
Formation of calcium monofluoride in graphite furnace molecular absorption spectrometry, part I: interference mechanisms of competitive metals Ga, Al, Ba, and Sr
Nil
Ozbek
 and
Suleyman
Akman
*
and
Suleyman
Akman
*
Istanbul Technical University, Faculty of Science and Letters, Department of Chemistry, 34469, Maslak, Istanbul, Turkey. E-mail: akmans@itu.edu.tr
First published on 22nd November 2017
Abstract
Fluorine can be determined via molecular absorption of CaF generated in a graphite furnace of a high resolution continuum source atomic absorption spectrometer (HR-CS AAS). In this study, the formation mechanisms of CaF in a graphite furnace were examined. To differentiate the condensed phase and gas phase interactions during CaF formation, Ca and F were mixed as well as separately pipetted onto a platform. The main mechanism for the formation of CaF was a gas phase combination reaction between Ca and F because CaF was formed irrespective of whether Ca and F were mixed or separated. When solid tea and Ca solution or solid Ca(NO3)2 were separately introduced on the platform, a significant CaF signal was observed. Secondly, the interference mechanisms of gallium, aluminum, barium, and strontium on the formation of CaF were researched. The interferents, along with Ca and F, were mixed or separately pipetted onto the platform in various combinations. The results revealed that F and the interferent reacted both in the condensed phase and gas phase. However, a gas phase reaction between F and the interferent metal played a dominant and final role. Because F was shared between Ca and the metallic interferent, the sensitivity for CaF was changed compared to matrix-free standards depending on the interferent and its concentration. In this case, F can accurately be determined only if matrix-matching standards or better are used when the analyte addition technique is applied.
Introduction
Because the atomic absorption resonance line of fluorine is in the vacuum-UV region, at approximately 95 nm, it cannot be determined by atomic absorption spectrometry (AAS). However, F has been successfully determined by high resolution-continuum source molecular absorption spectrometry (HR-CS MAS). For this purpose, a molecule-forming element such as Al, Ba, Ca, Ga, or Sr is added to the standards and samples to form a stable diatomic molecule of fluorine in the gas phase of the flame or graphite furnace. The molecular absorption of a diatomic molecule, namely AlF, BaF, CaF, GaF, or SrF, is then measured at one of its finely structured rotational lines.1–12Zaitseva and Pupyshev theoretically described the formation of gaseous SrF molecules using thermodynamic simulation for graphite furnace molecular absorption determination of fluorine.13 According to theoretical calculations, it was proposed that SrF(g) molecules were formed due to thermal decomposition of condensed SrF2 during vaporization. The same theoretical thermodynamical approach was studied by the same group for the formation mechanisms of CaF.14 Similar to SrF, it was proposed that CaF was formed upon decomposition of CaF2 in the gas phase during vaporization.
In our previous study, an experimental method was described for the explanation of the formation mechanism of SrF(g) molecules in a graphite furnace using mixed and separate introduction of a molecule-forming element and the analyte in the graphite furnace.15 It was assumed that when F and Sr were separated, they did not contact in the condensed phase, and thus, SrF should be formed upon a gas phase reaction between F and Sr. When they were injected together, both condensed phase and gas phase interactions may occur. By comparing the sensitivities for the two cases, it was possible to differentiate whether condensed phase or gas phase interactions were responsible for SrF formation. It was proposed that SrF molecules were formed upon gas phase combination reaction between Sr(g) and F(g). However, in real samples, matrix components unavoidably react with the molecule-forming element and the analyte both in the condensed and gas phases. If the effect of the interfering element on F concentration is negligible, aqueous standards can be used for calibration. Otherwise, non-spectral interferences become significant, resulting in non-negligible sensitivity loss. In this case, the analysis using aqueous calibrants is unavoidably wrong. This problem may be overcome and controlled by increasing the amounts of the molecule-forming element. If this is insufficient to eliminate the effects of competitive interference, or at least reduce it to a negligible level, then a matrix-matching calibration standard or standard (analyte) addition technique should be considered as an alternative solution.
To recognize and eliminate any interference in the determination of non-metals via molecular absorption of their diatomic molecules, the elucidation of interaction mechanisms is highly important. However, in the literature, the interference mechanisms of metals in the determination of F via molecular absorption of its diatomic molecules have not yet been systematically studied.
In this study, the formation mechanisms of CaF and the interference mechanisms of Al, Ba, Ga, and Sr on the determination of F via CaF were examined. For this purpose, Ca, F, and interferents were mixed as well as separately placed on the opposite sides of a solid sampler platform of a HR-CS GFAAS in various combinations, and molecular absorptions of CaF were compared.
