
Controllable synthesis of nanocrystals in droplet reactors
Liang-Jun
Pan
,
Jia-Wei
Tu
,
Hao-Tian
Ma
,
Yu-Jun
Yang
,
Zhi-Quan
Tian
,
Dai-Wen
Pang
and
Zhi-Ling
Zhang
 *
*
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, People's Republic of China. E-mail: zlzhang@whu.edu.cn; Fax: +0086 27 68754067; Tel: +0086 27 68756759
First published on 3rd November 2017
Abstract
In recent years, a broad range of nanocrystals have been synthesized in droplet-based microfluidic reactors which provide obvious advantages, such as accurate manipulation, better reproducibility and reliable automation. In this review, we initially introduce general concepts of droplet reactors followed by discussions of their main functional regions including droplet generation, mixing of reactants, reaction controlling, in situ monitoring, and reaction quenching. Subsequently, the enhanced mass and heat transport properties are discussed. Next, we focus on research frontiers including sequential multistep synthesis, intelligent synthesis, reliable scale-up synthesis, and interfacial synthesis. Finally, we end with an outlook on droplet reactors, especially highlighting some aspects such as large-scale production, the integrated process of synthesis and post-synthetic treatments, automated droplet reactors with in situ monitoring and optimizing algorithms, and rapidly developing strategies for interfacial synthesis.
1. Introduction
Nanocrystals (NCs, i.e., crystalline nanoparticles) have attracted intensive research interest due to their unique optical, electronic, magnetic and thermal properties.1 However, the fact that no two NCs are absolutely the same has prevented the deep understanding of some essential properties of NCs.2 Therefore, high-quality NCs with narrow size distribution, highly uniform shape, well-controlled surface chemistry, and other properties have always been desirable in this field.3–5 Over the past two decades, the rapid development of synthetic methods has provided better control over the nucleation and growth of NCs.6,7 Despite great progress in conventional batch reactions, nevertheless, superior control over the synthetic process still remains a challenge.In response to this challenge, the microfluidic reactor (microreactor) has emerged as a powerful platform for controllable synthesis of various colloidal nanomaterials, such as metals, metal oxides, and semiconductor quantum dots (QDs).8,9 In general, a microreactor can be defined as a reactor with channel diameters ranging from several tens of microns to one millimetre. Indeed, the small dimension allows one to achieve mixing, reaction and quenching in an extremely short time as a result of enhanced mass and heat transport.10,11 Therefore, the reaction time can be shortened to minutes or even seconds. In addition, better reproducibility for scaling up the production can be achieved because the precise and consistent control over flow minimizes potentially uncertain factors.12,13 More specifically, in contrast with complicated procedures in the batch method, the powerful control over flow provides a simplified and continuous process for sequential multistep reactions.14,15 Furthermore, real-time monitoring allows one to momentarily obtain information on NCs relating to size, shape, structure, composition and other properties. Hence, this offers the opportunity to investigate the mechanisms of NC nucleation and growth.9 Moreover, an integrated device with a self-optimizing control system enables completely automated or intelligent synthesis without manual intervention.16,17 All these advantages manifest the microreactor as a promising alternative to batch equipment. As a dominant branch of the microfluidic reactor, the droplet reactor especially plays an essential role in the synthesis of high-quality NCs.12,18
The droplet reactor offers significant advantages such as enhanced mass and heat transport, accurate manipulation, and reliable automation. Prior to discussion, we must mention previous reviews relating to NC synthesis in droplet reactors. In 2013, Nightingale et al.18 discussed NC synthesis in droplet reactors, with emphasis on high-temperature synthesis. Later, Phillips et al.8 summarized the advantages of the microfluidic reactor for NC synthesis, focusing on research directions including scale-up and in situ monitoring. Subsequently, detailed discussions of scalable synthesis of NCs in droplet reactors were also reviewed.12 Overall, these reviews have concentrated mainly on NC synthesis in microfluidic reactors or droplet reactors in terms of some advantages or challenges. Here we aim to provide a comprehensive summary of the recent progress in NC synthesis in droplet reactors. Our primary purposes in this review are to focus on the essential factors that control NC properties and to elucidate the superiorities of the droplet reactor. To this end, general concepts of the droplet reactor are presented. Also, the functional regions of the droplet reactor for NC synthesis are discussed and illustrated. In addition, enhanced mass and heat transport properties are summarized and discussed. Furthermore, detailed discussions of research frontiers associated with this topic are given.
Notably, the droplet reactor will be discussed in depth; nevertheless, single-phase continuous flow will be mentioned where appropriate. In addition, although the main discussions are relating to NCs, most of the concepts can also be applied to guiding the synthesis of other nanoparticles like polymers and liposomes.19 We believe that this review will provide the readers a deeper understanding of the underlying theoretical aspects and enable researchers to design their experiments more efficiently whether in laboratory investigations or further commercial processes.
2. General concepts of a microfluidic reactor
Materials for fabricating a microfluidic reactor
The fundamental step in microfluidic synthesis requires selection of proper materials and methods to fabricate a microreactor. In general, two kinds of different microreactors can be utilized for the synthesis of NCs. The first one is the chip-based microreactor, which can integrate multiple functions in a microfluidic chip with channel diameters ranging from tens to hundreds of micrometers.11 Considering the need of durability for a device, the materials used for the fabrication of a microfluidic chip should be chemically compatible with the reagents for synthesis. In addition, high-temperature and high-pressure conditions for some reactions should be considered in the selection of suitable materials. Currently, microfluidic chips are mainly fabricated using glass, silicon, and polymers.20 Glass and silicon have been widely used as fabricating materials in microfluidics for a long time. In general, they possess obvious advantages, such as thermal stability, solvent resistance, and high thermal conductivity.21 However, the high cost and complicated fabrication process have limited their potential for large-scale production. During the past decade, polymer-based replica molding has greatly promoted the development of microfluidics due to the convenient and cheap fabrication with many kinds of available materials. One of the most representative materials is polydimethylsiloxane (PDMS), which has optical transparency, gas permeability, and natural hydrophobicity. However, microfluidic devices made of PDMS have high-pressure intolerance, which is a major limitation in scale-up as higher flow rate is usually needed to increase the throughput. To solve the problem, Jeong et al.22 designed a three-dimensional (3D) monolithic PDMS device with 1000 parallel droplet generators for mass production of droplets at a rate of 1.5 L per hour. This device achieved the high flow rate and pressure required for the kilo-scale formation of droplets. Apart from PDMS, a large number of materials, including polymethylmethacrylate (PMMA), poly(vinyl chloride) (PVC), and fluorinated ethylene propylene (FEP), have become available for microfluidic fabrication. The choice of materials is mainly dependent on the desired properties. For example, FEP has excellent solvent resistance that enables use of various solvents.23The second is the tubing-based microreactor assembled from commercially available junction geometries (e.g., T-junction, Y-junction and cross-shaped junction) and tubing with channel inner diameters ranging from hundreds of micrometers to one millimeter. Generally, fabricating such a device involves materials including glass capillary and polymer tubing. Nightingale et al.24 designed a tubing-based droplet reactor using glass capillary and silicone tubing to form droplets. Also, polytetrafluoroethylene (PTFE) tubing was used as the reaction channel due to its chemical and high-temperature compatibility. Indeed, these devices enable the utilization of relatively larger-diameter channels even at millimeter scale than chip-based microchannels. Interestingly, this offers the opportunity to scale up the microfluidics, which is also known as millifluidics. To some extent, a millifluidic device shares similar fluid mechanics derived from a microfluidic reactor.12 Overall, the simple assembly of multiple modules makes it easy to scale up the reaction. Recently, the 3D-printing technique has become popular in microfluidic device fabrication due to its automation, assembly-free 3D fabrication, gradually decreasing cost, and rapidly improving throughput.25 For example, Zhang et al.26 designed 3D-printed droplet generators using photo-curing resin as the material based on a stereolithography (SLA) technique. Notably, the Young's modulus of the postcured resin was much higher than that of PDMS. Therefore, such a device could tolerate a higher flow rate and input pressure. In addition, the 3D-printed droplet generator can be connected to PTFE tubing for NC synthesis, which further expands the practicability of this technique.27 It should be noted that the 3D-printing technique enables accurate fabrication of complex structures; nevertheless, a higher resolution should be further developed to satisfy the need for the fabrication of small structures.
Single- versus multi-phase flow reactors
Compared with conventional batch methods, microreactors provide more effective control over the nucleation and growth of NCs. In 2003, Chan et al.28 demonstrated the controllable synthesis of CdSe QDs using a single-phase continuous flow reactor. Miscible streams of reagents were injected into a channel to form a laminar flow where mixing and reaction could occur. Due to the miniaturized fluid dimension and resultant short diffusion distance, the mixing within a microchannel was obviously more efficient than in a macroscale batch reactor.28,29 Therefore, NCs could be reproducibly prepared with size distributions comparable to those for batch syntheses. In addition, the microreactor offers the advantage of allowing monitoring of the nucleation and growth of NCs using in situ fluorescence detection, which is difficult for a conventional batch reactor.30However, the parabolic velocity profile of a laminar flow could result in residence time distribution and undesirable dispersion of particle properties (Fig. 1a).31 To some extent, this phenomenon could be partly overcome through using a higher flow rate and temperature.30 One more reliable solution to this issue is the utilization of a segmented multi-phase flow reactor (also known as droplet reactor). A representative example of the utility of the segmented flow reactor for synthesizing NCs was reported by Yen et al.,10 who designed a gas–liquid segmented flow reactor (Fig. 1b) for the synthesis of CdSe QDs. In this method, the introduction of the gas phase could not only eliminate the parabolic velocity profile of flow but also enhance mixing due to recirculation within each liquid slug. These advantages resulted in both a significant improvement in size distribution of NCs and shorter reaction time. Moreover, Lee et al.32 reported the formation of a gas–liquid segmented flow without gas delivery. By adding a volatile amine, gas bubbles could be generated in the single-phase continuous flow during heating. In addition to the gas phase, the immiscible liquid phase provides another way for flow segmentation (Fig. 1c). In 2004, Shestopalov et al.11 developed a droplet reactor for performing multistep reactions on a millisecond time scale. The reaction phase was dispersed and spontaneously broken up into droplets surrounded and separated by the immiscible inert carrier phase. Also, the reactants in droplets could be rapidly mixed as a result of recirculation. In addition, a droplet containing the reactants was allowed to react for a given time and then initiate the further stages of the reaction through introducing additional reactant into the droplet. Indeed, this strategy gives an insight into sequential multistep synthesis which is very useful in NC synthesis. In contrast with both single-phase continuous flow and gas–liquid segmented flow, the risk of channel clogging caused by reactant/product deposition on the channel wall is minimized due to the indirect contact of the reaction phase with the microchannel in a liquid–liquid segmented flow.11,33 In this review, droplet flows refer to all types of segmented flows including gas–liquid flow, liquid–liquid flow, and gas–liquid–liquid flow.
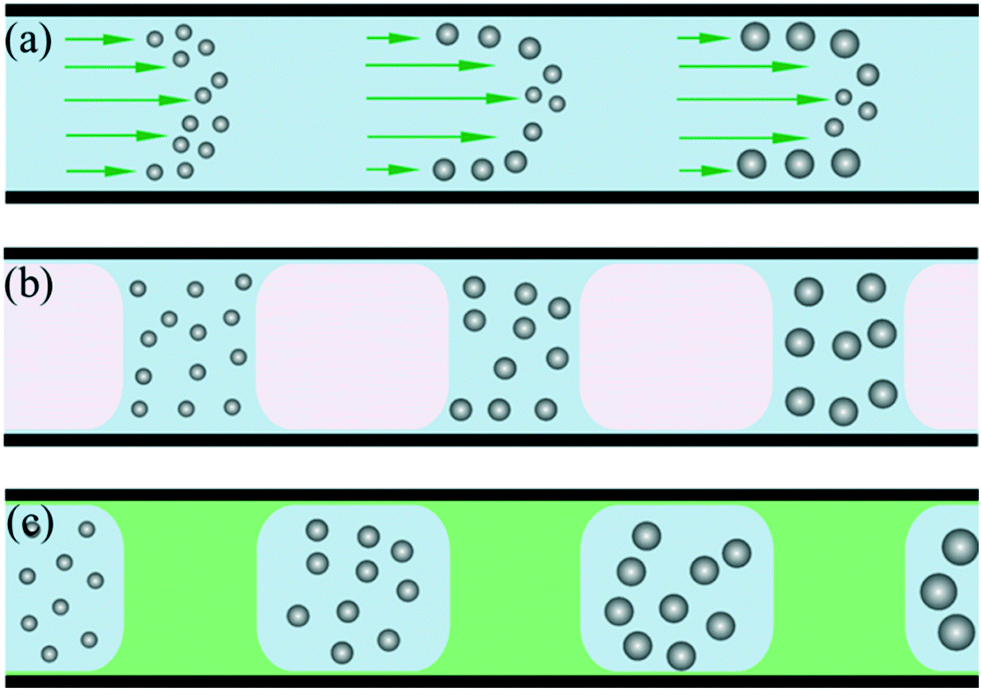 | ||
| Fig. 1 Three main flow patterns involved in the microfluidic channel for the synthesis of NCs. (a) Single-phase continuous flow. (b) Immiscible gas–liquid segmented flow. (c) Immiscible liquid–liquid segmented flow. | ||
Generally, low temperature synthesis tends to produce NCs with high defect densities. Thus, high temperature is usually used to anneal out crystal defects and improve crystalline quality.18 In 2005, Chan et al.34 reported a droplet reactor for the synthesis of CdSe NCs at high temperature (up to 240 °C) within a surface-modified glass chip. High-boiling-point octadecene (ODE) and perfluorinated polyether (PFPE) were used as the reaction phase and the carrier phase, respectively. However, the reaction was limited to approximately five hours as harsh operating conditions resulted in degradation of the surface coating and subsequent failure of the experiment. Therefore, the choice of suitable fabrication materials is crucial for NC synthesis. Notably, a reactor made from PTFE can be preferentially wetted by PFPE without surface coating and can tolerate high temperature. A representative example of the use of a PTFE channel was reported by Nightingale et al.,24 who synthesized CdSe NCs at temperatures of up to 250 °C using a reaction system similar to that of Chan et al.,34 but using PTFE tubing instead of a glass chip. The droplet reactor exhibited remarkable stability under high temperature, producing NCs with well-controlled emission spectra during a full day's synthesis.
3. Functional regions of a droplet reactor for the synthesis of NCs
In general, the droplet reactor for NC synthesis consists of four functional regions including droplet generation, mixing of reactants, reaction controlling, and reaction quenching (Fig. 2a). In addition, integrated in situ monitoring systems are usually preferred for screening the synthetic parameters (Fig. 2b). This section has been tailored to discuss the exquisite control over each functional region.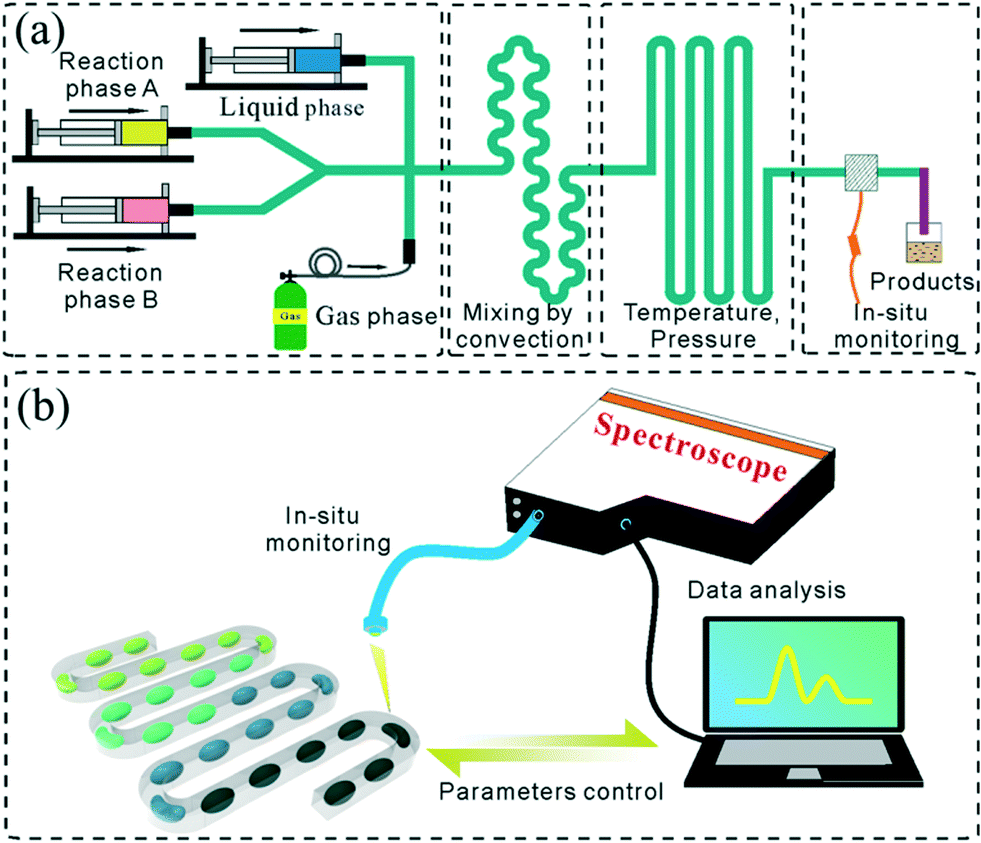 | ||
| Fig. 2 (a) Schematic view of a typical droplet reactor including different functional regions of droplet generation, mixing of reactants, reaction controlling, in situ monitoring and reaction quenching. (b) Schematic view of a droplet reactor with in situ monitoring and automated control system. | ||
Droplet generation
Precise and effective control over droplet generation is a fundamental step for the synthesis of NCs in droplet reactors. In general, methods for droplet generation can be either passive or active.35 Herein, we mainly focus on passive methods for generating monodisperse droplets in a hydrodynamically driven manner (e.g., T-junction,36 flow focusing,34 and co-flow geometries24). In passive methods, immiscible fluids meet at a junction where interface deformation and droplet breakup can occur. Generally, droplet formation is related to viscous force and interfacial tension, which can be characterized by the capillary number (Ca = Uμ/γ, where U is the velocity of the flow, μ is the viscosity of the fluid, and γ is the interfacial tension).37 The Ca is especially helpful in predicting droplet formation with different regimes including squeezing, dripping, and jetting.35 In general, at low Ca, squeezing mode occurs as droplets are larger than the channel size and highly uniform. As the Ca increases until the mode transforms from squeezing to dripping, droplets are smaller than channel size and uniform. As the Ca further increases, dripping to jetting transition can occur. However, jetting mode tends to form polydisperse droplets. Therefore, it is especially important to control droplet generation with proper modes by adjusting the flow rate and corresponding properties of fluids.To guarantee excellence for the synthesis of NCs in a droplet flow reactor, the proper carrier phase as well as reaction phase should be compatible with the reaction conditions and materials of the device. Inert solvents (e.g., silicone oil, mineral oil, and fluorocarbon oil) are commonly suitable for use as the carrier phases. Meanwhile, immiscible solvents including water, ethylene glycol,38 and ionic liquids39,40 are all suitable for use as the reaction phases. Depending on the type of reaction phase and carrier phase, surface coating of the channel might be necessary to ensure good wetting of the channel walls by the carrier phase.
Fluid pumping is an essential function in a microfluidic system. Using the relative pressure difference between the inlet and the outlet of the flow channel, the fluid flow can be facilitated along the microfluidic channel. For most of the reported studies, the liquid phase with a given flow rate can be precisely delivered into the microfluidic device by a syringe pump. Similarly, the gas can be pumped into the microfluidic channel via the syringe pump.10,41 In addition, the gaseous flow can also be delivered and monitored by either a mass flow controller42 or a gas pressure regulator.43 Notably, a syringe pump for fluid delivery provides the advantage of pumping with high precision and stability. However, the issue of the restocking of the limited amount of reagents makes it hard to scale up the reaction. In this regard, a piston pump44 and a peristaltic pump45,46 can satisfy the need for continuous droplet synthesis without any interruption. By using the fluidic capacitance element, the periodic and aperiodic flow disturbances in these pumping systems could be well overcome.47 To achieve high droplet formation rates, some new methods like parallelizing channels have been developed in recent years.48,49 This parallelization method offers an economically practicable strategy to achieve a high throughput as only more reactors or channels are needed while the control system and pumping device are still shared.
Mixing of reactants
The fast mixing of reagents within the microchannel is essential to the preparation of high-quality NCs. For a single-phase continuous flow reactor, different reagents can be brought together and mixed mainly depending on diffusion across laminar streams.50 However, mixing by diffusion has limited further applications to reactions with fast kinetics.51Rapid and controllable mixing of the reagents confined within the droplet allows controlling the reaction more exquisitely. While the droplets are passing through the channel, symmetric recirculation parallel to the flow direction emerges within the droplet due to boundary friction.21,52 Despite particular recirculation of the droplet, different mix-enhancing methods have been developed to further promote mixing. A representative method of the utility of chaotic advection was reported by Song et al.,53 who used a winding microfluidic channel to achieve fast passive mixing of droplets in submillisecond scale (Fig. 3a). In addition, mixing can be assisted actively by electrical field control. Frenz et al.54 demonstrated the precipitation process of magnetic iron oxide nanoparticles after fusion of droplet pairs by electrocoalescence. The reagents in the droplet pairs were homogenized in approximately 2 ms (Fig. 3b). Recently, Yesiloz et al.55 designed a microwave-based microfluidic mixer which enabled rapid mixing within the range of milliseconds due to the non-uniform distribution of an electrical field in the droplets. Furthermore, adding a periodic perturbation to the flow could also result in fast mixing. In that vein, Kim et al.56 demonstrated a tubing-based droplet mixing approach by periodically pinching the PTFE tubing with precision-tip tweezers (Fig. 3c). This approach allowed for fast mixing within one second.
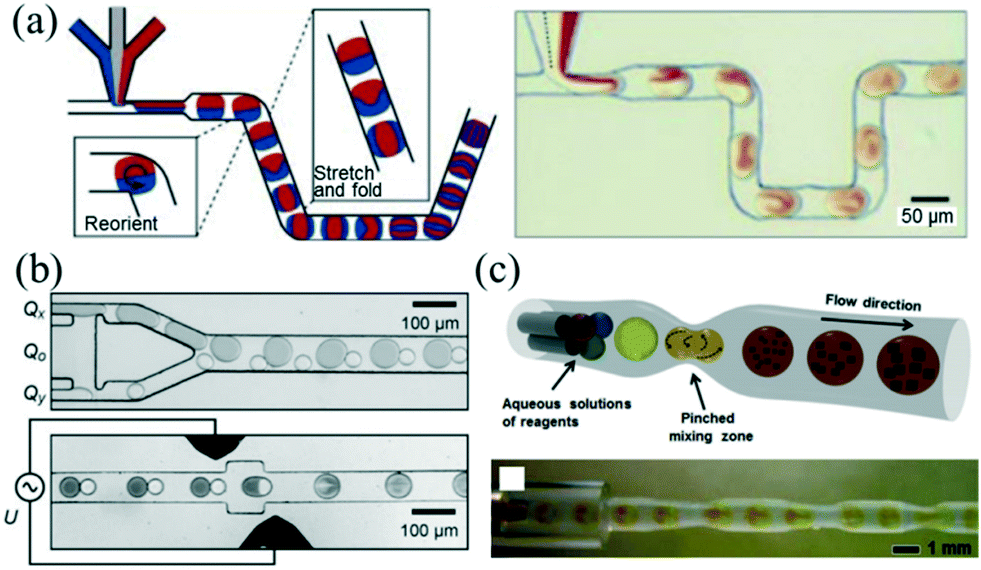 | ||
| Fig. 3 Three methods for promoting the mixing inside droplets. (a) Using a winding microfluidic channel to induce chaotic advection inside droplets. Reproduced from ref. 53, copyright 2003 American Institute of Physics. (b) Using in-line droplet fusion by electrocoalescence to induce chaotic advection inside droplets. Reproduced from ref. 54, copyright 2008 Wiley. (c) Using a periodically pinched PTFE tubing to induce chaotic advection inside droplets. Reproduced from ref. 56, copyright 2013 Wiley. | ||
For continuous multiple-step reactions, the droplet containing the reactant is allowed to react for a given time and then the second step of the reaction starts through introducing another reactant into the droplet.57 The reactant for the second step of the reaction was commonly injected directly into the droplets through a T-junction side channel. Notably, the new droplet may form rather than merge with existing droplets. When the side channel is preferentially wetted by the reactant phase, the above-mentioned phenomenon could be well avoided.58 Another injection method was developed by Nightingale et al.,42 who utilized a three-phase flow reactor containing the gas phase and two immiscible liquid phases to ensure reproducible dosing through direct injection of reactants. In their method, the gas phase guaranteed elimination of the formation of a new droplet while also allowing for uniform introduction of a new reactant into the existing droplet. For gas–liquid segmented flow, the new reactant could be directly injected into the channel and subsequently added to the reaction phase.15
Reaction controlling
An integrated microreactor enables exquisite control over the reaction conditions (e.g., temperature and pressure). Generally, the reaction temperature can be controlled via external block heaters10 and integrated internal thin-film heaters.34 For example, Yao et al.59 assembled a simple temperature control system consisting of an indium tin oxide (ITO) film-based heater, a PT-100 micro-thermistor, and a controlled voltage output device. Due to an automated temperature-feedback unit, this integrated device allows precise control of the temperature in a heated region. For the tubing-based droplet reactor, the desired temperature of the tubular channel can be controlled through simply immersing in a solvent bath24 or embedding onto a heated block.60 In addition to the above-mentioned methods, non-contact heaters like microwave dielectric heating have also been applied to both chip-based61 and tubing-based27 droplet reactors. For example, Koziej et al.61 demonstrated a microfluidic device with an integrated microwave heater that allowed dielectrical heating of droplets using time-varying electrical fields. This device allowed for crystallization of tungsten oxide nanoparticles within 64 ms, which was obviously shorter than the 15 min in a batch reactor.Flow reactions can be readily performed under high pressure conditions through using a high-pressure pump and a back-pressure regulator. Faustini et al.57 demonstrated a hydrothermal synthetic strategy for metal–organic framework (MOF) crystals in droplet reactors which acted as micrometer-scale autoclaves. High-quality MOF crystals could be produced within a few minutes. Furthermore, a recent work by Liu et al.62 indicated that crystallization time could be obviously reduced using pressurized solvents in a continuous flow reactor. In addition, when the pressure was sufficiently high, higher reaction temperature could be accessed under supercritical conditions that resulted in a narrower size distribution of NCs.63 Furthermore, gas pressure control is still of great significance for gas–liquid segmented flow especially involved in interfacial mass transport.43,64
In situ monitoring and reaction quenching
Currently, a droplet-based microfluidic platform has been coupled with a variety of spectroscopic monitoring systems (Fig. 2b) which can elucidate reaction mechanisms governing nucleation and growth.9 A fluorescence detection system is most widely used to couple with a droplet-based microfluidic platform due to its sensitivity in rapidly analyzing picoliter droplets and extracting information relating to fluorescent nanoparticles (e.g., QDs).59 In contrast, absorption spectroscopy can acquire information relating to both particle size and concentration. However, absorbance detection is more difficult due to the short path length. To couple ultraviolet/visible (UV/vis) absorption spectroscopy detection with the droplet microreactor, Yue et al.65 demonstrated two detection systems for in situ monitoring of the size and concentration of gold nanoparticles. The experiments revealed that droplet information could be determined at a high time resolution (2 ms). X-ray spectroscopy offers the opportunity to study the composition and structure of nanoparticles.66 Detailed advances in microfluidic platforms in conjunction with X-ray techniques are also covered in recent reviews.67Rapid quenching is also essential to some reactions with fast kinetics. The reaction can be quenched in the following ways including physical methods (e.g. fast cooling),10 and chemical methods (e.g. chemistry termination).11 Due to the highly efficient reaction quenching, the proper reaction time can be accurately controlled. The synthetic products can be collected for further post-synthetic treatments.
4. Enhanced mass and heat transport in a microfluidic reactor
Enhanced mass and heat transport can be achieved in a microfluidic reactor due to the small dimension of the channel. This section has been tailored to elucidate the mass and heat transport processes which are critical to NC nucleation and growth.Enhanced mass transport in a microfluidic reactor
Mass transport in the microfluidic system is mainly dominated by diffusion and convection. The Péclet number (Pe = convective transport rate/diffusive transport rate = wU/D, where w is the diameter of the channel, U is the velocity of flow, and D is the diffusion coefficient of the reagents) is often used to characterize mass transport resulting from diffusion and convection.53,68 The mixing time is related to the channel diameter and the flow rate. Therefore, the smaller the diameter of the reactor, the faster a uniform concentration of reactants across the channel can be achieved. This insight leads to the development of a micromixer in which the diffusion distance is minimized to obtain a shorter mixing time. In this regard, Sounart et al.50 investigated the spatially resolved process of nanoparticle nucleation and growth in the microchannel that combined two laminar flows consisting of cadmium and sulfur precursors. The diffusional mixing resulted in a supersaturated region at the boundary between the two streams and formation of CdS QDs. Also, nanoparticle growth between 0.4 s and 28 s was analyzed through monitoring the emission at different positions along the microchannel. The experimental results agreed with a diffusion-limited nucleation model with a rate-limited activation process. Mass transport in the segmented system is more complex as convection plays an important role in such a system. For example, the convective timescale can be defined as tconvection = 2L/U, where L is the length of the slug and U is the velocity of the flow.69 When the mixing time is on the same order of magnitude as tconvection and Pe > 1, convection is the dominant transport mechanism. In order to accelerate mixing, the timescale required for mass transport should be shortened. Thus, both higher flow rate and shorter length of the slug will decrease the mixing time.Chaotic advection inside a droplet can be induced to accelerate mixing on millisecond scale. This advantage offers the opportunity to accurately control the nucleation and growth of NCs. On the one hand, smaller nanoparticles with narrower size distribution could be produced due to the fast mixing and mass transport.54,70 On the other hand, the crystallization time could be reduced in the droplet due to the enhanced mass transport with a short diffusion distance.40 Notably, the fast mass transport also affects the seed-mediated heterogeneous nucleation and growth that enables the simple synthesis of NCs with complex nanostructures.12 On the basis of a seed-mediated approach, droplet reactors have been widely applied to the synthesis of shape-tunable NCs36,71 and core–shell structures.72,73
The introduction of a gas phase can enhance mixing due to recirculation inside the slugs.74 The recirculation results in homogeneous mixing in the droplet while also facilitating fast mass transport between the two phases. Khan et al.75 demonstrated a synthetic strategy for ultra-small Au NCs using N2 bubbles which could provide the space for the diffusion of H2 (Fig. 4a). The approach ensured that the dissolved gas concentration in the aqueous phase could never cross the threshold for bubble nucleation due to the presence of rapid mass transport across the gas–liquid interface (Fig. 4b). In addition, the fast mass transport from gas phase to liquid phase has also been utilized for synthesis of NCs that requires the presence of abundant gas to participate in the reaction. For example, Sebastian et al.43 demonstrated that anisotropic Pd nanorods could be produced using air as the segmentation gas. Indeed, the air bubbles provided an oxidative environment for oxidative etching, promoting the anisotropic growth of Pd. Recently, Sebastian et al.76 further investigated the effects of different gas phases on the synthesis of faceted Pt and Pd NCs with different shapes using a gas–liquid segmented microfluidic reactor. N2, O2, and CO were used to control the size and shape of NCs. For instance, switching the gas from O2 to CO permitted the changing in structure growth from 1D (nanorods) to 2D (nanosheets) due to the particular role of CO as the reductant and capping agent.
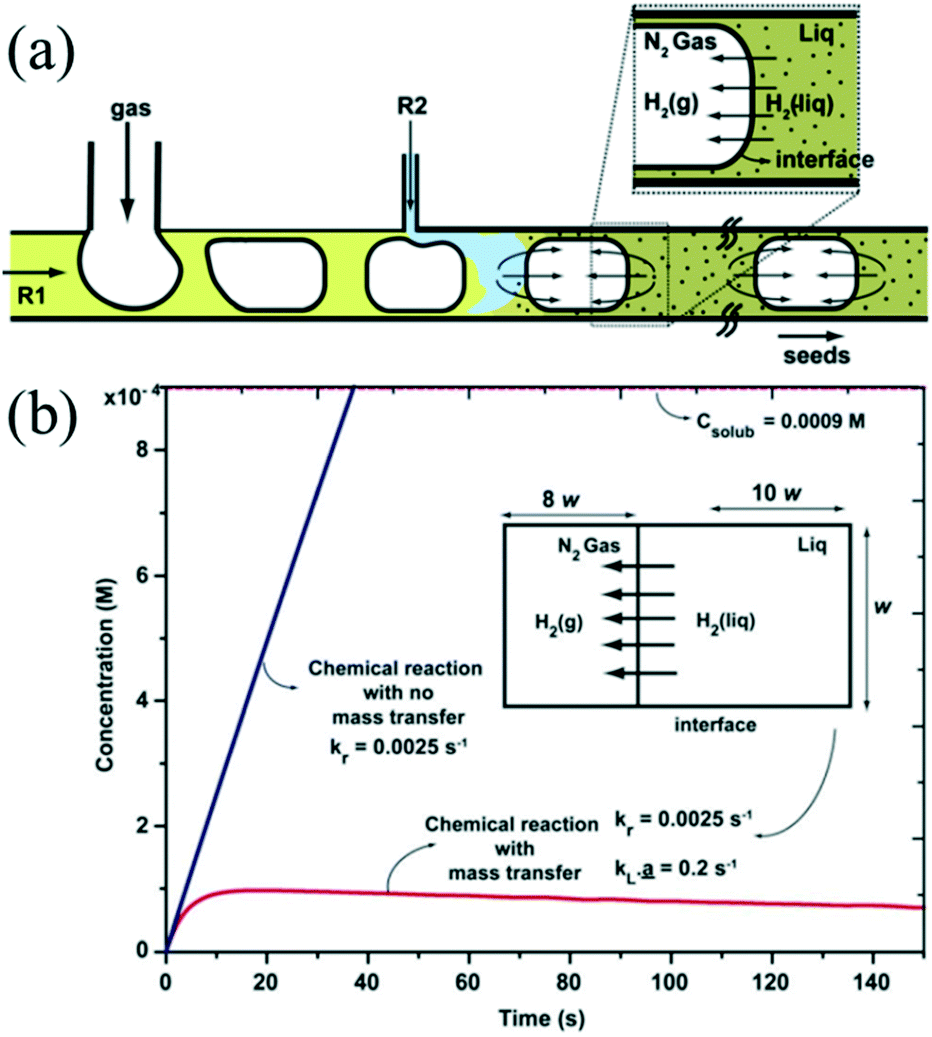 | ||
| Fig. 4 The fast mass transport across the gas–liquid interface. (a) Schematic showing the hydrogen gas transport in segmented gas–liquid flow. (b) Plots showing the time variation of hydrogen concentration in the liquid phase with and without mass transport into the gas phase. Reproduced from ref. 75, copyright 2012 Royal Society of Chemistry. | ||
Enhanced heat transport in a microfluidic reactor
The thermal Péclet number (Pe = convective transport rate/diffusive transport rate = wUρCp/k, where w is the diameter of the channel, U is the velocity of flow, ρ is the density of the fluid, Cp is the specific heat capacity of the fluid, and k is the thermal conductivity of the fluid) can be used to characterize heat transport resulting from fluid motion and conductive heat transport in the microfluidic environment.77 However, it should be noted that the investigations of heat transport in a microfluidic reactor are obviously less than those of mass transport due to the complexity of heat transport. Similar to the mass transport, reduced reactor dimension also allows increasing the heat transport rate and avoiding heat inhomogeneity resulting from local hot spots, which strongly affect the nucleation and growth of NCs but are difficult to control. For example, Yao et al.59 simulated the heat transport process of a droplet reactor using COMSOL Multiphysics. The results demonstrated that the moving droplet could be heated from 25 °C to 85 °C and could achieve temperature equilibrium in merely 18 ms. High heat-transport efficiency is likely to be an important reason for the fast growth rate of CdTe QDs (approximately 1 min). Generally, a microreactor provides better control over the temperature than a batch reactor due to the fast heat transport. On the one hand, enhanced heat transport allows the reaction to be initiated and quenched within seconds.78,79 LaGrow et al.78 investigated the nucleation and growth of Pt–Ni octahedral NCs using a continuous flow reactor. By quenching the reaction within seconds, the growth process could be well determined, from poorly crystalline nanoparticles (nucleation) at 5 seconds, to 1.5 nm NCs at 7.5 seconds, followed by growth into octahedral NCs at 20 seconds. In addition, heat removal may result in an interesting phenomenon. Kunal et al.27 recently reported the synthesis of Rh NCs in a droplet reactor using microwave heating. They found that the fluorous carrier phase, which had a poor microwave absorbance, allowed removal of heat away from droplets, favouring the formation of kinetically stable NC morphology. On the other hand, enhanced heat transport also improves the size distribution of NCs, especially when the synthetic process involves the thermal decomposition of precursors.80Generally, it is believed that temporal separation of nucleation and growth enables formation of NCs with a narrow size distribution.3 To this end, both the concentration of monomers and the reaction temperature should be considered in the synthetic process. Indeed, a droplet reactor with multiple temperature regions has been studied to control the process of nucleation and growth. Erdem et al.81 developed a droplet microreactor that integrated multiple isolated heated and cooled regions (Fig. 5a). The infrared (IR) camera image showed that nucleation and growth regions were thermally isolated (Fig. 5b). The authors also utilized this device to synthesize TiO2 nanoparticles by controlling temperatures and residence times for nucleation and growth (Fig. 5c–e). Pan et al.82 developed a strategy for the synthesis of PbS NCs using a dual-temperature-stage droplet reactor. The nucleation and growth temperatures were set in the range of 80–150 °C and 50–100 °C, respectively. The experiment revealed that NCs produced from the dual-stage flow reactor were superior to the products synthesized in the single-stage reactor.
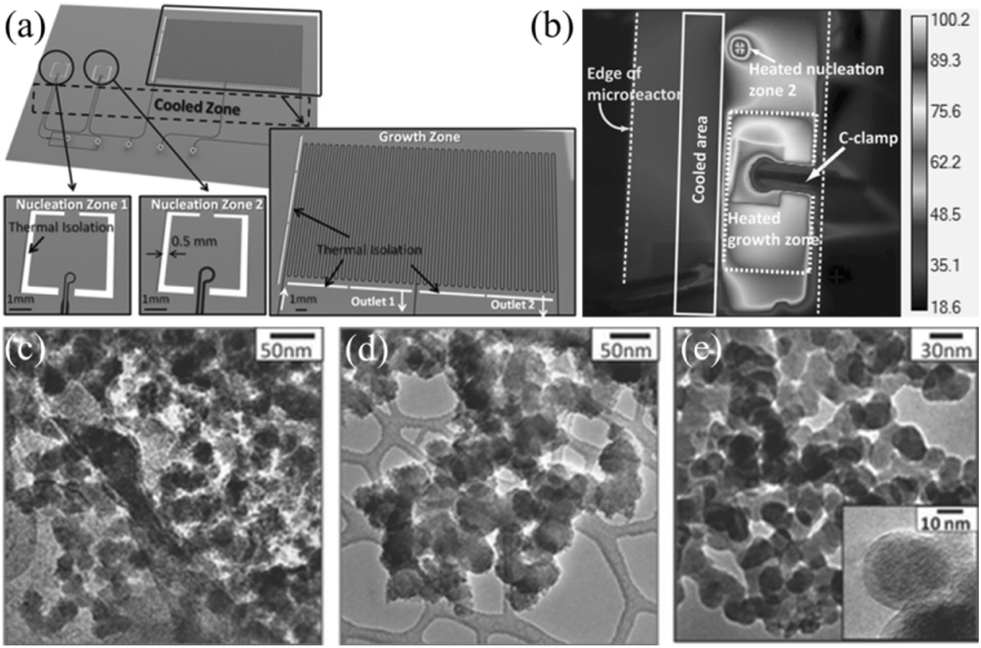 | ||
| Fig. 5 Droplet microreactor with multi-temperature regions for synthesis of TiO2 nanoparticles. (a) Schematic showing the nucleation and growth regions. (b) IR image of the heated and cooled zones. (c) TEM image of nanoparticles synthesized by heating at 100 °C for 6 s in the nucleation zone. (d) TEM image of nanoparticles synthesized by heating at 100 °C for 6 s in the nucleation zone and 80 °C for 120 s in half of the growth zone. (e) TEM image of nanoparticles synthesized by heating at 100 °C for 240 s. Reproduced from ref. 81, copyright 2014 Wiley. | ||
5. Research frontiers
The droplet reactor allows control of the product qualities since it can access experimental conditions/operations unavailable within the batch reactor. In addition, the advantages of high reproducibility and reliable automation manifest it as a promising alternative to batch synthesis. For these reasons, the droplet reactor has emerged as a powerful platform which has been increasingly utilized to synthesize various high-quality NCs.Sequential multistep reaction
The microfluidic system linearly transforms the temporal distribution into spatial distribution, thereby allowing multistep reactions successively along the length of the flow in the microchannel. This subsection has been tailored to focus on the multistep reaction, such as synthesis, functionalization and purification in a successive flow process.The seed-mediated approach has been extensively used to synthesize core–shell nanostructures. In order to control the shell thickness, the synthetic process requires a proper addition rate of raw materials to maintain a moderate growth of seeds. Khan et al.15 investigated the multistep coating of SiO2 by TiO2 in a gas–liquid segmented flow reactor. In their strategy, multistep addition using side channels enabled fast mixing of given amounts of raw materials and yielded shells with controlled thickness. Also, this method avoided both the secondary nucleation of TiO2 and the agglomeration of SiO2. Similar to the multistep coating above, larger NCs with narrow size distribution could be produced through repeatedly supplying additional precursors into the reaction system. Baek et al.83 presented a continuous microfluidic platform that allowed for sequential multistep injection of reagents for the synthesis of InP NCs. The successive injections favored the further growth of NCs while maintaining the homogeneous size distribution. Furthermore, Nightingale et al.42 investigated the multistep growth of CdSe NCs by repeated reagent addition in a five-stage droplet reactor that consisted of an initiation stage followed by four growth stages (Fig. 6a). The reaction temperature and residence time were well controlled in each stage. Compared with single-step reagent addition, the five separate reaction stages resulted in a more significant shift in the peak emission wavelength (Fig. 6b and c). In a recent report, Maceiczyk et al.84 used a two-stage droplet reactor for the synthesis of CdSe NCs. The experiment revealed that NCs with optimal properties were produced through both addition of precursors subsequent to the nucleation stage and control over the reaction temperature in the growth stage.
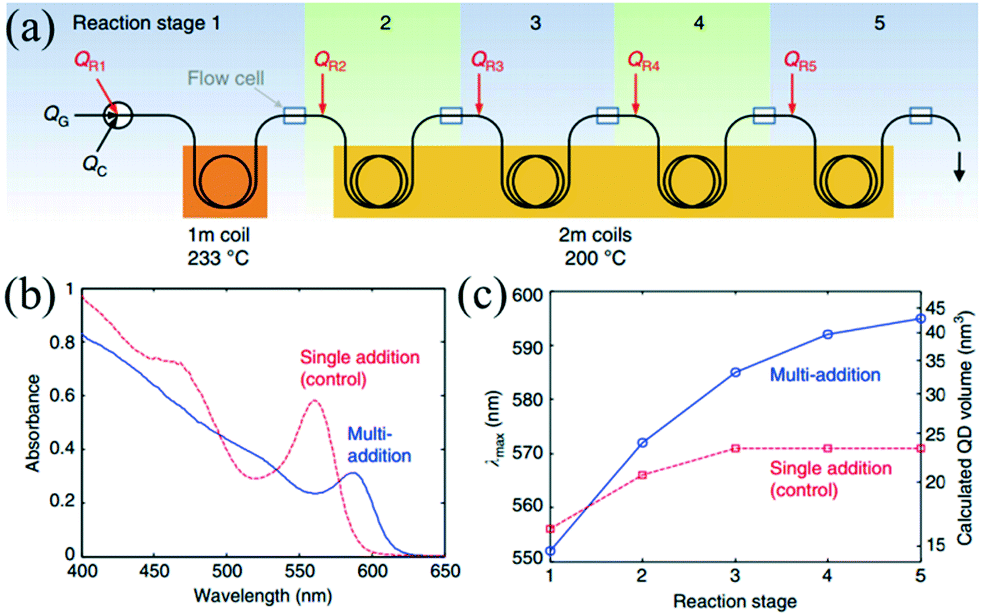 | ||
| Fig. 6 Controlled multistep synthesis in a droplet reactor. (a) Schematic showing a five-stage droplet reactor used for multistep growth of CdSe NCs by repeated reagent addition. (b) Absorption spectra of the as-prepared QDs for multiple- and single-step reagent addition. (c) Peak emission wavelength (λmax) vs. reaction stage for multiple- and single-step reagent addition. Reproduced from ref. 42, copyright 2014 Nature Publishing Group. | ||
Interestingly, a microfluidic platform allowed for direct synthesis of core nanoparticles followed by precise addition of the shell materials for further growth in a continuous process without manual manipulation. In this regard, Yang et al.85 reported the formation of CdSe NCs and ZnS passivation of the synthesized cores using a two-stage continuous flow reactor. Recently, such a strategy has been extended to the continuous synthesis of CuInS2/ZnS NCs without an intermission.86 Yashina et al.60 further investigated the synthetic process for CuInS2/ZnS NCs in a two-stage droplet reactor. This method provided separate control over the formation of a CuInS2 core followed by ZnS capping without the need for an intermediate purification process. By coupling with an in situ monitoring system, this approach also enabled fast screening of synthetic parameters including adjustment of molar ratio, reaction temperature, and reaction time.
The post-synthetic treatments involving several steps along with intermediate purification are usually very time-consuming and even hard to carry out. The direct coupling of several reaction steps successively in a continuous process allowed reduction of the reaction time and reagent consumption.87,88 Uson et al.89 presented a multistep strategy for the synthesis and functionalization of gold nanorods in a continuous flow process (Fig. 7). The strategy allowed integrating multiple reaction stages, such as precursor activation, seed formation, nanorod growth and PEGylation, into a single process with independent control over each stage. Notably, streamlined functionalization enabled a 100-fold reduction of PEG consumption compared with the conventional batch procedures. Subsequently, Jambovane et al.90 demonstrated the continuous synthesis and functionalization of MOFs in a droplet reactor. This method allowed for the combination of four steps into a single step that required obviously less time (1 hour) compared with time-consuming currently available batch procedures (several days).
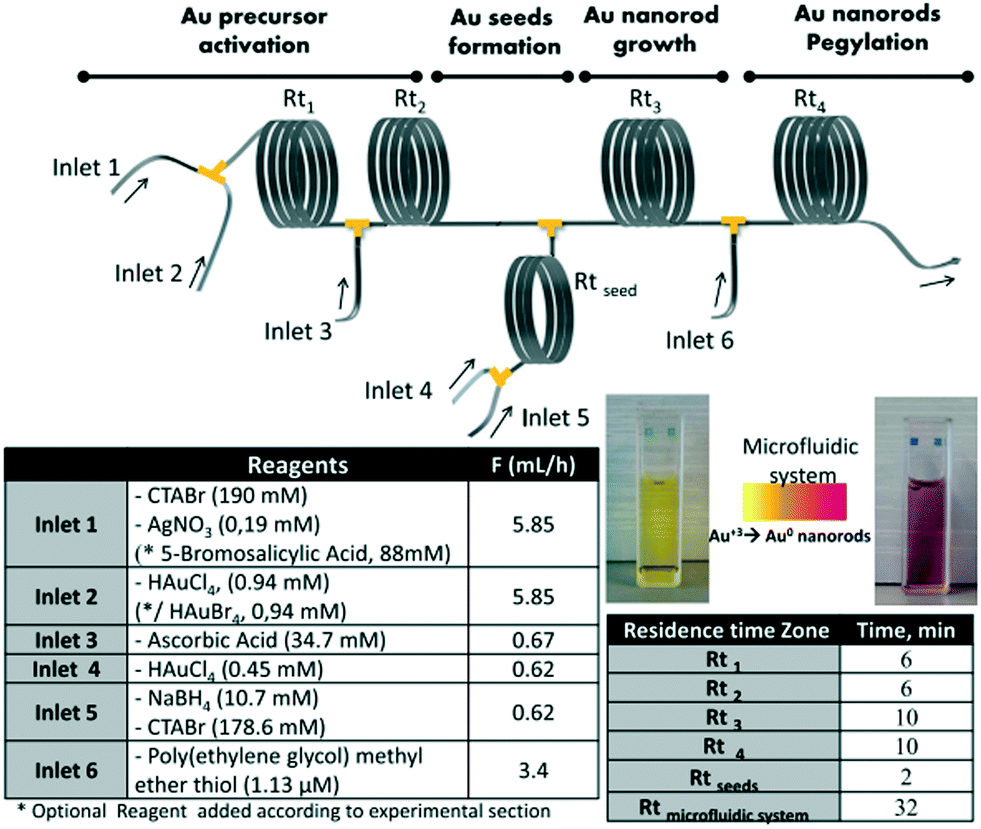 | ||
| Fig. 7 The sequential multistep synthesis of PEGylated gold nanorods in a continuous flow process. The reagents and flow rates of each inlet and the corresponding residence times are given. Reproduced from ref. 89, copyright 2016 Elsevier. | ||
Although the aforementioned strategies enabled combination of synthesis and functionalization into a continuous process, in situ purification was still a challenge for the post-synthetic treatments. Generally, a common strategy to obtain purified NCs from a solution involves solvent induced precipitation and centrifugation. However, the process not only consumes a lot of organic solvents but also impacts NC surface properties negatively.91 Shen et al.92 recently demonstrated a multi-stage purification strategy for both CdSe NCs in organic solvent and Au NCs in water using a continuous liquid–liquid-extraction process. The purified NCs could be obtained by the extraction of precursors into the other immiscible phase, followed by the phase separation using the membrane of different wettability for two solvents. This strategy significantly improved the purity of the samples without the high solvent consumption. For magnetic nanoparticles, magnetic separation in a microfluidic device has been proved an effective method to obtain cleaned product.93 In this regard, Ferraro et al.94 demonstrated sequential multistep reactions for the assembly of fluorescent and magnetic SiO2@γ-Fe2O3 using an automated microfluidic platform coupled with magnetic tweezers (Fig. 8a). The platform allowed combining different operations including mixing, flocculation, extraction, washing, redispersion, heating and colloidal assembly into a single system with the flexibility to control each stage separately (Fig. 8b). Notably, by merging the ferrofluid droplet with an acetone droplet, nanoparticles aggregated quickly at the magnetic tweezers, forming a cluster that could be extracted from the initial droplet and washed with subsequent droplets. In a word, in situ purification is a promising technique which not only brings the benefits of less consumption of time and solvent compared with conventional approaches but also enables a completely continuous production of purified NCs.
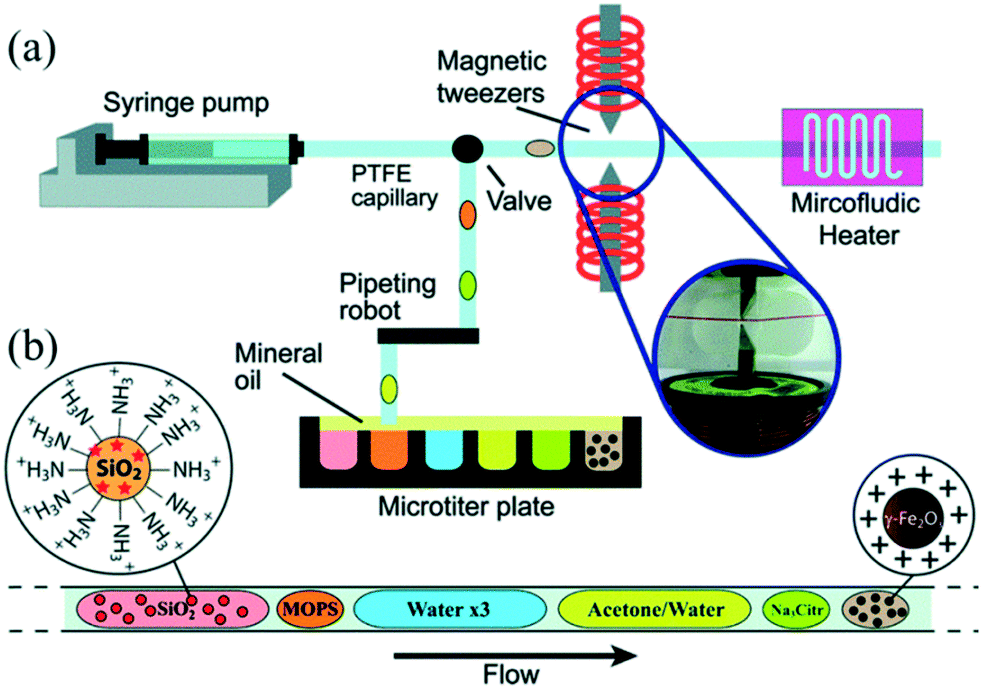 | ||
| Fig. 8 Droplet-based microfluidic platform for continuous operations on magnetic γ-Fe2O3 nanoparticles. (a) Schematic showing a microfluidic device for formation of droplets by alternatively extracting the water phase and the oil phase. Subsequently, these droplets were driven for ferrofluid manipulation. (b) Train of droplets for assembling reaction. Reproduced from ref. 94, copyright 2015 Royal Society of Chemistry. | ||
Intelligent synthesis
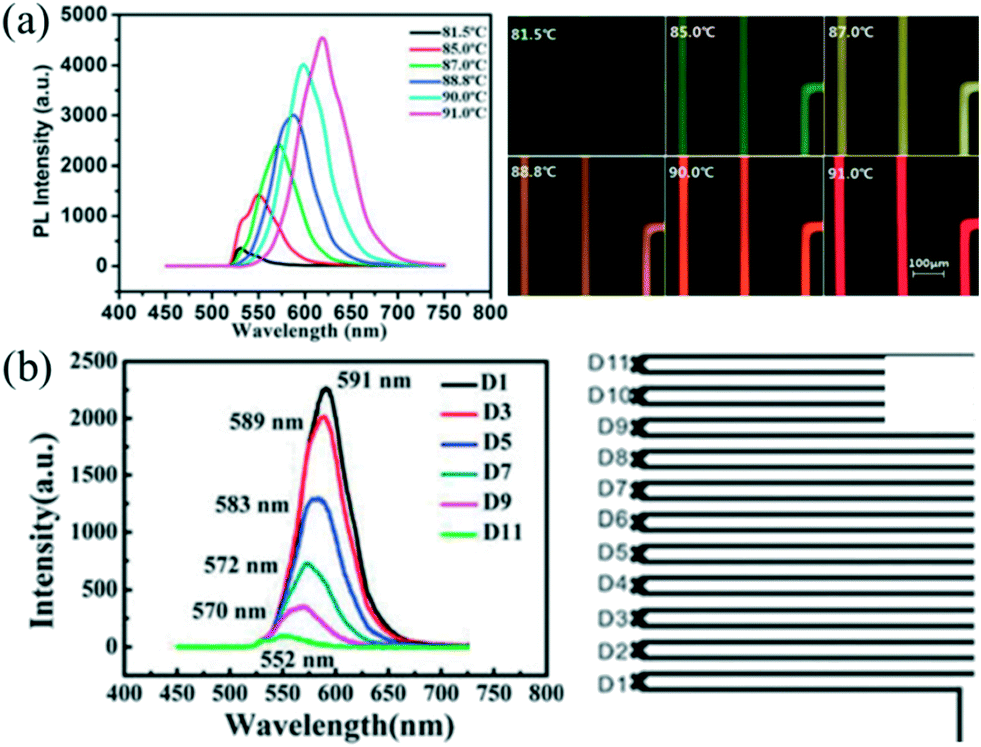
Automated droplet-based microfluidic platforms with integrated real-time monitoring systems and optimization algorithms allow not only rapid screening of synthetic parameters but also investigating the mechanism of NC nucleation and growth.9,16 This subsection has been tailored to focus on currently available in situ monitoring techniques and optimization algorithms for intelligent synthesis.Nowadays, in situ fluorescence detection has been extensively utilized to analyze QDs that have size-depending fluorescence properties including the peak wavelength and the full width at half-maximum (FWHM). Chan et al.28 firstly coupled a fluorescence monitoring device with a microfluidic chip for analyzing CdSe QDs. In their strategy, the fluorescence spectra of products were continuously collected at the end of the reactor. The extracted spectra could be analyzed to screen the reaction conditions. Indeed, such a strategy has been used and extended to the rapid optimization of synthetic parameters and preparation of high-quality QDs in a droplet reactor.38,95 Furthermore, the real-time monitoring system is helpful to study the reaction kinetics through probing different positions of the channel. Yao et al.59 investigated the effects of temperature and reaction time on ultrafast synthesis of CdTe QDs in picoliter droplets. A platform with an integrated microscopic imaging device and a portable optical fiber spectrometer allowed the collection of in situ fluorescence signals, offering a feasible method for probing and analyzing the dynamic growth process of CdTe QDs. Significantly, a series of QDs with different emission wavelengths could be synthesized through a slight change of 1–2 °C in the controlled reaction temperature (Fig. 9a). Compared with the batch experiment, the synthetic approach also showed an obvious decrease in the reaction time (approximately 1 min), which could be attributed to the fast mass and heat transport in such a droplet reactor. Moreover, the temporal evolution fluorescence spectra showed that emission peaks presented a significant red shift gradually increasing in peak intensity as the residence time became longer (Fig. 9b).
 | ||
| Fig. 9 Picoliter droplet reactor for the synthesis of CdTe QDs. (a) In situ fluorescence spectra and optical micrographs of formed QDs at different temperatures. (b) In situ fluorescence spectra of formed QDs at the different positions of the microchannel. Reproduced from ref. 59, copyright 2013 Royal Society of Chemistry. | ||
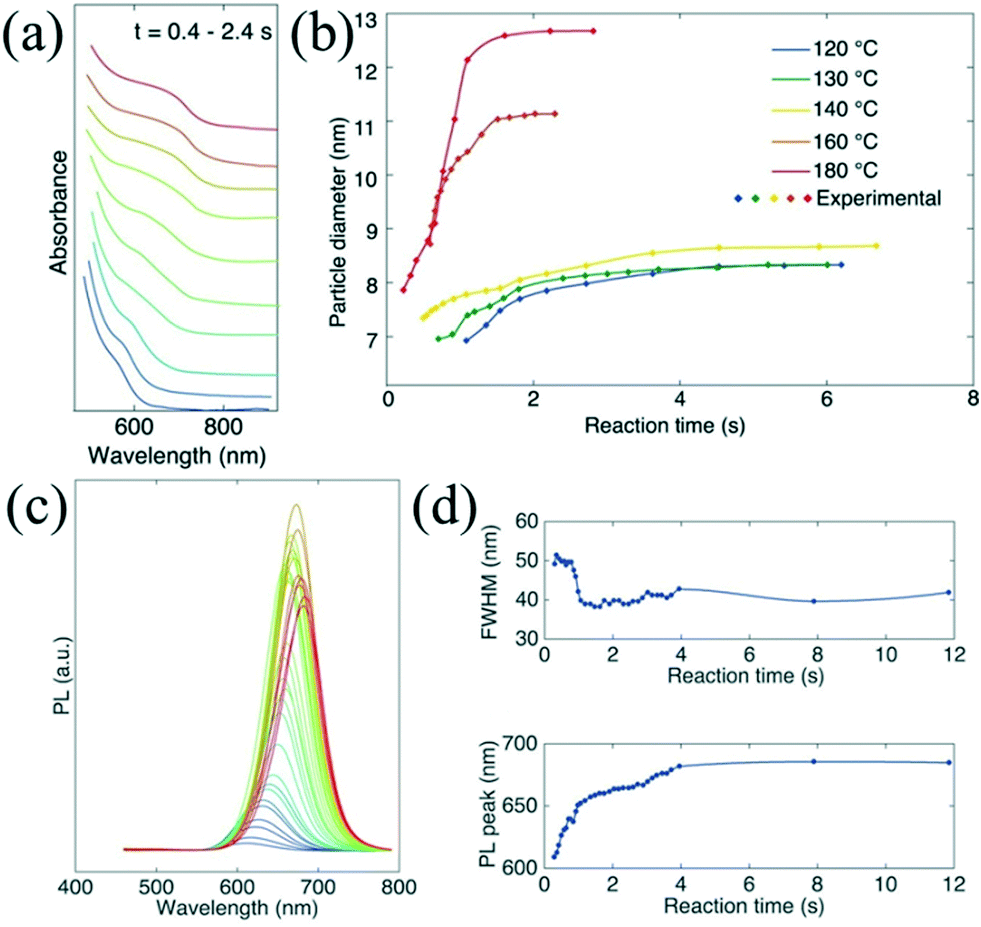
In situ absorption spectroscopy is especially valuable to acquire information relating to both size and concentration of nanoparticles, which can provide valuable information for nuclei during the initial stage of the reaction.9 Lignos et al.96 studied millisecond kinetics of PbS NC formation in a droplet reactor with integrated detection modules for on-line absorption and fluorescence spectra. Kinetic investigations suggested that the formation mechanism of PbS NCs could be divided into two stages during the early reaction time. In the first stage (<1 s), PbS nuclei with a constant size were continuously produced. Subsequently, the Ostwald ripening process for growth of the formed nuclei was dominant in the later stage. Recently, they have further extended the investigation strategy and presented the fast screening of synthetic parameters for the formation of cesium lead halide perovskites (CsPbX3, X = Cl, Br, I) NCs.97 They highlighted the kinetics study in the early stage of the reaction within the initial 0.4–2.4 s, which was not accessible in batch experiments (Fig. 10a and b). The temporal evolution of photoluminescence (PL) spectra suggested the narrowing of the FWHM during synthesis (Fig. 10c and d). Also, this platform allowed rapid optimization of the synthetic parameters including reaction time, reaction temperature, and molar ratio of precursors. A similar investigation method may also provide insight into the kinetic process for the formation of other ternary or quaternary NCs (e.g., cation exchange reaction).98 To gain further insight into the reaction mechanism, an oscillatory microprocessor was developed for rapid collection of 7500 spectral data within 10 min.99
 | ||
| Fig. 10 Droplet-based microfluidic platform for studying the early time kinetics of CsPbI3 NC synthesis. (a) Temporal evolution of in situ absorption spectra at 180 °C. (b) Temporal evolution of particle diameters at different temperatures. The sizes were estimated from bandgap energies according to the absorption spectra. (c and d) Temporal evolution of the PL emission spectra and corresponding FWHM and peak wavelength at 180 °C. Reproduced from ref. 97, copyright 2016 American Chemical Society. | ||
Currently, X-ray techniques including X-ray absorption spectroscopy (XAS) and small-angle X-ray scattering (SAXS) have been well combined with a microfluidic platform. XAS including X-ray absorption near-edge spectroscopy (XANES) and X-ray absorption fine structure spectroscopy (XAFS) can provide the composition and structural information.66 Chan et al.100 investigated the kinetics of a cation exchange reaction between CdSe NCs and Ag+ in a continuous flow using XAS. The experiment revealed the structural evolution of CdSe NCs to Ag2Se on the millisecond scale. Furthermore, information on the oxidation state could be obtained from XANES which was used to measure the conversion yield of Ag+ to Ag0 during the process for the formation of Ag nanoparticles.101 As an important complementary technique, in situ SAXS gives access to direct determination of the size, shape and concentration of particles.102,103 In this regard, Karim et al.104 used in situ SAXS and XAFS to investigate the formation mechanisms of Pd nanoparticles in a microfluidic reactor. SAXS results revealed a slow nucleation process that was evidenced by a continuous increase in the concentration of particles. XAFS results suggested an increase in Pd–P coordination in coincidence with the change in the growth rate. Recently, Pham et al.105 demonstrated the combination of SAXS and a droplet-based microfluidic platform to study protein crystallization. This platform offered the advantage of avoiding shooting a droplet interface that could produce a huge SAXS signal and then could lead to misinterpretations of the data. Overall, in situ X-ray studies are particularly helpful in investigating the rapid kinetics during the NC nucleation and growth.
Automated feedback enables efficient characterization of reaction and optimization of the synthetic conditions by using microfluidic technology with integrated an in situ monitoring system and an optimization algorithm.16,17 Detailed advances in the optimization process are also covered in recent reviews.106 Krishnadasan et al.107 firstly presented an intelligent strategy that used an automated microfluidic platform with a control algorithm for the synthesis of CdSe QDs. The acquired data were reduced to a dissatisfaction coefficient which defined the success of the reaction. Then, an optimization algorithm (stable noisy optimization by branch and fit, SNOBFIT) was used to intelligently change the synthetic parameters which minimized this coefficient, forming a product with optimized properties. Maceiczyk et al.108 further demonstrated the application of the Universal Kriging metamodeling algorithm for the synthesis of CdSe QDs in a droplet-based microfluidic platform. Such a system allowed screening multidimensional parameters including FWHM, fluorescence emission maximum and intensity. The experiment suggested that the algorithm could accurately predict reaction outcomes. In addition to optimization algorithms, an automated pump system with a control algorithm is also desirable to intelligently adjust the flow rates, which affect the residence time and precursor concentration ratios.109 Knauer et al.72 used programmed flow rate variations in combination with an automated droplet reactor for precise adjustment of the silver shell thickness during the synthesis of Au–Ag core–shell nanoparticles. This device screened the reactant concentration ratios while monitoring shell thickness by in situ UV/vis absorption spectroscopy. Moreover, with optimized reaction parameters and a computer-controlled programmable microfluidic platform, such algorithms are able to facilitate the scale-up for the industrialized production of NCs.110,111
Reliable scale-up synthesis
Nowadays, there is an increasing need to scale up the synthesis strategy for NCs owing to industrial and commercial demands. The formation of colloidal NCs is very sensitive to a change in reaction conditions such as reagent concentration, solution volume, and mixing intensity.13 Therefore, enlarging the reaction in batch reactors may be confronted with many challenges, which could result in a loss of product quality and poor batch-to-batch reproducibility.112 Droplet reactors provide obvious advantages over conventional batch reactors, such as accurate manipulation, better reproducibility, and reliable automation.12 Recent efforts to scale up the reaction in droplet reactors have mainly focused on operating parallel multi-channels and enlarging the size of the reactor.On the one hand, a parallelization approach has been widely applied to high-throughput screening.48,49 Yang et al.48 presented a droplet array generator with parallel multi-channels, which could generate 64 lines of droplets with 33 gradient concentrations in a microfluidic chip. This device allowed rapidly obtaining different ratios of Au/Ag nanoparticles. On the other hand, a parallelization method offers a feasible strategy to carry out multiple reactions simultaneously, thereby allowing for large-scale synthesis of NCs. In recent years, the PTFE tubing-based droplet reactor has been extensively utilized for high-temperature synthesis of nanomaterials.24,27,60 Nightingale et al.113 further developed a five-channel droplet reactor for large-scale synthesis of CdTe QDs at high temperature (Fig. 11a). In their strategy, the reaction phase and carrier phase were extracted from reservoirs and then split into five-way parallel streams by two passive flow dividers. Individual streams of each phase were merged at five separate T-junctions, generating parallel droplet streams. Interestingly, in contrast with pumping a limited amount of solution in a syringe, the pumps and valves were simultaneously used to extract fresh precursor solution, ensuring continuous synthesis without any interruption. Indeed, this nice design satisfies the need for scale-up while also taking full use of the advantage of a syringe pump with high precision and stability. Also, the carrier phase was spontaneously recycled by a high pressure liquid chromatography (HPLC) pump in a closed-loop manner. The fluids can be used to the best advantage that accords with a concept of sustainable synthesis. Consequently, they obtained 54.4 g of dry products after nine hours of production.
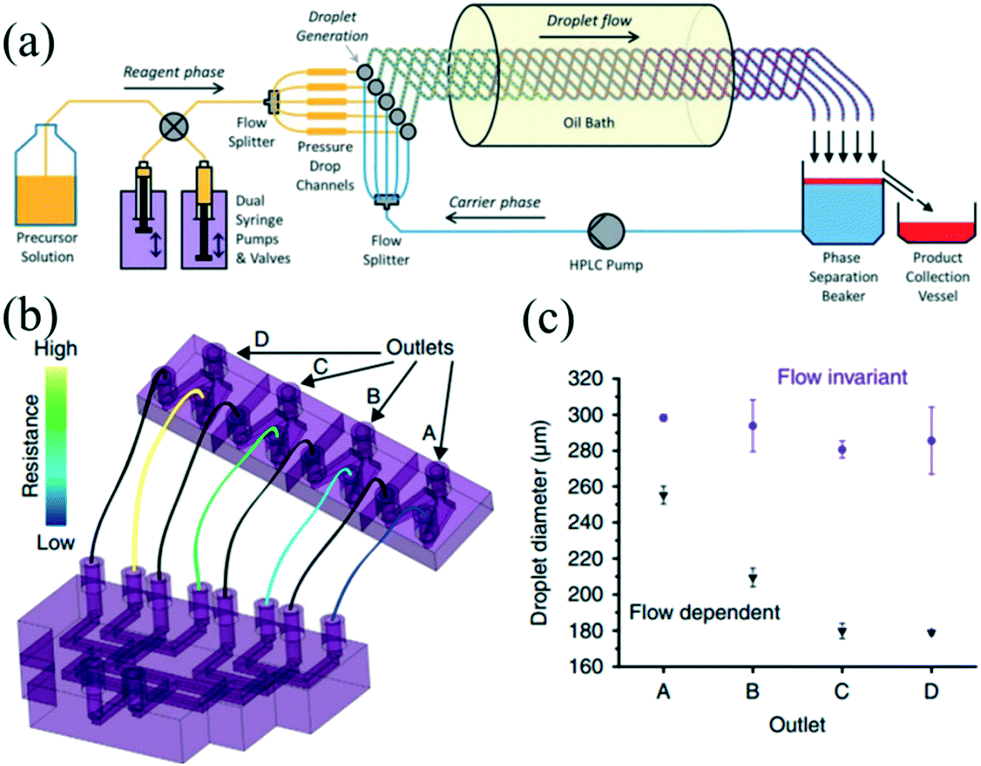 | ||
| Fig. 11 The parallelizing droplet generation devices for synthesis of NCs. (a) Schematic showing the multichannel droplet reactor for the large-scale synthesis of CdTe QDs. Reproduced from ref. 113, copyright 2013 Royal Society of Chemistry. (b) Schematic showing the parallel device that was robust to fluctuations in flow rate. (c) Droplet diameters generated by the four branches of the parallel channel. The carrier phase and reaction phase flow rates of 70 and 10 mL h−1 (purple circles) and 210 and 30 mL h−1 (black triangles) while performing within and beyond the flow invariant regime, respectively. Reproduced from ref. 115, Copyright 2016 Nature Publishing Group. | ||
The essential part in designing such a parallelizing device is the distributor section, which equalizes and controls the fluid flows over the multiple droplet generators.47,114 In a recent report, Riche and colleagues115 designed a novel parallel droplet reactor which was insensitive to fluctuations in flow rates (Fig. 11b). Notably, a 3D-printing technique was used to fabricate the complex channel structures. The droplet size in each branch was determined by the inner diameter of the outlet tubing rather than the geometry of dispersed phase inlet. Despite the intentional gradient of flow rates, the four-branched parallel channels could still generate droplets with similar size in each branch while operating in the flow invariant regime (Fig. 11c). The authors synthesized Pt nanoparticles using this device that achieved an approximately double yield compared with batch synthesis. In addition, a fluidic capacitance element has also been developed to overcome the flow disturbances in a parallelized fluid delivery system. For example, Yap et al.47 recently developed a fluidic circuit-based device for an eight-fold parallelized three-phase segmented-flow reactor. This method allowed elimination of the periodic and aperiodic flow disturbances resulting from the peristaltic pump and fluidic resistance. Also, the authors used this device for conducting a catalytic reaction with continuous recycling of the catalyst phase over five hours of operation. Recent efforts have also been dedicated to a monitoring system for high-throughput processing of droplets. For example, Conchouso and colleagues116 developed both capacitive and radio frequency (RF) resonator sensors, which could continuously monitor droplet formation in parallelization systems.
By enlarging the diameter of the channel, the reaction can be scaled up in the millifluidic reactor. Lohse et al.117 demonstrated a millifluidic device that allowed for gram-scale synthesis and functionalization of gold nanoparticles with controlled sizes and shapes. Similar methods have been used to produce various NCs, such as Ni,118 Cu2−xS,119 Pt–Ni,78 CdSe/CdS/ZnS,120 and Cu2ZnSnS4.121 Indeed, these synthetic methods enabled an increase in the throughput while also providing substantial control over the reaction. Also, the residence time required for some scaled-up reactions was nearly the same as that of the microfluidic process.88 In addition to the aforementioned single-phase flows, droplet flows in millifluidic scale have also been investigated. Li et al.122 studied the flow pattern of droplets in a flow-focusing millifluidic chip. This device was able to produce uniform droplets with similar controllability as in the case of microfluidic devices. Also, this device allowed achieving approximately 51 mL per hour volumetric productivity for Cu nanoparticle synthesis. Zhang et al.123 reported a milliliter-sized droplet reactor for the scalable synthesis of metal NCs. The device enabled scale-up while also ensuring NC uniformity in both size and shape. The authors also demonstrated that the volume of the reaction phase should not affect the shape of the NCs when rapid mixing was achieved in the droplets. Consequently, they produced NCs with controllable properties on a scale of 1–10 g per hour. Recently, Wong et al.124 presented a scaled-up strategy for the synthesis of Pd nanoparticles using a gas–liquid–liquid segmented millifluidic reactor. This strategy has achieved approximately 10 L per day volumetric productivity with consistent control over the quality of products during long-term synthesis. Overall, the throughput of a single-channel device can be obviously increased by enlarging the channel size. However, further scale-up may encounter the problems of poor mass and heat transport.
Interfacial synthesis
For a gas–liquid segmented flow, fast mass transport across the interface can affect nucleation and growth of NCs. For example, air bubbles could provide oxygen to participate in the process of oxidative etching.43,125 In addition to the synthesis involving self-nucleation only, seeded growth has also been studied. Zhang et al.41 demonstrated a synthetic process for the seed-mediated growth of Ag nanocubes in droplets separated by air. The oxygen in air promoted the formation of a reducing agent while also providing buffer space for the diffusion of NO resulting from the oxidative etching. Apart from O2 gas, other gases (e.g., CO)64,76,126 could also play important roles in the synthesis of NCs. Larrea et al.127 investigated the effect of different types of gas on the synthesis of iron oxide nanoparticles in gas–liquid segmented flow reactors (Fig. 12a). In their strategy, the materials' properties (e.g., size, shape, and crystalline structures) could be well controlled through simply changing the composition of the gas including inert (N2), oxidizing (O2), and reducing (CO, H2) gases (Fig. 12b). Consequently, the gas–liquid segmented flow reactor offered a flexible and easy approach to implement such a process to produce various NCs with particular properties.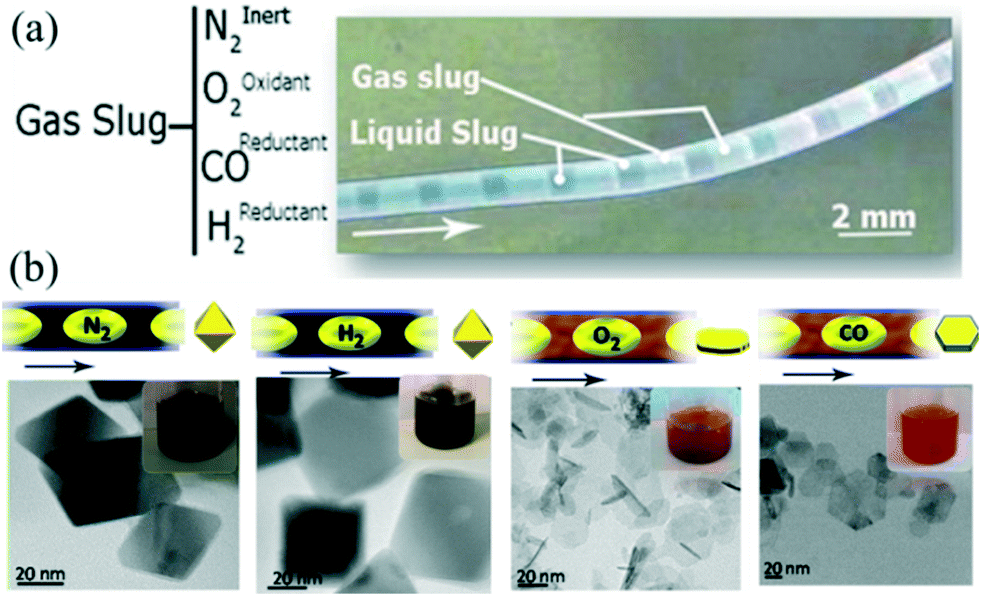 | ||
| Fig. 12 Synthesis of iron oxide nanostructures using gas–liquid interfacial mass transport. (a) Optical image of the segmented flow with different gases. (b) TEM images of magnetic nanoparticles synthesized using different gases. Reproduced from ref. 127, copyright 2015 American Chemical Society. | ||
The reagents reacting at the liquid–liquid (L–L) interface offer a convenient and versatile strategy for the preparation of nanomaterials.128 In this regard, Wang et al.129 presented a microfluidic approach for the synthesis of TiO2 nanoparticles using the reaction at a pinned L–L interface. Notably, the droplet reactor can provide higher surface-to-volume ratios and thus higher heat and mass transport rates. Recently, Zukas et al.130 have demonstrated a strategy for the interfacial synthesis of ZnO nanoparticles using a carrier phase of sodium hydroxide in 1-octanol and a droplet phase of aqueous zinc acetate. The reaction was initialized due to the fast mass transport across the interface. Notably, compared with the batch reaction, ZnO nanoparticles with narrower size distribution and controllable shape were produced in this strategy. More recently, Sachdev et al.131 presented a synthetic strategy for Au particles using the L–L interface of the emulsion droplet. By tuning the reaction parameters (e.g., concentration and droplet size), Au particles ranging from nanometer to micrometer sized spheres or platelets could be readily produced. Interestingly, the authors found that the presence of surfactants significantly affected the formation of different particles. This was consistent with a report by Bawazer et al.,132 who demonstrated that diverse surfactants acted as templates at the L–L interface, thereby accelerating the TiO2 crystallization.
Similarly, interfacial adsorption for colloidal NCs has also been studied since NCs have a tendency to be concentrated at the L–L interface. Zhang et al.133 studied the effects of interfacial adsorption on the synthesis of NCs in a droplet reactor. By eliminating the mixing zone and allowing the Ag nanocubes to be adsorbed onto the interface, interfacial adsorption was utilized to produce NCs with asymmetric shapes including Ag octahedra and Au–Ag nanocups. Also, interfacial adsorption could be mitigated or even avoided through introducing a proper surfactant (e.g., Triton X-100) to passivate the interface and form NCs with uniform sizes and controlled shapes. Furthermore, Hassan et al.134 presented a synthetic process for assembling asymmetrical nanohybrids using the L–L interface. This strategy offered a higher control over the interfacial assembly of gold nanoparticles (15 nm) and silica nanoparticles (160 nm) while also greatly reducing the reaction time compared with batch experiments. Also, the authors demonstrated that the enhanced transport of nanoparticles to the interface could be attributed to both flow recirculation inside and outside the droplet and the confinement effect.
6. Conclusions and outlook
In this review, we have provided a comprehensive summary of the recent progress of NC synthesis in droplet reactors, covering fundamental investigations from device fabrication, droplet generation, and essential properties of droplet reactors to a broad range of applications using various synthetic strategies. Overall, the past decade has witnessed significant advances in droplet reactors for the syntheses of high-quality NCs that have enabled studies of their fundamental properties and potential applications. Despite the great success in this emerging field, more efforts should be dedicated to further investigations. Therefore, it is necessary to point out key challenges which should be considered seriously to push the droplet reactor forward to be an irreplaceable tool for NC synthesis.The first challenge is involved in device fabrication and large-scale production. Indeed, the microfluidic reactor has achieved the parallelization of droplet generation. However, the issue of fluid distribution remains a challenge. A 3D-printing technique can be developed to fabricate small and complex channel structures, which can be used to control fluid distribution. In addition, the 3D-printing technique can be further developed to integrate industrial-scale interfaces and built-in control systems. These designs will bring the benefits of scale-up and provide powerful control over flow and product. Overall, the current scale-up is still limited to laboratory investigation which is a long way to the commercial-scale application.
The second challenge is related to multistep reactions, such as synthesis, functionalization and purification, in a successive flow process. Many efforts should be devoted to the simplification of fundamental functional units, including droplet generation, droplet merging, and phase separation. Moreover, an integrated process combining synthesis and post treatments in a continuous flow process deserves particular attention. Notably, in situ purification can be further developed in terms of some reported techniques including extraction92 and electrophoresis.136,137 We believe that purified NCs with specific functionalization can be directly obtained from raw materials in an integrated microfluidic reactor in the future.
The third challenge is associated with in situ monitoring techniques and optimizing algorithms. The droplet-based microfluidic system is expected to be a promising platform that allows investigation of the mechanism of NC nucleation and growth. To meet this demand, more efforts should be devoted to novel in situ monitoring techniques such as mass spectrometry,135,136 electron microscopy, and spectroscopic methods (e.g., photothermal interferometry137 and surface enhanced Raman spectroscopy138). In addition, more optimizing algorithms for NC synthesis should be developed to satisfy the need for automated feedback, which enables fast characterization of the reaction and optimization of the synthetic parameters.
Lastly, interfacial synthesis, which involves mass transport across the interface, should be further utilized to produce NCs with unique properties. Interestingly, interfacial synthesis can be rapidly quenched by phase separation, which may provide particular insight into kinetics study.139 In addition, multiple emulsions should be utilized to control NC properties at the interface by changing the composition of droplets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21375100, 21475099, 21535005).References
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- Y. N. Xia, X. H. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- C. D. Donega, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 CrossRef PubMed.
- J. van Embden, A. S. R. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS.
- T. W. Phillips, I. G. Lignos, R. M. Maceiczyk, A. J. deMello and J. C. deMello, Lab Chip, 2014, 14, 3172–3180 RSC.
- I. Lignos, R. Maceiczyk and A. J. deMello, Acc. Chem. Res., 2017, 50, 1248–1257 CrossRef CAS PubMed.
- B. K. H. Yen, A. Gunther, M. A. Schmidt, K. F. Jensen and M. G. Bawendi, Angew. Chem., Int. Ed., 2005, 44, 5447–5451 CrossRef CAS PubMed.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- G. Niu, A. Ruditskiy, M. Vara and Y. Xia, Chem. Soc. Rev., 2015, 44, 5806–5820 RSC.
- S. E. Skrabalak and R. L. Brutchey, Chem. Mater., 2016, 28, 1003–1005 CrossRef CAS.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
- S. A. Khan and K. F. Jensen, Adv. Mater., 2007, 19, 2556–2560 CrossRef CAS.
- R. M. Maceiczyk, I. G. Lignos and A. J. deMello, Curr. Opin. Chem. Eng., 2015, 8, 29–35 CrossRef.
- B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef CAS PubMed.
- A. M. Nightingale and J. C. deMello, Adv. Mater., 2013, 25, 1813–1821 CrossRef CAS PubMed.
- M. Q. Lu, A. Ozcelik, C. L. Grigsby, Y. H. Zhao, F. Guo, K. W. Leong and T. J. Huang, Nano Today, 2016, 11, 778–792 CrossRef CAS.
- K. N. Ren, J. H. Zhou and H. K. Wu, Acc. Chem. Res., 2013, 46, 2396–2406 CrossRef CAS PubMed.
- L. Shang, Y. Cheng and Y. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS PubMed.
- H. H. Jeong, V. R. Yelleswarapu, S. Yadavali, D. Issadore and D. Lee, Lab Chip, 2015, 15, 4387–4392 RSC.
- K. N. Ren, W. Dai, J. H. Zhou, J. Su and H. K. Wu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8162–8166 CrossRef CAS PubMed.
- A. M. Nightingale, S. H. Krishnadasan, D. Berhanu, X. Niu, C. Drury, R. McIntyre, E. Valsami-Jones and J. C. deMello, Lab Chip, 2011, 11, 1221–1227 RSC.
- N. Bhattacharjee, A. Urrios, S. Kanga and A. Folch, Lab Chip, 2016, 16, 1720–1742 RSC.
- J. M. Zhang, A. A. Aguirre-Pablo, E. Q. Li, U. Buttner and S. T. Thoroddsen, RSC Adv., 2016, 6, 81120–81129 RSC.
- P. Kunal, E. J. Roberts, C. T. Riche, K. Jarvis, N. Malrnstadt, R. L. Brutchey and S. M. Humphrey, Chem. Mater., 2017, 29, 4341–4350 CrossRef CAS.
- E. M. Chan, R. A. Mathies and A. P. Alivisatos, Nano Lett., 2003, 3, 199–201 CrossRef CAS.
- J. B. Edel, R. Fortt, J. C. deMello and A. J. deMello, Chem. Commun., 2002, 1136–1137 RSC.
- S. Krishnadasan, J. Tovilla, R. Vilar, A. J. deMello and J. C. deMello, J. Mater. Chem., 2004, 14, 2655–2660 RSC.
- S. A. Khan, A. Gunther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS PubMed.
- C. G. Lee, M. Uehara, H. Nakamura and H. Maeda, J. Chem. Eng. Jpn., 2008, 41, 644–648 CrossRef CAS.
- M. Schoenitz, L. Grundemann, W. Augustin and S. Scholl, Chem. Commun., 2015, 51, 8213–8228 RSC.
- E. M. Chan, A. P. Alivisatos and R. A. Mathies, J. Am. Chem. Soc., 2005, 127, 13854–13861 CrossRef CAS PubMed.
- P. Zhu and L. Wang, Lab Chip, 2017, 17, 34–75 RSC.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef CAS.
- Y. Shu, P. Jiang, D. W. Pang and Z. L. Zhang, Nanotechnology, 2015, 26, 275701 CrossRef PubMed.
- L. L. Lazarus, C. T. Riche, B. C. Marin, M. Gupta, N. Malmstadt and R. L. Brutchey, ACS Appl. Mater. Interfaces, 2012, 4, 3077–3083 CAS.
- P. H. Hoang, H. Park and D. P. Kim, J. Am. Chem. Soc., 2011, 133, 14765–14770 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Langmuir, 2013, 29, 15719–15725 CrossRef CAS PubMed.
- A. M. Nightingale, T. W. Phillips, J. H. Bannock and J. C. de Mello, Nat. Commun., 2014, 5, 3777 CAS.
- V. Sebastian, S. Basak and K. F. Jensen, AIChE J., 2016, 62, 373–380 CrossRef CAS.
- D. Arndt, J. Thoming and M. Baumer, Chem. Eng. J., 2013, 228, 1083–1091 CrossRef CAS.
- J. Knossalla, S. Mezzavilla and F. Schuth, New J. Chem., 2016, 40, 4361–4366 RSC.
- K. J. Kim, R. P. Oleksak, E. B. Hostetler, D. A. Peterson, P. Chandran, D. M. Schut, B. K. Paul, G. S. Herman and C. H. Chang, Cryst. Growth Des., 2014, 14, 5349–5355 CAS.
- S. K. Yap, W. K. Wong, N. X. Y. Ng and S. A. Khan, Chem. Eng. Sci., 2017, 169, 117–127 CrossRef CAS.
- C. G. Yang, Z. R. Xu, A. P. Lee and J. H. Wang, Lab Chip, 2013, 13, 2815–2820 RSC.
- B. H. Park, D. Kim, J. H. Jung, S. J. Oh, G. Choi, D. C. Lee and T. S. Seo, Sens. Actuators, B, 2015, 209, 927–933 CrossRef CAS.
- T. L. Sounart, P. A. Safier, J. A. Voigt, J. Hoyt, D. R. Tallant, C. M. Matzke and T. A. Michalske, Lab Chip, 2007, 7, 908–915 RSC.
- M. Thiele, A. Knauer, D. Malsch, A. Csaki, T. Henkel, J. M. Kohler and W. Fritzsche, Lab Chip, 2017, 17, 1487–1495 RSC.
- C. N. Baroud, F. Gallaire and R. Dangla, Lab Chip, 2010, 10, 2032–2045 RSC.
- H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts and R. F. Ismagilov, Appl. Phys. Lett., 2003, 83, 4664–4666 CrossRef CAS PubMed.
- L. Frenz, A. El Harrak, M. Pauly, S. Begin-Colin, A. D. Griffiths and J. C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS PubMed.
- G. Yesiloz, M. S. Boybay and C. L. Ren, Anal. Chem., 2017, 89, 1978–1984 CrossRef CAS PubMed.
- Y. H. Kim, L. Zhang, T. Yu, M. S. Jin, D. Qin and Y. N. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- M. Faustini, J. Kim, G. Y. Jeong, J. Y. Kim, H. R. Moon, W. S. Ahn and D. P. Kim, J. Am. Chem. Soc., 2013, 135, 14619–14626 CrossRef CAS PubMed.
- H. Song, H.-W. Li, M. S. Munson, T. G. Van Ha and R. F. Ismagilov, Anal. Chem., 2006, 78, 4839–4849 CrossRef CAS PubMed.
- S. Yao, Y. Shu, Y. J. Yang, X. Yu, D. W. Pang and Z. L. Zhang, Chem. Commun., 2013, 49, 7114–7116 RSC.
- A. Yashina, I. Lignos, S. Stavrakis, J. Choo and A. J. deMello, J. Mater. Chem. C, 2016, 4, 6401–6408 RSC.
- D. Koziej, C. Floryan, R. A. Sperling, A. J. Ehrlicher, D. Issadore, R. Westervelt and D. A. Weitz, Nanoscale, 2013, 5, 5468–5475 RSC.
- Z. Liu, K. Okabe, C. Anand, Y. Yonezawa, J. Zhu, H. Yamada, A. Endo, Y. Yanaba, T. Yoshikawa, K. Ohara, T. Okubo and T. Wakihara, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14267–14271 CrossRef CAS PubMed.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef CAS.
- V. Sebastian and K. F. Jensen, Nanoscale, 2016, 8, 15288–15295 RSC.
- J. Yue, F. H. Falke, J. C. Schouten and T. A. Nijhuis, Lab Chip, 2013, 13, 4855–4863 RSC.
- D. Koziej, Chem. Mater., 2016, 28, 2478–2490 CrossRef CAS.
- A. Ghazal, J. P. Lafleur, K. Mortensen, J. P. Kutter, L. Arleth and G. V. Jensen, Lab Chip, 2016, 16, 4263–4295 RSC.
- J. Atencia and D. J. Beebe, Nature, 2005, 437, 648–655 CrossRef CAS PubMed.
- L. Yang, M. J. Nieves-Remacha and K. F. Jensen, Chem. Eng. Sci., 2017, 169, 106–116 CrossRef CAS.
- M. Thiele, J. Z. E. Soh, A. Knauer, D. Malsch, O. Stranik, R. Muller, A. Csaki, T. Henkel, J. M. Kohler and W. Fritzsche, Chem. Eng. J., 2016, 288, 432–440 CrossRef CAS.
- A. Knauer, A. Csaki, F. Moller, C. Huhn, W. Fritzsche and J. M. Kohler, J. Phys. Chem. C, 2012, 116, 9251–9258 CAS.
- A. Knauer, A. Eisenhardt, S. Krischok and J. M. Koehler, Nanoscale, 2014, 6, 5230–5238 RSC.
- J. S. Santana, K. M. Koczkur and S. E. Skrabalak, Langmuir, 2017, 33, 6054–6061 CrossRef CAS PubMed.
- V. S. Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef PubMed.
- S. A. Khan and S. Duraiswamy, Lab Chip, 2012, 12, 1807–1812 RSC.
- V. Sebastian, C. D. Smith and K. F. Jensen, Nanoscale, 2016, 8, 7534–7543 RSC.
- K. S. Elvira, X. C. i. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- A. P. LaGrow, T. M. D. Besong, N. M. AlYami, K. Katsiev, D. H. Anjum, A. Abdelkader, P. Costa, V. M. Burlakov, A. Goriely and O. M. Bakr, Chem. Commun., 2017, 53, 2495–2498 RSC.
- D. T. Zhang, F. X. Wu, M. H. Peng, X. Y. Wang, D. G. Xia and G. S. Guo, J. Am. Chem. Soc., 2015, 137, 6263–6269 CrossRef CAS PubMed.
- T. Akdas, M. Haderlein, J. Walter, B. A. Zubiri, E. Spiecker and W. Peukert, RSC Adv., 2017, 7, 10057–10063 RSC.
- E. Y. Erdem, J. C. Cheng, F. M. Doyle and A. P. Pisano, Small, 2014, 10, 1076–1080 CrossRef CAS PubMed.
- J. Pan, A. O. El-Ballouli, L. Rollny, O. Voznyy, V. M. Burlakov, A. Goriely, E. H. Sargent and O. M. Bakr, ACS Nano, 2013, 7, 10158–10166 CrossRef CAS PubMed.
- J. Baek, P. M. Allen, M. G. Bawendi and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 627–630 CrossRef CAS PubMed.
- R. M. Maceiczyk, L. Bezinge and A. J. deMello, React. Chem. Eng., 2016, 1, 261–271 CAS.
- H. Yang, W. Luan, Z. Wan, S.-T. Tu, W. K. Yuan and Z. M. Wang, Cryst. Growth Des., 2009, 9, 4807–4813 CAS.
- S. Z. Tian, M. Fu, W. Hoheisel and L. Mleczko, Chem. Eng. J., 2016, 289, 365–373 CrossRef CAS.
- A. Abou-Hassan, R. Bazzi and V. Cabuil, Angew. Chem., Int. Ed., 2009, 48, 7180–7183 CrossRef CAS PubMed.
- L. Gomez, V. Sebastian, S. Irusta, A. Ibarra, M. Arruebo and J. Santamaria, Lab Chip, 2014, 14, 325–332 RSC.
- L. Uson, V. Sebastian, M. Arruebo and J. Santamaria, Chem. Eng. J., 2016, 285, 286–292 CrossRef CAS.
- S. R. Jambovane, S. K. Nune, R. T. Kelly, B. P. McGrail, Z. M. Wang, M. I. Nandasiri, S. Katipamula, C. Trader and H. T. Schaef, Sci. Rep., 2016, 6, 36657 CrossRef CAS PubMed.
- Y. Shen, M. Y. Gee and A. B. Greytak, Chem. Commun., 2017, 53, 827–841 RSC.
- Y. Shen, N. Weeranoppanant, L. Xi, Y. Chen, M. Lusardi, J. Imbrogno, M. G. Bawendi and K. F. Jensen, Nanoscale, 2017, 9, 7703–7707 RSC.
- F. C. Cabrera, A. F. Melo, J. C. de Souza, A. E. Job and F. N. Crespilho, Lab Chip, 2015, 15, 1835–1841 RSC.
- D. Ferraro, Y. Lin, B. Teste, D. Talbot, L. Malaquin, S. Descroix and A. Abou-Hassan, Chem. Commun., 2015, 51, 16904–16907 RSC.
- I. Lignos, L. Protesescu, S. Stavrakis, L. Piveteau, M. J. Speirs, M. A. Loi, M. V. Kovalenko and A. J. deMello, Chem. Mater., 2014, 26, 2975–2982 CrossRef CAS.
- I. Lignos, S. Stavrakis, A. Kilaj and A. J. deMello, Small, 2015, 11, 4009–4017 CrossRef CAS PubMed.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. deMello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef CAS PubMed.
- S. V. Kershaw, N. M. Abdelazim, Y. H. Zhao, A. S. Susha, O. Zhovtiuk, W. Y. Teoh and A. L. Rogach, Chem. Mater., 2017, 29, 2756–2768 CrossRef CAS.
- M. Abolhasani, C. W. Coley, L. S. Xie, O. Chen, M. G. Bawendi and K. F. Jensen, Chem. Mater., 2015, 27, 6131–6138 CrossRef CAS.
- E. M. Chan, M. A. Marcus, S. Fakra, M. ElNaggar, R. A. Mathies and A. P. Alivisatos, J. Phys. Chem. A, 2007, 111, 12210–12215 CrossRef CAS PubMed.
- J. E. Q. Quinsaat, A. Testino, S. Pin, T. Huthwelke, F. A. Nuesch, P. Bowen, H. Hofmann, C. Ludwig and D. M. Opris, J. Phys. Chem. C, 2014, 118, 11093–11103 CAS.
- J. Polte, R. Erler, A. F. Thunemann, S. Sokolov, T. T. Ahner, K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano, 2010, 4, 1076–1082 CrossRef CAS PubMed.
- J. Watt, B. G. Hance, R. S. Anderson and D. L. Huber, Chem. Mater., 2015, 27, 6442–6449 CrossRef CAS.
- A. M. Karim, N. Al Hasan, S. Ivanov, S. Siefert, R. T. Kelly, N. G. Hallfors, A. Benavidez, L. Kovarik, A. Jenkins, R. E. Winans and A. K. Datye, J. Phys. Chem. C, 2015, 119, 13257–13267 Search PubMed.
- N. Pham, D. Radajewski, A. Round, M. Brennich, P. Pernot, B. Biscans, F. Bonneté and S. Teychené, Anal. Chem., 2017, 89, 2282–2287 CrossRef CAS PubMed.
- K. M. Tibbetts, X. J. Feng and H. Rabitz, Phys. Chem. Chem. Phys., 2017, 19, 4266–4287 RSC.
- S. Krishnadasan, R. Brown, A. J. deMello and J. C. deMello, Lab Chip, 2007, 7, 1434–1441 RSC.
- R. M. Maceiczyk and A. J. deMello, J. Phys. Chem. C, 2014, 118, 20026–20033 CAS.
- M. Abolhasani, M. Singh, E. Kumacheva and A. Gunther, Lab Chip, 2012, 12, 4787–4795 RSC.
- K. Watanabe, Y. Orimoto, K. Nagano, K. Yamashita, M. Uehara, H. Nakamura, T. Furuya and H. Maeda, Chem. Eng. Sci., 2012, 75, 292–297 CrossRef CAS.
- B. Swain, M. H. Hong, L. Kang, B. S. Kim, N. H. Kim and C. G. Lee, Chem. Eng. J., 2017, 308, 311–321 CrossRef CAS.
- L. Zhang and Y. N. Xia, Adv. Mater., 2014, 26, 2600–2606 CrossRef CAS PubMed.
- A. M. Nightingale, J. H. Bannock, S. H. Krishnadasan, F. T. F. O'Mahony, S. A. Haque, J. Sloan, C. Drury, R. McIntyre and J. C. deMello, J. Mater. Chem. A, 2013, 1, 4067–4076 CAS.
- M. Hashimoto, S. S. Shevkoplyas, B. Zasońska, T. Szymborski, P. Garstecki and G. M. Whitesides, Small, 2008, 4, 1795–1805 CrossRef CAS PubMed.
- C. T. Riche, E. J. Roberts, M. Gupta, R. L. Brutchey and N. Malmstadt, Nat. Commun., 2016, 7, 10780 CrossRef CAS PubMed.
- D. Conchouso, G. McKerricher, A. Arevalo, D. Castro, A. Shamim and I. G. Foulds, Lab Chip, 2016, 16, 3210–3219 RSC.
- S. E. Lohse, J. R. Eller, S. T. Sivapalan, M. R. Plews and C. J. Murphy, ACS Nano, 2013, 7, 4135–4150 CrossRef CAS PubMed.
- E. J. Roberts, S. E. Habas, L. Wang, D. A. Ruddy, E. A. White, F. G. Baddour, M. B. Griffin, J. A. Schaidle, N. Malmstadt and R. L. Brutchey, ACS Sustainable Chem. Eng., 2017, 5, 632–639 CrossRef CAS.
- T. L. Cheung, L. Y. Hong, N. X. Rao, C. B. Yang, L. B. Wang, W. J. Lai, P. H. J. Chong, W. C. Law and K. T. Yong, Nanoscale, 2016, 8, 6609–6622 RSC.
- M. S. Naughton, V. Kumar, Y. Bonita, K. Deshpande and P. J. A. Kenis, Nanoscale, 2015, 7, 15895–15903 RSC.
- A. Shavel, D. Cadavid, M. Ibanez, A. Carrete and A. Cabot, J. Am. Chem. Soc., 2012, 134, 1438–1441 CrossRef CAS PubMed.
- Y. H. Li, D. G. Yamane, S. N. Li, S. Biswas, R. K. Reddy, J. S. Goettert, K. Nandakumar and C. Kumar, Chem. Eng. J., 2013, 217, 447–459 CrossRef CAS.
- L. Zhang, G. Niu, N. Lu, J. Wang, L. Tong, L. Wang, M. J. Kim and Y. Xia, Nano Lett., 2014, 14, 6626–6631 CrossRef CAS PubMed.
- W. K. Wong, S. K. Yap, Y. C. Lim, F. Pelletier, E. C. Corbos and S. A. Khan, React. Chem. Eng., 2017, 2, 636–641 CAS.
- H. Wang, G. Niu, M. Zhou, X. Wang, J. Park, S. Bao, M. Chi, Z. Cai and Y. Xia, ChemCatChem, 2016, 8, 1658–1664 CrossRef CAS.
- G. D. Niu, M. Zhou, X. Yang, J. Park, N. Lu, J. G. Wang, M. J. Kim, L. D. Wang and Y. N. Xia, Nano Lett., 2016, 16, 3850–3857 CrossRef CAS PubMed.
- A. Larrea, V. Sebastian, A. Ibarra, M. Arruebo and J. Santamaria, Chem. Mater., 2015, 27, 4254–4260 CrossRef CAS PubMed.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Chem. Rev., 2016, 116, 2141–2169 CrossRef CAS PubMed.
- H. Z. Wang, H. Nakamura, M. Uehara, M. Miyazaki and H. Maeda, Chem. Commun., 2002, 1462–1463 RSC.
- B. G. Zukas and N. R. Gupta, Ind. Eng. Chem. Res., 2017, 56, 7184–7191 CrossRef CAS.
- S. Sachdev, R. Maugi, J. D. Woolley, C. A. Kirk, Z. Zhou, S. D. Christie and M. Platt, Langmuir, 2017, 33, 5464–5472 CrossRef CAS PubMed.
- L. A. Bawazer, C. S. McNally, C. J. Empson, W. J. Marchant, T. P. Comyn, X. Niu, S. Cho, M. J. McPherson, B. P. Binks, A. deMello and F. C. Meldrum, Sci. Adv., 2016, 2, e1600567 Search PubMed.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Nano Lett., 2014, 14, 4189–4194 CrossRef CAS PubMed.
- N. Hassan, A. Stocco and A. Abou-Hassan, J. Phys. Chem. C, 2015, 119, 10758–10765 CAS.
- L. S. Xie, Y. Shen, D. Franke, V. Sebastian, M. G. Bawendi and K. F. Jensen, J. Am. Chem. Soc., 2016, 138, 13469–13472 CrossRef CAS PubMed.
- M. S. Jie, S. F. Mao, H. F. Li and J. M. Lin, Chin. Chem. Lett., 2017, 28, 1625–1630 CrossRef CAS.
- R. Maceiczyk, H. Shimizu, D. Muller, T. Kitamori and A. deMello, Anal. Chem., 2017, 89, 1994–1999 CrossRef CAS PubMed.
- A. R. Salmon, R. Esteban, R. W. Taylor, J. T. Hugall, C. A. Smith, G. Whyte, O. A. Scherman, J. Aizpurua, C. Abell and J. J. Baumberg, Small, 2016, 12, 1788–1796 CrossRef CAS PubMed.
- K. P. Nichols, R. R. Pompano, L. Li, A. V. Gelis and R. F. Ismagilov, J. Am. Chem. Soc., 2011, 133, 15721–15729 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |