (A) The reaction was carried out in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1 mM NADP+, 0.88 U mL−1 EtDH:D46G, 0.05 U mL−1 FLS:L482S, 0.98 U mL−1 NOX, 5.11 U mL−1 BDH:S199A, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, and 100 mM ethanol. (B) The reactions were carried out under the same conditions except that the substrate concentrations were 200–500 mM ethanol. These reactions were conducted at 30 °C for 6 h. Results are the means ± SD of three parallel replicates. | ||
2-Butanol production from ethanol
:1 were produced from butanone using BDH:S199A at 30 °C after 6 h.
Theoretically, two moles of ethanol are oxidized to generate two moles of reducing equivalents, which satisfies the need for the reduction of acetoin and butanone into 2,3-butanediol and 2-butanol in the artificial pathway (Fig. 1C). However, the DDH enzyme only catalyzed meso-2,3-butanediol into butanone, whereas (2R,3R)-butanediol and (2S,3S)-butanediol are not substrates for DDH (Fig. S4†). Thus, reducing equivalents would be excessive if the products from acetoin by BDH were other forms of 2,3-butanediol except meso-2,3-butanediol. Therefore, the configurations of acetoin and 2,3-butanediol were determined using a GC system equipped with a chiral column. Chiral analysis indicated that 2,3-butanediol generated from ethanol contained meso-2,3-butanediol (65%) and (2R,3R)-butanediol (35%) from (3S)-acetoin and (3R)-acetoin, which were produced from acetaldehyde using FLS:L482S (Fig. S14 and S15†). Considering that only meso-2,3-butanediol was converted into butanone by DDH,34 we used the same strategy for the 2-butanol production as the 2,3-butanediol production from ethanol by employing an NAD(P)H purge valve regulatory node to prevent the buildup of NADH (Fig. 1C).
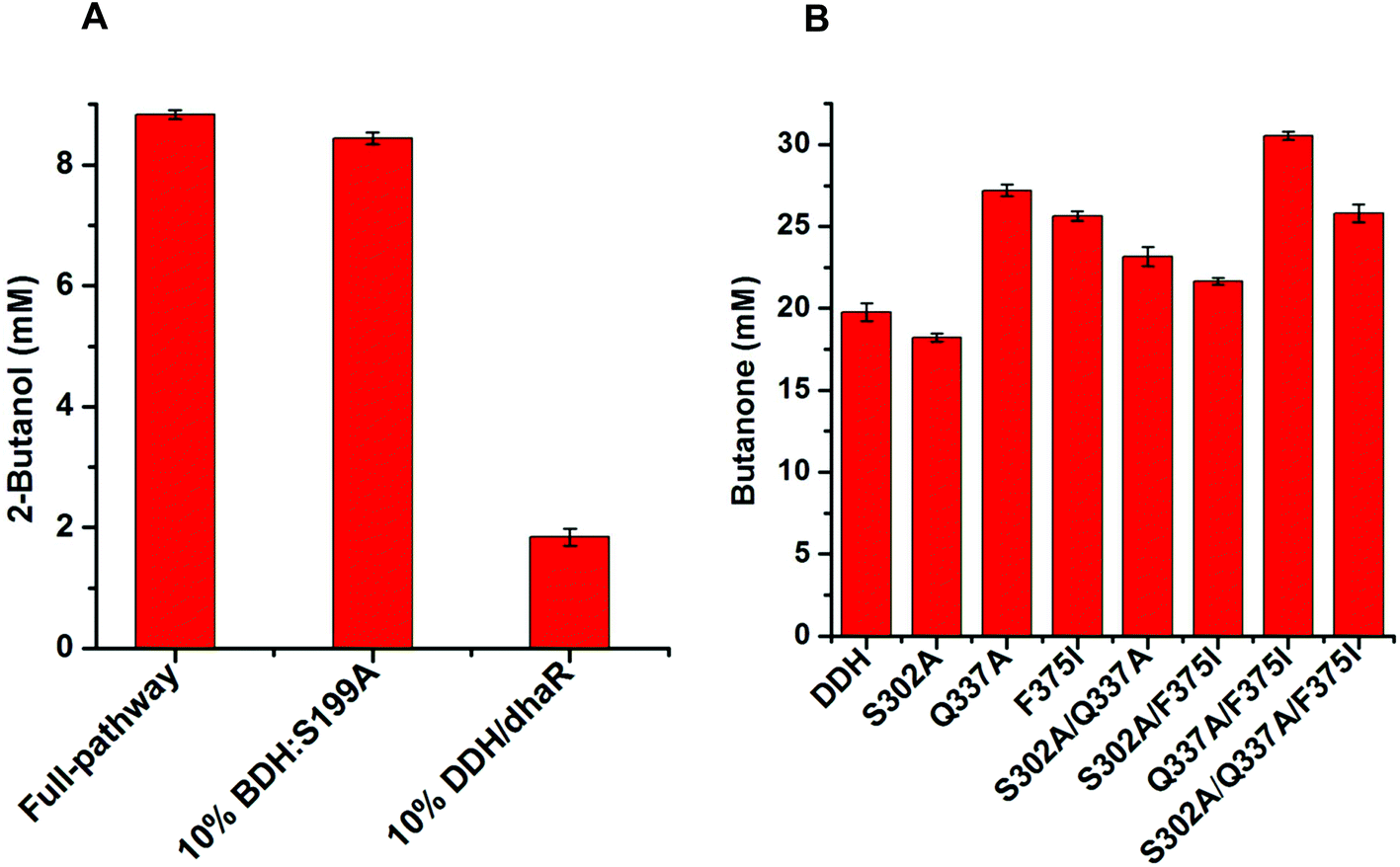 | ||
| Fig. 6 Analysis of the rate-limiting step in the conversion of ethanol to 2-butanol (A). Catalytic efficiency assays of the DDH enzyme and its variants with meso-2,3-butanediol (50 mM) as a substrate using the whole-cell biocatalytic method (B). Results are the means ± SD of three parallel replicates. | ||
