
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling precision manufacturing of active pharmaceutical ingredients: workflow for seeded cooling continuous crystallisations†
Cameron J.
Brown
 a,
Thomas
McGlone
a,
Stephanie
Yerdelen
a,
Vijay
Srirambhatla
a,
Fraser
Mabbott
a,
Rajesh
Gurung
a,
Maria
L. Briuglia
a,
Bilal
Ahmed
a,
Hector
Polyzois
b,
John
McGinty
a,
Francesca
Perciballi
a,
Dimitris
Fysikopoulos
c,
Pól
MacFhionnghaile
a,
Humera
Siddique
a,
Vishal
Raval
a,
Tomás S.
Harrington
d,
Antony D.
Vassileiou
a,
Murray
Robertson
a,
Elke
Prasad
a,
Andrea
Johnston
a,
Blair
Johnston
a,
Alison
Nordon
a,
Jagjit S.
Srai
d,
Gavin
Halbert
a,
Joop H.
ter Horst
a,
Chris J.
Price
a,
Chris D.
Rielly
c,
Jan
Sefcik
a and
Alastair J.
Florence
*a
a,
Thomas
McGlone
a,
Stephanie
Yerdelen
a,
Vijay
Srirambhatla
a,
Fraser
Mabbott
a,
Rajesh
Gurung
a,
Maria
L. Briuglia
a,
Bilal
Ahmed
a,
Hector
Polyzois
b,
John
McGinty
a,
Francesca
Perciballi
a,
Dimitris
Fysikopoulos
c,
Pól
MacFhionnghaile
a,
Humera
Siddique
a,
Vishal
Raval
a,
Tomás S.
Harrington
d,
Antony D.
Vassileiou
a,
Murray
Robertson
a,
Elke
Prasad
a,
Andrea
Johnston
a,
Blair
Johnston
a,
Alison
Nordon
a,
Jagjit S.
Srai
d,
Gavin
Halbert
a,
Joop H.
ter Horst
a,
Chris J.
Price
a,
Chris D.
Rielly
c,
Jan
Sefcik
a and
Alastair J.
Florence
*a
aEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, University of Strathclyde, Glasgow, UK. E-mail: alastair.florence@strath.ac.uk; Tel: +44 (0)141 485 4877
bNational Physical Laboratory (NPL) Scotland, University of Strathclyde, Glasgow, UK
cEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, Loughborough University, Loughborough, UK
dEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, University of Cambridge, Cambridge, UK
First published on 20th February 2018
Abstract
Continuous manufacturing is widely used for the production of commodity products. Currently, it is attracting increasing interest from the pharmaceutical industry and regulatory agencies as a means to provide a consistent supply of medicines. Crystallisation is a key operation in the isolation of the majority of pharmaceuticals and has been demonstrated in a continuous manner on a number of compounds using a range of processing technologies and scales. Whilst basic design principles for crystallisations and continuous processes are known, applying these in the context of rapid pharmaceutical process development with the associated constraints of speed to market and limited material availability is challenging. A systematic approach for continuous crystallisation process design is required to avoid the risk that decisions made on one aspect of the process conspire to make a later development step or steps, either for crystallisation or another unit operation, more difficult. In response to this industry challenge, an innovative system-wide approach to decision making has been developed to support rapid, systematic, and efficient continuous seeded cooling crystallisation process design. For continuous crystallisation, the goal is to develop and operate a robust, consistent process with tight control of particle attributes. Here, an innovative system-based workflow is presented that addresses this challenge. The aim, methodology, key decisions and output at each at stage are defined and a case study is presented demonstrating the successful application of the workflow for the rapid design of processes to produce kilo quantities of product with distinct, specified attributes suited to the pharmaceutical development environment. This work concludes with a vision for future applications of workflows in continuous manufacturing development to achieve rapid performance based design of pharmaceuticals.
Design, System, ApplicationThe design and operation of crystallisation processes is a complex subject when considering the range of phenomena such as nucleation, breakage/attrition, fouling, agglomeration, etc., that can occur in addition to crystal growth. This task is further complicated by limitations on process development time and material quantities. Here we present a systematic, rapid and efficient workflow approach to the design and operation of continuous crystallisation of active pharmaceutical ingredients (APIs). To achieve this the workflow contains: clearly described tasks, appropriate use of automation, minimisation of material usage and resource through experimental design approaches, estimated task time scales and pre-empting of common issues. Through this approach data driven decisions are made considering their system wide implication, allowing for API particles of specified attributes to be manufactured by first intent. This standardised approach provides the first steps towards pathways for predictive design of API manufacturing processes. |
1. Introduction
Crystallisation is a complex, multi-phase unit operation used in a wide range of manufacturing industries to achieve separation and purification of products.1,2 For the pharmaceutical industry, active pharmaceutical ingredient (API) crystallisation may be regarded as the first step in the formulation process with molecules stabilised within the crystal lattice throughout the subsequent processing steps until the crystal dissolves upon administration to the patient allowing the molecular form of the drug to be absorbed. As a consequence, crystallisation is a critical process step. The pharmaceutical industry is also placing increasing demands on crystallisation, for example, as drug structures become more complex they can be more challenging to crystallise; also, advanced formulations require tighter control of API particle attributes.3 Industry requires controlled crystallisations that consistently deliver appropriate chemical and solid phase purity coupled with a crystal size distribution and habit suitable for isolation and downstream processing. These multiple objectives must be met whilst attaining the highest possible yield at the lowest economic and environmental cost. The value of the global pharmaceutical industry exceeds one trillion dollars per year4 and the cost of drug substance manufacture represents around 150 billion dollars based on a cost of goods of 20–25%5 and the cost of drug substance representing around 65% of the cost of goods.6,7 Consequently, even modest percentage improvements in product consistency, and/or process yield can lead to significant cost savings.Crystallisation processes may be classified according to how supersaturation is generated;8 reactive, evaporative, anti-solvent and cooling crystallisation can be applied depending on the nature of the feed stream from the upstream process, be it a batch or continuous synthesis or workup step. For most drug substances, the crystal size distribution (CSD) is a critical quality attribute (CQA) impacting the drug product performance in the patient either directly or through its impact in subsequent processing steps. Consistent achievement of the required specification for each CQA is a prerequisite of the material being approved for clinical use. However, multiple factors including primary and secondary nucleation, growth, agglomeration, attrition and crystal breakage and encrustation can all influence the capability of a process to meet a specified CQA, e.g. particle size. Conventional approaches for obtaining consistent crystals with CQAs can struggle with batch-to-batch variability,9,10 particularly at manufacturing scale. In addition, batch configurations have associated scale-up challenges11,12 when moving from development to production.
Pharmaceutical regulatory agencies are driving the industry to adopt quality-by-design (QbD) methods13,14 to enhance quality and reduce variability. Process cost reductions and maximising operation efficiency are key drivers for change and continuous manufacturing (CM) is widely used to address these challenges in the production of commodity products. Recently CM is attracting increasing interest from pharmaceutical industry and regulatory agencies15,16 as a means to provide a reliable supply of medicines9,17 consistent with QbD. Continuous crystallisation has been demonstrated on a number of compounds18–21 using a range of processing technologies and scales that includes single and multiple stage continuous stirred reactors22–24 and near plug flow reactors, such as oscillatory baffled crystallisers,25 segmented flow26 and static mixers.18 In addition to the general benefits of continuous processing,9,24,27–29 it offers a means to enhance control of the physical properties of the crystalline product.30,31 Scale-up or scale-out can also be achieved with less effort and risk.20
Whilst basic design principles for crystallisations and for continuous processes are known, applying these in the context of a pharmaceutical development programme with the associated constraints of limited time and material availability is challenging. This is especially problematic when complex phenomena such as secondary nucleation, attrition, fouling or agglomeration significantly complicate the process. These effects frequently occur and can be highly unpredictable. Hence, the motivation for this work is to develop a rapid and efficient means of specifying process parameters, which deliver consistent API physical properties, defined by the CQAs, to optimise both product performance and processability.
Workflows for process development have been reported and examples are given in Table 1. However, here a novel, systematic, science-based process design workflow for the robust design of seeded cooling crystallisation for any API that identifies process parameters and pre-empts common late-stage failures modes is developed. Key considerations for the approach include:
| Workflow | Goal |
|---|---|
| 96-Well plates to define a set of scaleable crystallisation conditions32 | Conditions to produce 100 g of material for preclinical testing |
| High-throughput nanolitre scale workflow33 | Crystallisation, imaging, automated storage and structure determination of proteins |
| Experimental and modelling aspects of solid–liquid equilibrium, impurity inclusion, washing and deliquoring34 | Managing impurities in crystallisation processes |
| Preparing the first crystalline solids and solvent screening. Target particle size, purity, crystallisation vessel, particle morphology, filtration behaviour and scale-up35 | Development of new crystallisation processes |
| Experimental procedures for solubility, nucleation threshold and rate, growth rate and agglomeration or breakage36 | Batch or continuous industrial crystalliser design |
| Hierarchical procedure covering product and process engineering considering design specifications, variables and domain knowledge37 | Predictive models for solution crystallisations |
| Multiple dimensional populations, polymorphic transformations and hydrodynamics38 | Bottom-up and top-down modelling framework for batch cooling crystallisations |
| Estimation for a continuous manufacturing plant flowsheet from which technical and economic evaluations can be performed39 | APIs suitability for continuous processing |
| Initial screen stage for candidates unsuitable for continuous processing. Extended evaluation of continuous and hybrid configurations. Execution of the chosen configuration40 | Converting fine chemical processes from batch to continuous |
1. Clearly defined experimental and analytical tasks with a detailed approach to:
a. allow for transparent and systematic data driven decisions.
b. enable facile adaption as the fundamental knowledge or equipment base develops.
2. Appropriate use of lab automation and automated data processing to allow:
a. the process developer to focus on the most value adding steps, e.g. data interpretation and process selection;
b. full exploitation of the advantages of systematic and accurate data recording, archival and retrieval associated with automated platforms and digital infrastructure such as electronic laboratory notebooks and machine learning tools.
3. Minimise material usage and resource whilst maximising process understanding via design of experiment (DoE) and/or model-based approaches.
4. Allow the realistic estimations of timescales;
5. Pre-empt commonly encountered issues and embed their solutions to development tasks and decisions.
Here we present the outputs of a large multidisciplinary, industry demand-led research project that embraces these aspects to enable a ‘right first time’, science based approach to seeded cooling crystallisation design. The next section gives a description of the workflow and each of its stages. For each stage the aim, methodology, key decisions, output and challenges are described. Section 3 exemplifies the workflow using paracetamol (acetaminophen) as a model drug substance. Section 4 discusses broader applications of the workflow into areas of meta-analysis, business case decisions, formulation and drug product design. Finally, this article concludes with its vision of future applications of workflows in continuous manufacturing development.
2. Workflow aim and description
Fig. 1 and Table 2 detail the general steps in the workflow developed here, including the various paths between stages based on the outcome at specific decision points. This workflow provides a framework for designing a crystallisation process but by no means covers all eventualities. It is not designed to be followed rigorously but instead emphasises a way of thinking which leads towards holistic process design. In this workflow, the following assumptions are made: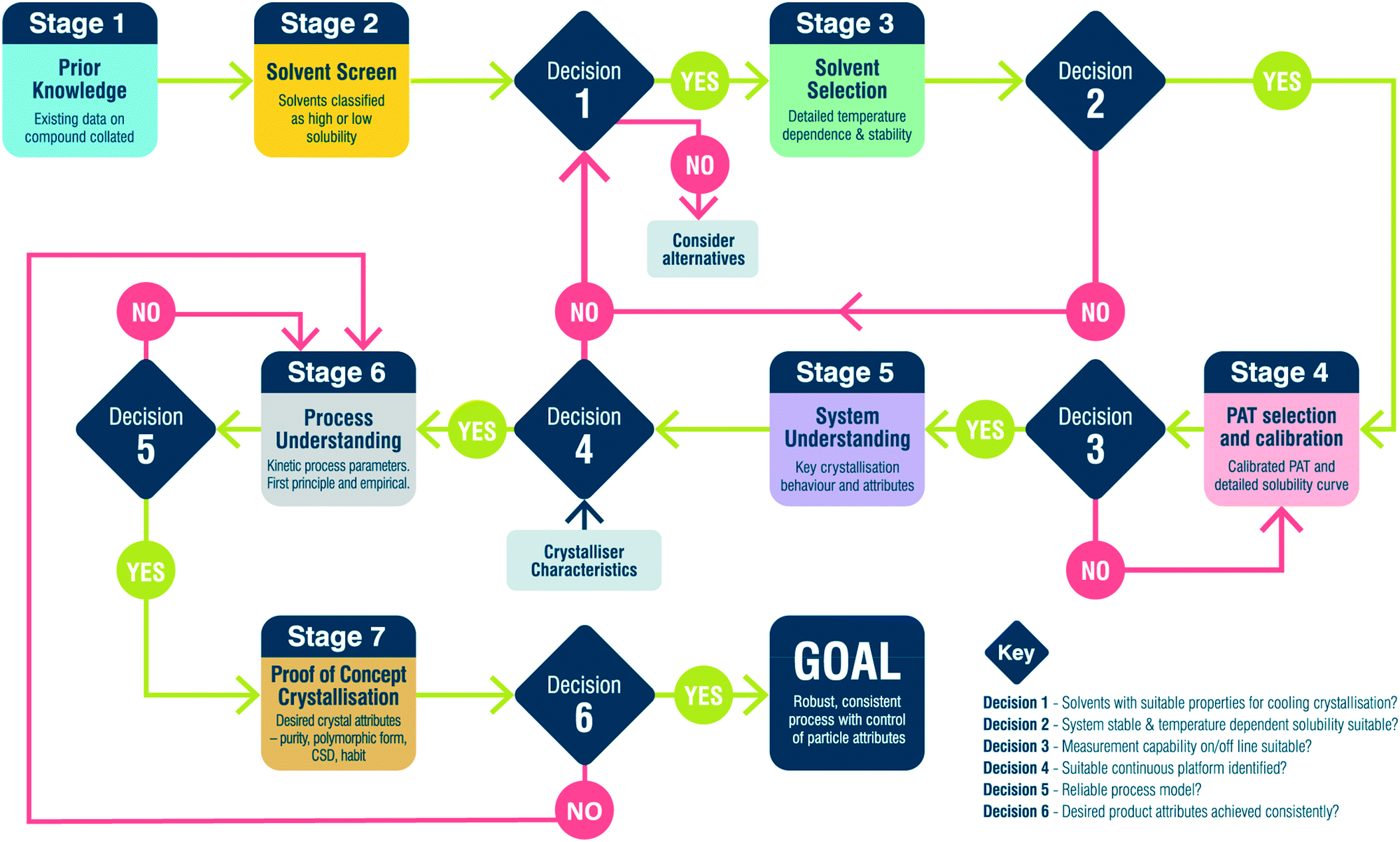 | ||
| Fig. 1 Workflow for seeded continuous crystallisation. | ||
| Description | Output | Equipment/tools | |
|---|---|---|---|
| Stage 1: prior knowledge | Collate prior knowledge of API | Existing data on API | Form screen, synthesis development, literature, XRD, DSC, NMR |
| Stage 2: solvent screen | Assess solubility in a library of process appropriate solvents | Solvents broadly classified | 1.5 mL vials, turbidity |
| Decision 1 | Do any solvents exhibit a temperature dependent solubility? | ||
| No: consider alternative isolation method (e.g. anti-solvent crystallisation) | |||
| Yes: move solvents with temperature dependence to stage 3 | |||
| Stage 3: solvent selection | Detailed solubility measurement and API stability in solution at elevated temperature | Solvent solubilities and selection criteria values | 5 mL vials, LS-MS, DSC, XRD |
| Decision 2 | Do any solvents meet selection criteria? | ||
| No: revisit output of decision 1 | |||
| Yes: move single solvent to stage 4 | |||
| Stage 4: PAT selection and calibration | Assess PAT suitability, calibrate and confirm solubility | Calibrated PAT method and detailed solubility curve | 100+ mL vessel, UV-Vis, FTIR, NIR, Raman |
| Decision 3 | Does the PAT calibration meet the required accuracy? | ||
| No: revisit PAT selection and calibration | |||
| Yes: move to stage 4 | |||
| Stage 5: system understanding | Assess relative importance of different processes (growth, nucleation, agglomeration etc.) | Understanding of API preferred crystallisation behaviour | Assorted test platforms and PAT |
| Decision 4 | Is the observed crystallisation behaviour compatible with continuous platforms? | ||
| No: revisit solvent screen and look at other solvents | |||
| Yes: select platform and move to stage 6 | |||
| Stage 6: process understanding | DoE and parameter estimation in specific chosen platform | Model of process | 100+ mL vessel, population balance, DoE, PAT |
| Decision 5 | Is the process model reliable? | ||
| No: revisit process understanding and perform further experiments | |||
| Yes: design continuous crystallisation and move to stage 7 | |||
| Stage 7: proof of concept crystallisation | Demonstrate continuous operation with design from decision 5 | Desired particle attributes – solid state, PSD, habit | MSMPR cascade, PFR and PAT |
| Decision 6 | Does the product match the desired particle attributes? | ||
| No: revisit process understanding and redesign crystallisation | |||
| Yes: workflow complete | |||
• Solid-state form predetermined. This workflow does not address the discovery and identification of different solid-state forms (polymorphs, solvates, hydrates etc.) but rather focuses on the production of a predetermined form. Checks are built into the workflow, particularly during solvent screening and selection to ensure the desired form is produced.
• Feed composition is constant. Impurities are known to have a significant impact on crystallisation processes and can influence the kinetics of nucleation and/or growth, leading to a change in particle morphology. This workflow assumes that the level and identity of impurities in the feed are constant and therefore does not discriminate any effects that changes in specific impurities may have on the crystallisation process. Consequently, material used for process development via this workflow must be consistent in terms of impurity profile and ideally from a single synthesis lot/batch. Recycling of API is possible however, checks must be included for increasing levels of known and new degradation products.
• Initial growth surface managed by seeding. Many methods are available for the generation of nuclei under supersaturation including, cooling, anti-solvent addition or through external fields, e.g. high shear or ultrasound. Whilst integrated approaches for manipulation and control of primary nucleation are of considerable interest, they present a significant challenge for robust, scalable control. Seeding is currently the industry norm providing a robust method for initiating crystallisation, controlling solid form and mitigation of fouling.
• Seed generation carried out through an auxiliary workflow. Assumption 3 dictates the need for seed crystals and it would be prudent to consider how those seed crystals are generated. This could be through, for example, wet milling, dry milling, micronisation or anti-solvent crystallisation. The generation of this seed material is outwith the scope of the present workflow.
2.1. Stage 1: prior knowledge
Stage 1 of the proposed workflow has three aims. Firstly, it aims to collate all available information on the compound to be taken through the workflow. This information can come from a range of sources, including, but not limited to: molecular design, synthetic route development, solid form screen, work-up development, previous published literature, analytical reports, physicochemical molecular properties and structural features of the API. Pertinent information at this stage will inform selection of analytical methods, indicate potential chemical incompatibilities and inform selection or exclusion of solvents for further investigation in stages 2 and 3. This may include solid-state landscape, physical form solubility and relative thermodynamic stabilities, solvate formation propensity, reactivity and impurity profiles. Data mining of internal and external databases, such as the Cambridge Structural Database,41 with structural or relevant process data for related molecules can also prove useful for prediction of crystallisation behaviour.42 Secondly, at this stage it is useful to collate reference characterisation data including XRPD, FTIR, Raman, DSC, NMR, LC-MS, etc. These will be used throughout the workflow to ensure the correct form and purity are achieved. Additionally, melting point or heat of fusion data from DSC can also prove useful for estimating an API's solubility.43 Finally, this stage sets the product CQAs, specified by drug product requirements, as the final goal for the workflow. These CQAs are set by the anticipated dose, scale of use, dose form and formulation.2.2. Stage 2: solvent screen and decision 1
This stage is concerned with guiding the selection of a suitable solvent for cooling crystallisation (Fig. 1). The aim is to take a broad library of solvents and rapidly identify only solvents that show desirable solubility to carry forward into stage 3. Here, this has been achieved by performing a dissolution screen of the compound in solvents from the library (details in ESI†). The choice of solvents for this screen can be narrowed by considering the information collated in stage 1 and neglecting any solvents that have shown any previous indication of an undesired polymorphic form, solvate formation or reactivity with the solute for example. Further restrictions may be placed on the solvent library to only include solvents listed in the International Conference on Harmonisation (ICH) classification for residual solvents45 as class 2 (solvents to be limited) or class 3 (solvents with low toxic potential). Additional restrictions on the solvent library could include: boiling point, viscosity, environmental impact46 and cost depending on relevant project constraints.Solubility selection criteria were based on the solubility magnitude and the solubility temperature dependence, both critical selection parameters for a cooling crystallisation. Specific solvent selection criteria chosen here are shown in Table 3. In decision 1 (Fig. 1), if suitable solvents are identified these are taken to stage 3. However, where no solvents demonstrate a suitable temperature dependence, cooling crystallisation in a single solvent system is unlikely to be feasible.
| Temperature | Solubility | |
|---|---|---|
| Low temperature threshold | 20 °C | <50 g L−1 |
| High temperature threshold | Solvent boiling point minus 10 °C | >50 g L−1 |
2.3. Stage 3: solvent selection and decision 2
Stage 3 (Fig. 1) is geared towards further narrowing the solvents selected using the selection criteria (Table 4) to inform selection of one or more candidate solvents to carry forward to crystallisation assessment in stage 4. Data from these experiments will also be used to extract information concerning the metastable zone width, agglomeration propensity and likelihood of fouling. This is done by preparing solutions of known concentration, C, and subjecting them to temperature cycles to determine the point of dissolution. Approximate solubility curves can then be produced by applying suitable fits (exponential or 2nd order polynomial47) to these points. From these curves, operating temperature range, yield and product solid fraction can be estimated to guide selection to the most appropriate choice. Here yield is defined as the mass theoretical yield, Yt, calculated from mass solubility data at initial, T0, and final temperature, Tf:48,49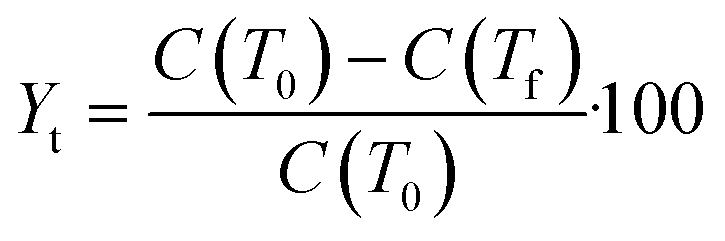 | (1) |
| Parameter | CPP | CQA | Reasoning |
|---|---|---|---|
| Yield, Yt | Feed concentration, final temperature | Yield < 90% likely to be economically unfeasible without recycle. Also dictates limits on saturated and final T values below | |
| Feed saturated temperature | Feed concentration | Solid state, purity, PSD | Higher operating T may require specific heat transfer fluids and extensive lagging to minimise heat loss. Higher T may also accelerate degradation |
| Final temperature | Final temperature | Solid state, purity, PSD | Low final T may require costly refrigerant systems and increase the complexity of washing and drying during isolation |
| Product solid mass fraction | Feed concentration, final temperature | PSD | Solid loadings (at final T) >25 w/w% are a challenge to maintain in a well-mixed state. Low loadings lead to handling excessive solvent volumes. Also affects upper boundary of saturated T |
| Agglomeration | Cooling rate, seed loading, seed size | Purity, PSD | Agglomerates complicate growth kinetics and reduce washing and drying efficiency downstream, potentially lowering purity |
| Fouling | Cooling rate | PSD | Fouling can prevent the system from reaching steady state, impacts on heat transfer surfaces, measurement probe windows, CQAs and potentially cause blockages |
| Solid state | Feed concentration, final temperature | Solid state | Formation of alternative forms is usually undesirable |
| Particle shape/crystal morphology | Feed concentration, final temperature, cooling rate | Purity, PSD | High aspect ratio particles can lead to poor filtration and bulk flow challenges downstream |
A chemical stability test of the compound in each solvent at an elevated temperature is also essential here as a further selection criterion. Similarly, mother liquors from the crystallisation portion of the temperature cycle should be analysed by LC, GC or NMR to indicate impurity rejection. Further considerations to solvent selection can also be incorporated here to aid selection, such as health and safety aspects and cost. A summary of the selection criteria is shown in Table 4 with an explanation of their relevance, i.e. which CQAs are influenced by each criteria and which critical process parameters (CPPs) control the criteria.
At decision point 2, any solvent that meets all criteria is carried on to stage 4. However, as with decision 1, if a single solvent does not meet the criteria set out then the short listed solvents from stage 2 should be re-evaluated to consider solvent mixtures. Further failure to meet criteria directs that an alternative crystallisation mode would be preferred, e.g. anti-solvent or evaporative.
1. There is often a necessary trade-off between resources spent and accuracy achieved. In this work, automated platforms for solubility measurement are exploited to minimise experimental time whilst maximising information obtained. Since a number of assumptions are made using automated platforms, validation of solubility curves is carried out in stage 4.
2. If solubility of a metastable form is to be investigated, there are additional challenges to be addressed, such as preventing form transitions within the time necessary for measurement.47,50,51
3. As crystallisation is predominantly used as a purification technique, the solubility of likely impurities from synthesis would be useful to know to allow for solvent selection based on impurity rejection. However, it would be time consuming to measure solubility for all relevant impurities in all solvents, see points 1 and 4.
4. Predictive and computational methods could be exploited at this stage to determine solubility without performing any experiments. There are a range of methods currently available, including COSMOtherm,52–54 SAFT,55 UNIQUAC,56 UNIFAC.57 In the context of pharmaceutical manufacturing, they do not currently provide sufficiently accurate quantitative predictions for a wide range of compounds. However, they are used to give early qualitative rankings of solvent/solute solubility.
5. Selection of a single solvent may prove challenging, as it has to deliver the desired yield, purity, solid state and morphology of crystals to be accepted. Therefore, a solvent mixture may be required to meet these requirements. Furthermore, the choice of solvent for crystallisation may be limited without consideration of the crystallisation process during synthetic route development.
2.4. Stage 4: process analytical technique selection, calibration and decision 3
The goal of stage 4 is to select appropriate process analytical techniques (PAT) for the in situ monitoring of the attributes of interest, including solute concentration, polymorphic form and particle size, during the crystallisation process. In situ analysis is favoured over off-line approaches as this enables monitoring and control in real-time, as long as the required sensitivity and accuracy of the in situ method can be assured. A range of optical spectroscopic techniques are available for in situ analysis including UV-visible, NIR, Mid-IR, and Raman. The different types of measurements available for each technique along with their key features are given in Table 5. In some cases, e.g. wide area illumination Raman spectrometry, techniques can be used to obtain information about both the liquid (solute concentration) and solid (polymorphic form and concentration) phases. In comparison, ATR, Mid-IR and UV-visible measurements can be used to measure selectively the liquid phase in the presence of particles owing to the short penetration depth of the evanescent wave. The key purpose of stage 4 is the selection of a process analytical technique that can be used for in situ measurement of the solute concentration. This is required to determine a solubility curve for the compound of interest in the solvent selected in stage 3 and for the monitoring and control of the continuous crystallisation process in subsequent stages (5 to 7) in the workflow.| Technique | Types of measurements | Key features |
|---|---|---|
| UV-visible | Transmission (weakly absorbing liquids) and ATR (strongly absorbing liquids) | Molecules must contain a suitable chromophore |
| Transflectance (liquids and slurries) | Can be very sensitive | |
| Reflectance (powders) | Compatible with silica fibre optics | |
| Gives rise to broad spectral bands ⇒ multivariate analysis often required | ||
| NIR | Transmission (liquids) | Spectra arise from combinations and overtones of X–H stretching vibrations |
| Transflectance (liquids and slurries) | Not particularly sensitive | |
| Reflectance (powders) | Compatible with silica fibre optics | |
| Spectra contain broad, overlapping peaks ⇒ multivariate analysis usually needed | ||
| Water absorbs strongly ⇒ good for measurement of low levels of water but not for analytes present in an aqueous process | ||
| MIR | ATR (liquids) | Good for molecular identification |
| Can be relatively sensitive | ||
| Need to use chalcogenide or silver halide fibre optics | ||
| Can be issues with a large background signal from solvents such as water | ||
| May be possible to use univariate or multivariate analysis | ||
| Raman | Reflectance – wide area (solids and slurries) | Not particularly sensitive |
| Reflectance – small area (liquids) | Good for molecular identification | |
| Commonly used laser wavelength of 785 nm compatible with silica fibre optics | ||
| Water is a poor Raman scatterer ⇒ good for analysis of organics in an aqueous solvent | ||
| Fluorescence can be a major problem | ||
| Good for distinguishing between different polymorphic forms (particularly THz Raman) | ||
| May be possible to use univariate or multivariate analysis | ||
Measurement of solute concentration requires construction of a calibration model relating the spectral response to the dependent variable. For development of a calibration model that is to be used across a range of temperatures, it is necessary to either remove the effects of temperature from the spectra or to include such effects in the model.58 Incorporation of the spectral variation arising from temperature into the calibration model can be achieved by subjecting solutions of different solute concentrations to a stepped heating/cooling temperature profile, and selection of spectra collected when the temperature was constant and hence, known. The model can then be validated using additional spectra collected of solutions of known concentration and temperature that were not used to build the calibration model. The choice of calibration model type is dependent on the complexity of the spectra. For example, if the solute gives rise to a single peak in the UV-visible region while the solvent does not absorb, then it may be possible to use a univariate model based on absorbance or peak area. In comparison, if the solute spectrum overlaps with that of the solvent, then use of a multivariate regression model such as partial least squares (PLS) is likely to be required. Further details of PAT calibration can be found elsewhere.59–61 The performance of the calibration model for prediction of solute concentration during a cooling crystallisation process can also be assessed via an off-line reference technique such as gravimetry or HPLC.
In summary, stage 4 evaluates the PAT tools required for the monitoring of the process of interest and the selection of those that can measure the required attributes with the required accuracy and precision. In decision 3, if a calibration model does not meet the required accuracy and precision during the process, then the calibration model may need to be re-evaluated after consideration of further pre-processing options (e.g. removal of the effects of temperature from the spectra using approaches such as loading space standardisation62) or another PAT technique (off-line if necessary) considered.
2. Selection of a PAT method must be guided by the desired accuracy of the measurement. A crystallisation system that exhibits a very narrow metastable zone width will require a PAT method with much higher accuracy and precision to operate the process in comparison to a system with a much wider zone width. Precision of in-line or in situ PAT is generally acceptable, while often lower than that of its off-line counterparts.
3. Whilst it is key to monitor the concentration of solute in solution during a process, it would also be desirable to monitor the concentration of impurities in situ. However, the concentration of impurities is usually below the detection limit of the optical spectroscopic techniques listed in Table 5 possibly with the exception of UV-visible spectrometry; although as the impurities are usually structurally related to the solute, then measurement of impurities can be difficult owing to spectral overlap with the much larger solute peak. Therefore, off-line or on-line UPLC, HPLC or GC may be required.
2.5. Stage 5: system understanding and decision 4
In order to guide development of specific process configurations and crystalliser designs, further understanding of key crystallisation responses for the selected solute/solvent system is required. A summary of these aspects to be assessed is given in Table 6.| Key aspect | Methodology | Rational | Product attribute |
|---|---|---|---|
| a In the absence of all other crystallisation processes. | |||
| Primary nucleation | Metastable zone width or induction time | Indication of the likelihood of nucleation. For a seeded crystallisation, the desired state is a wide metastable zone or long induction time that maximises the window for seed addition | Solid state, PSD |
| Secondary nucleation | Single crystal or bulk suspension | Indication of the supersaturation and agitation required for substantial numbers of secondary nuclei to be generated | PSD |
| Fouling | Induction time for nucleation on material surfaces | Presence of undesired nucleation and subsequent growth on surfaces can cause blockages, prevent the attainment of steady state and affect heat transfer | Solid state, PSD |
| Growth rate | Single crystal growth on hot stage microscopy or bulk crystal growth through image analysis | Crystal growth rates dictate the residence time required for crystals to grow to a target sizea and therefore the volume of the crystalliser | PSD, purity |
| Agglomeration | Image analysis of crystal suspensions | The presence of agglomerates affects PSD, particle shape and downstream processing such as filtration, washing and drying | PSD, purity |
Based on the assessment of these aspects one can develop understanding of suitable crystalliser configurations:
1. A broad metastable zone width and the requirement for high supersaturation to trigger secondary nucleation suggests that the system nucleates slowly. This is desired for a seeded crystallisation as it increases the design space across which seeds can be added. There are also practical considerations, seeding within a narrow metastable zone width (<2 °C) is likely to be difficult to perform reliably in practice.
2. Secondary nucleation tends to dominate in MSMPR at elevated supersaturation and quantification of secondary nucleation rate dependence of supersaturation is needed to understand the dynamics of the crystal size distribution. This also influences the yield of crystallisation via the surface area of crystals available for growth. A high secondary nucleation rate dependence on supersaturation indicates that it will be difficult to grow large crystals regardless of the equipment. Adding further vessels to a cascade can help but this may be offset by increased secondary nucleation associated with transferring material between vessels.
3. Growth rate at a specific supersaturation dictates residence time and thus crystalliser volume provided that the secondary nucleation rate is not significant. For given seed size and target product size, seed loading and hence seed feeding rate can be determined for desired mass throughput.2
4. Depending on the strength of the interparticle forces involved, agglomeration can be reduced through increasing shear rate levels. For example, if agglomeration propensity is found to be significant, an MSMPR might be preferred over a PFR due to the high-localised shear rates in the region close to the impeller which could break up agglomerates.63 However, this effect must be balanced with the increase in the secondary nucleation rate associated with more intense local shear.
5. Common PFR designs (Table 8) are generally tubular in nature and have a higher specific surface area in comparison to MSMPRs. Therefore, if an API was found to have a very short fouling induction time a PFR should be avoided due to the increased risk of blockage.
Under ideal seeding conditions (in terms of supersaturation, temperature, agitation) where both primary and secondary nucleation are negligible, the number of crystals is conserved during crystallisation.2 As a result, the crystal mass deposited through growth is evenly distributed across the seed crystals. Therefore, design equations that relate crystallisable mass, seed size and seed mass can be used to calculate the final crystal size. The mass ratio of seed or seed loading, Mseed, can be defined by:73
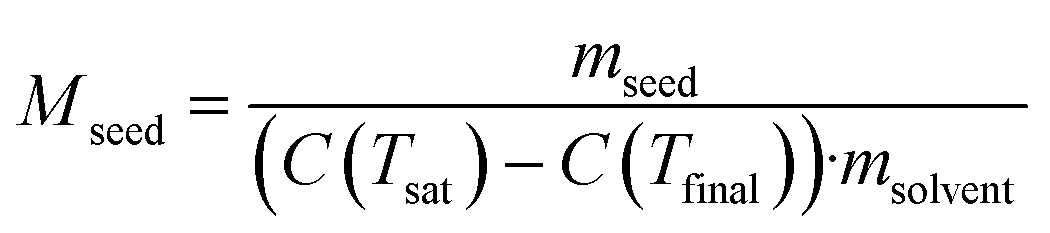 | (2) |
Under the assumptions that growth only occurs on the seeds, a monodispersed seed size and that the number of seeds is conserved, the product crystal size, Lp, can be related to the seed crystal size, Ls, by:2
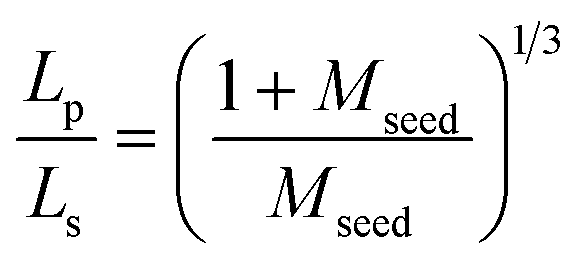 | (3) |
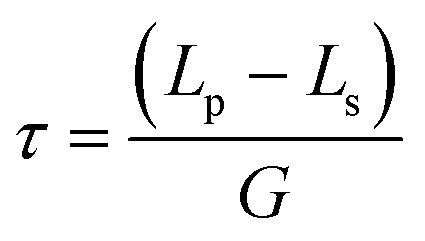 | (4) |
To aid in platform selection, Table 7 provides a comparison between MSMPR and PFR type crystallisers indicating which aspects should be considered. Furthermore, Table 8 describes the different methods of generating plug flow.
| MSMPR | PFR | |
|---|---|---|
| Theoretical yield | Lower in a single stage MSMPR where there will be some residual supersaturation compared to a PFR, especially where the final portion of the residence time is an isothermal hold. Introducing additional MSMPR stages can eliminate this difference | Equivalent to batch in a single pass (assuming sufficient residence time and equivalent cooling profile) |
| Design principle | Different approach to that of batch. System operates at fixed points of supersaturation | Relatively easy to convert time in batch to distance along a plug flow crystalliser |
| Familiarity/visuals | Commonly a single or cascade of stirred vessels. Looks visually similar to batch | Generally tubular geometry. Visually very different to batch |
| Cost of implementation | Lower cost if converted from existing batch (although vessel could be overly large). Similar cost to PFR if new installation | Likely to be high as a new installation. Lab scale can be inexpensive |
| Fouling/encrustation | Potential of encrusting around liquid levels but unlikely to block, relatively easy to remove encrustation | Higher likelihood of fouling leading to blockage that is potentially difficult to remove. Addressed by periodic pre-emptive cleaning |
| Cleaning in place | For both cases ca. 2 vessel volumes would be required to demonstrate cleanliness | |
| Achievable residence time | Function of number of stages, volume of each stage and net flow. Large flexible range. “Lab” scale up to 20 h | Function of length and net flow. Commercial units up to 5 h |
| Crystallisation kinetics | Flexible for a range of growth kinetics. Secondary nucleation can dominate due to impeller/crystal collision | Growth rate at an acceptable supersaturation and maximum residence time limit the maximum particle size attainable. Continuous cooling crystallisations in tubular geometries should always be seeded especially during start-up to prevent encrustation occurring at high supersaturations |
| Crystalliser volume | Single stages from 50 mL upwards have been demonstrated | Lab units exist from 60 mL to 10 L. Commercial units start at 1.25 L |
| Method | Description | Advantages | Disadvantages | Examples |
|---|---|---|---|---|
| Tanks in series | Cascade of stirred vessels with transfer between stages | Most common | Equipment intensive and reasonable number of tanks needed (>10) | Paracetamol,64 PABA65 |
| Turbulent tubular flow | High velocity flow through tubular section | Simple construction | Long lengths required for all but very fast crystallisations | Benzoic acid,66 glycine67 |
| Narrow bore laminar flow | Low velocity flow through small bore (<1 mm) | Simple construction | Narrow bore sizes can lead to blockages | L-Alanine68 |
| Oscillatory flow | Pulsations of flow superimposed on a net flow through periodically spaced orifices | Reduced length compared to turbulent tubular flow. Mixing conditions independent of net flow | Medium construction complexity. Oscillations can dampen over long lengths. Generation of oscillation can be mechanically complex | Lipoic acid – nicotinamide,69 salicylic acid,70L-glutamic acid,69 lactose31 |
| Static mixer tubes | Typically inserted in tubular vessels with short alternate pitched sections which “cut and dice” the fluid to promote radial flow | Low to medium construction complexity. No moving mechanical components | Readily available in short sections in a wide range of materials of construction | Ketoconazole, flufenamic acid, L-glutamic acid18 |
| Slug/segmented flow in tubes | Immiscible fluid added to stream to separate process into individual slugs | Low to medium construction complexity. No moving mechanical components. Each slug like a micro-crystalliser | Can have fouling/blockage issues. Spontaneous generation of slugs is within limited operating window | Acetylsalicylic acid,30 succinic acid,71 lysozyme72 |
Decision 4 (Fig. 1), selection of appropriate crystallisation platforms requires the measurement of key crystallisation parameters. These are linked to the characteristics (residence time, heat transfer, energy dissipation, shear rates, etc.) of the available crystallisers (Table 7). Using Table 6 and the explanations associated with decision 4, potential crystallisation platforms can be selected for further investigation in the workflow. If no crystallisation platform matches the requirements associated with a particular API/solvent pairing alternative crystalliser designs may need to be considered or the solvent selection made in stages 2 and 3 may be re-evaluated to identify solvents which are more compatible with the available crystallisation platforms.
2. The process developer needs to consider how well results from small-scale test beds translate to the larger scale continuous environment they are expected to mimic and consider this when selecting which crystallisation platforms to evaluate.
3. Given the range of measurements required at this stage a variety of scaled down test beds are of value. Therefore, at the scales used here, this stage is relatively costly in terms of both material and time resources.
2.6. Stage 6: process understanding and decision 5
Ideally process understanding should be performed in the continuous platform selected at the end of stage 5. However, depending on the specific crystalliser scale, the quantity of material required to perform this stage in a continuous platform may be prohibitive. If this is the case, then well-designed batch experiments should be employed which are in a crystalliser with similar hydrodynamic characteristics as the chosen continuous platform(s).A range of methods can potentially be used at this stage to develop an understanding of the crystallisation process in the chosen platform:
1. Design of Experiments (DoE). Commonly used to study the impact of various process parameters (seed loading, seed size, etc.) on the product attributes. This is followed by determination of the design space, which leads to achieving the desired product attributes. One downside of this approach is that for full deconvolution (i.e. a full factorial model) of all the process parameters the number of experiments required can be significant and impractical. Other DoE models are available74 which seek to reduce the number of experiments but can lead to compounding of process parameters.
2. Population balance modelling (PBM). With the estimation of kinetic parameters for the various crystallisation mechanisms (nucleation, growth, agglomeration) the development of a PBM can be possible. Many PBM implementations have been described and reported75 ranging from bespoke, freeware formulations implemented in MATLAB by MathWorks or commercial platforms such as gCRYSTAL by Process Systems Enterprise are currently the most common. Whichever model package is used, carefully designed experiments are required to achieve reliable estimation of the various parameters.76 Furthermore, model complexity increases as more model mechanisms are required to describe the process dynamics, for example to describe attrition and/or agglomeration kinetics.
In practice, a combination of both DoE and accurate experimental kinetic parameter estimation is an effective approach. Regardless of the approach adopted, the goal is the same: to develop a model that allows for the identification of the process parameters required for continuous operation to deliver product with the desired attributes. In decision 5, the chosen model should be validated by additional experiments under conditions that are within the explored design space but were not used as part of the original model development. If the model does not predict the validation experiment outcomes with suitable accuracy, then model development must be revisited.
In addition to the DoE and PBM methods detailed above, a third approach employing mechanistic model free control strategies could also be employed.77 This methodology would essentially merge stages 6 and 7 and utilise PAT signals in control loops for process parameters including flow rate, temperature, concentration, or seed addition rate to drive the process to the desired product attributes. A limitation of this approach is that it may be difficult to demonstrate understanding of the system behaviour for process validation, but may be more suitable for complex crystallisation systems where it could be difficult to make sufficiently accurate estimations of individual mechanism's parameters.
1. Accuracy of solute concentration measurement. Whilst an error of 5% in concentration measurement may be acceptable, how this error propagates through other parameters needs to be noted. For example, in systems with steep solubility curves the error in measurement of supersaturation will be much greater in reality.
2. Accuracy of particle size measurement. Samples are typically taken from the suspension and re-suspended for particle sizing. Careful method development is required to avoid inducing any breakage or agglomeration of particles such that the measured particles are not representative of the original sampled suspension.
3. Sampling. It is essential that all sampling methods employed ensure representative samples are available for measurement. To partially alleviate this, the PSD is commonly measured at the end of a batch crystallisation and the entire suspension sampled. However, without sampling over time, valuable information on the PSD trajectory during crystallisation is lost. Techniques such as focused beam reflectance measurement (FBRM) can be used to monitor the PSD trajectory directly in situ. However, processing of the chord length data captured is required to convert from the characteristic chord length to volume density PSD typically needed for PBMs.78,79
Model building itself is not without its challenges. The more crystallisation phenomena that occur (secondary nucleation, agglomeration, etc.) the more complex the model becomes and more parameters need to be fitted. However, the addition of extra parameters can lead to over-parameterisation. Therefore, aspects of sensitivity analysis are required to identify which parameters are the most important to be included.
2.7. Stage 7: proof of concept crystallisation and decision 6
Having developed in stage 6 a model that reliably represents the crystallisation behaviour of the system taking into account the process conditions within a specific platform, in stage 7, the aim is to deploy this model to design a suitable process to achieve the required product attributes and translate the design into practice. A range of methods for process design can be utilised depending on the type of model developed. For example, if the process model was developed purely through a design of experiments approach then identification of the continuous process parameters would be via identification of the optimal operating region or determination of the design space boundaries using experimental design software analysis. This would require setting upper and lower bounds for the process response (product attribute) values. In contrast, if the process model is based on a validated population balance relying on estimated kinetics then a global optimisation or Monte Carlo approach can be employed to run a range of simulations to identify the optimal process conditions. Alternatively, as in stage 6, a combination of population balance and DoE methods may also be employed.Once the desired process conditions have been selected and the crystallisation platform configuration determined, the equipment can be assembled in preparation for operation. The exact details of each configuration will vary depending on the selected platform (stage 5). However, other requirements that are common across all platforms are modules for delivery of a feed solution, delivery of a seed stream and collection of product. Basic requirements for these modules include: suitably sized feeding and receiving systems or vessels for extended continuous operation (or connection to appropriate upstream and downstream continuous unit operations), temperature controllers for vessels and pipework and monitoring and control of mass flow rate. In addition, appropriate safety measures such as bunding and head space inerting must be implemented. For a seeded crystallisation continuous delivery of a consistent stream of seeds is a critical requirement. Seeds could be supplied externally by a stream that is combined with the main feed stream. This stream consists of seed crystals (ideally non-agglomerated with a narrow PSD) suspended in saturated solution. The design of such a supply should consider:
1. Pipework diameter. Large enough to minimise blockage but small enough to maintain a high enough velocity to prevent settling/classification. This requirement also applies to flows in ancillary devices such as mass flow meters.
2. Pump type (if used). The use of pumps can be avoided via utilisation of pressure transfers.80 However, where pumps are used the selected pump must be able to transfer particles without causing crystal breakage. Peristaltic or valveless positive displacements pump are generally suitable for this application.
3. Mixing point design. The point at which the seed stream meets the main feed stream should be designed such that the seed crystals are dispersed as quickly and uniformly as possible into the bulk process volume. This is to ensure particles do not accumulate at this point and block the seed stream.
Instead of external seeding, seeds can also be generated in situ. Potential methods include: anti-solvent addition,81 ultrasound induced nucleation19,31 and high shear for example as generated in a rotor stator wet mill.82
A key challenge in constructing an MSMPR cascade is to ensure controlled transfer of slurry between stages. A range of possibilities are available, from pump transfer to pneumatic transfer under pressure or vacuum. Where pump transfer is used, as with externally seeding by slurry, pump type and design must be selected to minimise crystal breakage. A common disadvantage of pumps is that at low flow rates (i.e. lab scale <20 g min−1) settling of particles and blockages can occur. This can be partially addressed with periodic transfer.64,83 In this operating mode, pumps operate for a short period of time at high flow rates to transfer slurry under conditions where particle settling and segregation are minimised. This requires the use of programmable pumps to control the desired periodic cycle. An alternative embodiment of periodic flow is to operate a pump-around loop withdrawing material from the bottom valve of each vessel and returning it below the liquid surface. In this way, a representative stream of particles is recirculated. This flow can then be fed forwards to the next vessel using a timer-actuated ball valve positioned to allow the transferred material to drain freely into the next vessel in the cascade.
A further consideration when positioning inlet and outlet pipes is the minimisation of blockages and the avoidance of short-circuiting of fluid suspension that would broaden the product RTD. The inlet pipe can be positioned either sub-liquid surface or above the liquid surface. Sub-liquid introduction can lead to high local supersaturation although this can be minimised by locating the end of the pipe in or near to the impeller zone where the fluid velocity is highest and mixing fastest. However, blockages can still occur. Positioning the pipe end above the liquid surface removes the risk of high local supersaturation but “bearding” (where a crust forms around the pipe outlet) is likely to occur. Similar to the sub-surface introduction the pipe end should be positioned such that the liquid feed contacts the vessel contents at a position where it is rapidly mixed with the bulk. For example with a downwards pumping agitator, this may be close to the stirrer shaft.
The control strategy84 must also be selected at this point in crystallisation development. Classical feedback control strategies (closed loop) such as PID or cascade are suitable for controlling aspects with simple responses such as temperature or mass flow rate. These relatively straightforward loops can be used when deploying the lowest level of control to maintain process set-points. The set-point values are determined by the process model developed in stage 6. Whilst such control loops can maintain an operating process this offers no guarantee that the product attributes will not vary. The availability of real-time process analytical technologies (PAT) for solute concentration (via ATR-FTIR or ATR-UV) and particle size and shape (via FBRM or PVM) allow process parameters that are more directly linked to product attributes to be included in the process control algorithm. PAT can be exploited via a mechanistic model or a mechanism free approach (as mentioned in section 2.6).85 For model-based strategies the process model interprets the PAT signal and provides correction of the process parameters (i.e. change in temperature or mass flow). In contrast, in a model free strategy the PAT signal directly controls a process parameter. Hybrid or multi-level strategies are also possible which are composed of aspects from model-based, model-free and closed loop approaches.
Once a crystallisation process has been designed, operated and material produced, decision 6 evaluates the extent to which the product achieved the attributes selected in stage 1, in addition to product consistency and process robustness. If neither of these criteria are met, then further process understanding is required and the workflow returns to stage 6 to develop the process model further and redesign/optimise the crystallisation. If the criteria set for the product and process are met, then the objective of the workflow is complete.
2. Lack of standardisation. Currently there is no standard MSMPR design, although partial guidelines do exist,87 and PFR platforms vary among commercial suppliers. As a result, implementations of continuous crystallisations can vary between sites even for the same platform. Consequently, without documented guidelines learning processes are repeated across sites. This also extends to equipment interfaces, ancillaries and control systems leading to bespoke systems.12
3. Case study: paracetamol (acetaminophen)
3.1. Materials
Paracetamol (acetaminophen, meets USP specification, 98–102%) was purchased from Sigma-Aldrich and used without further purification. All solvents were purchased from either VWR International or Sigma-Aldrich and while the exact grade varied between solvents, purity was always > 99.5% and used without further purification.3.2. Stage 1: prior knowledge
Paracetamol as a model compound is well characterised and reported in the literature. For the purpose of this case study chemical and physical purity of the material as supplied by Sigma Aldrich was confirmed as paracetamol form I by a complementary suite of techniques including XRPD (X-ray powder diffraction), DSC (differential scanning calorimetry) and NMR (nuclear magnetic resonance). Key data including crystal structure and reference patterns from each technique is presented in the ESI.† Solvates of paracetamol have been reported in 1,4-dixoane88 and pyridine.89 Therefore, these solvents were excluded from the solvent screen as the target polymorph was the monoclinic form I.3.3. Stage 2: solvent screen
Solvents used for the screen were restricted to class 2 (solvents to be limited) and 3 (solvents with low toxic potential) solvents from the ICH classification45 for residual solvents. Additionally, from the information collated in stage 1, 1,4-dioxane and pyridine were removed from the solvent list due to avoid the risk of solvate formation.88,891. Hold at 20 °C (room temperature) for 1 h
2. Heat to 10 °C below the boiling point of solvent (elevated temperature) at a rate of 5 °C min−1
3. Hold at the elevated temperature for 1 h
Agitation was provided by a 7 mm stirrer bar at 800 rpm throughout the experiment. At the end of each hold period the transmission of each vial was recorded. This methodology was repeated 4 times for each solvent. Based on the transmission recorded each solvent was classified according to Table 9.
| Room temperature (R.T.) | Elevated temperature (E.T.) | |||
|---|---|---|---|---|
| Transmission | >95% | <95% | >95% | <95% |
| Classification | R.T. high solubility | R.T. low solubility | E.T. high solubility | E.T. low solubility |
| Room temperature low solubility | Room temperature high solubility | |
|---|---|---|
| Elevated temperature low solubility | 2-Methoxy-2-methylpropane, anisole, butyl acetate, chlorobenzene, chloroform, cyclohexane, dichloromethane, diethyl ether, ethyl acetate, heptane, hexane, isobutylacetate, o,m,p-xylene, pentane, toluene, trichloroethylene | |
| Elevated temperature high solubility | 1-pentanol, 2-methyl-1-propanol, 3-methyl-1-butanol, acetonitrile, butyl acetate, methyl acetate, methyl isobutyl ketone, nitromethane, propyl acetate, water | 1,2-Dimethoxyethane, 1-butanol, 1-propanol, 2-butanol, 2-butanone, 2-ethoxyethanol, 2-methoxyethanol, 2-propanol, acetic acid, acetone, DMSO, ethanol, ethylene glycol, formamide, methanol, N,N-dimethylacetamide, N,N-dimethylformamide, N-methyl-2-pyrrolidone, tetrahydrofuran |
Acetonitrile and nitromethane are ICH class 2, also met the criteria but were not considered further in stage 3 due to the additional considerations required for residual solvent levels during isolation.
3.4. Stage 3: solvent selection
With the initial list of solvents reduced to 8 in stage 2, these were each studied further to measure their full temperature dependent solubility curve to inform decision 2.1. Cool to 5 °C (in case dissolution was below ambient conditions), hold for 1 h
2. Heat to 10 °C below the solvent boiling point (elevated temperature) at a rate of 0.1 °C min−1, hold at the elevated temperature for 1 h
3. Cool to 5 °C at a rate of 0.1 °C min−1
Agitation was provided by a 10 mm stirrer bar at 800 rpm throughout the experiment. The clear point (point of complete dissolution upon heating) was defined as the temperature during the heating cycle at which the transmission reached 100% with respect to a sample of the test solvent measured at ambient temperature. Similarly, the cloud point (temperature at which crystals are detected upon cooling) was defined as the temperature during the cooling cycle at which the transmission deviated from 100%.
| C = a·exp(b·T) | (5) |
| ΔTsd = 0.693/b | (6) |
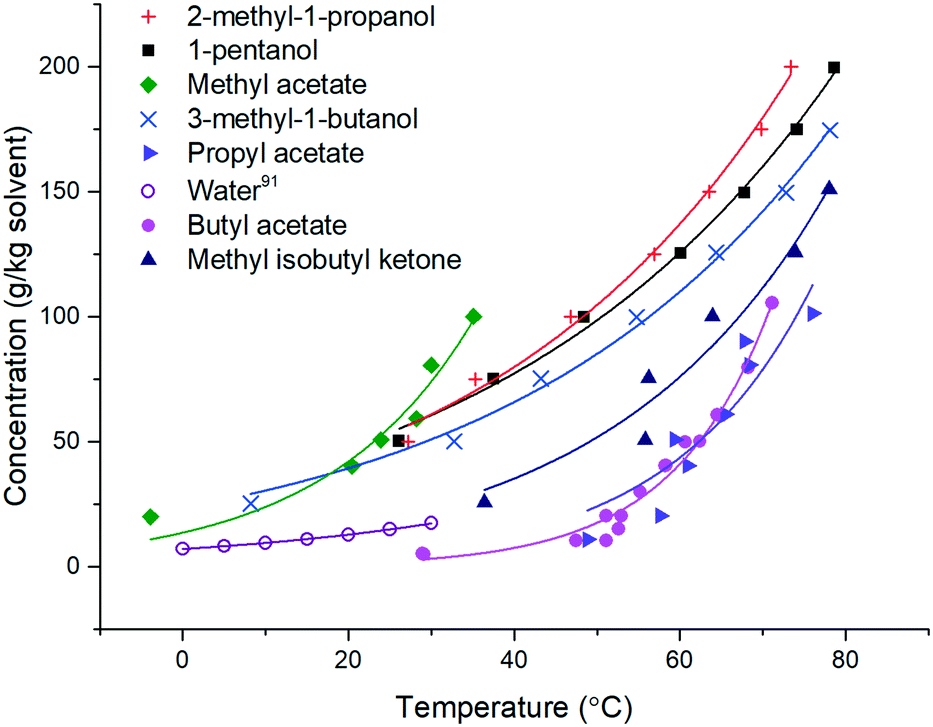 | ||
| Fig. 2 Solubility curves for stage 3 solvents. Water solubility.91 | ||
For the solvents presented in Fig. 2, the solubility was found to double every 19.3 ± 7.9 °C. This is comparable to the qualitative heuristic “Black's rule” which states that solubility generally doubles every 20 °C.90
Building upon the solubility fits, Table 11 provides a summary of the 8 stage 3 solvents against the solvent selection criteria previously described in Table 4. For all solvents 5 °C was chosen as the lower temperature as from a practical perspective this allows water to be used as a heat transfer fluid in the heater/chillers. The saturated temperature was chosen as the temperature required to achieve >90% yield or the solvent boiling point minus 10 °C, whichever is lowest, whilst maintaining the product solid mass fraction less than or equal to 0.25 to enable processability. With these points defined, the theoretical yield operating between these two points is defined by eqn (1). From the difference between C(T0) and C(Tf), the mass fraction of solids present in the product and an estimated throughput for a given solvent flow rate (50 g min−1) were also calculated (Table 11). Visual checks were made in each sample for encrustation (fouling) and oiling out. Other important phenomena associated with nucleation and growth were assessed based on MSZW and final particle size. Growth rates could also be assessed through image analysis of the in situ particle images with acknowledgment of the limits of an experiment conducted at an 8 mL scale.
| 1-pentanol | 2-Methyl-1-propanol | 3-Methyl-1-butanol | Butyl acetate | Methyl acetate | MIBK | Propyl acetate | Water | |
|---|---|---|---|---|---|---|---|---|
| a Product solid mass fraction limited to 0.25 for processability. b Throughput assuming 50 g min−1 solvent net flow. | ||||||||
| Boiling point (°C) | 137 | 108 | 131 | 126 | 57 | 117 | 102 | 100 |
| Saturated temp. (°C) | 100 | 93 | 100 | 70 | 47 | 88 | 70 | 90 |
| Final temp. (°C) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Yield, Yt (%) | 91 | 91 | 93 | 99 | 81 | 98 | >99 | 92 |
| Product solid mass fractiona | 0.25 | 0.25 | 0.25 | 0.07 | 0.11 | 0.25 | 0.08 | 0.09 |
| Throughput (kg h−1)b | 0.75 | 0.75 | 0.75 | 0.22 | 0.32 | 0.75 | 0.23 | 0.28 |
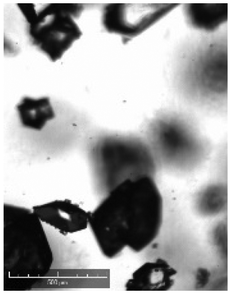
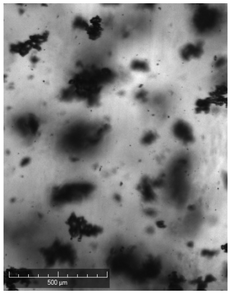
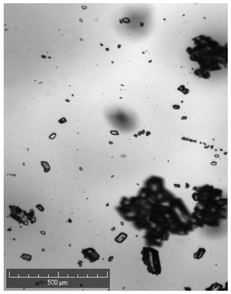
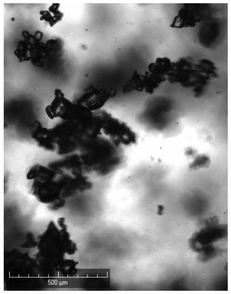
| Comments | Observed droplets on cooling | Observed fouling on vial | Agglomerated particles | Agglomerated particles | Agglomerated particles | Wetting issues | ||
| Image |
|

|
|

|
|
|
|
|
When compared against the selection criteria (Table 4), butyl acetate, methyl acetate, MIBK, propyl acetate and water were considered unattractive due to the consistent occurrence of crystal agglomeration. Of the 3 remaining solvents, 1-pentanol was excluded from further consideration due to droplet formation (oiling out) upon cooling. 3-Methyl-1-butanol was selected in preference to 2-methy-1-propanol on the basis that 3-methyl-1-butanol delivered a slightly higher yield (93% vs. 91%) and readily formed large crystals that would be easier to recover in comparison to 2-methyl-1-propanol.
3.5. Stage 4: PAT selection and calibration
With the final solvent chosen from stage 3 as 3-methyl-1-butanol (isoamyl alcohol or isopentanol), an optical spectroscopic technique was selected for in situ measurement of solute concentration. This also enabled the determination of the solubility-temperature curve for paracetamol in 3-methyl-1-butanol in the range 0 to 100 °C as well as providing a means of in situ monitoring and affecting control of the crystallisation process in stages 5 to 7 of the workflow.Measurement and control of solute concentration is vital in crystallisation and techniques that may be influenced by particle scattering (e.g. Raman backscattering) were not considered further here. Both UV-visible and IR spectrometries can be used in conjunction with an ATR probe to measure the concentration of paracetamol in 3-methyl-1-butanol solutions and suspension. Although 3-methyl-1-butanol absorbs in the IR but not in the UV-visible region, IR was selected in this case as the software used for spectral acquisition (see below) was fully integrated into the control system simplifying execution of subsequent workflow stages.
For the calibration experiments, a series of fixed concentrations of paracetamol in 3-methyl-1-butanol solvent were prepared at ca. 105 °C and transferred to the OptiMax™ workstation. A stepped cooling profile, driven by the iControl software, was completed using 15 °C steps over the range 105 to 0 °C. Evaporation of solvent at high temperature was minimised by use of a reflux condenser. Six paracetamol concentrations were included in the calibration set: 10, 50, 90, 125, 200, 275 g per kg solvent. Two further experiments were performed to validate the calibration model: 60 g kg−1 solvent at 20 °C and 180 g per kg solvent at 65 °C. All experiments were held at a constant temperature for a period of ca. 30–70 min.
To measure the solubility curve for paracetamol in 3-methyl-1-butanol, a paracetamol–solvent slurry of 300 g per kg solvent was prepared and a stepped heating profile (5 °C min−1 ramps and 60 min hold periods) was completed with excess solids present (confirmed by FBRM total counts) throughout to ensure a saturated supernatant.
All spectral data were processed using GRAMS V9 software (Thermo) which was also used to generate the PLS models via the GramsIQ chemometrics add-on. Only those spectra that were acquired when the temperature had stabilised were used to construct the calibration model. All spectra were baseline corrected and the 1700–800 cm−1 region was selected. Both the spectral and solute concentration data were mean centred prior to construction of the PLS model. The optimum PLS model was selected on the basis of the accuracy of the predictions obtained for the two validation experiments. The optimum calibration model employed 6 latent variables and R2 was 0.99966. The calibration model was then used to determine the solubility curve for paracetamol in 3-methyl-1-butanol from IR spectra acquired when the temperature was constant during the hold periods.
The temperature dependent solubility curve obtained for paracetamol in 3-methyl-1-butanol by in situ IR spectrometry is shown in Fig. 3. The plot obtained is in reasonable agreement with the data obtained using the Crystalline platform shown in Fig. 2.
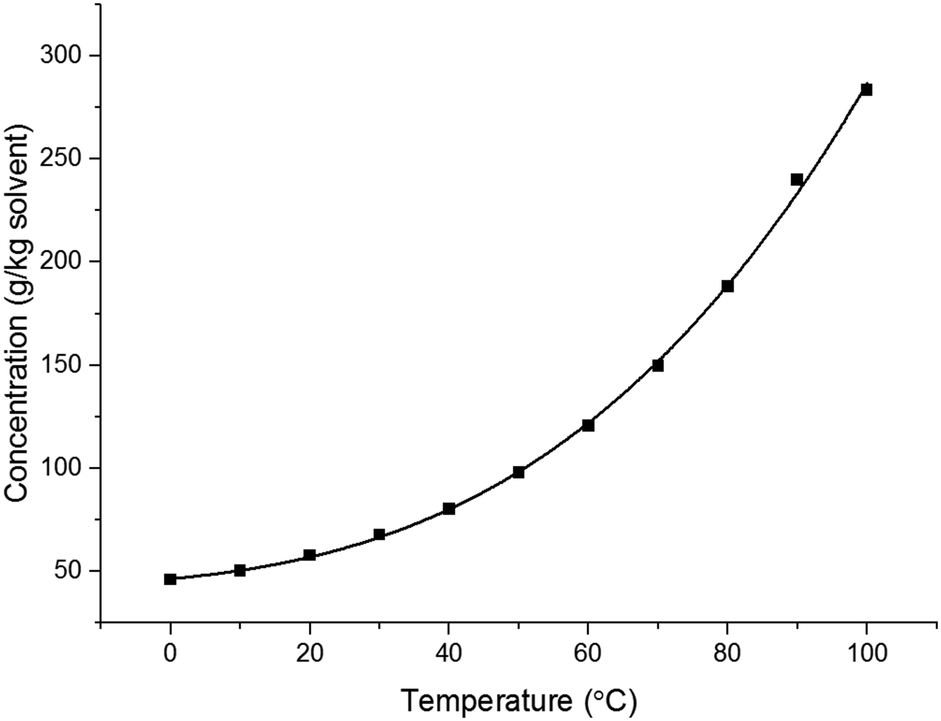 | ||
| Fig. 3 Temperature dependent solubility profile of paracetamol in 3-methyl-1-butanol. | ||
3.6. Stage 5: system understanding
To inform platform selection a series of assessments were carried out of processes that may impact the performance of seeded cooling crystallisations (primary and secondary nucleation), alter the resulting particle attributes (agglomeration) or continuous process performance (fouling). Crystal growth rates were also quantified to guide the crystallisation process design, residence time selection and final crystal size distribution.1. 1 L stirred tank reactor (Mettler Toledo OptiMax™ workstation), equipped with a Hastelloy® Pt100 temperature sensor. Agitation was provided by a 45° pitched blade turbine rotating at 350 rpm.
2. Moving fluid oscillatory baffled crystalliser (MF-OBC)92 of 125 mL capacity, equipped with a PTFE coated Pt100 temperature sensor. Tube diameter was 15 mm with a baffle orifice diameter of 7 mm and baffle spacing = 22 mm. Oscillation was provided by a 25 mm diameter PEEK piston using frequency and amplitude of 1.5 Hz and 20 mm, respectively.
3. Moving baffle oscillatory baffled crystalliser (MB-OBC)92 of 120 mL capacity, equipped with a PTFE coated Pt100 temperature sensor. Vessel diameter was 24 mm with baffles 23 mm in diameter fitted with a single 10 mm orifice and spaced 37 mm apart. Baffles were connected via stainless steel 316 L support rods. Oscillation of the baffle string was at 1.5 Hz and 20 mm amplitude.
The operating conditions for the scale up crystallisers were selected to maintain similar mixing intensities corresponding to a volume average energy dissipation rate of 0.160 W kg−1 (based on the minimum suspended speed for the expected particle size in the Optimax crystalliser). This was achieved through control of stirrer rotation rate in the stirred tank and oscillation frequency and amplitude in the MF-OBC and MB-OBC crystallisers.
For Crystal16 MSZW assessment, sample compositions of 100, 117, 130 and 148 g per kg solvent of paracetamol in 1 mL of 3-methyl-1-butanol were used. 1.5 mL glass vials were tightly sealed with a lid and para-film to avoid solvent evaporation and placed in the Crystal16. The heating and cooling rates between 10 °C to 80 °C were set to 0.5 °C min−1. Samples were stirred with a controlled stirring speed of 700 rpm, using a PTFE coated magnetic stirring bar.
A FBRM probe with iC FBRM V4.3 was used in each crystalliser configuration to track nucleation. Five fixed concentrations of paracetamol in 3-methyl-1-butanol; 50, 80, 120, 160, 200 g per kg solvent were prepared and heated to 85 °C, then held for 1 h to ensure complete dissolution. Cooling was then applied at 1 or 0.16 °C min−1 (0.16 °C min−1 represents the slowest linear cooling rate achievable in the available crystallisation platforms useable in stage 7). The nucleation temperature was taken as the temperature at which the number of FBRM particle counts in the sub 10 μm range exceeded 100 per s. Comparison of the average MSZW over the five concentrations measured in these crystallisers along with Crystal16 and Crystalline experiments in stage 3 are shown in Table 12. The temperatures at which nucleation was observed under the selected conditions are shown in Fig. 9. Across all crystallisers, the narrowest MSZW was observed in the MB-OBC for concentrations <120 g per kg solvent and in the 1.5 mL stirred vessels at concentrations >120 g per kg solvent. Taking a conservative approach using the narrowest MSZWs, Fig. 9 shows the primary nucleation threshold, 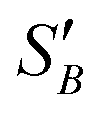 .
.
| Cooling rate | Average metastable zone width (°C) | |||
|---|---|---|---|---|
| 1 °C min−1 | 0.5 °C min−1 | 0.16 °C min−1 | 0.10 °C min−1 | |
| Crystal16 | — | 32.1 ± 4.1 | — | — |
| Crystalline | — | — | — | 28.8 ± 20.6 |
| Optimax | 28.7 ± 0.8 | — | 20.9 ± 6.9 | — |
| MF-OBC | 28.6 ± 18.1 | — | 19.9 ± 2.4 | — |
| MB-OBC | 18.5 ± 8.1 | — | 22.9 ± 14.6 | — |
Four stock solutions with various concentrations were prepared. Complete dissolution was verified by heating the samples to 20 °C above the saturation temperatures whilst stirring at 500 rpm on a stirrer hot plate. Each stock solution was quickly filtered to minimise the amount of heterogeneous particles or residual seed present and transferred into a pre-warmed bottle. The warm, filtered stock solutions were then divided into 3 mL vials through a pre-warmed syringe. A magnetic stirrer bar was added to each vial and the vials were tightly closed with a lid before they were located in the Crystalline workstation at a stirrer speed of 700 rpm. At each supersaturation a seeded and unseeded experiment (serving as controls) was carried out.
8 vials were placed inside the instrument per run and solutions heated to 20 °C above their saturation temperatures and maintained at these temperatures for 20 min to ensure complete dissolution prior to cooling. At the highest temperature the solutions were undersaturated and a cooling profile of 5 °C min−1 was applied generating each target supersaturation ratio. The solutions were prepared with compositions such that the required supersaturation ratio (S = 1.9, 2.0, 2.1 and 2.2) was attained at a target temperature T = 20 °C.
Vials were removed from the instrument once the solutions reached the desired temperature. One vial was seeded with a single crystal of form I paracetamol. The second vial was not seeded with any crystal but in every other respect, it received the same treatment as the seeded sample. The vials were replaced in the Crystalline platform which was taken as t0. Time tn indicates the time from which primary nucleation can be expected to occur. Based on the MSZW values, sufficiently low supersaturations were chosen to prevent primary nucleation occurring. This allowed enough time after the desired temperature was reached to seed the supersaturated clear solution. This was also checked by performing a parallel unseeded experiment to each single crystal seeded test. Therefore, effects caused by primary and secondary nucleation can be distinguished.
Fig. 4a shows the results for seeded experiments at different supersaturation ratios. For the secondary nucleation rate measurements, a threshold of 10 particles was used to define the delay time before secondary nucleation occurs. When the count per frame reached over 200 particles, the particle count is not reliable because the particle suspension is too dense and it is not possible to identify single particles reliably.
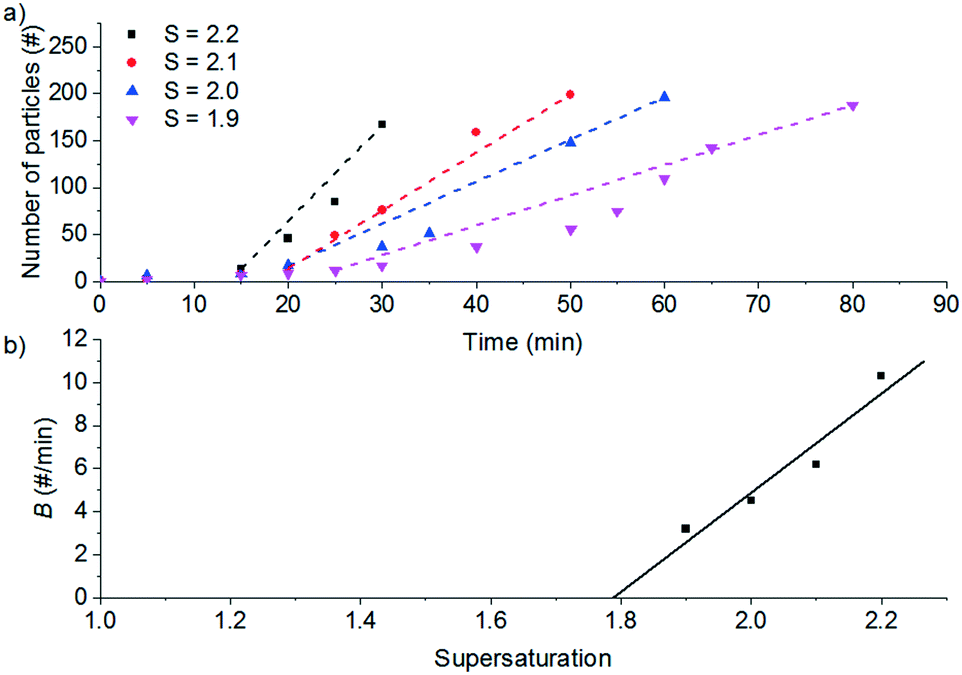 | ||
| Fig. 4 a) Particle count, N, in arbitrary units against time for seeded secondary nucleation experiments at different supersaturation ratios. b) Observed secondary nucleation rate, B, in arbitrary units as a function of the supersaturation in the seeded experiments. | ||
From the slope of the fitted lines in Fig. 4a, the secondary nucleation rate, B, in arbitrary units can be obtained and is shown as a function of supersaturation in Fig. 4b. The measured secondary nucleation increases with supersaturation: for S = 2.2 secondary nucleation is 3 times faster than that at S = 1.9. Extrapolating a linear fit to these supersaturation dependent secondary nucleation rates allows the determination of a supersaturation threshold, 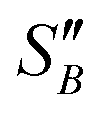 = 1.79, at which the secondary nucleation rate, B, is zero. As the supersaturation required for primary nucleation changes with solubility, the secondary nucleation supersaturation threshold would also be expected to change. To estimate this, the ratio between the primary, secondary nucleation thresholds and solubility was defined as:
= 1.79, at which the secondary nucleation rate, B, is zero. As the supersaturation required for primary nucleation changes with solubility, the secondary nucleation supersaturation threshold would also be expected to change. To estimate this, the ratio between the primary, secondary nucleation thresholds and solubility was defined as:
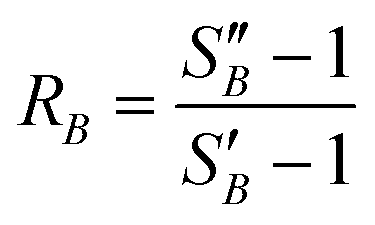 | (7) |
Using the values for 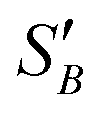 (2.17) and
(2.17) and 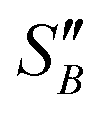 (1.78) at 20 °C, RB was found to be 0.667. A rearrangement of eqn (7) was then used to calculate
(1.78) at 20 °C, RB was found to be 0.667. A rearrangement of eqn (7) was then used to calculate 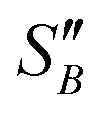 over the full working temperature range in Fig. 9.
over the full working temperature range in Fig. 9.
Paracetamol and 3-methyl-1-butanol stock solutions were prepared (66.5 g per kg solvent) which corresponds to a saturation temperature of 40 °C. This was heated until fully dissolved and 250 mL of the resulting solution was added to the circulation vessel. The solution was circulated around the fouling platform at 40 °C i.e. undersaturated. Once temperature determined by all of the Pt100 sensors had stabilised at a constant value, the circulating heat transfer fluid was cooled. Local supersaturation was generated on the crystallisation solution side of the material coupon by cooling the heat transfer side. It is assumed coolant entry temperature is identical to the local surface temperature of the material coupon. Once the cold stream was initiated it was assumed that supersaturation at the MOC coupon was constant. Images were recorded every 30 s using a monochrome camera and IC capture software. Solution and coolant flow rates were both kept at 50 mL min−1 throughout all experiments, as was the circulating solution temperature at 40 °C. Local supersaturations (dictated by the coolant temperatures of 26.4, 20.7 and 15.9 °C) investigated were S = 1.50, 1.75 and 2.00, respectively, for both Hastelloy® C276 and borosilicate glass. Fouling induction times and example images collected are shown in Table 13 and Fig. 5, respectively.
| Local supersaturation | Fouling induction time (min) | |
|---|---|---|
| Hastelloy® C276 | Borosilicate | |
| 1.50 | No fouling after 12 hours | No fouling after 12 hours |
| 1.75 | 278 | No fouling after 12 hours |
| 2.00 | 107 | No fouling after 12 hours |
 | ||
| Fig. 5 Sequential fouling images for a) borosilicate (0, 205 and 602 min) and b) Hastelloy® C276 (0, 107 and 653 min) at S = 2.00. | ||
From the results in Table 13, the fouling threshold can be defined as S = 1.75 for Hastelloy® C276. Assuming this constant S across the operating temperature range the fouling threshold is shown in Fig. 9. It can be seen that the fouling threshold intersects the conservative primary nucleation threshold at 30 °C. This is unsurprising given the close relationship been fouling induction time and MSZW.93 Therefore, for temperatures >30 °C it was assumed that the fouling threshold was equal to the primary nucleation threshold.
| Crystal growth temperature (°C) | Relative supersaturation, S |
|---|---|
| 25 | 1.05 |
| 25 | 1.25 |
| 50 | 1.05 |
| 50 | 1.25 |
Solutions of paracetamol in 3-methyl-1-butanol were prepared which had the desired relative superstation, S, at each target temperature (Table 14). These solutions were maintained at 20 °C above the saturation temperature for 1 h to ensure complete dissolution. A single crystal was placed in the slide well of a Linkam (LTS420) microscopy hot stage and held at the desired crystal growth temperature leading up to the experiment. This was done to ensure the single crystal and Linkam stage were at the desired temperature from the start of the experiment. Prior to the experiment, a 1 mL syringe with 0.1 μm filter was kept in an incubator <5 °C above the desired temperature. This was done to minimise the impact of momentary cooling when transferring the supersaturated solution to the microscope hot stage. Once the stage and syringe were at the desired temperatures, the solution was quickly cooled to the target temperature and the solution transferred into the slide well. This contained the thermally equilibrated test crystal. Imaging commenced immediately after the supersaturated solution was added. When a complete sequence of images had been collected, the crystal growth rate was determined by manually measuring the crystal dimensions in successive images in the sequence of frames. Typical crystal face measurements and their change with time are shown in Fig. 6. The range of crystal face growth rates observed are summarised in Table 16.
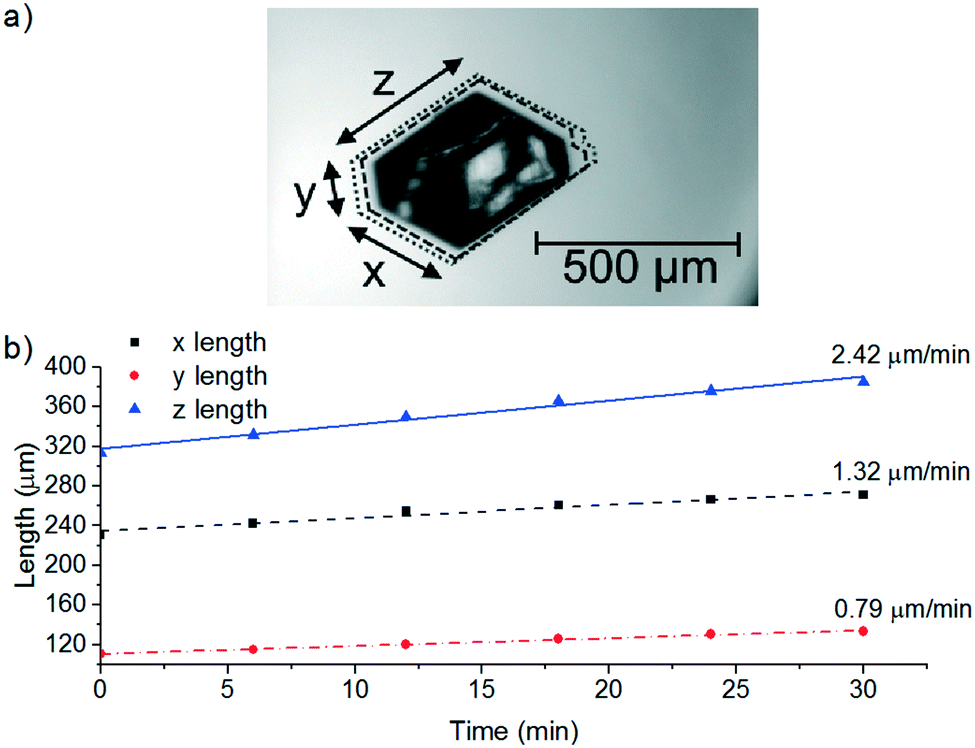 | ||
| Fig. 6 Typical a) microscopy images of single crystal growth and b) crystal face lengths as a function of time for initial S = 1.25 at 50 °C. | ||
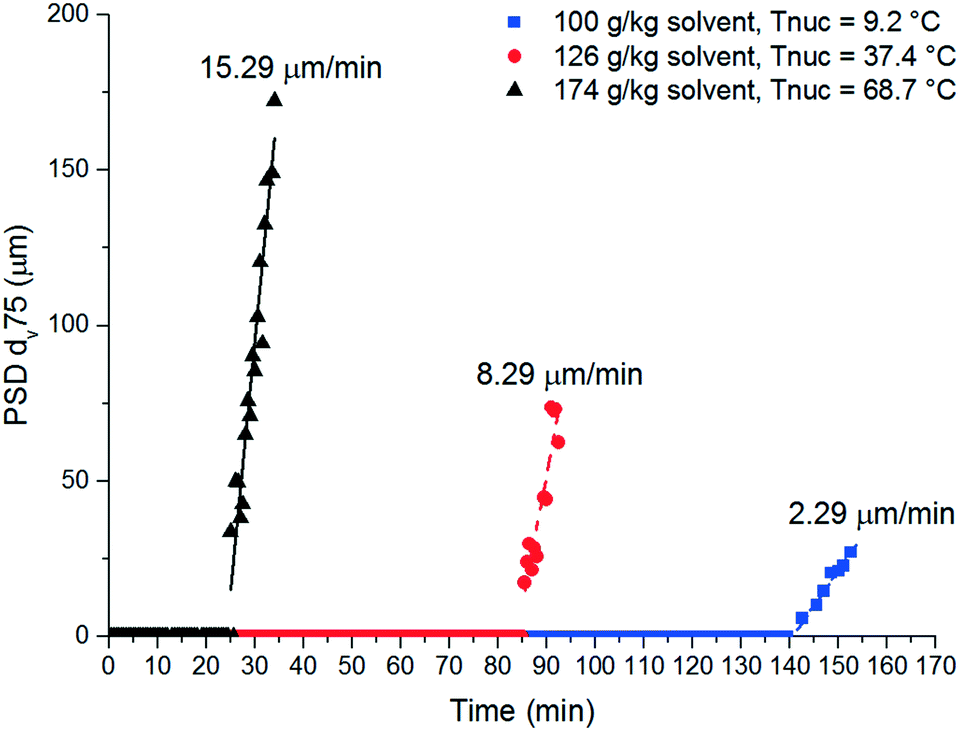 | ||
| Fig. 7 Example growth rates from Crystalline PSD. | ||
| Supersaturation, S | Seeding temperature (°C) | Agitation rate (rpm) |
|---|---|---|
| 1.05 | 38.1 | 250 |
| 1.10 | 36.3 | 250 |
| 1.05 | 38.1 | 400 |
| 1.10 | 36.3 | 400 |
| 1.50 | 24.1 | 400 |
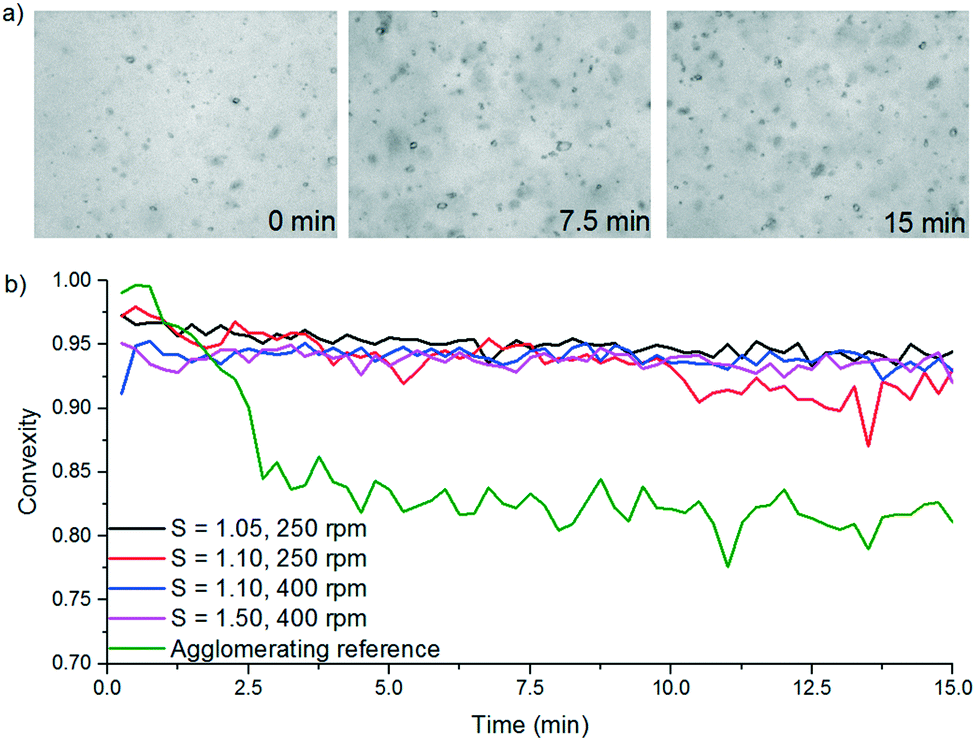 | ||
| Fig. 8 a) Example PVM images for S = 1.50 at 400 rpm experiment and b) trends of particle convexity distribution mode. Agglomerating reference represents the trend for a strongly agglomerating system (acetone95). | ||
| Mechanism | Measure | Result |
|---|---|---|
| Primary nucleation | Narrowest metastable zone width | 18.5 °C |
| Secondary nucleation | Secondary nucleation threshold | S = 1.79 at 20 °C |
| Fouling | Fouling threshold | S = 1.50 |
| Growth rate | Single crystal face | 0.79 to 2.42 μm min−1 |
| Bulk crystal | 2.26 to 19.97 μm min−1 | |
| Agglomeration | Particle convexity distribution mode change | Negligible |
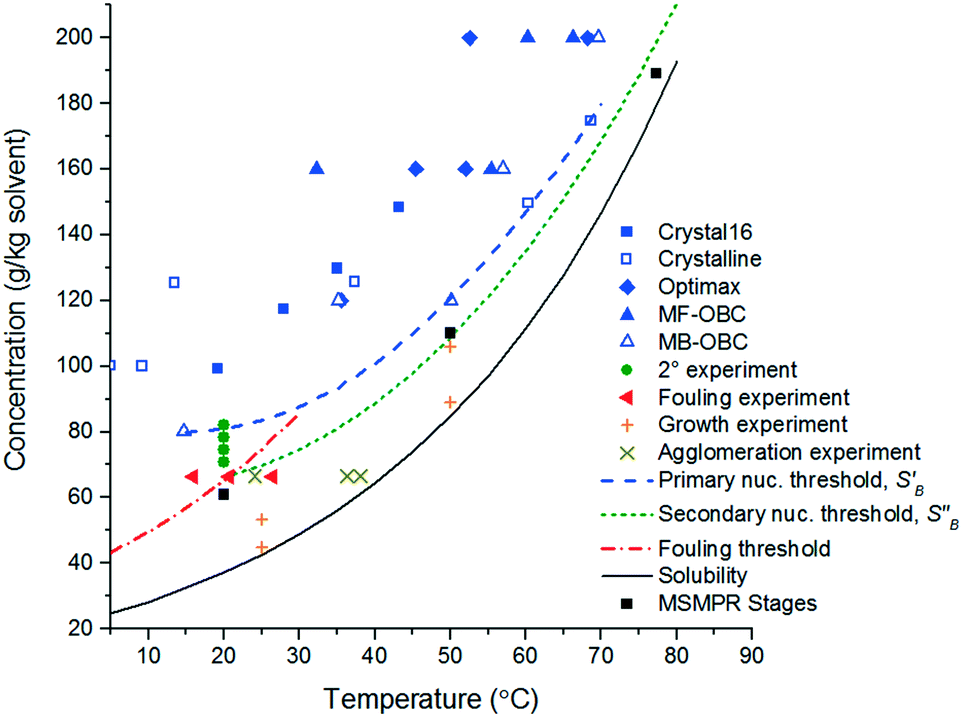 | ||
| Fig. 9 Operating space in the phase: cloud points (Crystal16, Crystalline, Optimax, MF-OBC and MB-OBC platforms), secondary nucleation experiments and corresponding threshold, fouling experiments and corresponding threshold, growth and agglomeration experimental conditions. | ||
1. Nucleation kinetics are slow. Both primary and secondary nucleation kinetic measures indicate a high level of supersaturation is required for the formation of new particles.
2. Fouling poses little risk at moderate supersaturations.
3. Growth rates are in such a range that they could be easily controlled by S.
4. Agglomeration propensity is low.
As stated in section 2.5 none of these statements rule out a specific crystallisation platform. For example:
1. Metastable zone widths are suitably wide to allow for seeding in all platforms.
2. Fouling poses little risk therefore a PFR could be used with minimum risk of blockage, assuming operation at appropriate levels of supersaturation.
3. Agglomeration was not observed therefore specific levels of shear above that for uniform mixing and particle suspension are not required to disperse agglomerates.
4. Growth rates would require residence times and crystalliser volumes that are compatible with the available laboratory scale PFR and MSMPR systems.
Hence for the case of paracetamol in 3-methyl-1-butonal, crystallisation in a wide range of platforms, covering MSMPR and PFR, would be possible. For the demonstration purposes of this case study an MSMPR cascade platform was chosen as the focus for the later stages of the workflow.
Estimation of the required platform volume is required to ensure suitable equipment selection and configuration. Assuming the slowest bulk crystal growth rate of 2.26 μm min−1, a net flow rate of 12.5 mL min−1 (lowest of available pumps) using eqn (2) to (4) the product size, required residence time and crystalliser volume were estimated, as a function of seed loading and seed size (Table 17). The crystallisation process design is refined as part of stage 6 once an appropriate model has been developed.
| M seed | L s = 10 μm | L s = 20 μm | ||||
|---|---|---|---|---|---|---|
| L p (μm) | τ (min) | V (ml) | L p (μm) | τ (min) | V (ml) | |
| 0.2 | 18 | 3.6 | 45 | 36 | 7.2 | 90 |
| 0.1 | 22 | 5.4 | 68 | 44 | 10.8 | 135 |
| 0.05 | 28 | 7.8 | 97 | 55 | 15.6 | 195 |
| 0.01 | 47 | 16.2 | 202 | 93 | 32.4 | 405 |
| 0.005 | 59 | 21.5 | 269 | 117 | 430 | 537 |
3.7. Stage 6: process understanding
The aim of stage 6 was to estimate kinetic parameters for use in a population balance model, which will be used in subsequent stages to identify suitable operating conditions.1. Cooled to 5 °C at the fastest possible rate.
2. Held at 5 °C for 2 h to allow for solid/liquid equilibrium.
3. Heated to 85 °C over a 16 h period.
4. Held for a further 2 h period.
Agitation was fixed at 600 rpm (0.053 W kg−1) supplied by a 45 mm pitched blade turbine (PBT). This step enables the IR absorbance of saturated solution as a function of temperatures to be determined and modelled. The solution was then cooled to 80 °C minus a temperature offset (either 5 or 8 °C for S = 1.08 and 1.20, respectively, to operate within the thresholds in Fig. 9). Once this temperature was reached the required seed mass was charged as dry seed into the vessel and the supersaturation control was then started using the absorbance-temperature model. 5 °C was set as the lower bound for the temperature. Seeds were prepared by sieving of ball milled commercial paracetamol. Two seed sizes, with dv50 of 40 and 100 μm, were utilised in addition to two seed loadings, Mseed, of 0.019 and 0.036. On completion of each experiment, the contents of the vessel were filtered and the cake washed twice with 2 cake volumes of chilled diethyl ether. The wet cake was then transferred to a vacuum oven for drying. Once dry, particle size distributions of the product were measured by laser diffraction (Malvern Mastersizer 3000) using isooctane as a dispersing fluid (due to its negligible solubility and wetting properties of paracetamol).
For parameter estimation, a model of the Optimax vessel was built in gCrystal 4.2.0 using an MSMPR unit operation configured in batch mode. Time invariant controls for the model included mass of crystals in slurry, liquid composition, PSD location parameter and PSD standard deviation. Temperature profiles were input as piecewise linear controls by approximating the temperature profile determined by the supersaturation control to a series of linear segments.
To prevent agglomeration affecting the estimation of growth parameters, only the concentration profile monitored by ReactIR, and quantified by the previously developed calibration model (section 3.5), was used as measured data to fit against. Kinetic parameters were the growth rate constant, kg, activation energy, EA,g, and order with respect to supersaturation, g, for a power law relationship given by eqn (8) and (9). It was assumed that no nucleation took place within this seeded batch experiment.
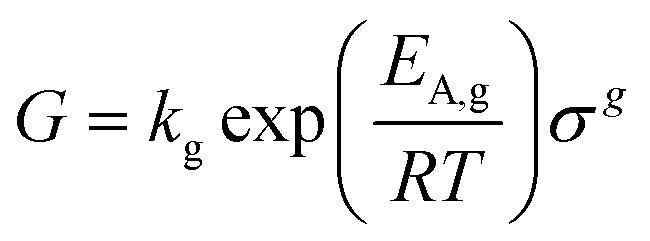 | (8) |
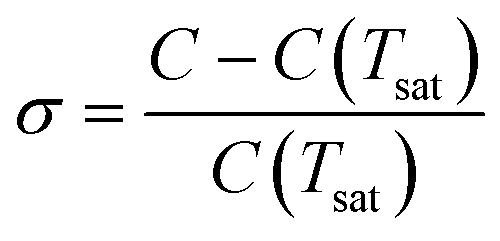 | (9) |
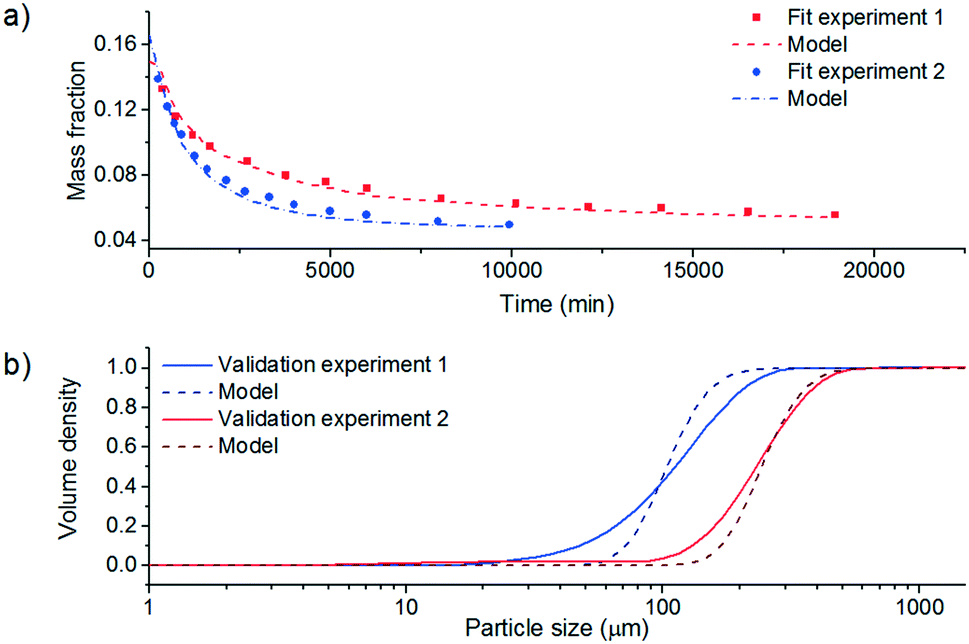 | ||
| Fig. 10 Comparison of experimental and model a) concentration profiles and b) product particle size distributions. | ||
| Model parameter | Value | Units |
|---|---|---|
| Activation energy, EA,g | 0 | J mol−1 |
| Growth rate constant, kg | 3.64 × 10−4 | m s−1 |
| Order with respect to supersaturation, g | 4.127 | |
| Weighted residual | 2.616 | |
| χ 2 value (95%) | 38.885 |
To validate these growth parameters and the mechanistic assumptions used the model and experimental PSDs of the product particles were compared for a pair of additional validation experiments (Fig. 10b). There is generally good agreement between the dv50 results, indicating that the growth parameters accurately represent the system. However, the experimental PSDs do show more small particles in the distribution than is predicted. This may be due to attrition, secondary nucleation or breakage taking place in the process or that particle breakage is induced by the particle sizing method. However, these model deviations were judged acceptable in light of the selected target specification and the model was sufficiently accurate to inform the remaining workflow stages. Where required, further investigations to determine additional kinetic parameters could be undertaken and various approaches have been described.76
3.8. Stage 7: proof of concept crystallisation
To implement the proof of concept crystallisation, consideration must be given to how seed crystals are generated and introduced to the process. As stated above, this is not the focus of this study, however a brief outline of the ancillary workflow used is provided. A rotor stator wet mill (RSWM) (MagicLab, IKA) was used for the generation of seed crystals in a recycle configuration based on a single MSMPR stage (Fig. 11). This method has been described elsewhere.82 Detailed description of the characterisation, modelling and parameter estimation for this unit will be covered in a follow up publication. The key parameter in this method is that the temperature of the single MSMPR stage controls the crystallisable mass and therefore the number of seeds that can be generated. For ease of operation, this seed generation unit was integrated with the other MSMPR stages. An integrated flowsheet was also produced in gCrystal 4.2.0 covering both the seed generation and crystal growth stages. | ||
| Fig. 11 Process and instrumentation diagram of configuration for stage 7. | ||
 | ||
| Fig. 12 Example a) response curve and b) design space showing probability of failure for a 110 μm product size. | ||
From the design space probabilities, the configuration to produce particles with a dv50 of 110 μm, whilst considering the previously determined constraints on stage supersaturation, was identified as stage volumes of 1500 mL, net flow rate of 100 g min−1 and stage temperatures of 77.3, 50 and 20 °C, respectively. Operating points of these MSMPR stages are shown in Fig. 9 to compare against the operating threshold for primary nucleation, secondary nucleation and fouling.
Equipment was configured to deliver the selected conditions (Fig. 11). The equipment consisted of a Microinnova Engineering GmBH miniflow plant acting as a feed unit (combining thermostat (Lauda, Proline 855), delivery pump with feedback control from a Coriolis mass flow meter (Siemens)), three 2 L glass reaction vessels (Radleys) each with a working volume of 1.5 L, three thermostat circulators (Lauda 420), a rotor stator wet mill (MagicLab, IKA) and two lab filter dryers (Powder Systems Ltd, Maxi Lab). Process monitoring was achieved through the mass flow meter, Pt100 probe in each vessel for temperature control, FBRM probe in all crystallisation vessels and a ReactIR15 in the 3rd crystallisation vessel. Continuous material transfer between crystallisation stages was maintained through application of positive pressure (N2 at 0.1 bar) to the first stage. Agitation in all crystallisation stages was provided by retreat curve impellers operated at 600 rpm and aided by three equally spaced baffles.
Preparation of the plant for start-up consisted of the following stages:
1. 25 L of feed solution was prepared in the feed vessel at a concentration of 188 g per kg solvent of paracetamol in 3-methyl-1-butanol. This solution was heated to 90 °C and maintained at this temperature until complete dissolution had occurred.
2. During this period, trace heating on the pipework and associated components (in-line filter, pump, mass flow meter) from the feed vessel to the first crystallisation vessel was commenced with a set point of 100 °C.
3. The crystalliser thermostats for each stage were set to their required temperatures of; 77.3, 50 and 20 °C, respectively.
4. Product valve, V-4 was set to recycle back to the feed vessel.
5. Each vessel was initially empty and un-pressurised.
Start-up was performed by transferring feed solution at 100 g min−1 (as controlled by mass flow meter and pump) into the seed generation stage. Once this stage was approximately 50% full the RSWM was switched on to trigger nucleation. Filling of this vessel continued until the 100% level (1.5 L) was reached at which point positive pressure was applied to enable material transfer to crystallisation stages 1 and 2. Once stage 2 reached its maximum level, material was transferred back to the feed vessel via a 3-way valve (V-4, Fig. 11). This recycle configuration allowed the cascade to reach steady state without consuming material. Once a steady state was achieved, as indicated by the PAT instruments showing minimum variance for 2 residence times, V-4 was turned to start product collection in the pair of pressure filter driers operating in duty and stand-by mode. Filtration was performed once 10 L of product suspension was collected. The filter cake was washed three times with two cake volumes of diethyl ether with agitation between washes to maximise removal of 3-methyl-1-butanol. Drying was performed under vacuum at 40 °C for 3 h. Diethyl ether was selected as a wash solvent based on the low solubility of paracetamol (stage 2 screen), miscibility with 3-methyl-1-butanol and low boiling point. A total of 3 continuous campaigns were performed to produce 6.5 kg of paracetamol over 6 filtration lots.
Fig. 13 shows the process trends collected from FBRM during one of the continuous campaigns. This highlights the dynamics of the start-up process showing steady state was not obtained until approximately 350 min. Optimisation of start-up conditions to minimise time to steady state was not in the scope of the current study. Operation in recycle continued until 440 min to ensure steady state operation was achieved. Once steady state was obtained and the process operated in single pass mode, the process trends remain relatively constant for the remaining 250 min of operation (the time taken to deplete the feed vessel). With a total mean residence time (across all three crystallisers) of approximately 45 min this would indicate a requirement to operate for 6 to 7 residence times before steady state was achieved. This is comparable to the generally accepted 7 to 10 residence times required for MSMPRs to reach steady state.96 From the FBRM measurements in Fig. 13, there is an increase of approximately 20 μm in the median size between stages 1 and 2 at steady state. In addition, Fig. 13b also shows the chord length distribution for the seeds generated.
 | ||
| Fig. 13 a) Example process trends from proof of concept crystallisation and b) chord length distributions across each MSMPR stage. | ||
Fig. 14 shows the results of particle size and shape analysis (measured by Malvern Instruments Morphologi G3) for the six filtration lots (A–F). In particular, Fig. 14a highlights the variation of PSD across the filtration lots in comparison to the average across all lots and the model predicted PSD from stage 6 of the workflow. Average dv50 for the lots was found to be 112.0 μm with a standard deviation of 16.2 μm. Similarly, the average span of the distributions was 1.66 with a standard deviation of 0.18. In comparison to the model predicted target size of a dv50 of 110 μm with a span of 1.53 it is concluded that the modelling framework was sufficient to accurately represent the process conditions and that the process configuration was operated to closely match the ideal conditions identified by the model. Fig. 14b gives the particle shape distributions relating to the elongation and circularity of the particles. These are features that are not captured by the 1-dimensional population balance model used to predict the process. As with the PSDs it can be seen that there is little variation across the filtration lots with median elongation and circularity values of 0.298 ± 0.013 and 0.849 ± 0.013, respectively. The values of both these measures suggest that particles are of low aspect ratio shape with low surface roughness. This is reflected in sample particles shown in Fig. 14c. It should be noted that the lot PSDs and average PSD is broader than that of the model predicted PSD. This could be for a number of reasons including:
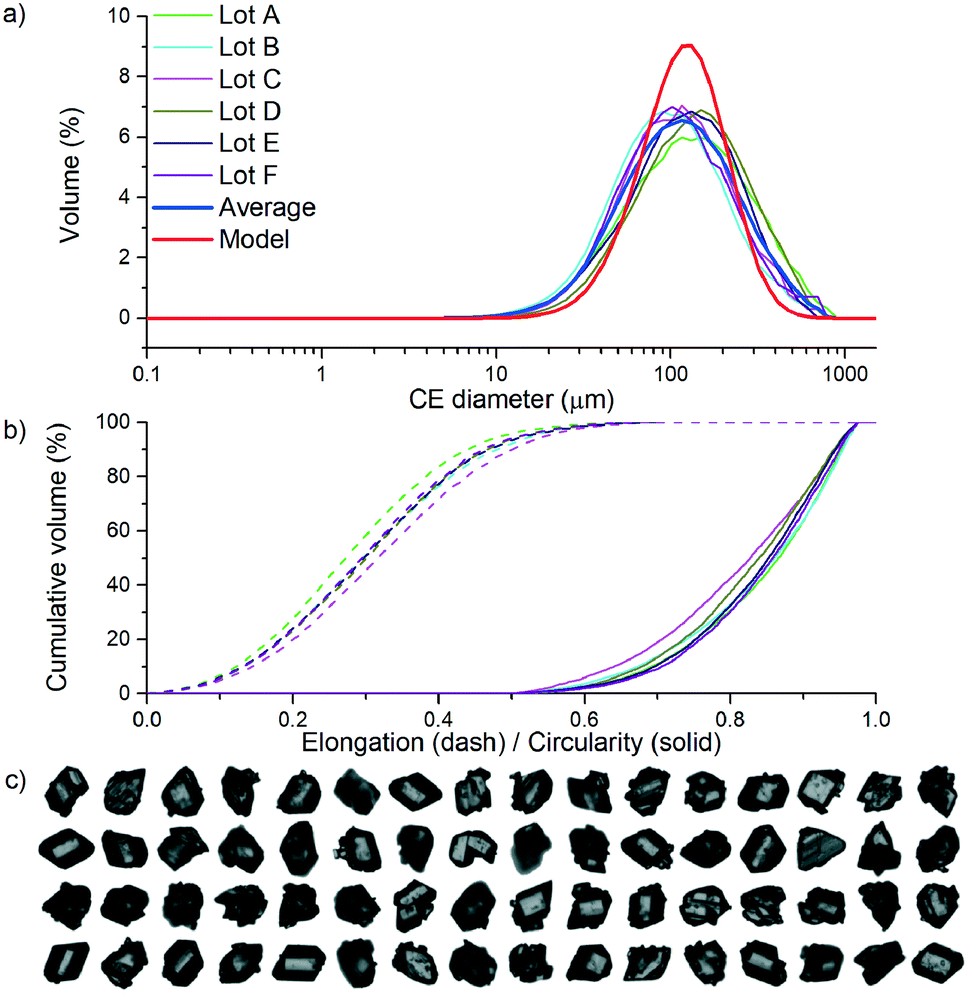 | ||
| Fig. 14 Comparison of product lot a) particle size distributions, b) particle elongation and circularity distributions and c) sample particle images. | ||
1. Unaccounted for crystallisation mechanism(s). As the experimental PSDs are broader in both directions this would indicated that both attrition and agglomeration may need to be accounted for. These mechanisms could be present in the crystalliser itself or could be induced during filtration and drying or occur during the PSD measurement method. A further explanation is that there could be a dispersion of growth rates within the crystallisation. Further investigation into these mechanisms would be merited where tighter control of the PSD is warranted.
2. Broader residence time distribution. The model assumed ideal uniform mixing throughout the process. However, despite the MSMPR being baffled to ensure it was as well mixed it is conceivable that product removal between vessels was not representative leading to a broader residence time distribution. As a result, particles would experience a wider range of growth times leading to a broader PSD.
In addition to the particle size analysis, additional tests were performed to give an indication of the product lots' performance in downstream unit operations. This included measurement of the product powder bulk and tapped density determined according to USP 616 Method I (Autotap, Quantachrome Instruments). The Hausner ratio was calculated according to USP 1174 and is shown in Fig. 15. Values ranging from 1.14 to 1.21 indicated that the product lots' flow character ranges from good to fair.
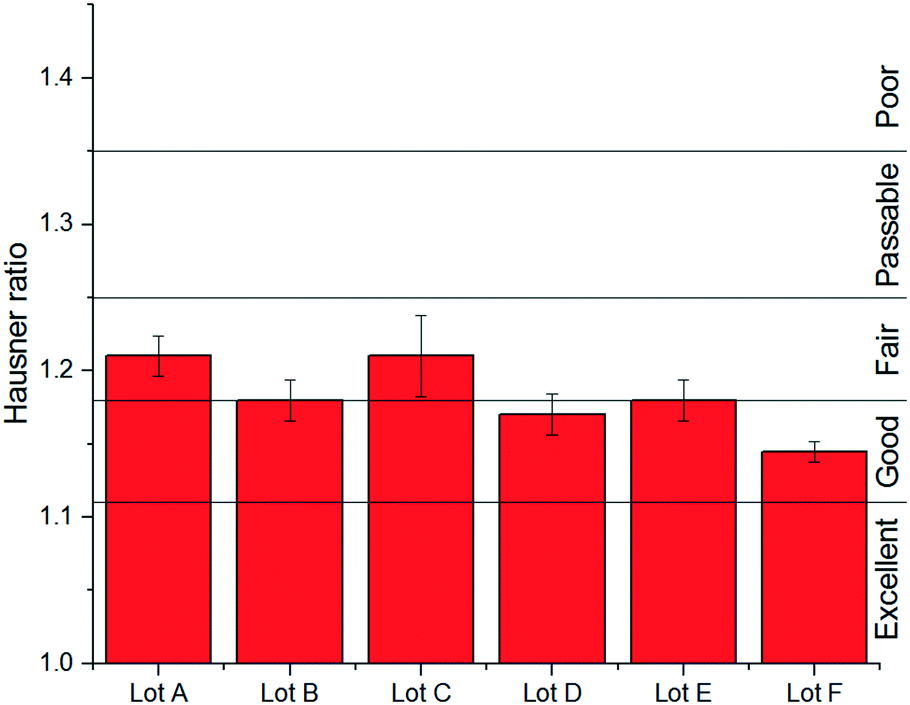 | ||
| Fig. 15 Comparison of Hausner ratio of product lots. | ||
3.9. Summary
Through the application of a systematic workflow for the design of a seeded paracetamol crystallisation process, this case study has demonstrated the selection of a suitable crystallisation solvent (3-methyl-1-butanol) which achieves a high yield whilst maintaining the desired solid form and minimising issues such as agglomeration or fouling. Suitable PAT was also identified to monitor the crystallisation process with appropriate accuracy (on-line FTIR-ATR). An array of testbeds was used to study crystallisation behaviour across a range of crystallisation platforms. MSMPR and PFR crystallisers were identified as suitable. An MSMPR platform was selected as an example for this case study. Based on this platform a mechanistic model capturing the crystal growth kinetics was developed. Utilising this model a range of process configurations were investigated covering; number of stages, stage volume, stage temperature and net flow rate. An integrated crystalliser configuration consisting of a RSWM with MSMPR for seed generation and 2 stage MSMPR with a mean residence time of 45 min was selected as suitable for the production of particles with a dv50 of 110 μm. Finally, based on this proposed configuration a proof of concept crystallisation was performed operating at the set points pinpointed. This proof of concept covered three continuous campaigns over 12.5 h leading to the production of 6.5 kg of paracetamol with a dv50 of 112 ± 16 μm. Furthermore, characterisation of the product lots revealed that the product particles showed good to fair flow character.This methodology was also deployed to produce an additional 2 size bands of paracetamol (dv50s of 50 and 70 μm, respectively) in the same solvent system but in different process configurations, details of these will follow in subsequent publications.
4. Applications of a workflow
4.1. Meta-analysis/machine learning
With an increasing emphasis placed on efficiency, control and mechanistic understanding, model-based tools are seeing greater utility in pharmaceutical manufacturing.97–99 In the context of continuous crystallisation, a wealth of in-line and off-line data sources exist which can potentially be used to construct models. A key use of machine learning models here, given the constraints on time and materials, would be to predict experimental outcomes, reducing the number of experiments that must be carried out in the laboratory. However, machine learning approaches require at least a modest-sized dataset for training before they can make accurate predictions. Since experimental outcomes in this area tend to be highly dependent on many precise operating parameters used, a pre-existing and exactly matching dataset is often unavailable. The implementation of a systematic workflow creates powerful new opportunities for data mining and meta-analysis of experimental results. With access to consistent, fully contextualised data from multiple APIs across all workflow stages, it becomes possible to explore inter-experimental relationships.For the case study described, limited meta-analytical work could be demonstrated since experimental data points were only generated for paracetamol. However, a simple example is presented to illustrate the current capabilities and limitations of predictive modelling when working without pre-existing experimental data. MSZW data for each solvent short-listed at stage 3 were chosen, since this kinetic measurement is highly sensitive to experimental conditions.100,101 A binary response was created of the data: “yes” for data points where a metastable zone was detected over the course of the experiment (regardless of width) and “no” for those where no metastable zone was detected. For each solvent, molecular descriptors were calculated using MOE.102 Descriptors were then filtered for correlation with a threshold of 0.6 before separating the data into training and testing sets. A total of 52 data points were available (several concentrations per solvent): 28 were assigned to the training set and the rest to the testing set, maintaining an even spread of data points per solvent and data points per concentration across both sets.
The randomForest package within R103 was used to train a random forest (RF) model. For information on the RF method, the reader is referred to a selection of publications.104–106 The model was then used to predict the detection of a metastable zone in test set experiments, achieving 91.7% accuracy (22 of 24 cases correct). Fig. 16 shows a multi-dimensional scaling (MDS) plot of the test set's proximities. In brief, this is a spatial representation of how often cases received the same classification in the model. Ideally, all cases of each class would be highly proximal, appearing in tight clusters (one per class) in MDS space. Even with a small dataset, this RF model performed well; this is because the question it provided predictions for was tailored around the available data. It was necessarily a highly specific question, namely whether a metastable zone will be detected for a given concentration of paracetamol under the precise experimental conditions used throughout. Changes to any experimental parameters, including the identity of the solute, would be out of the model's scope, requiring it to be retrained with a different dataset. This limited the utility of the model to intra-experimental predictions, i.e. further data points beyond the 28 that had to be found experimentally.
 | ||
| Fig. 16 Multi-dimensional scaling of RF prediction proximities. | ||
The above scenario is prevalent in the context of continuous crystallisation: while great quantities of experimental data are generated, they often cannot be combined and repurposed to construct models due to the vast number of varying parameters associated with them. Following a consistent, well-defined workflow lays the foundations necessary for the collection of systematic, comparable experimental data. Each successive compound progressing through it augments the knowledge base, enabling the mining of data points from previous workflow iterations and the construction of cross-API models (cf. the above example model constrained to only a single API). A source of reliable and consistent data, facilitated by a systematic workflow, is an essential platform towards the ultimate goal of predictive crystallisability versus manufacturability.
4.2. Future supply chain design
The emergence of both new technologies and therapy areas has the potential for dramatically changing the pharmaceutical manufacturing and supply chain landscape.107,108 One key research area involves the design of more agile continuous processing based supply chains, in order to manage future demand requirements of increased product complexity, more product variety, shorter product lifecycles, and smaller drug volumes.109This section summarises how a crystallisation workflow may be utilised to inform the design of future pharmaceutical supply chains. The workflow serves as an important asset in the development of a standardised data acquisition, analysis and reporting system, and may help identify emerging patterns and clusters of drug products that may benefit from similar supply chain design and reconfiguration opportunities in the future (in better understanding the design space, accelerating and de-risking development, and reducing costs). In conjunction with a supply chain analytical framework previously reported,108 recent studies have shown network design activities integrating outputs from the crystallisation workflow to evaluate potential benefits and opportunities for case-specific integrated end-to-end supply chain scenarios.107 The integrated process is summarised in Fig. 17.
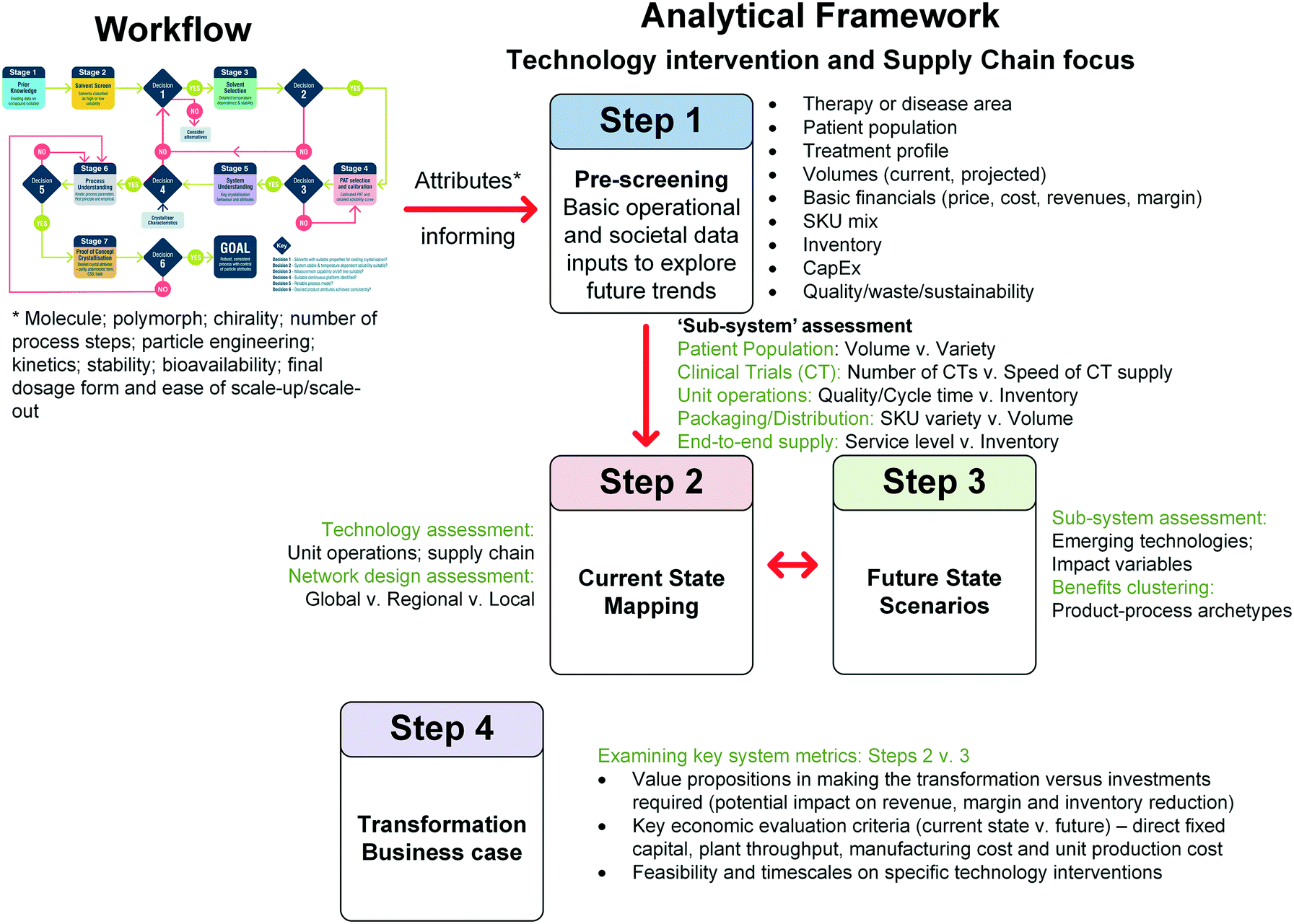 | ||
| Fig. 17 Workflow attributes informing supply chain design. | ||
In addition to informing workflow activities, process and network designers are using outputs from stage 1 of the crystallisation workflow for rapid product assessment and continuous process selection. In addition to yield, quality, form, purity, and consistency, ten critical ‘attributes’ – in terms of technology feasibility and chemistry are considered at this pre-screening step,107 with a focus on complexity and area(s) of opportunity in the context of ‘continuous’ processing:
• Molecule
• Polymorph
• Chirality
• Number of process steps
• Particle engineering
• Kinetics
• Stability
• Bioavailability
• Final dosage form
• Ease of scale-up/scale-out
Crystallisation workflow outputs also inform the mapping of the ‘current state’, in terms of technologies, unit operations and the supply chain. The first current state mapping exercise identifies those unit operations where an existing batch production process may be ‘pre-disposed’ to a series of continuous technologies (in terms of current state and future potential), namely, in synthesis; purification; isolation; formulation and packaging. While evidence exists that continuous processing delivers financial benefits for single-purpose plants, a business case for transformation assessing the resultant impact across the end-to-end (E2E) supply chain is needed for such a technology to be better quantified, and for the upstream and downstream linkages to emerging continuous process technologies (e.g. in synthesis and work-up, filtration, drying, secondary processing) to be effectively exploited. The second current state mapping exercise examines network performance where, crystallisation workflow outputs involving technology interventions inform 15 impact variables, namely: inventory; lead-time to supply; lead-time to market; scale-up (going into); volume flexibility (mix and volume); process control (including reliability and safety); quality (purity); yield; IP protection and extension (including issue of counterfeits); cost (process, packaging and transport); investment cost (incorporating financial impacts and return on investment); fiscal/tax; environmental impact; viability/adaptability and asset utilisation. For these mapping exercises, the specific implications of defined continuous technology developments and readiness levels may be assessed using outputs from the crystallisation workflow.
Finally, in this section, assessment of future state models generally explores several potential future options prior to a final decision, with alternative states based on emerging process and production technologies that are still yet to be fully developed. Critically, these alternative ‘states’ can be informed/modelled using crystallisation workflow outputs. Stages 2–7 of the crystallisation workflow can also support a performance assessment of several potential technological choices involving batch, continuous or hybrid routes, providing a benchmark – in terms of potential yield and purity, and evaluating various scenarios, which may involve alternative scale production footprints.
Future work in this area will focus on digital supply chains and the integration of crystallisation-type workflows to enable the rapid digital design of products and processes, to develop design rules for manufacturing and supply chains, and construct a library of functions to match (virtual) products to their optimal supply chain design.
4.3. Formulation and drug product
Quality by Design (QbD) principles have been used to advance product and process quality in industry and have also been adopted by the U.S. Food and Drug Administration (FDA) and other regulatory agencies for the discovery, development, and manufacture of drugs.110 Under a QbD approach, product and process performance characteristics are scientifically designed to meet specific objectives, not merely empirically derived from performance of test batches. The formulation development of a drug product is based on drug substance attributes and a target product profile (TPP), detailing (amongst other product attributes) the desired dosage form, dose range, release profile and shelf life. Ticehurst and Marziano111 described that the internal structure (solid form selection) and particle engineering of a drug substance are interconnected to key API properties which in turn can affect drug product attributes (Fig. 18). Both the TPP and the drug substance attributes will influence the choice of excipients for the drug product formulation, the processability of the drug substance and the resulting formulation in downstream processes (flowability, blending, compressibility), stability of formulation/drug product as well as the product performance (disintegration, dissolution, bioavailability).112 | ||
| Fig. 18 The link between solid form selection (internal structure), particle engineering and key active pharmaceutical ingredient properties and drug product attributes. Copyright (2015) Wiley. Used with permission from Ticehurst, M. D. and Marziano, I. (2015), Integration of active pharmaceutical ingredient solid form selection and particle engineering into drug product design. J. Pharm. Pharmacol., 67, 782–802. | ||
The systematic workflow approach for cooling crystallisation design enables QbD and therefore should enable downstream processes in pharmaceutical drug development to work with consistently high quality material and avoid the costly variability often experienced in drug substances in batch processes.92 The resulting well characterised continuous crystallisation process, based on data driven decisions, will inform downstream product development processes with much more detail regarding the drug substance characteristics and particle engineering possibilities, which in turn may allow the formulator to design processes that can directly ‘dial a particle’ with the desired attributes. This information may also allow for early identification of appropriate excipient selection and processing routes, focusing formulation/drug product development activities in a much more specific manner. Whilst significant progress has been made in multi-scale modelling, further developments of the underlying materials science and process physics will be required to realise this vision.
5. Conclusions and vision
Through this work, we have demonstrated how a systematic workflow for the design of a seeded continuous cooling crystallisation can be used to design and operate a robust process to meet a specific target size by first intent. To achieve this aim, the workflow comprises clearly defined tasks that allow for data driven decisions. Lab automation and design of experiment approaches have been shown to minimise the resource requirements in both time and materials. To achieve this robustness, common crystallisation issues such as agglomeration and fouling are identified at an early stage by considering them during the solvent selection phase allowing them to be avoided without significant rework. This first development and demonstration of the workflow took 2 researchers 12 months with significant additional ad hoc support from other researchers and required 5 kg of paracetamol. Subsequent demonstrations with other APIs have taken a single researcher 3 months with 500 g to complete (to be reported in follow-up articles).This workflow addresses a specific type of crystallisation process with prescribed assumptions (solid state, impurity profile, etc.). Therefore, the future direction for this research is the development of workflows to address the limitations of the current workflow (such as an unseeded version of the cooling crystallisation and a workflow for the generation of seed crystals), alternative crystallisations (anti-solvent, evaporative, reactive, etc.) and to develop workflows for other unit operations (filtration, washing, drying, formulation, etc.). In some cases, these workflows will share common stages, e.g. solvent screening and selection, to maximise development efficiency. An overarching workflow for selection of overall process architecture is also a desirable goal. To enable a step change in pharmaceutical manufacturing this higher-level pathway could use product specifications and predictive tools to be able to identify the required unit operations, select appropriate workflows for process design and operate all unit operations from fundamental molecular descriptors.
Nomenclature and abbreviations
| Term | Description |
| a | Solubility parameter 1 |
| b | Solubility parameter 2 |
| B | Secondary nucleation rate (unit: # per min) |
| C | Concentration (unit: g per kg solvent) |
| C(Tf) | Solute concentration at final isolation temperature (unit: g per kg solvent) |
| C(T0) | Solute concentration at saturation temperature (unit: g per kg solvent) |
| E A,g | Activation energy (unit: J mol−1) |
| g | Order with respect to supersaturation |
| G | Growth rate (unit: μm min−1) |
| k g | Growth rate constant (unit: m s−1) |
| L p | Product crystal size (unit: μm) |
| L s | Seed crystal size (unit: μm) |
| m seed | Mass flow rate of seed crystals (unit: g min−1) |
| m solvent | Mass flow rate of solvent (unit: kg min−1) |
| M seed | Mass ratio of seed or seed loading |
| N | Particle count (unit: #) |
| R B | Primary and secondary nucleation boundary ratio |
| S | Supersaturation ratio |
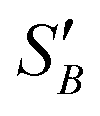
| Primary nucleation supersaturation boundary |
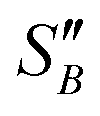
| Secondary nucleation supersaturation boundary |
| T | Temperature (unit: °C) |
| t 0 | Time vial is placed into crystalline (unit: min) |
| t n | Time primary nucleation is expected (unit: min) |
| Y t | Mass theoretical yield (unit: %) |
| Greek letters | |
| σ | Relative supersaturation |
| τ | Residence time (unit: min) |
| ΔTsd | Temperature change required to double solubility (unit: °C) |
| Abbreviation | |
| API | Active pharmaceutical ingredient |
| ATR | Attenuated total reflectance |
| CM | Continuous manufacturing |
| CPP | Critical process parameter |
| CQA | Critical quality attribute |
| CSD | Crystal size distribution |
| DoE | Design of experiments |
| DSC | Differential scanning calorimetry |
| E.T. | Elevated temperature |
| E2E | End to end |
| FBRM | Focused beam reflectance measurement |
| FTIR | Fourier transform infrared spectroscopy |
| GC | Gas chromatography |
| HPLC | High performance liquid chromatography |
| ICH | International conference on harmonisation |
| LC-MS | Liquid chromatography-mass spectroscopy |
| MB-OBC | Moving baffle oscillatory baffled crystalliser |
| MDS | Multidimensional scale |
| MF-OBC | Moving fluid oscillatory baffled crystalliser |
| MIR | Mid infrared |
| MOC | Material of construction |
| MOE | Molecular operating environment |
| MSMPR | Mixed suspension, mixed product removal |
| MSZW | Metastable zone width |
| NIR | Near infrared spectroscopy |
| NMR | Nuclear magnetic resonance spectroscopy |
| PABA | para-Aminobenzoic acid |
| PAT | Process analytical technology |
| PBM | Population balance model |
| PBT | Pitched blade turbine |
| PFR | Plug flow reactor |
| PLS | Partial least squares |
| PSD | Particle size distribution |
| QbD | Quality by design |
| R.T. | Room temperature |
| RF | Random forest |
| RSWM | Rotor stator wet mill |
| RTD | Residence time distribution |
| SKU | Stock keeping unit |
| TPP | Target product profile |
| UPLC | Ultra performance liquid chromatography |
| USP | United States Pharmacopeia |
| UV-vis | Ultraviolet-visible spectroscopy |
| XRD | X-Ray diffraction |
| XRPD | X-Ray powder diffraction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank EPSRC, Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation (Grant Ref EP/I033459/1) and Computationally Designed Templates for Exquisite Control of Polymorphic Form (Grant Ref EP/K039229/1) for funding this work. The authors would like to acknowledge that this work was carried out in the CMAC National Facility supported by UKRPIF (UK Research Partnership Fund) award from the Higher Education Funding Council for England (HEFCE) (Grant Ref HH13054). All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at http://dx.doi.org/10.15129/37e8aaf0-4153-4e7a-b00a-13daa26310de. The authors would also like to thank the following for their support and assistance throughout this work: Niall Mitchell (Process Systems Enterprise), Jon Goode (Mettler Toledo), Ian Haley (Mettler Toledo), John Mulgrew (CMAC), Helen Feilden (CMAC), Natalia Dabrowska (CMAC), Ian Houson (CMAC) and the CMAC Tier 1 companies Technical Committee.References
- J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887–895 CAS.
- J. W. Mullin, Crystallization, Elsevier Science, 2001 Search PubMed.
- F. Kesisoglou and Y. Wu, AAPS J., 2008, 10, 516–525 CrossRef CAS PubMed.
- Global Pharmaceutical Industry - Statistics and Facts, https://www.statista.com/topics/1764/global-pharmaceutical-industry/, (accessed 3rd Nov, 2016).
- P. Basu, G. Joglekar, S. Rai, P. Suresh and J. Vernon, J. Pharm. Innov., 2008, 3, 30–40 CrossRef.
- G. Karris, How will changes in India and China affect API outsourcing?, http://www.contractpharma.com/issues/2002-09/view_features/api-manufacturing, (accessed 3rd Nov, 2016).
- The Metamorphosis of Manufacturing. From art to science, http://www-935.ibm.com/services/us/imc/pdf/ge510-4034-metamorphosis-of-manufacturing.pdf, (accessed 3rd Nov, 2016).
- J. H. ter Horst, C. Schmidt and J. Ulrich, in Handbook of Crystal Growth, Elsevier, Boston, 2015, 2nd edn, pp. 1317–1349, DOI:10.1016/B978-0-444-63303-3.00032-8.
- I. R. Baxendale, R. D. Braatz, B. K. Hodnett, K. F. Jensen, M. D. Johnson, P. Sharratt, J.-P. Sherlock and A. J. Florence, J. Pharm. Sci., 2015, 104, 781–791 CrossRef CAS PubMed.
- Z. K. Nagy, Comput. Chem. Eng., 2009, 33, 1685–1691 CrossRef CAS.
- W. Beckmann, Crystallization: Basic Concepts and Industrial Applications, Wiley, 2013 Search PubMed.
- T. Page, H. Dubina, G. Fillipi, R. Guidat, S. Patnaik, P. Poechlauer, P. Shering, M. Guinn, P. McDonnell and C. Johnston, J. Pharm. Sci., 2015, 104, 821–831 CrossRef CAS PubMed.
- G. Allison, Y. T. Cain, C. Cooney, T. Garcia, T. G. Bizjak, O. Holte, N. Jagota, B. Komas, E. Korakianiti, D. Kourti, R. Madurawe, E. Morefield, F. Montgomery, M. Nasr, W. Randolph, J.-L. Robert, D. Rudd and D. Zezza, J. Pharm. Sci., 2015, 104, 803–812 CrossRef CAS PubMed.
- L. X. Yu, Pharm. Res., 2008, 25, 781–791 CrossRef CAS PubMed.
- L. X. Yu, Continuous Manufacturing Has a Strong Impact on Drug Quality, FDA Voice, 2016, vol. 2016 Search PubMed.
- S. Chatterjee, presented in part at the IFPAC Annual Meeting, Baltimore, 2012 Search PubMed.
- C. Badman and B. L. Trout, J. Pharm. Sci., 2015, 104, 779–780 CrossRef CAS PubMed.
- A. J. Alvarez and A. S. Myerson, Cryst. Growth Des., 2010, 10, 2219–2228 CAS.
- R. J. P. Eder, S. Schrank, M. O. Besenhard, E. Roblegg, H. Gruber-Woelfler and J. G. Khinast, Cryst. Growth Des., 2012, 12, 4733–4738 CAS.
- L. Zhao, V. Raval, N. E. B. Briggs, R. M. Bhardwaj, T. McGlone, I. D. H. Oswald and A. J. Florence, CrystEngComm, 2014, 16, 5769–5780 RSC.
- H. Zhang, R. Lakerveld, P. L. Heider, M. Tao, M. Su, C. J. Testa, A. N. D'Antonio, P. I. Barton, R. D. Braatz, B. L. Trout, A. S. Myerson, K. F. Jensen and J. M. B. Evans, Cryst. Growth Des., 2014, 14, 2148–2157 CAS.
- A. J. Alvarez, A. Singh and A. S. Myerson, Cryst. Growth Des., 2011, 11, 4392–4400 CAS.
- S. Ferguson, F. Ortner, J. Quon, L. Peeva, A. Livingston, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2014, 14, 617–627 CAS.
- S. D. Schaber, D. I. Gerogiorgis, R. Ramachandran, J. M. B. Evans, P. I. Barton and B. L. Trout, Ind. Eng. Chem. Res., 2011, 50, 10083–10092 CrossRef CAS.
- S. Lawton, G. Steele, P. Shering, L. Zhao, I. Laird and X.-W. Ni, Org. Process Res. Dev., 2009, 13, 1357–1363 CrossRef CAS.
- R. Vacassy, J. Lemaître, H. Hofmann and J. H. Gerlings, AIChE J., 2000, 46, 1241–1252 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- K. Plumb, Chem. Eng. Res. Des., 2005, 83, 730–738 CrossRef CAS.
- R. J. P. Eder, S. Radl, E. Schmitt, S. Innerhofer, M. Maier, H. Gruber-Woelfler and J. G. Khinast, Cryst. Growth Des., 2010, 10, 2247–2257 CAS.
- H. Siddique, C. J. Brown, I. Houson and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1871–1881 CrossRef CAS.
- K. M. Wells, D. A. Beauchamp, A. Fermier, W. Wu, S. Mehrman, S. Youells, Z. Hu, X. Li, M. J. Macielag, G. Bignan, G. Xu, B.-P. Zhao, L. Huang, J. Weber, M. D. Gaul and R. K. Russell, Cryst. Growth Des., 2013, 13, 4635–4641 CAS.
- T. S. Walter, J. M. Diprose, C. J. Mayo, C. Siebold, M. G. Pickford, L. Carter, G. C. Sutton, N. S. Berrow, J. Brown, I. M. Berry, G. B. E. Stewart-Jones, J. M. Grimes, D. K. Stammers, R. M. Esnouf, E. Y. Jones, R. J. Owens, D. I. Stuart and K. Harlos, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 651–657 CrossRef PubMed.
- Y. S. Cheng, K. W. Lam, K. M. Ng and C. Wibowo, AIChE J., 2010, 56, 633–649 CAS.
- H.-H. Tung, E. L. Paul, M. Midler and J. A. McCauley, in Crystallization of Organic Compounds, John Wiley & Sons, Inc., 2008, ch. 11, pp. 235–277, DOI:10.1002/9780470447796.
- A. Rashid, E. White, T. Howes, J. Litster and I. Marziano, Org. Process Res. Dev., 2017, 21, 187–194 CrossRef CAS.
- S. K. Bermingham, A design procedure and predictive models for solution crystallisation processes, Delft University of Technology, TU Delft, 2003 Search PubMed.
- L. J. Shaikh, A. H. Bari, V. V. Ranade and A. B. Pandit, Ind. Eng. Chem. Res., 2015, 54, 10539–10548 CrossRef CAS.
- H. G. Jolliffe and D. Gerogiorgis, Chem. Eng. Res. Des., 2015, 97, 175–191 CrossRef CAS.
- S. K. Teoh, C. Rathi and P. Sharratt, Org. Process Res. Dev., 2016, 20, 414–431 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- R. M. Bhardwaj, A. Johnston, B. F. Johnston and A. J. Florence, CrystEngComm, 2015, 17, 4272–4275 RSC.
- F. S. Mortimer, J. Am. Chem. Soc., 1922, 44, 1416–1429 CrossRef CAS.
- M. Robertson, T. McGlone, A. Johnston, A. J. Florence, B. Johnston, J. Dziewierz, C. Tachtatzis, A. Cleary, A. Gachagan, I. Andonovic and J. Sefcik, Proceedings of ACM International Conference on Multimedia Retrieval (ICMR), Shanghai, China, 2015 Search PubMed.
- International Conference on Harmonisation, Impurities: Guideline for Residual solvents Q3C(R5), 2011 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- S. Black, L. Dang, C. Liu and H. Wei, Org. Process Res. Dev., 2013, 17, 486–492 CrossRef CAS.
- W. Beckmann, Org. Process Res. Dev., 2000, 4, 372–383 CrossRef CAS.
- N. Doki, N. Kubota, A. Sato, M. Yokota, O. Hamada and F. Masumi, AIChE J., 1999, 45, 2527–2533 CrossRef CAS.
- G. Milosovich, J. Pharm. Sci., 1964, 53, 484–487 CrossRef CAS PubMed.
- M. A. O'Mahony, D. M. Croker, Å. C. Rasmuson, S. Veesler and B. K. Hodnett, Org. Process Res. Dev., 2013, 17, 512–518 CrossRef.
- L. Di, P. V. Fish and T. Mano, Drug Discovery Today, 2012, 17, 486–495 CrossRef CAS PubMed.
- A. Klamt, F. Eckert, M. Hornig, M. E. Beck and T. Bürger, J. Comput. Chem., 2002, 23, 275–281 CrossRef CAS PubMed.
- C. Loschen and A. Klamt, J. Pharm. Pharmacol., 2015, 67, 803–811 CrossRef CAS PubMed.
- T. Spyriouni, X. Krokidis and I. G. Economou, Fluid Phase Equilib., 2011, 302, 331–337 CrossRef CAS.
- M. Mirmehrabi and S. Rohani, Can. J. Chem. Eng., 2004, 82, 335–342 CrossRef CAS.
- S. Gracin, T. Brinck and Å. C. Rasmuson, Ind. Eng. Chem. Res., 2002, 41, 5114–5124 CrossRef CAS.
- T. Chen and E. Martin, J. Chemom., 2007, 21, 198–207 CrossRef CAS.
- P. Billot, M. Couty and P. Hosek, Org. Process Res. Dev., 2010, 14, 511–523 CrossRef CAS.
- S. S. Kadam, A. Mesbah, E. van der Windt and H. J. M. Kramer, Chem. Eng. Res. Des., 2011, 89, 995–1005 CrossRef CAS.
- E. Simone, A. N. Saleemi and Z. K. Nagy, Chem. Eng. Res. Des., 2014, 92, 594–611 CrossRef CAS.
- Z.-P. Chen, J. Morris and E. Martin, Anal. Chem., 2005, 77, 1376–1384 CrossRef CAS PubMed.
- C. M. Chew, R. I. Ristic, R. D. Dennehy and J. J. De Yoreo, Cryst. Growth Des., 2004, 4, 1045–1052 CAS.
- G. Power, G. Hou, V. K. Kamaraju, G. Morris, Y. Zhao and B. Glennon, Chem. Eng. Sci., 2015, 133, 125–139 CrossRef CAS.
- T.-T. C. Lai, J. Cornevin, S. Ferguson, N. Li, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2015, 15, 3374–3382 CAS.
- S. Ferguson, G. Morris, H. Hao, M. Barrett and B. Glennon, Chem. Eng. Sci., 2013, 104, 44–54 CrossRef CAS.
- T. Tari, Z. Fekete, P. Szabó-Révész and Z. Aigner, Int. J. Pharm., 2015, 478, 96–102 CrossRef CAS PubMed.
- L. Hohmann, R. Gorny, O. Klaas, J. Ahlert, K. Wohlgemuth and N. Kockmann, Chem. Eng. Technol., 2016, 39, 1268–1280 CrossRef CAS.
- L. Zhao, V. Raval, N. E. B. Briggs, R. M. Bhardwaj, T. McGlone, I. D. H. Oswald and A. J. Florence, CrystEngComm, 2014, 16, 5769–5780 RSC.
- C. J. Brown, J. A. Adelakun and X.-W. Ni, Chem. Eng. Process.: Process Intesif., 2015, 97, 180–186 CrossRef CAS.
- K. Robertson, P.-B. Flandrin, A. R. Klapwijk and C. C. Wilson, Cryst. Growth Des., 2016, 16, 4759–4764 CAS.
- P. Neugebauer and J. G. Khinast, Cryst. Growth Des., 2015, 15, 1089–1095 CAS.
- O. Narducci, A. G. Jones and E. Kougoulos, Org. Process Res. Dev., 2011, 15, 974–980 CrossRef CAS.
- L. Eriksson, Design of Experiments: Principles and Applications, Umetrics Academy, 2008 Search PubMed.
- H. M. Omar and S. Rohani, Cryst. Growth Des., 2017, 17, 4028–4041 CAS.
- J.-F. Pérez-Calvo, S. S. Kadam and H. J. M. Kramer, AIChE J., 2016, 62, 3992–4012 CrossRef.
- Z. K. Nagy and R. D. Braatz, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 55–75 CrossRef CAS PubMed.
- O. S. Agimelen, P. Hamilton, I. Haley, A. Nordon, M. Vasile, J. Sefcik and A. J. Mulholland, Chem. Eng. Sci., 2015, 123, 629–640 CrossRef CAS.
- O. S. Agimelen, A. Jawor-Baczynska, J. McGinty, J. Dziewierz, C. Tachtatzis, A. Cleary, I. Haley, C. Michie, I. Andonovic, J. Sefcik and A. J. Mulholland, Chem. Eng. Sci., 2016, 144, 87–100 CrossRef CAS.
- Y. Cui, M. O'Mahony, J. J. Jaramillo, T. Stelzer and A. S. Myerson, Org. Process Res. Dev., 2016, 20, 1276–1282 CrossRef CAS.
- N. E. B. Briggs, U. Schacht, V. Raval, T. McGlone, J. Sefcik and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1903–1911 CrossRef CAS.
- Y. Yang, L. Song, T. Gao and Z. K. Nagy, Cryst. Growth Des., 2015, 15, 5879–5885 CAS.
- K. A. Powell, A. N. Saleemi, C. D. Rielly and Z. K. Nagy, Chem. Eng. Process.: Process Intesif., 2015, 97, 195–212 CrossRef CAS.
- A. S. Myerson, M. Krumme, M. Nasr, H. Thomas and R. D. Braatz, J. Pharm. Sci., 2015, 104, 832–839 CrossRef CAS PubMed.
- Z. K. Nagy, G. Fevotte, H. Kramer and L. L. Simon, Chem. Eng. Res. Des., 2013, 91, 1903–1922 CrossRef CAS.
- R. R. McKeown, J. T. Wertman and P. C. Dell'Orco, in Chemical Engineering in the Pharmaceutical Industry, John Wiley & Sons, Inc., 2010, ch. 13, pp. 213–247, DOI:10.1002/9780470882221.
- A. G. Jones, SPS DR17. (Harwell/Warren Spring: Separation Process Service), 1984, vol. 40 Search PubMed.
- R. M. Vrcelj, N. I. B. Clark, A. R. Kennedy, D. B. Sheen, E. E. A. Shepherd and J. N. Sherwood, J. Pharm. Sci., 2003, 92, 2069–2073 CrossRef CAS PubMed.
- B. A. Zakharov, A. G. Ogienko, A. S. Yunoshev, A. I. Ancharov and E. V. Boldyreva, CrystEngComm, 2015, 17, 7543–7550 RSC.
- F. L. Muller, M. Fielding and S. Black, Org. Process Res. Dev., 2009, 13, 1315–1321 CrossRef CAS.
- R. A. Granberg and Å. C. Rasmuson, J. Chem. Eng. Data, 1999, 44, 1391–1395 CrossRef CAS.
- T. McGlone, N. E. B. Briggs, C. A. Clark, C. J. Brown, J. Sefcik and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1186–1202 CrossRef CAS.
- J. Nývlt and F. Veverka, Cryst. Res. Technol., 1997, 32, 773–781 CrossRef.
- C. Borchert and K. Sundmacher, Chem. Eng. Technol., 2011, 34, 545–556 CrossRef CAS.
- E. M. Ålander, M. S. Uusi-Penttilä and Å. C. Rasmuson, Ind. Eng. Chem. Res., 2003, 43, 629–637 CrossRef.
- A. D. Randolph and M. A. Larson, Theory of particulate processes: analysis and techniques of continuous crystallization, Academic Press, 1988 Search PubMed.
- E. Tomba, P. Facco, F. Bezzo and M. Barolo, Int. J. Pharm., 2013, 457, 283–297 CrossRef CAS PubMed.
- A. Burggraeve, T. Monteyne, C. Vervaet, J. P. Remon and T. D. Beer, Eur. J. Pharm. Biopharm., 2013, 83, 2–15 CrossRef CAS PubMed.
- D. Rossetti, S. Squartini and S. Collura, in Advances in Neural Networks: Computational Intelligence for ICT, ed. S. Bassis, A. Esposito, F. C. Morabito and E. Pasero, Springer International Publishing, Cham, 2016, pp. 335–343, DOI:10.1007/978-3-319-33747-0_33.
- S. S. Kadam, S. A. Kulkarni, R. Coloma Ribera, A. I. Stankiewicz, J. H. ter Horst and H. J. M. Kramer, Chem. Eng. Sci., 2012, 72, 10–19 CrossRef CAS.
- F. L. Nordstrom, M. Svard and A. C. Rasmuson, CrystEngComm, 2013, 15, 7285–7297 RSC.
- Molecular Operating Environment (MOE), Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2017 Search PubMed.
- R Development Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2008, ISBN 3-900051-07-0, http://www.R-project.org Search PubMed.
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, J. Chem. Inf. Comput. Sci., 2003, 43, 1947–1958 CrossRef CAS PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- J. B. O. Mitchell, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 468–481 CrossRef CAS PubMed.
- T. S. Harrington, M. A. Phillips and J. S. Srai, Int. J. Prod. Res., 2016, 1–17, DOI:10.1080/00207543.2016.1221541.
- J. S. Srai, T. Harrington, L. Alinaghian and M. Phillips, Chem. Eng. Process.: Process Intesif., 2015, 97, 248–258 CrossRef CAS.
- J. S. Srai, C. Badman, M. Krumme, M. Futran and C. Johnston, J. Pharm. Sci., 2015, 104, 840–849 CrossRef CAS PubMed.
- International Conference on Harmonisation, Pharmaceutical Development Q8(R2), 2009 Search PubMed.
- M. D. Ticehurst and I. Marziano, J. Pharm. Pharmacol., 2015, 67, 782–802 CrossRef CAS PubMed.
- R. G. Iacocca, C. L. Burcham and L. R. Hilden, J. Pharm. Sci., 2010, 99, 51–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00096k |
| This journal is © The Royal Society of Chemistry 2018 |