Experimental
Instrumentation and reagents
All measurements were carried out using a ContrAA 700 Analytik Jena (Germany) high resolution continuum source graphite furnace atomic absorption spectrometer (HR-CS GFAAS) equipped with a 300 W xenon short-arc lamp (XBO 301, GLE, Germany) and manual solid sampler. Argon (99.99%) was used as a purge gas. The molecular absorbance for CaF was measured at 606.440 nm (integration of 3 pixels, CP ± 1 pixel). The graphite furnace program used for the determination of fluorine in this study is given in Table 1. All chemicals were of analytical grade (Merck, Germany). The F stock solution (1000 mg L−1) was prepared from NaF (Merck, Germany) and further diluted with ultrapure water daily (TKA Wasseraufbereitungsysteme GmbH, Niederelbert, Germany). Al, Ba, Ca, Ga, and Sr were prepared from their nitrates. Solutions were manually injected on the platforms using a 5 μL Eppendorf pipettor.| Step | Temperature, °C | Ramp, °C s−1 | Hold, s |
|---|---|---|---|
| Drying | 80 | 6 | 20 |
| Pyrolysis | 350 | 50 | 20 |
| Pyrolysis | 850 | 300 | 10 |
| Gas adaption | 850 | 0 | 5 |
| Molecule formation (volatilization) | 2200 | 1200 | 5 |
| Cleaning | 2450 | 500 | 4 |
Procedure
Different experimental procedures were applied to determine the formation mechanism of CaF and the effects of various interferents on CaF formation. In order to study the CaF formation mechanism, (i) Ca and F were mixed on the platform or (ii) separately injected on opposite sides of the platform. However, to elucidate the interference mechanisms, (iii) “Ca + F + interferent (Al, Ba, Ga, Sr)” were mixed on the platform, (iv) “Ca + F” were mixed on one side of the platform whereas interferent was pipetted on the other side, and (v) Ca was pipetted on one side, and “interferent + F” were on the other side of the platform. To successfully perform separate injections, all liquids were pipetted onto solid sampling platforms, which were manually transferred in and out of the furnace. All injections were performed with 5 μL of solution. Manual injections with minimum volumes on solid sampling platforms were applied to facilitate and effectively control the separation of drops. The drying process was observed from a monitor by means of a camera focused on the inside of the furnace. If the separately introduced solutions touched during the drying step, the results were discarded. All evaluations were made according to absorbance values.In addition, Ca solution and a solid tea (Camellia sinensis) sample as well as very small solid particles of Ca and sodium fluoride were separately placed on opposite sides of the platform cavity. Finally, calcium was deposited in a graphite tube, and then F was introduced using a new calcium-free platform. It should be stated that although the interferents, Ca and F, were prepared from their salts and were present as ionic forms in solutions, their valances are not indicated on their symbols in the text.
Results and discussion
Experiments to determine CaF formation mechanisms
In the literature, a specially designed dual cavity platform, which had two separate cavities instead of one, was used to differentiate the interferences due to gas phase combination reactions and condensed phase reactions between various analytes and interferents (mostly chlorides) or their decomposition products.16–19 For this purpose, the analyte was pipetted into one cavity and the interferent was pipetted into the other cavity of the platform, and the results were compared with those found for the mixture of the interferent and the analyte. It was assumed that when the analyte and interferent were separately pipetted into the different cavities, condensed phase interactions were prevented and any interference originated from gas phase reactions. The same strategy was applied to elucidate the CaF formation as well as interference mechanisms of some elements by pipetting Ca, F, and interferent together or separately in different combinations on the platform.For this purpose, (i) Ca was manually injected on top of F or (ii) Ca and F were introduced separately on opposite sides of a solid sampling platform, which are mentioned as “mixed case” and “separated case”, respectively, in the current study. It was assumed that when Ca and F were mixed, both condensed phase reactions and gas phase reactions between Ca and F likely occurred, whereas when Ca and F were separated, only gas phase interactions could occur. The separate injections of drops without touching were enabled by manually pipetting minimum volumes of solutions (5 μL) on a solid sampler platform. To ensure that the Ca and F drops were dried without contacting, the drying process was always observed from the monitor of the instrument by means of the camera. When Ca and F were pipetted separately, the sensitivity for CaF was reduced only 15% compared to the mixed case. This indicates that the CaF was dominantly or completely formed upon gas phase reactions. When Ca and F were mixed, CaF2 should be unavoidably formed. During the volatilization (molecule-forming step) step, CaF is formed from either the separation of one F from CaF2 (CaF2(s/g) → CaF(g) + F(g)), or most likely, the decomposition of CaF2 or other Ca and F species to their atoms and recombination of Ca and F to form CaF in the gas phase (Ca(g) + F(g) → CaF(g)). The condensed phase reactions that form precursors of Ca and F are beyond the scope of this study.
Ca(g) and F(g) should be formed upon atomization of Ca and F species in the volatilization step. Because the amounts of Na+ and F− are much lower than those of Ca2+ and NO3−, at the end of the pyrolysis step, most of the Ca is found as CaO generated upon thermal decomposition of calcium nitrate. Therefore, most likely, the precursors of Ca(g) and F(g) in the gas phase are CaO and CaF2. With an excess of Ca, it can be assumed that the contribution of Na to CaF formation is negligible.
When Ca and F were separately pipetted onto opposite sides of the platform, Ca and F were individually atomized and made contact only in the gas phase. Therefore, the formation of CaF2 and thus the first alternative, i.e., decomposition of CaF2(s/g), is unlikely, whereas the second alternative, i.e., Ca(g) + F(g) → CaF(g), is valid for mixed and separated cases.
With an excess of Ca(g), the CaF formation reaction is forwarded according to pseudo-first order kinetics and depends only on the concentration of F. However, when experiments (i) and (ii) were performed with excessive F using 0.05 μg of Ca2+ and 10 μg of F−, again, CaF spectra were observed for both mixed and separated cases as well. For the mixed case, the amount of CaF2 to be precipitated should theoretically be the same as that obtained from 0.05 μg of F− and 10 μg of Ca2+. The likely explanation is that a portion of F reacts with Na to form NaF as well.
To provide better separation between Ca and F as well as to prevent their contact by surface tension in the volatilization step, a solid tea powder, which is known to contain high F concentration, and 5 μL of Ca solution was mixed or separately placed on the platform. When tea and Ca were separated, a CaF peak was repeatedly observed, and the absorbance was only 20% lower than that for the mixed case. The same experiment was repeated using solid tea as a fluorine source and a very tiny amount of solid Ca(NO3)2 containing approximately 10 μg of Ca, and a significant CaF peak was obtained once more. It may be assumed that CaF is formed upon the interaction of Ca and F in tea leaves. However, when only tea leaves were placed on the platform without an additional calcium source, no CaF signal was observed by itself. Moreover, barely visible solid NaF and Ca(NO3)2 particles were separately put on opposite sides of the platform. It can be expected that the solid particles surely did not touch, nor did a volatile compound form during drying and pyrolysis. As expected, a huge unfinished CaF signal was observed due to the high amount of F, which supports the role of a gas phase combination reaction in the formation of CaF. In all those experiments, the separation of Ca and F on the platform in the condensed phase was guaranteed. The dominant role of a gas phase reaction between Ca and F to form CaF was evident because no contact between Ca and F was required prior to volatilization (molecular formation) step.
It may be thought that even if F and Ca were well separated, F may vaporize during the pyrolysis step and interact with Ca on the other side of the platform upon the gas/condensed phase reaction. In the literature, it was shown that if hydrated chlorides, e.g., NiCl2·6H2O, are formed in the drying step, HCl was generated by thermal hydrolysis at elevated pyrolysis temperatures and reacted with the analyte on the other side of platform upon the gas/condensed phase reaction, and chlorides of the analytes were formed.18,20 This reaction was not observed in the case of non-hydrated chlorides, e.g., NaCl.19 Therefore, there is no reason for F, prepared from NaF (MP: 993 °C), to vaporize and become subject to thermal hydrolysis and, in fact, to melt at the temperatures of the pyrolysis step to react with Ca species on the side of the platform. To provide further proof, pyrolysis curves were prepared between 300 °C and 850 °C for pipetting of Ca and F together and separately. When F and Ca were separated, if F had interacted with Ca on the other side prior to the volatilization (molecular formation) step, then the behaviours of the pyrolysis curves for the two cases would have been different and sensitivities for the separated case would have changed with temperature. As shown in Fig. 1, the sensitivities for CaF for the mixed case and separated case did not significantly change, and the pyrolysis curves were almost parallel. This indicates that no losses and/or no interaction occurred below 850 °C for the mixed and separated cases.
 | ||
| Fig. 1 Pyrolysis (on the left) and molecule formation (on the right) curves for (i) and separate pipettings of Ca and F on the platform (ii) (F: 0.05 μg, Ca: 10 μg). Tvap: 2200 °C for (i), and Tpy: 850 °C for (ii). | ||
It may also be thought that the CaF peaks for the separated case originated from the reaction of F with the residual fractions of Ca collected in the furnace. When only F was introduced on a platform that was previously used, a negligible CaF signal around the baseline level was observed, which proved that the CaF peak could not have formed from any residual Ca remaining from the previous cycle.