Computational analysis showed that the proton donor residue E171 of the double mutant DDH (Q337A/F375I) (1.9 Å) comes into contact with the substrate 2,3-butanediol more strongly than the wild-type (2.6 Å) (Fig. S13A and B†). E171 showed more hydrogen bond contacts with the substrate in the double mutant compared to the wild-type. Long-range simulation (100 ns) results also showed that the substrate had stronger interaction networks including hydrogen bonding and water bridges with E171 in the double mutant compared to that in the wild-type (Fig. S13C and D†).
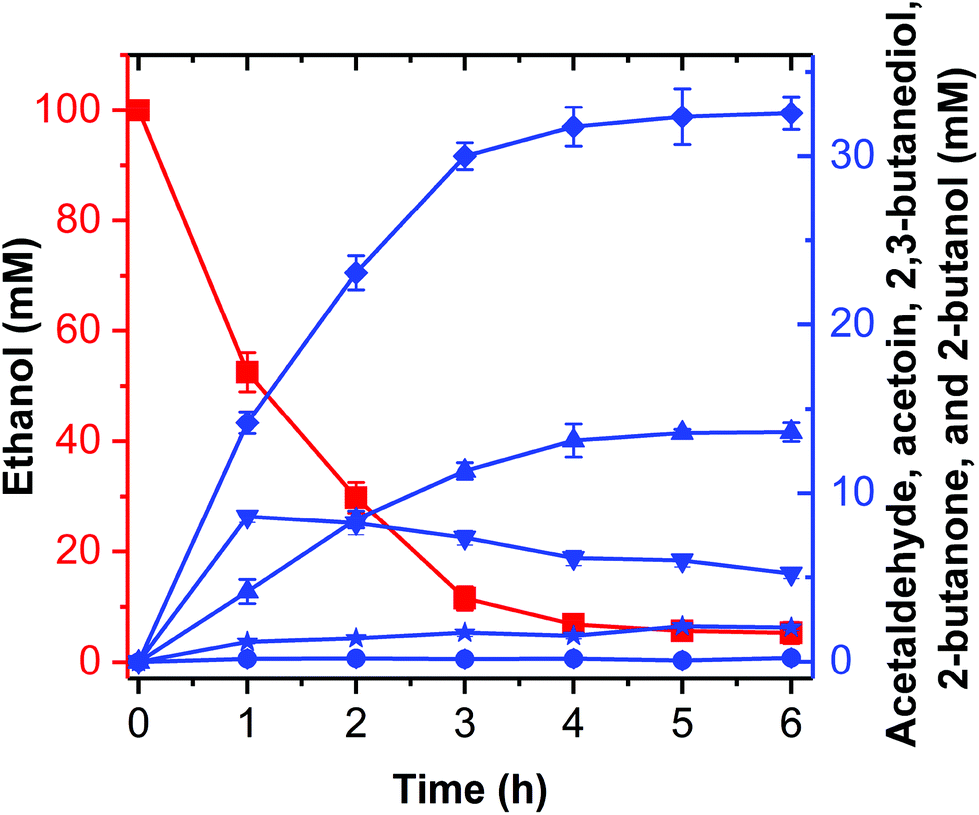 | ||
| Fig. 7 Conversion of ethanol into 2-butanol by the artificial reaction cascade. Ethanol (squares), acetaldehyde (inverted triangles), acetoin (circles), 2,3-butanediol (diamonds), butanone (stars), and 2-butanol (upright triangles). The reaction was carried out in a 0.5 mL reaction mixture containing 50 mM HEPES buffer (pH 8.0), 1 mM NAD+, 1 mM NADP+, 0.88 U mL−1 EtDH:D46G, 0.05 U mL−1 FLS:L482S, 0.98 U mL−1 NOX, 5.11 U mL−1 BDH:S199A, 0.01 U mL−1 DDH:Q337A/F375I, 0.2 mg mL−1 dhaR, 0.1 mM TPP, 1 mM Mg2+, 1 mM DTT, 20% DMSO, 1 mM coenzyme B12, 100 mM ATP and 100 mM ethanol. The reaction was conducted at 30 °C for 6 h. Results are the means ± SD of three parallel replicates. | ||
Recyclability of cascade reactions
The recyclability of the catalytic system is an attractive parameter that dictates the economic feasibility of using these enzymes in the cell-free bio-system. To evaluate the recyclability of the catalytic system, the enzymes for acetoin production (EtDH, FLS:L482S, and NOX) and 2,3-butanediol production (EtDH:D46G, FLS:L482S, BDH:S199A, and NOX) were immobilized on glutaraldehyde-functionalized silicon oxide nanoparticles.37,38 Immobilized enzymes showed more than 90% immobilization efficiency. Acetoin production by the immobilized enzymes was evaluated and a recyclability test of the catalytic system was performed (Fig. 8). Enzymes immobilized on silicon oxide nanoparticles exhibited 94% of their initial product concentrations, even after 10 cycles of reuse to produce acetoin. This indicates that there was no significant decrease in the acetoin production using the immobilized enzymes during repeated use. Additionally, the enzymes immobilized on silicon oxide nanoparticles were stable because of the covalent linkages between the enzymes and silicon oxide nanoparticles. Similarly, the 2,3-butanediol production by immobilized enzymes and recyclability of the catalytic system were evaluated (Fig. 8). Here, the immobilized enzymes exhibited 73% of their initial 2,3-butanediol production activity after 10 cycles of reuse. Upon immobilization, biocatalysts catalyzed the production of acetoin and 2,3-butanediol for many cycles without significant loss of activity. Hence, the immobilized catalytic system can be efficiently recycled for industrial production of acetoin and 2,3-butanediol. Unfortunately, we were not able to produce 2-butanol under the same experimental conditions because of the low stability of DDH.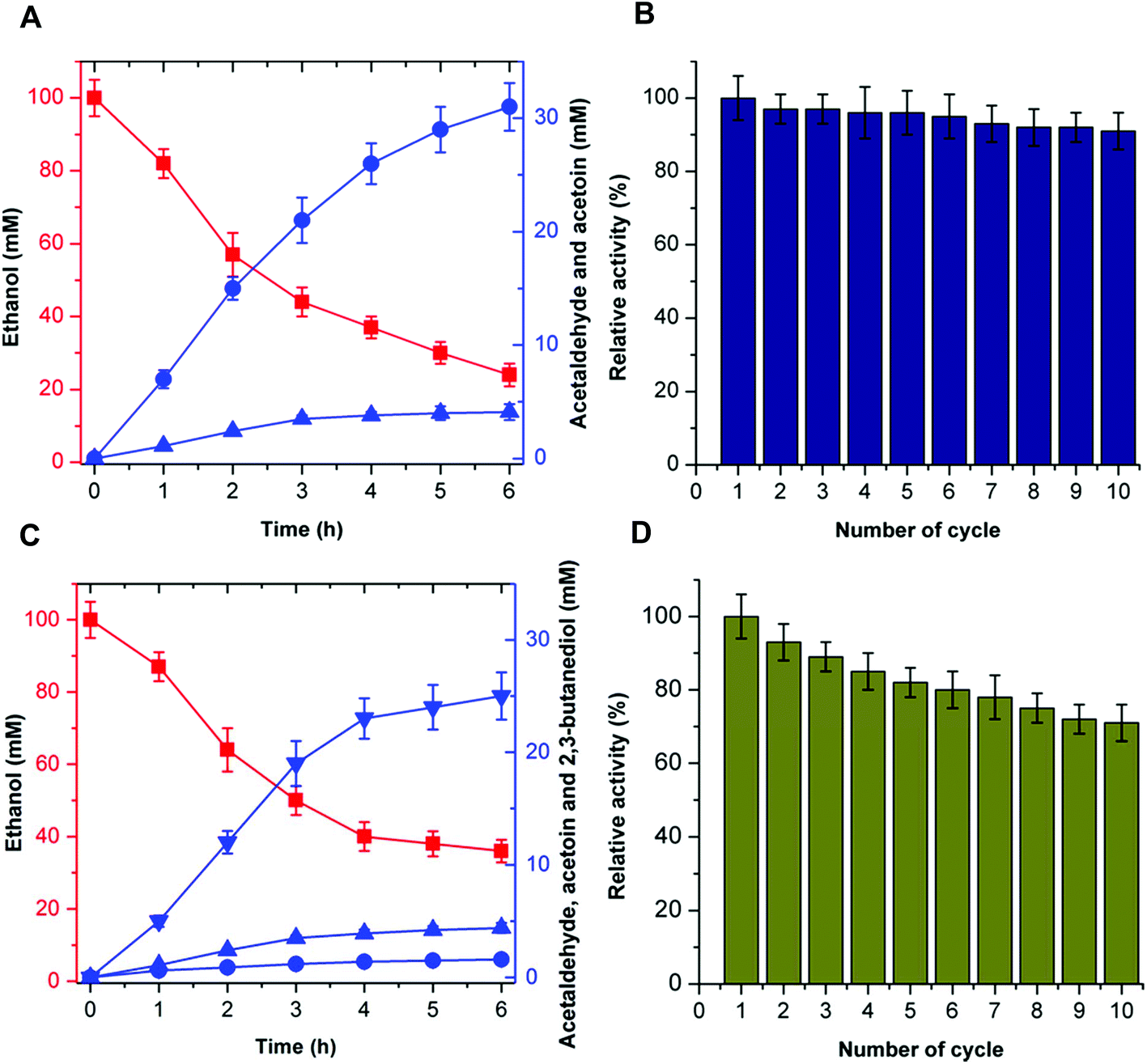 | ||
| Fig. 8 Recyclability of the catalytic system. (A) Conversion of ethanol into acetoin by the artificial cascade reaction using immobilized enzymes. Ethanol (squares), acetaldehyde (upright triangles), and acetoin (circles). (B) Reusability of the immobilized enzymes to produce acetoin from ethanol. (C) Conversion of ethanol into 2,3-butanediol by the artificial reaction cascade using immobilized enzymes. Ethanol (squares), acetaldehyde (upright triangles), acetoin (circles), and 2,3-butanediol (inverted triangles). (D) Reusability of the immobilized enzymes to produce 2,3-butanediol from ethanol. The product concentration of the immobilized enzyme catalyzed reaction after the first cycle was set at 100%. Acetoin and 2,3-butanediol concentration were determined using a gas chromatograph system equipped with a chiral column (Supelco β-DEX™ 120, 30 m length, 0.25 mm inner diameter). | ||
In recent years, acetoin, 2,3-butanediol, and 2-butanol as platform chemicals have gained more attention because of their increasing market demands. The derivatives of 2,3-butanediol have a potential global market of approximately 32 million tons per annum.29 As a precursor of 2,3-butanediol, acetoin has been classified as one of the 30 platform chemicals that are given priority for development and utilization by the US Department of Energy, and its market demand has reached more than 10 kilotons per annum,39 while the market capacity of bio-butanol is approximately 5 million tons per annum.40 Previous studies have shown that acetoin and 2,3-butanediol could be produced by microbial fermentation or chemical synthesis.39,41 Compared to chemical synthesis, microbial fermentation for acetoin and 2,3-butanediol production has attracted great attention due to its mild conditions. However, during the fermentation process, many metabolic byproducts are inevitably produced, resulting in difficulty in product isolation and high purification cost. In addition, the resulting microbial biomass causes environmental problems. Commercially available 2-butanol is primarily obtained industrially by the hydration of 1-butene using sulfuric acid as a catalyst. However, the harsh reaction conditions result in a higher cost for the 2-butanol production and potential environmental pollution. Compared to chemical synthesis and microbial fermentation, the artificial multi-enzyme pathway in the current work provides an environmentally-friendly, fewer byproduct-forming, and recyclable process with high conversion efficiency, thus supporting the viability of producing acetoin, 2,3-butanediol, and 2-butanol from ethanol by CFME.
Conclusions
In conclusion, we developed an artificial multi-enzyme pathway capable of upgrading ethanol to C4 compounds (Table 3). High activity (>88% conversion) was obtained for acetoin or 2,3-butanediol production from ethanol. Additionally, the artificial synthetic pathway showed potential for butanone and 2-butanol production, although their yields were relatively low because of the low activity and stability of the DDH enzyme. This environmentally friendly novel approach can be used to upgrade bio-ethanol to acetoin, 2,3-butanediol, and 2-butanol. Ongoing efforts are focused on developing modified FLS and DDH enzymes with high catalytic efficiency as well as identifying new enzymes. Overall, this artificial CFME approach is a promising and highly selective strategy for ethanol-to-C4 compound conversion using our novel cascade enzyme system.| Product (mM) | Enzymes (U mL−1) | Chemicals (mM) | Yield (%) | Reaction time (h) |
|---|---|---|---|---|
| All reactions were performed in 50 mM HEPES buffer (pH 8.0) and 20% DMSO using 100 mM EtOH as a substrate at 30 °C. | ||||
| Acetoin (44.39) | EtDH (1.06), FLS:L482S (0.05), NOX (0.98) | NAD+ (1), TPP (0.1), Mg2+ (1), DTT (1) | 88.78 | 4 |
| 2,3-BD (44.14) | EtDH:D46G (0.88), FLS:L482S (0.05), NOX (0.98), BDH:S199A (5.11) | NAD+ (1), NADP+ (1), TPP (0.1), Mg2+ (1), DTT (1) | 88.28 | 5 |
| 2-Butanol (13.62) | EtDH:D46G (0.88), FLS:L482S (0.05), NOX (0.98), BDH:S199A (5.11), DDH:Q337A/F375I (0.01) | NAD+ (1), NADP+ (1), TPP (0.1), Mg2+ (1), DTT (1), coenzyme B12 (1), ATP (100) | 27.24 | 6 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 81673542), the New Century Excellent Talents Supporting Plan of the Provincial Education Department of Fujian Province of China (no. K8015056A), the Development Platform of Edible Fungi Industry in Fujian Province (no. K5114001A), and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2017R1A2B3011676, 2017R1A4A1014806, 2013 M3A6A8073184).References
- Z. Chen, Y. Wu, J. H. Huang and D. H. Liu, Bioresour. Technol., 2015, 197, 260 CrossRef CAS PubMed.
- H. Latif, A. A. Zeidan, A. T. Nielsen and K. Zengler, Curr. Opin. Biotechnol., 2014, 27, 79 CrossRef CAS PubMed.
- A. S. Heeres, C. S. Picone, L. A. van der Wielen, R. L. Cunha and M. C. Cuellar, Trends Biotechnol., 2014, 32, 221 CrossRef CAS PubMed.
- C. Gao, Z. Li, L. J. Zhang, C. Wang, K. Li, C. Q. Ma and P. Xu, Green Chem., 2015, 17, 804 RSC.
- M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS PubMed.
- K. N. Tseng, S. Lin, J. W. Kampf and N. K. Szymczak, Chem. Commun., 2016, 52, 2901 RSC.
- S. Manzetti and O. Andersen, Fuel, 2015, 140, 293 CrossRef CAS.
- P. Dürre, Biotechnol. J., 2007, 12, 1525 CrossRef PubMed.
- V. García, J. Päkkilä, H. Ojamo, E. Muurinen and R. L. Keiski, Renewable Sustainable Energy Rev., 2011, 2, 964 CrossRef.
- E. I. Lan and J. C. Liao, Bioresour. Technol., 2013, 135, 339 CrossRef CAS PubMed.
- P. P. Peralta-Yahya, F. Zhang, S. B. Del Cardayre and J. D. Keasling, Nature, 2012, 7411, 320 CrossRef PubMed.
- L. X. Li, K. Li, Y. Wang, C. Chen, Y. Q. Xu, L. J. Zhang, B. B. Han, C. Gao, F. Tao, C. Q. Ma and P. Xu, Metab. Eng., 2016, 28, 19 CrossRef PubMed.
- L. Y. Zhang, Y. L. Yang, J. A. Sun, Y. L. Shen, D. Z. Wei, J. W. Zhu and J. Chu, Bioresour. Technol., 2010, 101, 1961 CrossRef CAS PubMed.
- N. Li, W. J. Yan, P. J. Yang, H. X. Zhang, Z. J. Wang, J. F. Zheng, S. P. Jia and Z. P. Zhu, Green Chem., 2016, 18, 6029 RSC.
- P. Ghiaci, J. Norbeck and C. Larsson, PLoS One, 2014, 9, e102774 Search PubMed.
- D. Cai, Y. Wang, C. J. Chen, P. Y. Qin, Q. Miao, C. W. Zhang, P. Li and T. W. Tan, Bioresour. Technol., 2016, 211, 704 CrossRef CAS PubMed.
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52 CrossRef CAS.
- A. Baez, K. M. Cho and J. C. Liao, Appl. Microbiol. Biotechnol., 2011, 90, 1681 CrossRef CAS PubMed.
- C. R. Shen, E. I. Lan, Y. Dekishima, A. Baez, K. M. Cho and J. C. Liao, Appl. Environ. Microbiol., 2011, 77, 2905 CrossRef CAS PubMed.
- B. R. Oh, S. Y. Heo, S. M. Lee, W. K. Hong, J. M. Park, Y. R. Jung, D. H. Kim, J. H. Sohn, J. W. Seo and C. H. Kim, Biotechnol. Lett., 2014, 36, 57 CrossRef CAS PubMed.
- Q. M. Dudley, A. S. Karim and M. C. Jewett, Biotechnol. J., 2015, 10, 69 CrossRef CAS PubMed.
- W. D. Fessner, New Biotechnol., 2015, 32, 658 CrossRef CAS PubMed.
- C. You, H. Chen, S. Myung, N. Sathitsuksanoh, H. Ma, X. Z. Zhang, J. Li and Y. H. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7182 CrossRef CAS PubMed.
- J. S. Martín del Campo, J. Rollin, S. Myung, Y. Chun, S. Chandrayan, R. Patiño, M. W. Adams and Y. H. Zhang, Angew. Chem., Int. Ed., 2013, 52, 4587 CrossRef PubMed.
- S. Myung, J. Rollin, C. You, F. Sun, S. Chandrayan, M. W. Adams and Y. H. Zhang, Metab. Eng., 2014, 24, 70 CrossRef CAS PubMed.
- T. Y. Wu, C. T. Chen, J. T. Liu, I. W. Bogorad, R. Damoiseaux and J. C. Liao, Appl. Microbiol. Biotechnol., 2016, 100, 1 CrossRef PubMed.
- J. Siegel, A. L. Smith, S. Poust, A. Wargacki, A. Bar-Even, C. Louw, B. Shen, C. Eiben, H. Tran, E. Noor, J. Gallaher, J. Bale, Y. Yoshikuni, M. Gelb, J. Keasling, B. Stoddard, M. Lidstrom and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3704 CAS.
- M. Köpke, M. L. Gerth, D. J. Maddock, A. P. Mueller, F. M. Liew and S. D. Simpson, Appl. Environ. Microbiol., 2014, 80, 3394 CrossRef PubMed.
- M. Yamanishi, K. Kinoshita, M. Fukuoka, T. Saito, A. Tanokuchi, Y. Ikeda, H. Obayashi, K. Mori, N. Shibata, T. Tobimatsu and T. Toraya, FEBS J., 2012, 279, 793 CrossRef CAS PubMed.
- Y. W. Zhang, M. K. Tiwari, H. Gao, S. S. Dhiman, M. Jeya and J. K. Lee, Enzyme Microb. Technol., 2012, 50, 255 CrossRef CAS PubMed.
- M. Seyfried, R. Daniel and G. Gottschalk, J. Bacteriol., 1996, 178, 5793 CrossRef CAS PubMed.
- J. Bendl, J. Stourac, E. Sebestova, O. Vavra, M. Musil, J. Brezovsky and J. Damborsky, Nucleic Acids Res., 2016, 44, 479 CrossRef PubMed.
- Z. Chen, H. Sun, J. H. Huang, Y. Wu and D. H. Liu, PLoS One, 2015, 10, e0140508 Search PubMed.
- D. J. Maddock, W. M. Patrick and M. L. Gerth, Protein Eng., Des. Sel., 2015, 28, 251 CrossRef CAS PubMed.
- S. Kwak, Y. C. Park and J. H. Seo, Bioresour. Technol., 2013, 135, 432 CrossRef CAS PubMed.
- S. K. S. Patel, S. H. Choi, Y. C. Kang and J. K. Lee, Nanoscale, 2016, 8, 6728 RSC.
- S. K. S. Patel, S. H. Choi, Y. C. Kang and J. K. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 2213 CAS.
- L. J. Zhang, Q. Y. Liu, Y. S. Ge, L. X. Li, C. Gao, P. Xu and C. Q. Ma, Green Chem., 2015, 18, 1560 RSC.
- G. R. Ding, B. Meng, R. L. Chen and J. H. Chen, Chem. Ind., 2016, 34, 37 Search PubMed.
- Y. Q. Xu, H. P. Chu, C. Gao, F. Tao, Z. K. Zhou, K. Li, L. X. Li, C. Q. Ma and P. Xu, Metab. Eng., 2014, 23, 22 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02898a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |