
An instant responsive polymer driven by anisotropy of crystal phases†
Heng
Deng‡
,
Yuan
Dong‡
 ,
Cheng
Zhang
,
Yunchao
Xie
,
Chi
Zhang
and
Jian
Lin
*
,
Cheng
Zhang
,
Yunchao
Xie
,
Chi
Zhang
and
Jian
Lin
*
Department of Mechanical & Aerospace Engineering, University of Missouri, Columbia, Missouri 65211, USA. E-mail: LinJian@missouri.edu
First published on 22nd November 2017
Abstract
We report a new type of responsive polymer film which is composed of pure poly(vinylidene fluoride) (PVDF) and is prepared using a simple solution-casting method. This responsive PVDF film possesses an anisotropic spatial distribution of the α phase and β phase, resulting in a fast response rate, high sensitivity, and robust responsiveness to organic vapors. Experimental and computational studies illustrate that α-phase PVDF exhibits a much stronger swelling ability in acetone vapor than β-phase PVDF. Anisotropic swelling induces internal stresses, offering a driving force for macroscopic shape transformation. This newly unveiled mechanism endows the material with new functionalities for fabricating devices such as a magnetically controlled soft tumbler and a chemi-mechanical sensor. Furthermore, crystal phase anisotropy in the PVDF film can be easily programmed into origami patterns by direct laser writing (DLW) for driving reversible three-dimensional (3D) shape transformation of the film.
Conceptual insightsResponsive materials with the ability to rapidly and reversibly change their shapes in response to external stimuli have great potential for applications in soft robotics, metamaterials and microelectromechanical (MEMS) systems. Despite much progress in this field, all the past reports have focused on tuning either the chemical functional groups or the physical density of building blocks in single- and multi-layered systems to enable them to possess anisotropic properties which lead to different responses to external stimuli. However, these methods often involve complicated and dedicated processes such as the synthesis of polymers with specific chemical functional groups, precise control of the cross-linking degrees, and numerous patterning and deposition steps in the single-layered systems, which could lessen their widespread applications. Herein, we report an unprecedented responsive mechanism in a single-layered poly(vinylidene fluoride) (PVDF) film, in which a shape change is driven by the anisotropy of crystal phases. Using this new responsive mechanism, a highly sensitive PVDF actuator could be fabricated using a simple solution-casting method, and a complex origami pattern on a PVDF film could be easily programmed using cheap CO2 laser writing. Our current report will broaden the knowledge in this research field and will inspire a new design philosophy for fabricating smart and responsive materials for application in sensors, bioelectronics, artificial muscles, soft robotics and so on. |
Introduction
Enthusiasm for constructing responsive materials is inspired by ubiquitous shape transformation phenomena in plants and animals and has continued to grow in recent decades.1,2 The materials that are capable of shape transformation by translating environmental stimuli (heat, light, electricity, chemical vapors, and pH) into desirable macroscopic shape changes are attracting burgeoning interest in emerging fields such as energy harvesting, metamaterials, soft robotics, sensors, and multifunctional bio-scaffolds. The shape transformation of most of the synthetic responsive materials is driven by anisotropic volume changes, which result from inhomogeneous swelling,3 thermal expansion,4 crystallization,5 or molecular reconfiguration6 upon external stimuli. Therefore, the quintessence of fabricating such materials lies in generating anisotropy in them. To achieve such anisotropy, the most straightforward approach is to laminate them into bilayered or multilayered structures. Due to their anisotropic properties, each layer exhibits a distinct volume change when responding to the same stimuli.7 As a result, the mismatch in their volume changes generates internal stresses at the interfaces, offering driving forces for macroscopic shape changes. This approach has been employed in metallic bilayered strips with different thermal expansion properties,8 multi-polymeric systems with different swelling abilities,3,9–11 metallic-polymeric bilayers,12 graphene/graphene oxide bilayers,13 and ceramic bilayers.14 Even though a large number of multilayered responsive materials have already been reported, the existing ones exhibit serious limitations. For instance, in the bilayered or multilayered responsive materials, stress induced by the anisotropy volume change in the interfaces of the layers can lead to delamination of the layers.15Inherent limitations in the bilayered or multilayered responsive materials have inspired the development of single-layered ones. Among them, single-layered responsive polymers are among the most important ones whose responsive shape change mechanisms can be attributed to their anisotropic chemical or physical properties, which can be achieved by creating different levels of crosslinking or adding extrinsic heterogeneous fillers into the polymer matrix. For example, vertical and lateral gradients of crosslinking degrees in the single-layered polymers result in anisotropic chemical properties.16–18 Extrinsic fillers such as heterogeneous polymer components,19,20 cellulose fibers,21 magnetic particles,15,22 and even swellable organic solvents23 can lead to anisotropic physical properties in the single-layered polymers. These distinct components result in distinguished responses to the stimuli. More recently, researchers have reported responsive shape changes driven by gradients of electrostatic complexation in a porous polymer film24 or actuated by anisotropic crystalline orders along the thickness direction of a polymer film.25 Although these anisotropic properties endow the single-layered polymers with responses to certain stimuli, they either show limited programmable freedom24,25 or involve complicated and dedicated processes such as the synthesis of polymers with specific chemical functional groups, precise control of the cross-linking degree, and numerous patterning and deposition steps,16–22 all of which restrict their widespread applications. Therefore, a single-layered responsive polymer driven by a new type of anisotropy that can be easily fabricated with a high freedom of programming is strongly desired.
Herein, we report an unprecedented responsive mechanism in a single-layered poly(vinylidene fluoride) (PVDF) film, whose shape change is driven by the anisotropy of crystal phases. The PVDF film prepared by a solution-cast method exhibits an anisotropic distribution of crystal phases on the top and bottom sides, in which the top one possesses a higher ratio of α phase PVDF (α-PVDF) than the bottom one (Fig. 1a). In contrast, the bottom side owns a higher ratio of β phase PVDF (β-PVDF). The resulting PVDF film with crystal phase anisotropy is extremely sensitive to organic vapors and shows an instant response rate that is comparable to a state-of-art superfast responsive polymer.24 Furthermore, experimental and computational studies illustrate that the α-PVDF has a much stronger absorption ability toward acetone than the β-PVDF, which results in a higher swelling volume on the side having a higher ratio of α-PVDF, causing the film to fold from the side with a higher ratio of α-PVDF to the other side with a lower one (Fig. 1a). Even though different piezoelectric or ferroelectric properties of the α-PVDF and the β-PVDF have been well studied,26,27 their distinct absorption properties have not yet been reported, let alone studying the resulting shape-changing behaviors. Motivated by this new responsive mechanism, we demonstrated functional devices such as a magnetically controlled soft tumbler and a chemi-mechanical sensor. Furthermore, the crystal phase anisotropy can be easily programmed in both vertical and lateral directions of the PVDF films by a direct laser writing (DLW) technique which has the advantages of low cost, high efficiency, and flexible designability.23,28–30 In the process, some domains in the β-PVDF film were selectively converted into α-phase domains via localized laser heat to form an origami pattern consisting of different phases (Fig. 1b). The programmed phase anisotropy enables the formation of a controlled swelling field, which endows the patterned PVDF film with a reversible shape transformation to form a complex 3D structure when stimulated by chemical vapors.
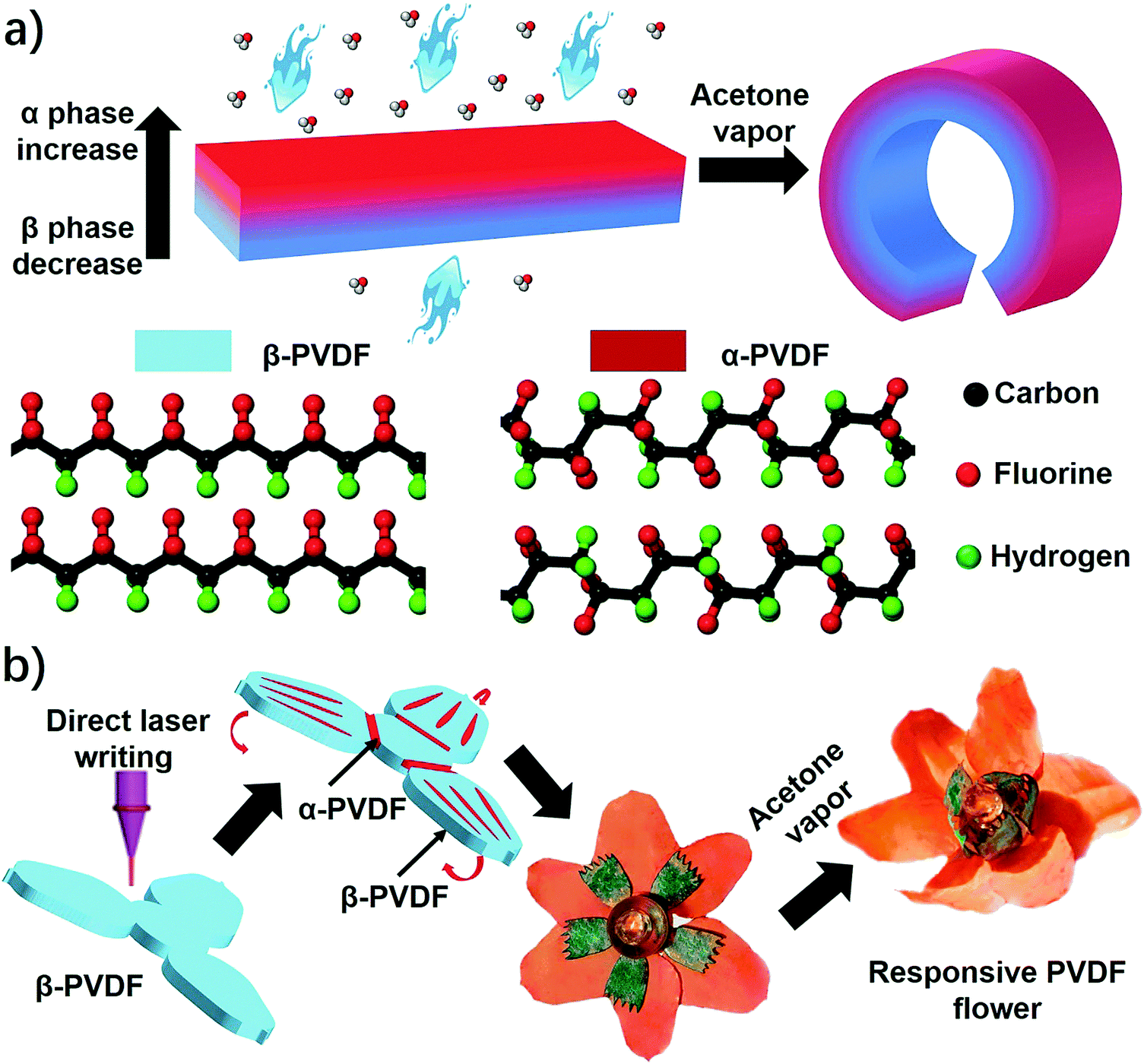 | ||
| Fig. 1 (a) A scheme of a PVDF film with anisotropic distribution of α and β phases and their curving response upon exposure to acetone vapor. (b) A scheme of using direct laser writing to fabricate origami patterns consisting of α- and β-phase domains in β-PVDF films by mimicking frilly textures in natural petals for driving 3D shape formation of a blooming PVDF flower. | ||
Results and discussion
Superfast and highly sensitive actuation of PVDF films
This study begins with the fabrication of PVDF films via a solution-cast method. First, commercial PVDF powder was dissolved in DMF to form a 20 wt% solution. Then the PVDF solution was spin coated to form thin films, followed by drying until all the solvent was evaporated. This process only involves solvent evaporation and recrystallization of the PVDF, and thus the fabricated films have the same chemical composition. We obtained two types of PVDF films when using different heating temperatures to evaporate the solvent. The PVDF film obtained at 120 °C is highly transparent, while the PVDF film dried at room temperature is almost opaque (Fig. S1, ESI†). The difference in transparency is a result of different surface morphologies caused by different solvent evaporation rates.31 The SEM images show that the transparent PVDF film has both smooth top and bottom surfaces, while the top and bottom surfaces of the opaque PVDF film show opposite surface morphologies (Fig. S2 and S3, ESI†). It should be noted that the porous structure itself does not cause the response of the polymer film to stimuli, but only accelerates the actuation speed by providing quick access of the stimuli to the pores of the film.24 Then we studied their responsive behaviors when exposed to acetone vapor. To facilitate the observation a red dye (FD&C, Red 40) was added to the PVDF films. The results show that it does not affect the responsive behaviors of the films (Fig. S4, ESI†). As shown in Fig. 2a and Fig. S4 (ESI†), when a transparent PVDF strip is exposed to acetone vapor (30 kPa, 25 °C), it bends quickly into a closed loop in 0.2 s and further into a multiple-round coil in just 0.4 s (Movie S1, ESI†). When exposed to air, this multiple-round coil recovers its flat shape in 0.8 s. This reversible shape change is very robust and no fatigue has been observed after hundreds of cycles. The transparent PVDF strip exhibits a maximum bending curvature of 32 cm−1 in 0.4 s when exposed to acetone vapor (Fig. 2b). This superfast response rate is comparable to that of a previously reported polymer actuator.24 Even though increased thickness decreases the response rate, the PVDF films still show a fast response to acetone vapor compared with the start-of-art responsive films (Fig. S5, ESI†).24 Interestingly, the transparent PVDF strip always folds from the top side interfaced with the air to the bottom side interfaced with the fabrication substrate as indicated in Fig. 1a. In contrast to the transparent PVDF strip, the opaque PVDF strip is almost inert to acetone vapor (Fig. S6, ESI†).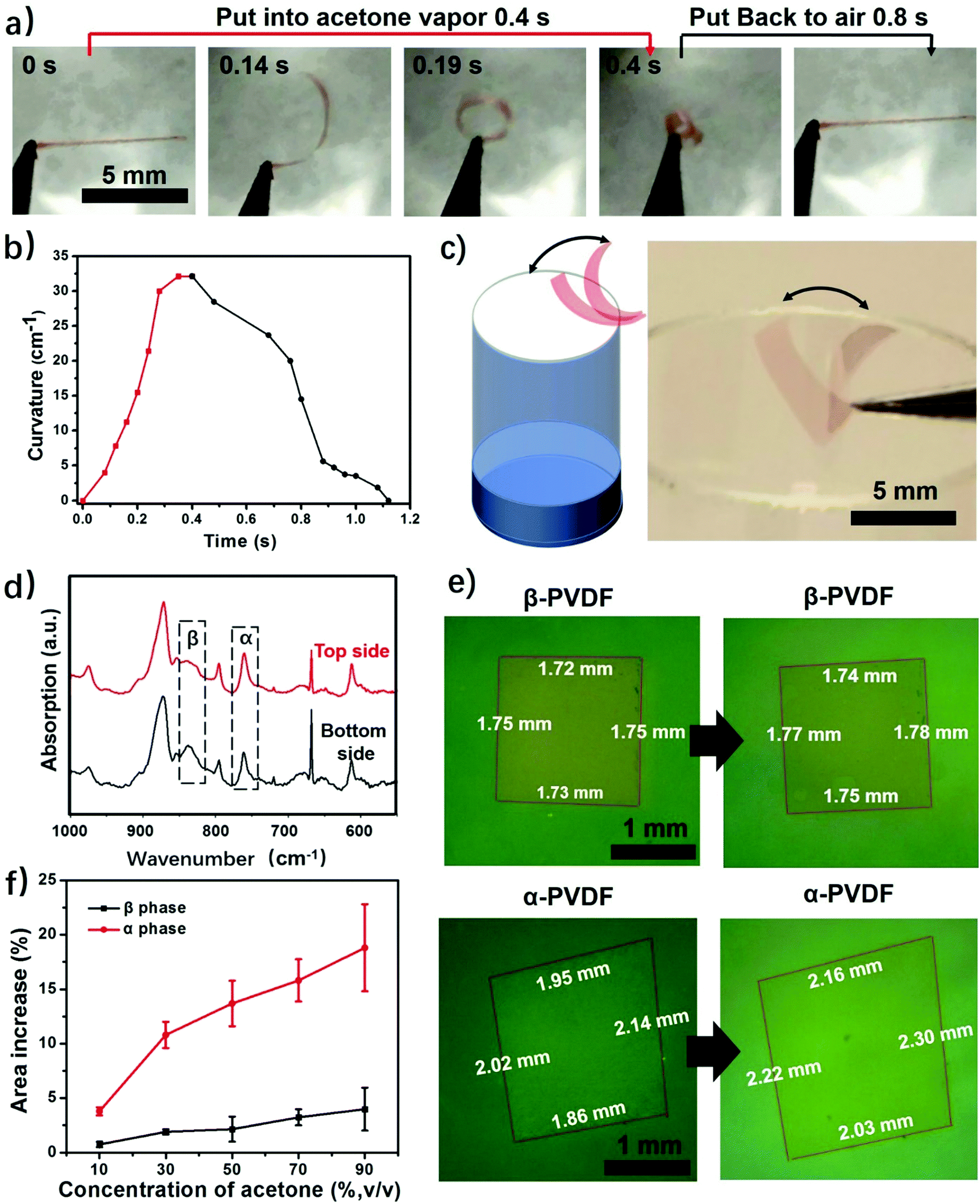 | ||
| Fig. 2 (a) A top-view photograph showing a responsive process of a transparent PVDF strip (1 mm × 7 mm × 3 μm). (b) Plot of curvatures versus time for the transparent PVDF strip. (c) Scheme and photograph of an autonomously wiggling PVDF strip when responding to the fluctuations of acetone concentration at the interface of the acetone vapor and the air. (d) FTIR spectra of top and bottom sides of a transparent PVDF film. (e) Optical images of homogeneous β-phase PVDF and α-phase PVDF films before and after being placed in 88 wt% acetone solution. (f) Area expansion of homogeneous β-phase PVDF and α-phase PVDF films in acetone solution with different acetone concentrations. | ||
This transparent PVDF film exhibits not only an instant response rate but also a high sensitivity to acetone vapor. Experiments conducted in aqueous acetone solutions with various concentrations show that the transparent PVDF strip still responds to 16 wt% acetone solution with an acetone vapor pressure of ≈1.71 kPa (Fig. S7, ESI†), even though the response rate decreases as the acetone concentration decreases. Moreover, as illustrated in Fig. 2a, the high sensitivity of the transparent PVDF film is also manifested in its fast recovery rate when the stimulus is absent. The characteristics of a superfast response and high sensitivity endow the films with the ability of autonomous actuation at the interface of the acetone vapor and the air where the acetone concentration is fluctuating (Fig. 2c and Movie S2, ESI†). The process is that when the transparent PVDF film absorbs even a little amount of acetone vapor near the bottleneck it bends up, making the film leave the location with a high acetone vapor concentration and be exposed to the air atmosphere. In turn, it recovers to its original location due to desorption of the acetone vapor, resulting in autonomous actuation. In contrast, the opaque PVDF film, which is inert to the acetone vapor, remains static at the bottleneck. Besides responding to the acetone vapor, the transparent PVDF films also respond to other organic vapors. As shown in Fig. S8 (ESI†), they show responses to dichloromethane (DCM), acetone, tetrahydrofuran (THF), and butanone with a response time ranging from 0.5 s to 1.2 s and maximum bending curvatures ranging from 4 cm−1 to 23 cm−1, indicating a possibility of using them as responsive components for chemi-mechanical sensors.
Responsive mechanism
Although transparent PVDF and opaque PVDF films share the same chemical composition, they exhibit distinguished responses to organic vapors. To unveil the mechanism accounting for these responsive behaviors, Fourier transform infrared spectroscopy with attenuated total reflectance (FTIR-ATR) was used to investigate the top and bottom surfaces of these two types of PVDF films. The top and bottom surfaces of the films are shown in Fig. S1 (ESI†). The top surface of the transparent PVDF film shows a higher intensity at the peak of 765 cm−1 and a lower intensity at the peak of 835 cm−1 than the bottom surface (Fig. 2d). According to the previous literature,26,32 the peaks at 765 cm−1 (CF2 skeletal bending) and 839 cm−1 (CH2 rocking) can be attributed to the density of the α phase and the β phase, respectively. By integrating the areas of these two peaks, the ratio of the α phase in the top surface is determined to be ∼80%, which is higher than that of the bottom one (∼58%). However, both surfaces of the opaque PVDF film possess almost pure β phase (Fig. S9, ESI†). A transparent PVDF film with anisotropic phase distribution (the phase-anisotropy PVDF film) and an opaque PVDF film with a homogeneous β phase (β-PVDF film) are thought to result from different solvent evaporation rates.33 The link of the anisotropic phase distribution in the phase-anisotropy PVDF film and the resulting bending response inspires a hypothesis that the α-PVDF and β-PVDF may have different swelling abilities toward acetone. To test this hypothesis, a homogeneous α-PVDF film was fabricated by heating a homogeneous β-PVDF film at the Curie temperature of 170 °C.34 The detailed fabrication of the films is described in the Method section. As expected, due to crystal homogeneity on both sides of these films (Fig. S10, ESI†), they exhibit a negligible bending response to the acetone vapor. But they show distinguished area expansions (Fig. 2e). For instance, when they were immersed in acetone solution with a concentration of 88 wt%, the homogeneous α-PVDF film expands by 21%, while the homogeneous β-PVDF film exhibited only 4% expansion. Further testing of the area expansions of both types of films in acetone solution at various acetone concentrations shows that the expansion increases with the concentration of acetone, and the homogeneous α-PVDF films exhibit an area expansion >4 times those of the homogeneous β-PVDF films (Fig. 2f), indicating a much stronger swelling ability of the homogeneous α-PVDF films in acetone. To the best of our knowledge, such a distinct swelling ability of α-PVDF and β-PVDF has not been reported, which inspired us to further investigate the mechanism using molecular dynamics (MD) simulation.Our and others’ simulation results show that at an atomistic level, absorption of small molecules to their host matrix is usually determined by their affinity and mobility in the matrix.23,24,35 To understand the absorption mechanism, we verified the following two hypotheses in this study: (1) the affinity of acetone toward α-PVDF crystals is stronger than that toward β-PVDF crystals; (2) acetone exhibits higher diffusivity on α-PVDF crystals than on β-PVDF crystals. To test these hypotheses, we built α-PVDF and β-PVDF crystals sandwiched by acetone–water mixtures with various concentrations (Fig. 3a and Fig. S11, ESI†). The detailed simulation set-up is illustrated in the ESI.† In the simulation, intermolecular energies caused by interactions of the α- and β-PVDF crystals with acetone (EPA) and water (EPW) were calculated, where EPW serves as a reference for comparison.24 The ratios of EPA over EPW for the α-PVDF and β-PVDF crystals in acetone solution with different concentrations are shown in Fig. 3b. The figure shows that the EPA/EPW values for the α-PVDF crystals increase from 2.5 to 11.5 as the concentration increases from 0% to 80 wt%, and are >4 times those for the β-PVDF crystals at the same concentrations, indicating that the interaction of acetone with the α-PVDF crystals is much stronger than that with the β-PVDF crystals.36 In other words, acetone molecules exhibit a stronger affinity towards α-PVDF than towards β-PVDF. This conclusion is also reflected by the number densities of equilibrium acetone molecules around the α-PVDF and β-PVDF crystals in 40 wt% acetone solution. In Fig. 3c, we can observe peaks appearing at ∼0.6 nm, indicating the layering behavior of acetone molecules around the PVDF surfaces.37 The peak intensity of acetone around the α-PVDF crystal is 50% higher than that around the β-PVDF crystal, indicating that at this distance more acetone molecules are gathered around the α-PVDF crystal. Excessive adsorption coefficients (Table S1, ESI†) show that more acetone molecules are absorbed around the α-PVDF than the β-PVDF crystal. These results verify the hypothesis that the α-PVDF crystals have a stronger affinity to acetone molecules than the β-PVDF crystals.
 | ||
| Fig. 3 (a) System construction and a snapshot of an α-PVDF crystal sandwiched by 40 wt% aqueous acetone solution. Atoms in the PVDF crystal are colored black (carbon), green (fluorine) and white (hydrogen). Water molecules are colored blue. Acetone molecules are colored red. Hydrogen atoms in water and acetone are not shown. (b) The ratios of intermolecular energies caused by the interaction of the α- and β-PVDF crystals with acetone (EPA) and water (EPW) in acetone solution with different acetone concentrations. (c) The number densities of acetone molecules around the α- and β-PVDF crystals in 40 wt% acetone solution after 10 ns simulation. (d) The MSD of acetone molecules on surfaces of α- and β-PVDF crystals. | ||
To validate the hypothesis that acetone exhibits a larger diffusivity on the α-PVDF crystals than on the β-PVDF crystals, we calculated the mean squared displacement (MSD) of acetone molecules in the region of −7.5 Å < z < 7.5 Å. This region is equivalent to a space defined by two planes located at a distance of 2.5 Å from the surfaces of the α-PVDF and β-PVDF crystals. Here, both PVDF crystals were placed in pure acetone systems. For details, please refer to the ESI.† As shown in Fig. 4d, the diffusivity of acetone molecules on α-PVDF crystals is calculated to be 1.63 × 10−5 cm2 s−1, which is 27% higher than that on β-PVDF crystals (1.28 × 10−5 cm2 s−1), confirming the second hypothesis. The aforementioned results indicate that synergetic effects of a stronger affinity and larger diffusivity make the acetone molecules be more easily absorbed to the α-PVDF crystals than to the β-PVDF crystals, which explains the greater swelling ability of the α-PVDF crystals than that of the β-PVDF crystals in acetone. Based on these results, it is reasonable to infer that anisotropy of the crystal phases in the phase-anisotropy PVDF film results in different swelling volumes in the top and bottom sides, causing the film to fold from the side with a higher ratio of α-PVDF to the other side with a lower one, which is in good agreement with the experimental results.
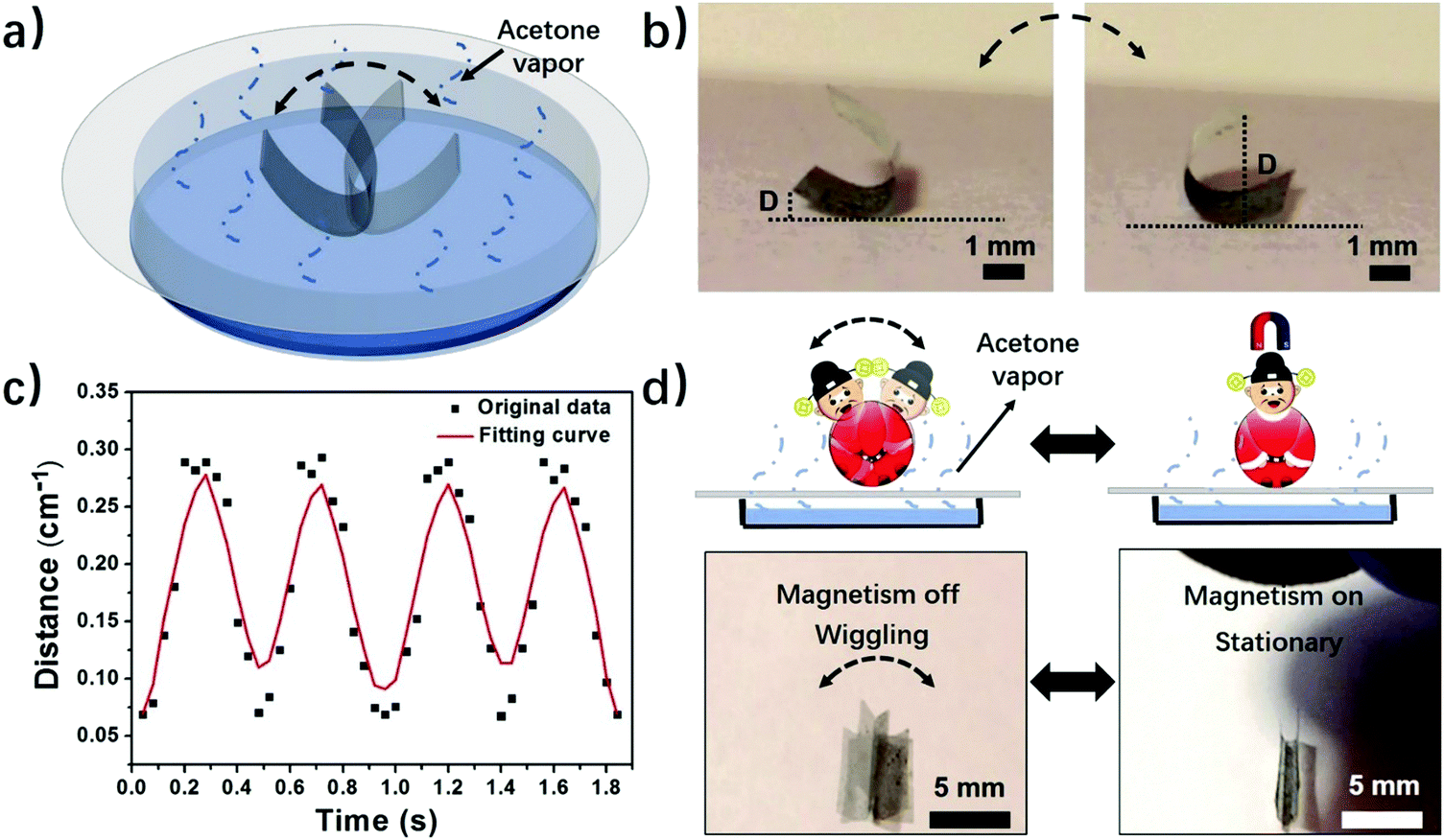 | ||
| Fig. 4 (a) Scheme of a PVDF tumbler autonomously wiggling in acetone vapor with a concentration gradient in the evaporation pathway. (b) Optical images of a PVDF tumbler autonomously wiggling in acetone vapor. “D” is the perpendicular distance from the edge of the film to the substrate. (c) Plot of “D” versus time, showing periodically wiggling movements. (d) Optical images of a PVDF tumbler with wiggling movements controlled by a magnet. | ||
Functional devices: a magnetically controlled tumbler and a chemi-mechanical sensor
Motivated by the experimental and computational results, we further explore applications of the phase-anisotropy PVDF film with anisotropic distribution of crystal phases in soft robotics and electronics. When the phase-anisotropy PVDF film was placed on a porous paper substrate covering the acetone solution it exhibited continuous locomotion (Fig. S12 and Movie S3, ESI†). This movement can be caused by the concentration gradient of the acetone vapor along the evaporation pathway. In contrast, in a closed chamber saturated with acetone vapor, the film remains static (Fig. S13, ESI†). To demonstrate a controlled movement, we fabricated a magnetically controlled soft tumbler by mixing nickel microparticles in the phase-anisotropy PVDF film (in the Method section). Instead of random locomotion, the resulting film exhibits a tumbler-like wiggling movement (Fig. 4a and Movie S4, ESI†). The mechanism is that when placed on the paper, the bottom side of the film absorbs more acetone than the top side, resulting in curving from the bottom toward the top. When the film is curved, its center of gravity rises, making it mechanically unstable (Fig. S14, ESI†). However, due to an increased weight from the added Ni particles, the acetone vapor is not strong enough to flip the film over to the other side. As a result, the curved film wiggles from one side to the other in the acetone vapor with a concentration gradient (Fig. 4b). To further investigate the wiggling behavior of this magnetically controlled tumbler, the perpendicular distance (D) from the edge of the film to the substrate, as indicated in Fig. 4b, was recorded as a function of time. As shown in Fig. 4c, the wiggling movement of the tumbler is periodic with a frequency of 2 Hz. Moreover, due to mixing of Ni particles, the film also responds to a magnetic field (Fig. 4d). When a magnet is placed above the wiggling films, the wiggling movement is stopped (Movie S4, ESI†). When the magnet is removed, the film recovers its tumbler-like wiggling movement. In this case, the magnet serves as a remote switch to control the movement of the soft tumbler. In addition to a soft tumbler, a chemi-mechanical sensor was also demonstrated. The sensor was fabricated by integrating laser-induced graphene (LIG)30 in the matrix of the phase-anisotropy PVDF (Fig. S15a, ESI†). In this case, the PVDF acts as an active swelling layer while the LIG acts as a passive layer, meanwhile, serving as an electrical sensing element for the sensor. When exposed to acetone vapor the PVDF swells, causing interfacial stresses among the network of PVDF and LIG. Thus, the composite film is stretched, resulting in an increase of the electrical resistance of the LIG (Fig. S15b, ESI†). The resistance change has a quite linear relationship with the acetone concentration (Fig. S15c, ESI†), suggesting a sensing behavior of the device.Programming crystal phases in PVDF films by direct laser writing
As shown above, the anisotropic phase distribution results in the shape transformation of the phase-anisotropy PVDF film. However, its fabrication by the solution-casting method possesses just one phase anisotropy along the vertical thickness direction, thus only resulting in a global bending response (Fig. 1a and 2a). To achieve a more sophisticated shape transformation, it is critical to locally and precisely program the phase anisotropy in both the lateral and vertical directions of the PVDF film (Fig. 5a). As we know that β-PVDF can be converted to α-PVDF near its melting point or the Curie temperature,34 if transient and localized heat can be applied to the homogeneous β-PVDF film, origami patterns consisting of α- and β-phase domains can be formed, thereby directing a complex 3D shape transformation. Motivated by this hypothesis, we employed the DLW technique, which was demonstrated by us to pattern anisotropy in a responsive polymer gel film,23 to locally pattern α-phase domains in a homogeneous β-PVDF film with a high spatial resolution for fabricating origami phase patterns.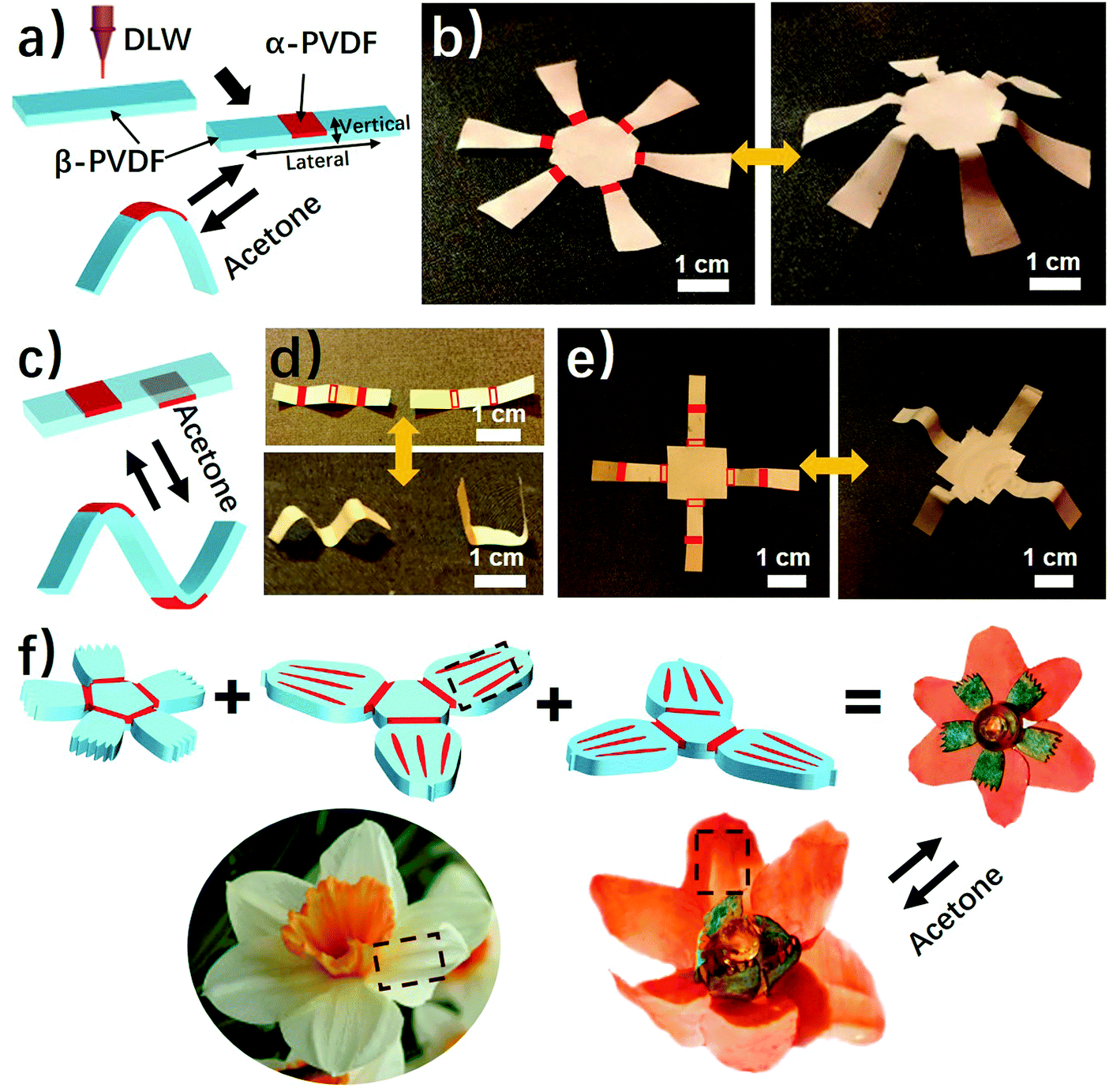 | ||
| Fig. 5 (a) Scheme of using DLW to pattern α-phase domains in a homogeneous β-PVDF film. (b) A photograph of the 3D structure transformed from a laser-patterned origami film. Red colored solid rectangles indicate α-phase domains created upon laser irradiation. (c) A scheme of laser patterning on both sides of PVDF, resulting in bidirectional folding. (d) A “MU” logo transformed from a β-PVDF film with laser patterning on both sides. Red-colored solid rectangles represent laser-irradiated domains on the top side of the β-PVDF film; and red colored framed hollow rectangles indicate laser-irradiated domains on the bottom side of the β-PVDF film. (e) A 3D structure transformed from a β-PVDF film with laser patterning on both sides. (f) A PVDF blooming flower evolving from assembled multilayered PVDF films with each layer having origami patterns consisting of α- and β-phase origami patterns. The origami patterns mimic frilly textures in natural petals as indicated in the dotted boxes. | ||
When a CO2 laser irradiates certain areas of a homogeneous β-PVDF film, the exposed areas have a greatly increased ratio of α-phase PVDF, as confirmed by the FTIR spectra (Fig. S16, ESI†). However, unexposed areas still own a negligible ratio of α-phase PVDF, which suggests that the laser implements confined thermal heating for phase conversion. Thus, laser-irradiated areas have more α-phase than the non-irradiated areas, creating crystal phase anisotropy in the film (Fig. 5a). The α-phase domains serve as active creases to direct the folding of the PVDF film (Fig. S17, ESI†), in which the folding angle of the PVDF films could be controlled by adjusting the width of the active creases (Fig. S18, ESI†). Please note that the folding direction is from the laser-irradiated side to the non-irradiated side, which is in good agreement with the proposed mechanism. Then active creases forming origami patterns were realized to achieve a more complex shape reconfiguration. As shown in Fig. 5b and Fig. S19 and S20 (ESI†), a series of active creases were patterned to direct the PVDF films to fold into different 3D scaffolds (Movie S5, ESI†). Besides programming on one side of the PVDF film, the DLW technique enables active creases to be patterned on the other side to realize different folding directions for building more complex structures (Fig. 5c). As shown in Fig. 5d and e, 3D structures such as the “MU” logo were achieved by involving bidirectional folding creases on the PVDF films. Finally, to demonstrate the high flexible designability of DLW, more complex origami creases mimicking the frilly textures in natural petals were patterned in three different PVDF films (Fig. 5f). Then these films were assembled into the desired geometry which evolved into a blossoming PVDF flower in acetone vapor (Movie S6, ESI†).
Conclusion
In summary, we report an unprecedented responsive mechanism in a single-layered poly(vinylidene fluoride) (PVDF) film, in which a responsive shape change is driven by the anisotropy of crystal phases. The resulting film with a spatial distribution of α- and β-phases exhibits a fast response rate, high sensitivity, and robust responsiveness to organic solvents, e.g. acetone. Experimental and simulation results show that the responsive shape change results from the distinct absorption abilities of α-PVDF and β-PVDF toward acetone. The newly unveiled mechanism endows the materials with new functionalities for fabricating devices such as a magnetically controlled soft tumbler and a chemi-mechanical sensor. Moreover, the phase anisotropy in the PVDF film can be easily programmed by the DLW technique for driving complex 3D shape transformations from their 2D counterparts. A responsive shape change controlled by crystal phase anisotropy broadens knowledge on the responsive mechanism, which will inspire researchers to further investigate a great variety of polymeric materials for applications in sensing, artificial muscles, soft robotics and so on.Methods
Fabrication of different phase-dominated PVDF films
2 g of PVDF powder (Kynar Flex® 761) was dispersed in 8 g of N,N-dimethylformamide (DMF) in a 20-ml bottle and then heated at 80 °C with vigorous magnetic stirring until complete dissolution of the PVDF powder. The PVDF/DMF solution was then cooled to room temperature and ready for use. 1 ml of the solution was dropped onto aluminum sheets, followed by spin coating at 3000 rpm for 30 s. The resulting PVDF/DMF films were then heated on a hotplate at 120 °C until complete evaporation of the DMF solvent to get transparent PVDF films. To prepare homogeneous β-phase PVDF films, wet films were dried at room temperature. Homogeneous α-phase PVDF films were obtained by heating homogeneous β-phase PVDF films at 170 °C for 10 min.Fabrication of Ni/PVDF films
After the PVDF/DMF solution was prepared, 2 g of Ni powder was added to the solution followed by vigorous stirring until the Ni powder was homogeneously dispersed in the solution. The Ni/PVDF/DMF mixture solution was spin-coated onto aluminum sheets at 3000 rpm for 30 s. The resulting Ni/PVDF/DMF films were then heated on a hotplate at 120 °C until complete evaporation of the DMF solvent to obtain Ni/PVDF films.Fabrication of LIG/PVDF films
Commercial PI tape was carbonized into LIG by direct laser writing (DLW) using a desktop CO2 laser system. The pulse width of the laser was ∼14 μs. The laser power was fixed at 12 W during processing. The LIG patterns were first designed using AutoCAD, and then DXF files were loaded onto the software that controls the movement of the laser beam. Then the prepared PVDF/DMF solution was spin-coated at 3000 rpm for 30 s on the surface of the LIG/PI film. The whole film was then kept in a vacuum box for 1 min to let the PVDF/DMF penetrate into the pores of the LIG. After that, the PVDF/DMF soaked LIG/PI film was heated on a hotplate at 120 °C to evaporate the DMF solvent. Then, the LIG/PVDF film was peeled from the PI tape and cut into the desired shapes. Finally, Cu electrodes were adhered to the LIG with silver ink to realize an electrical connection.Fabrication of origami patterns with anisotropic phase distribution in PVDF films
To program crystal anisotropy in the PVDF films, first, homogeneous β-PVDF films were prepared according to the previously illustrated method. DLW was then conducted on the homogeneous β-PVDF films using a desktop CO2 laser system in scanning mode. The pulse width of the laser is ∼14 μs. The laser power was fixed at 6.6 W during processing. Origami patterns were first designed using AutoCAD, and then DXF files were loaded onto the software that controls the movement of the laser beam. After the laser treatment, the areas irradiated by the laser were converted into α-PVDF.Characterization
FTIR was performed using a Thermo Nicolet Avatar 360 FT-IR spectrometer (Thermo Electron Inc. USA). In the FITR spectra, peaks at 765 cm−1 and 839 cm−1 were used to calculate the densities of the α-phase and β-phase in the PVDF films, respectively. The relative ratio between the α-phase and β-phase can be calculated by integrating these two peaks. Morphology characterization of the PVDF films was conducted on a Hitachi S-4800 SEM. The thicknesses of the PVDF films were measured using an optical profilometer (Vecco NT 9109). Optical images showing self-folding behaviors of the β-PVDF and α-PVDF films were taken using a microscope (MU130, AmScope). Graphical analysis was done using the software of AmScope. The videos and photographs of the responsive behaviors of the PVDF films were taken using an iPhone 6.Data and material availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data related to this paper may be requested from the authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the University of Missouri-Columbia start-up fund, the University of Missouri System Research Board, Oak Ridge Associated Universities (ORAU), Ralph E. Powe Junior Faculty Award, the NASA Missouri Space Consortium (Project: 00049784), the NSF IGERT program (Award number: 1069091), and the NSF SBIR program (Award number: 1648003). The computations were performed on the HPC resources at the University of Missouri Bioinformatics Consortium (UMBC) supported in part by NSF (award number: 1429294).References
- Y. Liu, J. Genzer and M. D. Dickey, Prog. Polym. Sci., 2016, 52, 79–106 CrossRef.
- L. Ionov, Adv. Funct. Mater., 2013, 23, 4555–4570 CrossRef CAS.
- G. Stoychev, N. Puretskiy and L. Ionov, Soft Matter, 2011, 7, 3277–3279 RSC.
- J. Luo, R. Huang, J. He, Y. Q. Fu, A. Flewitt, S. Spearing, N. Fleck and W. Milne, Sens. Actuators, A, 2006, 132, 346–353 CrossRef CAS.
- M. Behl, K. Kratz, J. Zotzmann, U. Nöchel and A. Lendlein, Adv. Mater., 2013, 25, 4466–4469 CrossRef CAS PubMed.
- T. Ube, K. Kawasaki and T. Ikeda, Adv. Mater., 2016, 28, 8212–8217 CrossRef CAS PubMed.
- R. Kempaiah and Z. Nie, J. Mater. Chem. B, 2014, 2, 2357–2368 RSC.
- S. Timoshenko, JOSA, 1925, 11, 233–255 CrossRef CAS.
- J. H. Na, A. A. Evans, J. Bae, M. C. Chiappelli, C. D. Santangelo, R. J. Lang, T. C. Hull and R. C. Hayward, Adv. Mater., 2015, 27, 79–85 CrossRef CAS PubMed.
- W. S. Wong, M. Li, D. R. Nisbet, V. S. Craig, Z. Wang and A. Tricoli, Sci. Adv., 2016, 2, e1600417 Search PubMed.
- L. Zhang, P. e. Naumov, X. Du, Z. Hu and J. Wang, Adv. Mater., 2017, 29, 1702231 CrossRef PubMed.
- T. S. Kelby, M. Wang and W. T. Huck, Adv. Funct. Mater., 2011, 21, 652–657 CrossRef CAS.
- J. Mu, C. Hou, H. Wang, Y. Li, Q. Zhang and M. Zhu, Sci. Adv., 2015, 1, e1500533 Search PubMed.
- R. K. Gogoi and K. Raidongia, Adv. Mater., 2017, 29, 1701164 CrossRef PubMed.
- R. M. Erb, J. S. Sander, R. Grisch and A. R. Studart, Nat. Commun., 2013, 4, 1712 CrossRef PubMed.
- J. Kim, J. A. Hanna, M. Byun, C. D. Santangelo and R. C. Hayward, Science, 2012, 335, 1201–1205 CrossRef CAS PubMed.
- M. Jamal, A. M. Zarafshar and D. H. Gracias, Nat. Commun., 2011, 2, 527 CrossRef PubMed.
- E. Palleau, D. Morales, M. D. Dickey and O. D. Velev, Nat. Commun., 2013, 4, 2257 Search PubMed.
- Z. L. Wu, M. Moshe, J. Greener, H. Therien-Aubin, Z. Nie, E. Sharon and E. Kumacheva, Nat. Commun., 2013, 4, 1586 CrossRef PubMed.
- H. l. Thérien-Aubin, Z. L. Wu, Z. Nie and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 4834–4839 CrossRef PubMed.
- A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef PubMed.
- H.-W. Huang, M. S. Sakar, A. J. Petruska, S. Pané and B. J. Nelson, Nat. Commun., 2016, 7, 12263 CrossRef CAS PubMed.
- H. Deng, Y. Dong, J.-W. Su, C. Zhang, Y. Xie, C. Zhang, M. R. Maschmann, Y. Lin and J. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 30900–30908 CAS.
- Q. Zhao, J. W. Dunlop, X. Qiu, F. Huang, Z. Zhang, J. Heyda, J. Dzubiella, M. Antonietti and J. Yuan, Nat. Commun., 2014, 5, 4293 Search PubMed.
- H. Arazoe, D. Miyajima, K. Akaike, F. Araoka, E. Sato, T. Hikima, M. Kawamoto and T. Aida, Nat. Mater., 2016, 15, 1084–1089 CrossRef CAS PubMed.
- J. Li, M. Yang, X. Sun, X. Yang, J. Xue, C. Zhu, H. Liu and Y. Xia, Angew. Chem., Int. Ed., 2016, 128, 14032–14036 CrossRef.
- S. K. Karan, R. Bera, S. Paria, A. K. Das, S. Maiti, A. Maitra and B. B. Khatua, Adv. Energy Mater., 2016, 6, 1601016 CrossRef.
- C. Zhang, Y. Xie, H. Deng, T. Tumlin, C. Zhang, J. W. Su, P. Yu and J. Lin, Small, 2017, 13, 1604197 CrossRef PubMed.
- H. Deng, C. Zhang, Y. Xie, T. Tumlin, L. Giri, S. P. Karna and J. Lin, J. Mater. Chem. A, 2016, 4, 6824–6830 CAS.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- M. Li, I. Katsouras, C. Piliego, G. Glasser, I. Lieberwirth, P. W. Blom and D. M. de Leeuw, J. Mater. Chem. C, 2013, 1, 7695–7702 RSC.
- A. Salimi and A. Yousefi, Polym. Test., 2003, 22, 699–704 CrossRef CAS.
- D. L. Chinaglia, R. Gregorio, J. C. Stefanello, R. A. Pisani Altafim, W. Wirges, F. Wang and R. Gerhard, J. Appl. Polym. Sci., 2010, 116, 785–791 CAS.
- P. J. Ratri and K. Tashiro, Polym. J., 2013, 45, 1107 CrossRef CAS.
- W. Fang, L. Zhang and J. Jiang, J. Phys. Chem. C, 2011, 115, 14123–14130 CAS.
- R. Parthasarathi, K. Balamurugan, J. Shi, V. Subramanian, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2015, 119, 14339–14349 CrossRef CAS PubMed.
- A. P. Willard and D. Chandler, J. Phys. Chem. B, 2010, 114, 1954–1958 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) is available. See DOI: 10.1039/c7mh00854f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |