
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotonic structuring improves the colour purity of rare-earth nanophosphors†
Dongling
Geng
,
Elena
Cabello-Olmo
 ,
Gabriel
Lozano
* and
Hernán
Míguez
*
,
Gabriel
Lozano
* and
Hernán
Míguez
*
Institute of Materials Science of Seville, Consejo Superior de Investigaciones Científicas (CSIC)-Univesidad de Sevilla (US), Américo Vespucio 49, 41092, Seville, Spain. E-mail: g.lozano@csic.es; h.miguez@csic.es
First published on 17th April 2018
Abstract
Nanophosphor integration in an optical cavity allows unprecedented control over both the chromaticity and the directionality of the emitted light, without modifying the chemical composition of the emitters or compromising their efficiency. Our approach opens a route towards the development of nanoscale photonics based solid state lighting.
Conceptual insightsWe demonstrate that an optical cavity can render a thin layer of rare-earth nanocrystals into a directional source of light with high colour purity. Rare earth nanocrystals or nanophosphors are central for many applications related to the generation of light because these nanomaterials feature exceptional thermal and chemical stability. However, such stability brings along an intrinsic complexity to alter their emission properties. The most common route to control the luminescence spectrum of nanophosphors is modifying their chemical composition. By doing so, enhanced colour gamut or purity is achieved at the expense of deteriorating the overall efficiency of the emitter. Herein, we demonstrate that the interplay between photon resonant modes and the natural emission of nanophosphors integrated in an optical cavity allows unprecedented control over both the chromaticity and the directionality of the emitted light, without modifying the chemical composition of the emitters. |
Light emitting materials based on inorganic matrices doped with rare earth (RE) cations, i.e. RE phosphors, feature robust photoemission with an outstanding thermal and chemical stability.1–4 All this makes them central in a variety of applications including bio-imaging and labelling, artificial lighting, solar cells or displays.5–13 In the particular case of full-colour displays comprising an array of dimmable pixels based on red (R), green (G) and blue (B) phosphors, the image quality is related to the chromaticity: the higher the colour purity of the individual pixels, the larger the accuracy with which an image can be displayed.14 In order to improve the image definition, the most widely employed approach consists of the chemical tuning of RGB emitters to achieve more saturated colours.15–17 This task is arduous, since chemical management often brings along a large distortion of the crystal structure, which may result in a deterioration of the phosphor stability.
An alternative that arose as a potential solution to these limitations was the development of RE-doped nanocrystals, also known as RE nanophosphors. The advent of these nanomaterials offered new opportunities for the realization of semi-transparent full-colour display devices with ultra-high resolution.18–20 However, due to their low crystallinity and high density of surface defects, nanophosphors feature low brightness, which has hindered their commercial use. In order to improve the photoemission of RE nanophosphors, strategies consisting of the use of core–shell architectures to reduce the quenching from surface defects and thus improve the quantum yield,21,22 or co-doping with sensitizers that provide efficient energy transfer to the activator to increase the brightness have been demonstrated.23–25 However, such approximations are not universal, being not suitable for arbitrary combinations of matrix and dopant. Furthermore they could even entail cross relaxation and concentration quenching processes, which ultimately deteriorate the performance of the emitter.26 The most widely selected activator for the vast majority of red phosphors employed in commercial displays is the trivalent europium cation (Eu3+).27,28 Many attempts have been made to improve the colour purity of Eu3+ nanophosphors, including different synthetic routes to modify the surface chemistry of the nanoscrystals. So far, the success has been fairly limited.29–31
In this work, we demonstrate a novel way to mould the colour purity and angular distribution of the photoemission of nanophosphors following an approach based on photonic tuning, rather than on customary chemical management, which has been recently proven to yield luminescent coatings with tuneable emission.32 Herein, we show that a thin layer of RE nanocrystals can behave as a directional source of light with high colour purity. In order to achieve a colour saturated directional emitter based on GdVO4:Eu3+ nanophosphors, which we choose as an example to prove our concept, a photonic crystal (PC) structure was designed and simulated first to extract the structural parameters required to achieve the targeted goal. Then, following this guide, an optical cavity, consisting of a middle layer of GdVO4:Eu3+ nanophosphors, was sandwiched between two photonic multilayers based on UV transparent ZrO2 and SiO2 layers. By doing so, we prevent UV wavelengths from being absorbed by the materials comprising the multilayers, interfering with the photoexcitation process. We demonstrate that such a photonic structure serves not only to enhance the emitted colour purity by an order of magnitude while reinforcing the overall intensity, but also to extraordinarily improve its directionality, with most of the emission being concentrated in a 30° cone around the perpendicular direction to the slab.
Among the many matrices used to host Eu3+, lanthanide orthovanadate compounds that crystallize in the zircon-type structure are interesting hosts for RE activators since their cationic sites present a size comparable to the radius of the dopant, unusual magnetic characteristics and efficient energy transfer between VO43− and Eu3+, which enable the effective excitation of the rare earth cation.33,34 Briefly, the space group symmetry of GdVO4 is I41/amd, Z = 4. As shown in Fig. 1a, its crystal structure is built from chains of alternating edge-sharing VO4 tetrahedra and GdO8 bisdisphenoids extending parallel to the c axis and joined laterally by edge-sharing bisdisphenoids.35 The preparation method and the experimental conditions under which nanophosphors are synthesized determine their morphology and size. In fact, nanorods, nanowires or nanoparticles with the same crystalline phase can be prepared by controlling the synthesis conditions.36–38 GdVO4:Eu3+ nanophosphors dispersed in methanol were prepared by a facile solvothermal route and polyacrylic acid as the functionalization agent, which not only has an important effect on the morphological characteristics of the final nanocrystals but also increases the colloidal stability of the suspension.39 A representative TEM micrograph of the as-prepared nanophosphors is shown in Fig. 1b. As it can be seen, non-aggregated nanoparticles with a quasi-spherical morphology, a mean size of 37 nm and a size distribution of 21 nm were obtained. We have also estimated the crystallite size from the XRD pattern shown in Fig. 1(c) and obtained a similar value (34 nm). These quantities are in fair agreement with those found in previous reports.40,41 On account of the same valence states and the similar ionic radii, the introduced Eu3+ ions replace Gd3+ ions of the host matrix and occupy the single site with D2d as the local site symmetry. The XRD pattern of the GdVO4:Eu3+ nanoparticles is shown in Fig. 1c. All the diffraction peaks of the sample coincide well with the standard pattern of GdVO4, whose diffraction peak positions are indicated with green arrows. No obvious shifting of the peaks or other impurity phases can be detected at the current doping level. The results reveal that the introduction of the activators (Eu3+ ions) has negligible influence on the crystal structure of the host matrix and Eu3+ ions were thus fully integrated in the GdVO4 lattice by replacing the Gd3+ ions. Fig. 1d displays the photoluminescence spectra of the GdVO4:Eu3+ suspension. The excitation spectrum (black line in Fig. 1d) exhibits a broad and intense band centred at 276 nm corresponding to the absorption of VO43− ions. Upon UV excitation, the energy absorbed through the GdVO4 host is efficiently transferred to the Eu3+ centers. Subsequently, the characteristic narrow emission peaks in the region between 500 and 725 nm, which clearly show the typical relaxation processes of Eu3+ ions, were observed, as displayed in Fig. 1d (red line). The major emission peaks at around 615 nm (electric dipole transition: 5D0 → 7F2) prevail over the emission line at 592 nm (magnetic dipole transition: 5D0 → 7F1) due to the absence of the inversion symmetry at the Eu3+ lattice site (D2d).37,38
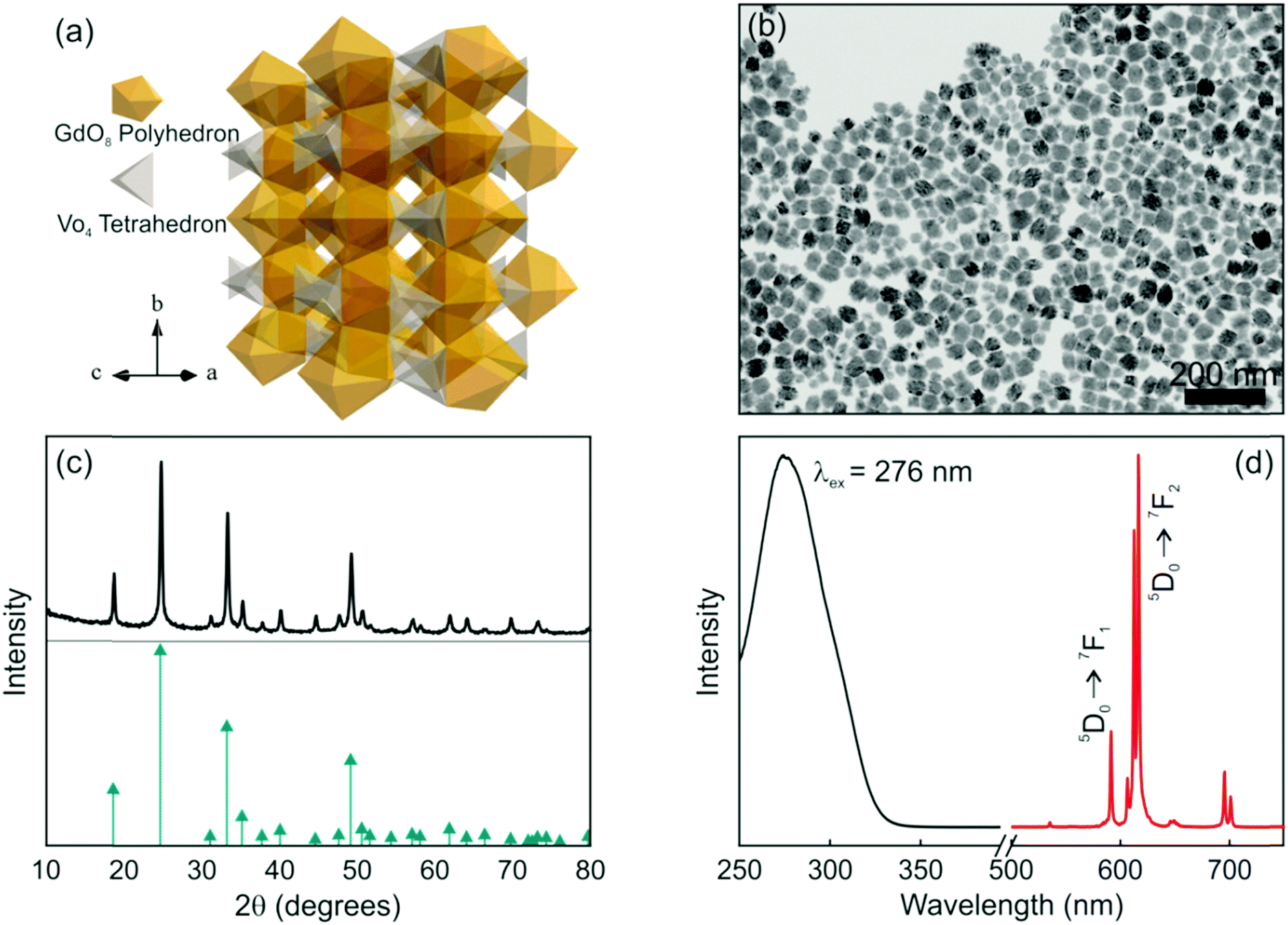 | ||
| Fig. 1 (a) Crystal structure of the GdVO4 matrix, and (b) TEM image and (c) X-ray diffraction pattern of the GdVO4:Eu3+ nanophosphors along with the JCPDs card No. 86-0996 (green arrows). (d) Excitation and emission spectra of GdVO4:Eu3+ nanophosphors in methanol. | ||
In order to investigate the interplay of photonic resonances and the spectral properties of GdVO4:Eu3+ nanophosphors, we first performed a theoretical study of the optical response of an optical cavity integrating a layer of the nanophosphors, as depicted in Fig. 2a, where a model of the proposed structure is plotted. The simulated GdVO4:Eu3+ nanophosphor layer is sandwiched between two multilayers made of alternate coatings of SiO2 and ZrO2 to form an optical resonator. These materials are chosen according to the requirements imposed by the need to excite the nanophosphors in the UV region as well as by their refractive index contrast, large enough as to give rise to strong cavity resonances with a relatively small number of layers. The wavelength dependent refractive index curves for the SiO2 and ZrO2 layers employed were estimated from experimental data using a Cauchy formula (see the ESI†). The thickness of each component layer in the structure was determined by an optimization code based on a genetic algorithm to maximize the spectral overlap of the optical resonances with the range where the dominant emission peak of Eu3+ ions occurs. In this process, we have taken into account that the higher diffraction orders of the structure should not block the photoexcitation (maximum at λ = 276 nm). Thus, a structure in which the second diffraction order has been suppressed was attained (see the ESI†).42 The estimated thicknesses of the ZrO2, SiO2 and nanophoshor layers were 87 nm, 109 nm and 200 nm, respectively. Note that these values fulfil the condition nSdS ∼ nZdZ, where nS (nZ) and dS (dZ) represent the refractive index and layer thickness of SiO2 (ZrO2), respectively. Fig. 2b shows the spatial distribution and spectral dependence of the electric field intensity across the 1DPC resonator when light impinges at normal incidence on its surface.
 | ||
| Fig. 2 (a) Model of the proposed layered structure that includes the devised dimensions of the layers involved. (b) Calculated spatial (vertical axis) profile and spectral (horizontal axis) distribution of the normalized electric field intensity across the section of the multilayer resonator, assuming a plane wave propagating along the direction perpendicular to the multilayer surface (θ = 0°). (c) Simulated angle dependent reflectance and (d) normalized electric field intensity integrated in the area where the nanophosphors are distributed within the optical multilayer. Vertical (horizontal) lines in (b) ((c) and (d)) indicate the spectral positions of the main emission lines of Eu3+ cations, i.e. 592 nm (orange), 613 nm and 617 nm (red). | ||
Different optical resonances can be identified as bright spots within the multilayer structure. The design is such that the light intensity is concentrated within the cavity for those frequencies for which the natural emission of the nanophosphors is maximum (λ ≈ 615 nm). The calculated angular dependence of the reflectance of the modelled structure is also displayed in Fig. 2c. A blue shift of the optical resonance mode, identified as the darker region in the map, takes place as the sample is tilted. Consistently, the spectral position of the resonances expected for light field intensity within the structure also blue shifts, as shown in Fig. 2d. These two effects, i.e., light field localization and angular dependence of the spectral resonances, are at the origin of the spectral and directional control over the emission properties of the GdVO4:Eu3+ layer that in principle can be achieved through integration in 1DPCs.
Following the guidance provided by the results of the simulations, we attempted the experimental realization of the optical resonator described in Fig. 2. In this regard, there are two main factors that can prevent the achievement of a high optical quality multilayer structure. One is the loss of mechanical stability when the total thickness of the multilayer is too large: tensile stress between the substrate and the film increases with thickness and hence with the number of layers, and might eventually result in the emergence of cracks. The second potentially limiting aspect is the formation of defects during layer deposition, which could give rise to undesired diffuse scattering and the loss of transparency of the stack. A critical step is the formation of the layer of nanophosphors, which are on average much larger and irregular than the other particles used to build the resonator. In order to shape them as a uniform film of optical quality (i.e., transparent) that can be incorporated into a photonic structure, we make use of our previous experience with the integration of GdVO4:Dy3+ nanophosphors by solution processing, which could be cast into well-defined visible transparent emitting films via spin coating.32 In this case, we could successfully prepare transparent GdVO4:Eu3+ nanophosphor films by dip coating, which allows even finer control over the thickness than spin-coating. Hence, integrity and uniformity at a large scale was achieved, as confirmed by both structural and optical characterization. Fig. 3a shows an image obtained using FESEM of the cross section of this structure. Good homogeneity of the layer thicknesses, with sharp interfaces between them, can be clearly observed. The integration of the nanophosphor layer into the one-dimensional multilayer (see the ESI†) breaks the transitional symmetry of the lattice and produces a resonant mode that can be identified as a dip in the reflectance spectrum of the final structure, as shown in Fig. 3b. Simulation of the reflectance spectrum performed using as input the structural parameters extracted from the FESEM image closely matches the experimental curve. A nanophosphor layer sandwiched between two ZrO2 films coated on a zero-fluorescence glass with a quantum yield of 50.4% was selected as the reference. The photoemission spectra of both the sample and reference when excited at λ = 276 nm are displayed in Fig. 3c. The luminescence peaks of the optical resonator present a two-fold enhancement with respect to the reference, while the rest of the emission lines are significantly diminished. Indeed, the ratio between the intensity of the peak associated with the 5D0 → 7F2 (red) and 5D0 → 7F1 (orange) transitions in GdVO4:Eu3+ is approximately 5. However, when a designed photonic environment surrounds the same layer of the emitting material, this red-to-orange (R/O) ratio becomes as large as ∼50. Fig. 3(d) shows the time dependent PL of a layer of nanophosphors and the same layer of nanophosphors integrated in a resonator (orange and blue symbols, respectively). Fittings, shown as solid lines, were done assuming the sum of two log-normal distributions of decay rates (see the ESI†). Short and long decay distributions are associated with the transition of Eu3+ cations that occupy crystal lattices in the bulk or close to the surface of the nanoparticle, respectively. Our results show that the change in the photonic environment provided by the resonator does not alter the dynamics of Eu3+ transitions since the average lifetime of the nanophosphors in the layer remains barely unaltered (0.79 ms) when the emitters are integrated into the resonator, the quantum yield of the material also remaining unchanged. This shows how the integration of nanophosphors into optical resonators yields a structure whose emission presents higher colour purity with a preserved high quantum yield.
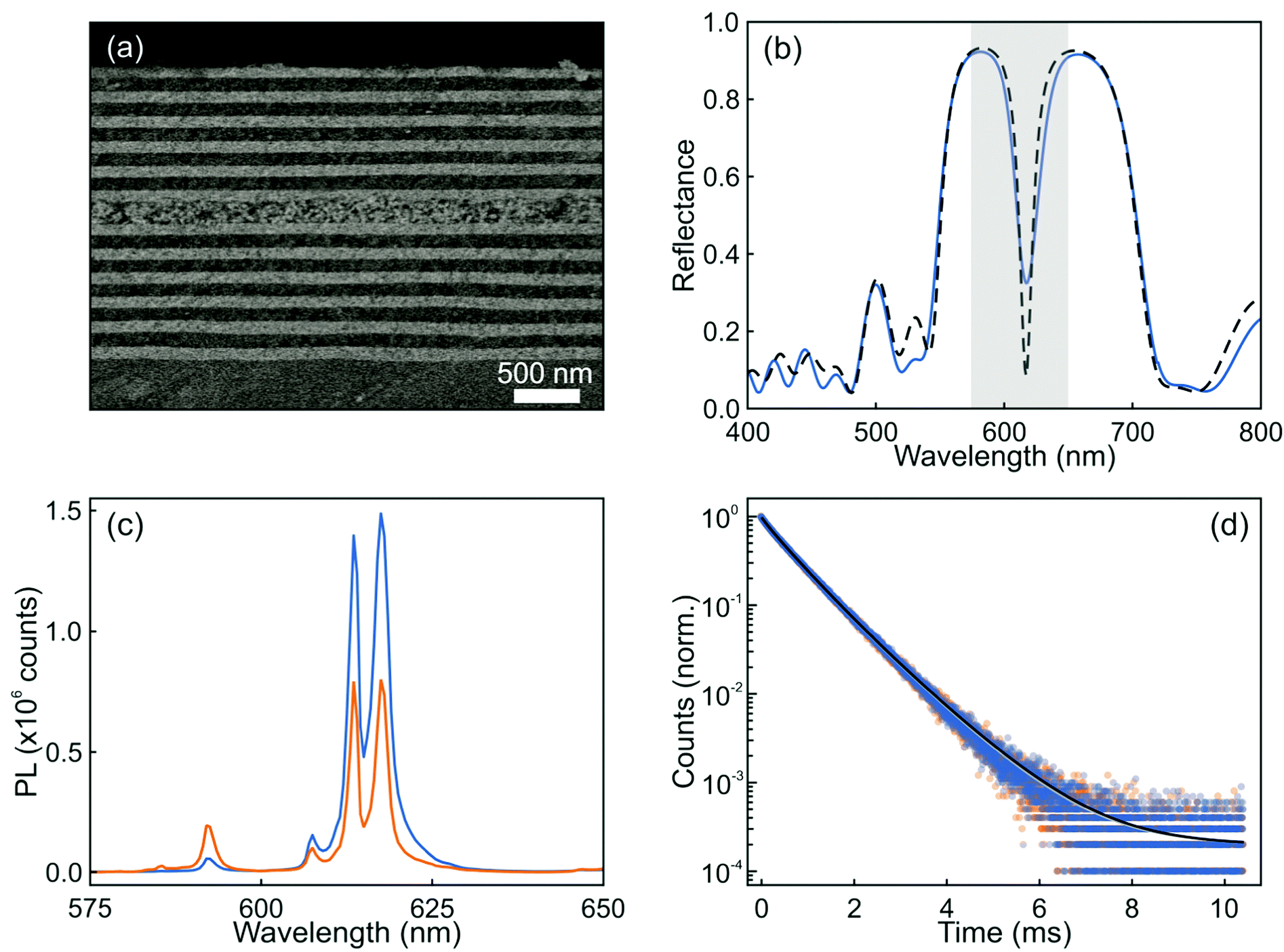 | ||
| Fig. 3 FESEM image of the cross section (a) and the corresponding reflectance spectrum (b) of an optical resonator integrating a layer of GdVO4:Eu3+ nanophosphors (blue solid line). The simulated spectrum is also shown for the sake of comparison (black dashed line). (c) Normal direction emission spectra of a nanophosphor layer (orange) and the same layer integrated in an optical resonator (blue). (d) Time dependence photoluminescence of Eu3+ cations in a reference layer (orange symbols) and integrated in the resonator (blue symbols) together with their corresponding fits to a sum of two log-normal distributions of decay rates (grey and black solid lines, respectively). | ||
While the photoemission from a layer of nanophosphors shows an angle independent chromaticity, the luminescence radiated from a similar layer integrated into a photonic resonator displays strong angular dependence. These results can be inferred from the angular dispersion of the spectral position of the resonant mode, as shown in Fig. 2c and d, which mediates the coupling between the light radiated by the phosphor and the propagating modes in a specific direction. The evolution of the experimental reflectance and the emission intensity distribution maps for the resonator were plotted with an angular resolution of 5°, as shown in Fig. 4a and b, respectively. As expected from the simulations, as the angle increases, the spectral location of the reflectance dip gradually shifts to shorter spectral values. This causes the intensities of the luminescence peaks attributed to the 5D0 → 7F2 transition of Eu3+ ions to be strongly enhanced at low view angles, but lowered at higher ones, as shown in Fig. 4b, due to the mismatch between the emission peak and the cavity resonant mode. On the other hand, the emission intensity of the orange peak originated from the 5D0 → 7F1 transition of Eu3+ is enhanced at higher angles, since the resonant mode is then spectrally closer to the corresponding emission peak (592 nm) of the nanophosphors. In contrast, it can be seen in Fig. 4c that a single nanophosphor layer behaves as a Lambertian emitter, with the photoemission being practically insensitive to variations of the view angle. Overall, the light emitted by the photonic structure embedding the nanophosphor layer behaves as a directional emitter in which most of the light is beamed within a cone of ±30° around its surface normal.
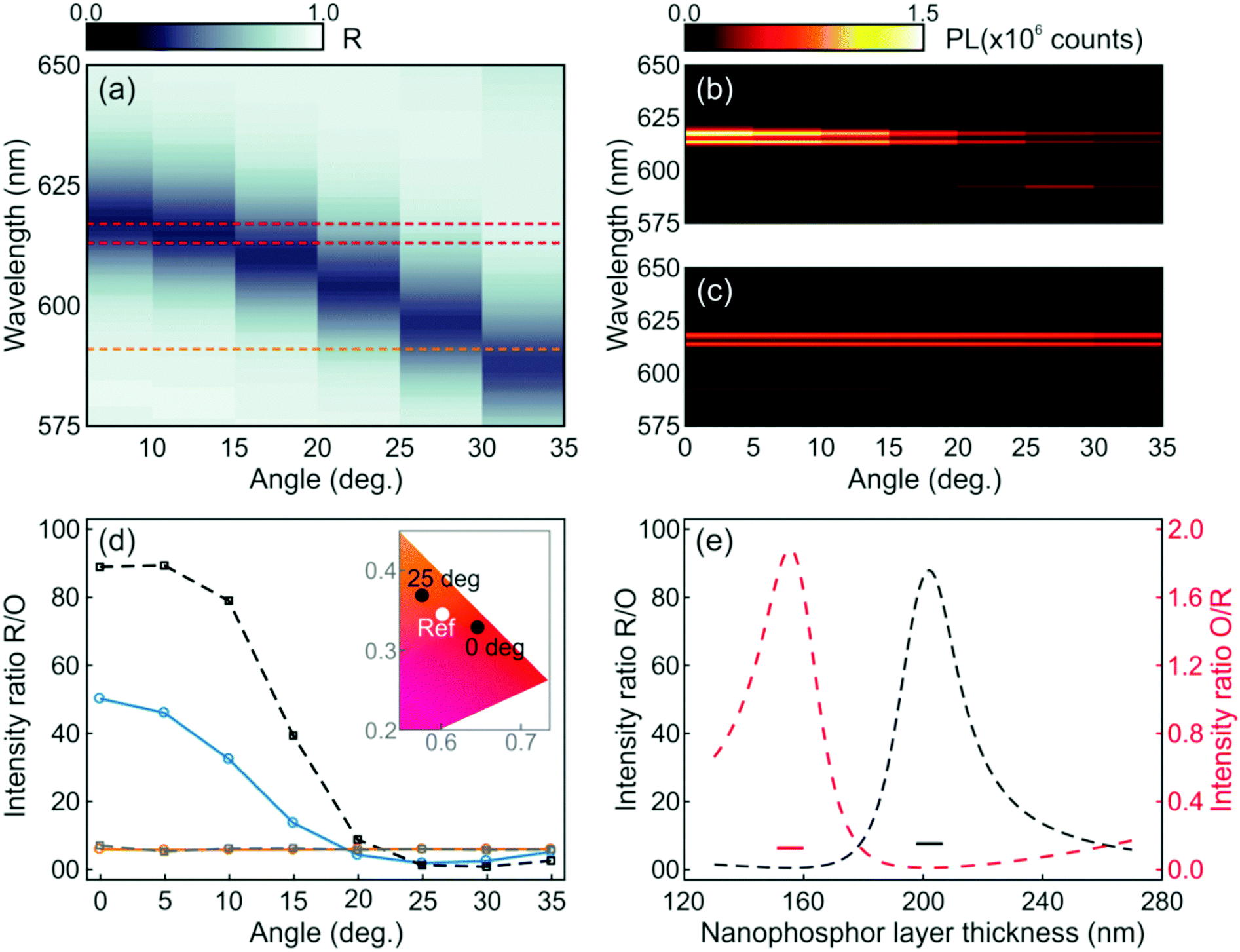 | ||
| Fig. 4 Angular dependence of (a) the reflectance and (b) the photoemission of an optical resonator integrating a layer of GdVO4:Eu3+ nanophosphors. (c) The angular variation of the photoemission of the reference is also shown for the sake of comparison. Horizontal dashed lines in (a) indicate the spectral positions of the main emission lines of Eu3+ cations, i.e. 592 nm (orange), 613 nm and 617 nm (red). (d and e) Analysis of the colour purity. (d) Experimental red-to-orange integrated photoemission peak intensity ratio for the resonator (blue solid line) and the reference (orange solid line). For the sake of comparison, we plot the theoretical red-to-orange ratio attained by spatially and spectrally integrating the electric field intensity multiplied by the emission spectrum of the reference for both a simulated resonator (black dashed line) and a reference (gray dashed line). The inset shows the dependence of CIE chromaticity coordinates with the direction of observation in the case of the resonator (black dots), while the reference remains stable (white dot). (e) Calculated dependence of the red-to-orange (black dashed line) and orange-to-red (red dashed line) ratios, estimated at normal incidence, versus the thickness of the nanophosphor layer in the resonator. The black and red short lines correspond to the red-to-orange and orange-to red ratios of the reference with associated nanophosphor thickness. | ||
In order to quantify the angular variation of chromaticity, we estimate the ratio of the experimental red and orange photoemissions for the resonator (blue solid line) and the reference (orange solid line), as shown in Fig. 4d. For the sake of comparison, we plot the theoretical R/O ratio attained by integrating spectrally and spatially, within the region where the nanophosphors sit, the electric field intensity multiplied by the emission spectrum of the reference when the nanophosphors are integrated to the optical resonator (black dashed line) and sandwiched between ZrO2 in the reference layer (gray dashed line). Although experimental and simulated results follow the same trend (see the ESI†), the actual R/O values are lower than expected, which is likely due to imperfections in the sample, as the small differences between the calculated and measured reflectance spectra displayed in Fig. 3b indicate. Coordinates for both the resonator and reference on a CIE chromaticity diagram are plotted in the inset. Please notice that while the reference values remain stable at (0.6017 and 0.3443) at all angles, the resonator ones shift from (0.6447 and 0.3275) to (0.5756 and 0.3678) as the angle of detection changes from 0° to 25° with respect to the direction perpendicular to the multilayer.
The emission chromaticity of the resonator also shows a strong dependence on the structural parameters. To illustrate this, we plot the calculated R/O (black dashed line) and orange-to-red (O/R, red dashed line) photoluminescence intensity ratios in Fig. 4e. Values for single nanophosphor layers are also provided as a reference (short lines). In the simulation, the thickness of the nanophosphor layer varied from 130 nm to 270 nm while the periodicity of the 1DPC is maintained. R/O and O/R quotients reach their maxima when the nanophosphor layer thicknesses are 202 nm and 155 nm, respectively. In contrast, the values of R/O and O/R of the reference are almost constant regardless of the thickness of the nanophosphor layer.
In summary, monodisperse rare earth nanophosphors were integrated into an ultraviolet transparent one dimensional photonic crystal structure to form an optical resonator. Following a precise computational design, the cavity resonances were devised to match selected emission peaks of the nanophosphors, with the aim of enhancing them while suppressing others, hence increasing the purity of the emitted light color. The chromaticity was enhanced by an order of magnitude at targeted wavelengths, which shows the potential that photonic structures might have to optimize the performance and improve the quality of phosphor based optical devices. Furthermore, directional control over the photoluminescence of the resonator was also achieved. The red-to-orange emission intensity ratio from the Eu3+ cations showed a strong dependence on the detection angle and the thickness of the nanophosphor layer embedded in the resonator. Overall, our results prove that the emission colour of nanophosphors can be adjusted by means of the photonic environment, without modifying their chemical composition or degrading their efficiency.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013/ERC Grant Agreement 307081, POLIGHT), the European Union's Horizon 2020 research and innovation programme (ERC-2016-StG Grant Agreement 715832, NANOPHOM), and the Spanish Ministry of Economy and Competitiveness under Grant No. MAT2014 54852-R. DG acknowledges the funding from the Marie Curie Action MSCA-IF-2014-657434 corresponding to the European Union's Horizon 2020 Programme.Notes and references
- T. Hirai, T. Hirano and I. Komasawa, J. Mater. Chem., 2000, 10, 2306–2310 RSC.
- G. Pan, H. Song, X. Bai, L. Fan, H. Yu, Q. Dai, B. Dong, R. Qin, S. Li, S. Lu, X. Ren and H. Zhao, J. Phys. Chem. C, 2007, 111, 12472–12477 CAS.
- C. Liu, W. Zhou, R. Shi, L. Lin, R. Zhou, J. Chen, Z. Li and H. Liang, J. Mater. Chem. C, 2017, 5, 9012–9020 RSC.
- Y. Zhang, J. Xu, Q. Cui and B. Yang, Sci. Rep., 2017, 7, 42464 CrossRef CAS PubMed.
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS PubMed.
- Y. Zhang, Z. Yu, J. Li, Y. Ao, J. Xue, Z. Zeng, X. Yang and T. T. Y. Tan, ACS Nano, 2017, 11, 2846–2857 CrossRef CAS PubMed.
- K. Dong, E. Ju, J. Liu, X. Han, J. Ren and X. Qu, Nanoscale, 2014, 6, 12042–12049 RSC.
- H. Lian, Y. Dai, D. Yang, Z. Cheng, C. Li, Z. Hou, M. Shang and J. Lin, Nanoscale, 2014, 6, 9703–9712 RSC.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- Y. Zhong, Z. Ma, S. Zhu, J. Yue, M. Zhang, A. L. Antaris, J. Yuan, R. Cui, H. Wan, Y. Zhou, W. Wang, N. F. Huang, J. Luo, Z. Hu and H. Dai, Nat. Commun., 2017, 8, 737 CrossRef PubMed.
- V. Kumar, O. M. Ntwaeaborwa, T. Soga, V. Dutta and H. C. Swart, ACS Photonics, 2017, 4, 2613–2637 CrossRef CAS.
- X. Liu, J. Zou and J. Lin, J. Electrochem. Soc., 2009, 156, P43–P47 CrossRef CAS.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199 CAS.
- G. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 7099–7131 RSC.
- W. S. Song, K. H. Lee, Y. R. Do and H. Yang, Adv. Funct. Mater., 2012, 22, 1885–1893 CrossRef CAS.
- S. Thakur and A. K. Gathania, J. Electron. Mater., 2015, 44, 3444–3449 CrossRef CAS.
- K. N. Venkatachalaiah, H. Nagabhushana, G. P. Darshan, R. B. Basavaraj, B. D. Prasad and S. C. Sharma, Mater. Res. Bull., 2017, 94, 442–455 CrossRef CAS.
- T. Samuel, C. S. Kamal, S. Ravipati, B. P. Ajayi, V. Veeraiah, V. Sudarsan and K. R. Rao, Opt. Mater., 2017, 69, 230–237 CrossRef CAS.
- C. S. Kamal, T. K. V. Rao, P. Reddy, K. Sujatha, B. P. Ajayi, J. B. Jasinski and K. R. Rao, RSC Adv., 2017, 7, 9724–9731 RSC.
- S. V. Kershaw, L. Jing, X. Huang, M. Gao and A. L. Rogach, Mater. Horiz., 2017, 4, 155–205 RSC.
- H. Zhu and D. Zuo, J. Phys. Chem. C, 2009, 113, 10402–10406 CAS.
- U. Rambabu, N. R. Munirathnam, S. Chatterjee, B. S. Reddy and S.-D. Han, Ceram. Int., 2013, 39, 4801–4811 CrossRef CAS.
- S. Nigam, V. Sudarsan, R. K. Vatsa, J. Ghattak and P. V. Satyam, J. Phys. Chem. C, 2009, 113, 8750–8755 CAS.
- M. N. Luwang, R. S. Ningthoujam, S. K. Srivastava and R. K. Vatsa, J. Am. Chem. Soc., 2011, 133, 2998–3004 CrossRef CAS PubMed.
- N. K. Sahu, N. Shanta Singh, R. S. Ningthoujam and D. Bahadur, ACS Photonics, 2014, 1, 337–346 CrossRef CAS.
- G. Blasse, J. Lumin., 1970, 1-2, 766–777 CrossRef.
- A. Daud, H. Futaki, K. Ohmi, S. Tanaka and H. Kobayashi, J. Soc. Inf. Disp., 1996, 4, 193–196 CrossRef.
- J.-G. Li, X. Li, X. Sun and T. Ishigaki, J. Phys. Chem. C, 2008, 112, 11707–11716 CAS.
- U. Rambabu and S.-D. Han, RSC Adv., 2013, 3, 1368–1379 RSC.
- S. Srivastava, A. Mondal, N. K. Sahu, S. K. Behera and B. B. Nayak, RSC Adv., 2015, 5, 11009–11012 RSC.
- S. Saha, S. Das, U. K. Ghorai, N. Mazumder, D. Ganguly and K. K. Chattopadhyay, J. Phys. Chem. C, 2015, 119, 16824–16835 CAS.
- D. Geng, G. Lozano, M. E. Calvo, N. O. Núñez, A. I. Becerro, M. Ocaña and H. Míguez, Adv. Opt. Mater., 2017, 5, 1700099 CrossRef.
- H. Cong, H. Zhang, B. Yao, W. Yu, X. Zhao, J. Wang and G. Zhang, Cryst. Growth Des., 2010, 10, 4389–4400 CAS.
- H. Wang, O. Odawara and H. Wada, Sci. Rep., 2016, 6, 20507 CrossRef CAS PubMed.
- B. C. Chakoumakos, M. M. Abraham and L. A. Boatner, J. Solid State Chem., 1994, 109, 197–202 CrossRef CAS.
- M. Gu, Q. Liu, S. Mao, D. Mao and C. Chang, Cryst. Growth Des., 2008, 8, 1422–1425 CAS.
- Y. Zheng, H. You, G. Jia, K. Liu, Y. Song, M. Yang and H. Zhang, Cryst. Growth Des., 2009, 9, 5101–5107 CAS.
- A. Szczeszak, T. Grzyb, Z. Śniadecki, N. Andrzejewska, S. Lis, M. Matczak, G. Nowaczyk, S. Jurga and B. Idzikowski, Inorg. Chem., 2014, 53, 12243–12252 CrossRef CAS PubMed.
- N. O. Nuñez, S. Rivera, D. Alcantara, J. M. de la Fuente, J. Garcia-Sevillano and M. Ocaña, Dalton Trans., 2013, 42, 10725–10734 RSC.
- N. S. Singh, R. S. Ningthoujam, G. Phaomei, S. D. Singh, A. Vinu and R. K. Vatsa, Dalton Trans., 2012, 41, 4404–4412 RSC.
- Y. Liu, G. Liu, J. Wang, X. Dong and W. Yu, New J. Chem., 2015, 39, 8282–8290 RSC.
- O. Sánchez-Sobrado, M. E. Calvo and H. Míguez, J. Mater. Chem., 2010, 20, 8240–8246 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, spectral dependent refractive index of ZrO2 and SiO2, SEM images and reflectance of photonic multilayers and optical resonators, time dependent photoluminescence measurements and their fits, and angular dependent emission intensity of red and orange peaks. See DOI: 10.1039/c8mh00123e |
| This journal is © The Royal Society of Chemistry 2018 |