Finally, in another set of experiments, only Ca was pipetted onto the platform, and the graphite furnace was run according to the program given in Table 1 without the cleaning step. It was assumed that a part of Ca was atomized and remained on/in the wall of the graphite tube and platform (presumably as atomic or carbide forms and/or intercalation compounds). A huge atomic absorption signal for Ca was observed when the empty furnace was run. After Ca was deposited in the furnace, F was introduced to the furnace on a new unused platform, and a significant CaF signal was observed that once again proved the role of the gas phase combination reaction in the formation mechanism of CaF. Because there was no Ca on the new platform, any condensed phase interaction between Ca and F to form CaF2 was unlikely. It can be assumed that CaF signal should be generated upon gas phase reaction between Ca vaporized from the graphite wall and F, and that CaF2 was not required as a precursor for CaF formation. When F was pipetted into the furnace, which was previously cleaned by a ‘cleaning step’, a CaF signal was not observed (or only a signal at blank level was observed). Pereira et al. investigated the formation of CaBr by separating Ca and Br in the furnace using a different method. For this purpose, they pipetted the Ca solution into the graphite tube under the platform, whereas the Br solution was pipetted onto the platform, and CaBr was observed in the gas phase.21 We performed a similar experiment using Ca and F, and a CaF peak was clearly detected. This proves one more time that when Ca and F were separated, CaF was formed in the gas phase. They proposed that NaBr melted (MP: 747 °C) and migrated towards the Ca, resulting in a condensed phase interaction. However, the melting point of NaF is much higher (993 °C), and it does not melt and migrate to the Ca on the other side.
All those experimental designs and results clearly showed that irrespective of the precursors of Ca and F, gas phase reactions played a decisive role in the formation of CaF in the graphite furnace. It should be noted that when Ca and F were mixed, the condensed phase reactions and formation of CaF2 were not eliminated. However, the final step in the CaF formation mechanism was a gas phase reaction.
Interference of Ga on CaF
Gallium has been used to determine F via GaF by HR CS AAS.12,22 The limit of detection (LOD) value for GaF is the lowest of all diatomic fluorine molecules. It can be expected that Ga favourably interacts with F to form GaF, and it should be a severe interferent. To elucidate the interference mechanisms of Ga on CaF, in addition to mixed and separated introduction of Ca and F on the platform, three new sets of experiments were carried out, namely, (iii) Ca + F + Ga were mixed on the platform, (iv) Ca + F was mixed on one side, and Ga (as the nitrate) was on the other side, and (v) Ca was on one side, and Ga + F were on the other side. The amount of F used in this study was always 0.05 μg (5 μL of 10 μg mL−1), whereas the amount of Ca used was 10 μg (5 μL of 2000 μg mL−1) and the amount of Ga used was 5 μg (5 μL of 1000 μg mL−1). The results were compared with those obtained for the mixture of Ca and F. The time-resolved molecular absorption signals for CaF obtained upon introduction of F, Ca, and Ga in different combinations on the platform are depicted in Fig. 2.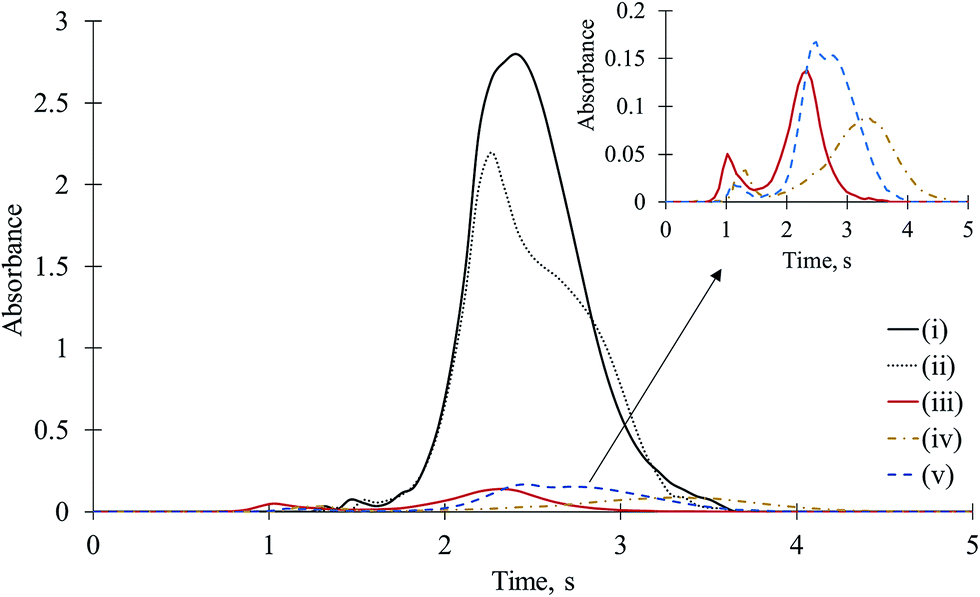 | ||
| Fig. 2 Time-resolved molecular absorption signals for CaF for various combinations of Ca, F, and Ga on the platform: Ca was pipetted on top of F (i), Ca and F were separately pipetted (ii), Ca + F + Ga were mixed on the platform (iii), Ca was on one side and Ga + F were on the other side of the platform (iv), Ca + F was mixed on one side and Ga was on the other side of the platform (v) (F: 0.05 μg, Ca: 10 μg, Ga: 5 μg). The small figure depicts the enlarged spectra (iii)–(v). | ||
In the cases of (iii)–(v), the CaF sensitivity was dramatically reduced. For example, in the case of (iii), i.e., for the mixture of 0.05 μg of F, 10 μg of Ca, and 5 μg of Ga, the CaF sensitivity was reduced approximately 20 times (almost completely). In the presence of 10 μg of Ga, absolutely no CaF signal (2% of the matrix-free CaF) was observed. In the case of (iv), although Ga and the mixture of Ca + F were separated and dried without contacting (as observed by means of a built-in camera), the CaF sensitivity was once again reduced 15 times (5 to 20 times depending on the Ga amount between 2.5 to 10 μg) to practically nothing.
When Ca and F were mixed, if CaF2 formed, the vaporization of F during pyrolysis was not expected. Therefore, the interaction of F and Ga should occur in the gas phase during the vaporization step. For case (iv), although Ca + F were on one side whereas Ga was on the other side of the platform, the CaF signal was severely suppressed as well. Prior to the molecular formation step, vaporization of any compound formed between Ga and F is not expected. Therefore, formation of any compound between Ca and F during the drying and pyrolysis steps due to gas/condensed phase reaction is unlikely. This was a very important experiment to prove the role of the gas phase reaction for CaF formation. When Ca and F were mixed on one side of the platform and Ga was separately introduced on the other side, F and Ga interacted only upon their gas phase combination reaction, resulting in sensitivity loss for CaF. However, when Ga was mixed with F on one side and Ca on the other side, CaF was still formed (even if at very low amounts), again which can be attributed to a partial gas phase combination reaction between Ca and F. Because Ga and F were intimately in contact, as expected, the suppressive effect of Ga was more dramatic. It may be suspected that the interference effect of Ga on CaF was due to residual Ga on the platform and tube that remained from previous experiments. Therefore, Ga was pipetted onto the side of a new (unused) platform, and Ca and F were pipetted onto the other side. The platform was inserted into a new graphite tube. The CaF peak was again dramatically suppressed, which indicated that the effect of Ga on CaF could not have originated from the residual Ga in the atomizer. Finally, Ga was atomized using the CaF formation conditions (Table 1), and atomic absorption signal was observed at the Ga wavelength. The time at which Ga atoms appeared coincided with that of the CaF molecular absorption signal, which supports the gas phase interaction between Ga and F atoms to form GaF. Obviously, F reacts with Ga or Ca in the gas phase. However, even at optimized conditions for CaF, F is much more favorably bound to Ga rather than to Ca.
Interference of Al on CaF
Aluminum is used for the determination of F in HR CS MAS via AlF as well and it is more abundantly found in samples compared to gallium.1,23 The series of experimental designs to explain the effects of Ga on CaF formation, namely, (iii), (iv), and (v), were performed with Al, too. The time-resolved molecular absorption signals for CaF obtained after introduction of F, Ca, and Al in different combinations are depicted in Fig. 3. When Al + Ca + F were mixed on the platform (iii) or Ca and F were mixed on one side and Al was introduced to the other side of the platform cavity (iv) or Al was mixed with F at one side and Ca was pipetted onto the other side of the platform cavity (v), the absorbances for CaF were dramatically reduced. In the cases of (iii)–(v), the CaF absorbances obtained from 0.05 μg of F, 20 μg of Ca, and 2.5 to 20 μg of Al were reduced 3 to 15 times (30% to 7% of matrix-free CaF), depending on the concentration of Al and combination design of Al, Ca, and F. Aluminum, which is above Ga on the periodic table, suppressed the CaF absorbances in all cases, as well. However, the effect of Ga on the CaF signal was more dramatic than that of the same amount of Al. For example, for the mixture of 0.05 μg of F, 10 μg of Ca, and 5 μg of Al, the CaF signal was reduced 9 times (11% of the CaF obtained from the mixture of Ca and F), whereas in the presence of the same amount of Ga, the CaF signal was reduced almost 20 times. Moreover, the atomic mass of Ga is 2.5 times higher than Al, which indicates that the number of Ga moles are less than Al. Therefore, when the number of moles were considered, the effect of Ga on CaF was much more significant. When Ca, F, and Al were mixed on the platform (iii), an unseen synergy influenced the evaluation of CaF. However, we hesitate to make speculation regarding its causes. The suppression of Al on CaF sensitivity was clearly observed when Ca + F and Al were separately introduced on a new (unused) tube and platform (for only one experiment). This indicates that the interference effect of Al on CaF was not due to the residual Al on the tube and platform surfaces remaining from previous experiments.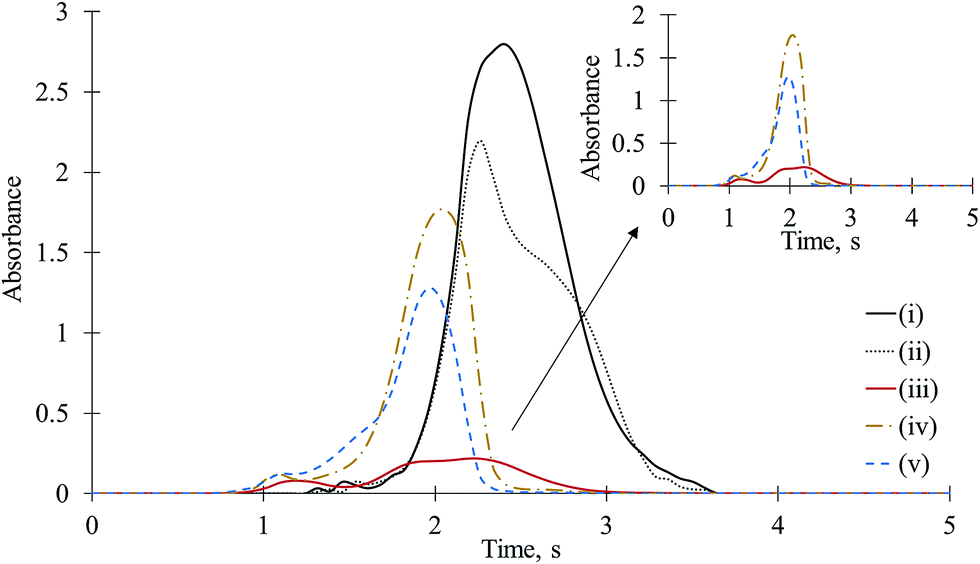 | ||
| Fig. 3 Time-resolved molecular absorption signals for CaF for various combinations of F, Ca, and Al on the platform: Ca was pipetted on top of F (i), Ca and F were separately pipetted (ii) Ca + F + Al were mixed on the platform (iii), Ca was on one side and Al + F were on the other side of the platform (iv), Ca + F was mixed on one side and Al was on the other side of the platform (v) (F: 0.05 μg, Ca: 10 μg, Al: 5 μg). The small figure depicts the enlarged spectra (iii)–(v). | ||
Interference of Ba and Sr on CaF
Again, experimental designs (iii), (iv), and (v) were performed using 2.5 to 20 μg of Ba or Sr. At higher amounts of interferents, the CaF signal did not return to the baseline in a reasonable time. Therefore, the effects of Sr and Ba were researched up to 20 μg. It is likely that Ba or Sr delays the volatilization of Ca and Cl as well as the formation of CaF. The time-resolved molecular absorption signals for CaF obtained with different combinations of 0.05 μg of F, 10 μg of Ca, and 20 μg of Ba on the platform are depicted in Fig. 4. Because the interference effects and signals for Ba and Sr were quite similar, the time-resolved absorbance signals for CaF in the presence of Sr were not given. The suppression effect of barium on CaF began above 10 μg of Ba. At low amounts, Ba caused even positive effects on CaF. In the presence of 2.5 and 5 μg of Ba, CaF signals were enhanced 15 to 10%, but at higher amounts, CaF sensitivity decreased depending on the amount of interferent. When Ca (10 μg) + Ba (20 μg) + F (0.01 μg) were mixed, the CaF absorbance was suppressed by approximately 50%, whereas when Ba was separated from Ca + F on the platform, CaF sensitivity decreased by 30%. Obviously, Ba (or Sr) has both positive and negative effects on CaF. | ||
| Fig. 4 Time-resolved molecular absorption signals for CaF for various combinations of F, Ca, and Ba on the platform: Ca was pipetted on top of F (i), Ca and F were separately pipetted on the platform (ii), Ca + F + Ba were mixed on the platform (iii), Ca was on one side and Ba + F were on the other side of the platform (iv), Ca + F were mixed on one side and Ba was pipetted on the other side of the platform (v) (F: 0.05 μg, Ca: 10 μg, Ba: 20 μg). | ||
The effects of interferents should be compared according to their number of moles rather than grams because the interacting amount of interferent is its number of moles. The atomic masses of interferents increase in the following order: Ba (137 g mol−1) > Sr (87 g mol−1) ≈ Ga (70 g mol−1) ≫ Al (27 g mol−1). Based on the numbers of moles, the suppressive effects of Ga and Al on CaF were much higher than those of Ba because as previously mentioned, for mixed cases, although 20 μg (0.146 μmol) of Ba reduced the CaF signal to 50% of that of the matrix-free CaF, 10 μg (0.143 μmol) of Ga and 5 μg (0.185 μmol) of Al reduced the CaF signal to 2% and 11% of the original value, respectively. Based on the number of moles, Ba was more effective than Ca because in the presence of 0.146 μmol of Ba and 0.25 μmol of Ca, the CaF signal was reduced by almost 50% of that of the matrix-free CaF. The interfering behavior of Sr was similar to that of Ba; therefore, it will not be mentioned here. However, it should be noted that the suppression of Sr was the lowest of all with respect to both the number of moles and the weight.
The effects of 5 μg of Ga, 2.5 μg of Al, 20 μg of Sr, and 20 μg of Ba on the relative sensitivity of CaF obtained from the mixing of 10 μg of Ca and 0.05 μg of F on the platform are summarized in Table 2.
| Experimental protocol | Experimental scheme | Relative absorbancesa | |||
|---|---|---|---|---|---|
| Al | Ba | Ga | Sr | ||
| 5 μg | 20 μg | 5 μg | 20 μg | ||
| a Relative absorbances are given compared to CaF absorbance obtained when Ca and F were mixed on the platform (case i). When Ca and F were separately pipetted, (case ii), the relative absorbance was 0.84. The relative standard deviations were not higher than 10%. b Int represents Al, Ba, Ga, and Sr. | |||||
| (iii) (Ca + F + Intb) mixed |
|
0.11 | 0.46 | <0.1 | 0.91 |
| (iv) (Ca + F) and Int separately |
|
0.44 | 0.75 | <0.1 | 0.94 |
| (v) Ca and (F + Int) separately |
|
<0.1 | 0.65 | <0.1 | 0.83 |
Ba and Sr as well as Ca are in the 2nd (2A) group, whereas Ga and Al are in the 13th (3A) group of the periodic table. It seems that the competition of Ca with interferents to form diatomic fluoride compounds in the gas phase is related to group number. The tendencies of Ca, Ba, and Sr towards F are similar, whereas the 13th group elements (even at low amounts) reduced the CaF signals much more dramatically compared to Ba and Sr. The most likely causes of different suppressing effects of various metals on CaF are (i) bond dissociation energies of diatomic molecules formed between the interferent and F, and (ii) the appearance times of F and metal atoms in the gas phase. The bond dissociation energies of GaF, AlF, BaF, and SrF are approximately 600 ± 50 kJ mol−1. In other words, the differences in bond dissociation energies change in a small range, whereas the interferences of Ga and Al compared to those of Ba and Sr were much higher than expectations according to only bond dissociation energies.
It is likely that many other parameters should influence the degree of interference as well. For example, if the appearance of their highest concentrations in the gas phase of the graphite furnace coincide, the number of F atoms interacting with metal and thus reducing interferences should be higher. In addition, the solubility of metal fluorides is an important parameter in the condensed phase (this is valid for mixed cases). It should be noted that not only dissociation and recombination reactions but many complicated and unpredictable reactions among Ca, F, and the metal of the interferent, and, in fact, non-metal atoms of the interferent, may occur in the condensed phase and gas phase, e.g., a collision-induced dissociation mechanism cannot be ruled out. It was principally shown that gas phase interactions among Ca, F, and interferent constituents play an important role in CaF formation and interference. The effect of temperature on the magnitude of signal suppression by concomitants should be significative. However, in this study, the effect of temperature on the degree of interference was not researched. All experiments were carried out at a volatilization temperature optimized to obtain interference-free CaF.
Conclusion
It was proved that even if formation of a compound between Ca and F, namely CaF2, is prevented by separating Ca and F on the platform, CaF is generated in the gas phase of a graphite furnace. Therefore, it can be proposed that irrespective of whether Ca and F are reacted to form a compound in the condensed phase, namely CaF2, the final step in the formation of CaF is a gas phase combination reaction between Ca and F. In the presence of an interfering metal (Al, Ba, Ga, or Sr), an interaction between F and the metal causes a reduction in CaF sensitivity because F is bound to the interferent and less F remains for Ca. This competition depends on the type of metal and its concentration, which, strictly speaking, would be the ratio of Ca to the interferent. Even if the interferent is separately pipetted from the mixture of Ca + F on the opposite sides of the platform, an interaction and sensitivity reduction occur as well. In this study, the effects of Al, Ba, Ga, and Sr were researched. However, F is a very reactive element, and there is no reason that similar interferences would not occur if other elements were present in real samples. By adding a sufficient amount of Ca and optimizing the experimental conditions, the interferences can be controlled and determinations using aqueous standards can be performed. If the effects of matrix components are still non-negligibly significant, then either matrix-matching standards or the analyte addition technique is unavoidable.Conflicts of interest
There are no conflicts of interest to declare.References
- N. Ozbek and S. Akman, Talanta, 2012, 94, 246–250 CrossRef CAS PubMed.
- N. Ozbek and S. Akman, Spectrochim. Acta, Part B, 2012, 69, 32–37 CrossRef CAS.
- N. Ozbek and S. Akman, Food Chem., 2013, 138, 650–654 CrossRef CAS PubMed.
- N. Ozbek and S. Akman, Microchem. J., 2014, 117, 111–115 CrossRef CAS.
- N. Ozbek and S. Akman, LWT--Food Sci. Technol., 2015, 61, 112–116 CrossRef CAS.
- N. Ozbek and S. Akman, Food Chem., 2016, 211, 180–184 CrossRef CAS PubMed.
- N. Ozbek and S. Akman, Food Anal. Methods, 2016, 9, 2925–2932 CrossRef.
- N. Ozbek, H. Baltaci and A. Baysal, Environ. Sci. Pollut. Res., 2016, 23, 13169–13177 CrossRef CAS PubMed.
- A. R. Borges, Á. T. Duarte, M. d. L. Potes, M. M. Silva, M. G. R. Vale and B. Welz, Microchem. J., 2016, 124, 410–415 CrossRef CAS.
- A. R. Borges, L. L. Francois, B. Welz, E. Carasek and M. G. R. Vale, J. Anal. At. Spectrom., 2014, 29, 1564–1569 RSC.
- W. Boschetti, M. B. Dessuy, A. H. Pizzato and M. G. R. Vale, Microchem. J., 2017, 130, 276–280 CrossRef CAS.
- H. Gleisner, B. Welz and J. W. Einax, Spectrochim. Acta, Part B, 2010, 65, 864–869 CrossRef.
- P. Zaitseva, A. Pupyshev and I. A. Kurmachev, Analytics and Control, 2014, 18, 287–301, DOI:10.15826/analitika.2014.18.3.005.
- P. V. Zaitceva and A. A. Pupyshev, Analytics and Control, 2016, 20, 34–40, DOI:10.15826/analitika.2015.20.1.003.
- N. Ozbek and S. Akman, Anal. Sci., 2013, 29, 741–746 CrossRef CAS PubMed.
- S. Akman and G. Doner, Spectrochim. Acta, Part B, 1996, 51, 1163–1167 CrossRef.
- S. Akman, B. Welz and N. Tokman, Spectrochim. Acta, Part B, 2005, 60, 1349–1356 CrossRef.
- B. Welz, S. Akman and G. Schlemmer, Analyst, 1985, 110, 459–465 RSC.
- B. Welz, S. Akman and G. Schlemmer, J. Anal. At. Spectrom., 1987, 2, 793–799 RSC.
- B. Welz, H. Becker-Ross, S. Florek and U. Heitmann, High Resolution Continuum Source AAS, Wiley-VC, Weinhein, 2005 Search PubMed.
- É. R. Pereira, I. N. B. Castilho, B. Welz, J. S. Gois, D. L. G. Borges, E. Carasek and J. B. de Andrade, Spectrochim. Acta, Part B, 2014, 96, 33–39 CrossRef.
- M. Kruger, M. D. Huang, H. Becker-Ross, S. Florek, I. Ott and R. Gust, Spectrochim. Acta, Part B, 2012, 69, 50–55 CrossRef.
- S. Bucker, V. Hoffmann and J. Acker, Curr. Anal. Chem., 2014, 10, 426–434 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |