
Probing nano-patterned peptide self-organisation at the aqueous graphene interface†
Zak E.
Hughes
 and
Tiffany R.
Walsh
*
and
Tiffany R.
Walsh
*
Institute for Frontier Materials, Deakin University, Geelong, VIC 3216, Australia. E-mail: tiffany.walsh@deakin.edu.au
First published on 27th November 2017
Abstract
The peptide sequence GrBP5, IMVTESSDYSSY, is found experimentally to bind to graphene, and ex situ atomic force microscopy indicates the formation of an ordered over-layer on graphite. However, under aqueous conditions neither the molecular conformations of the adsorbed peptide chains, nor the molecular-level spatial ordering of the over-layer, has been directly resolved. Here, we use advanced molecular dynamics simulations of GrBP5, and related mutant sequences, to elucidate the adsorbed structures of both the peptide and the adsorbed peptide over-layer at the aqueous graphene interface. In agreement with a previous hypothesis, we find GrBP5 binds at the aqueous graphene interface chiefly via the tyrosine-rich C-terminal region. Our simulations of the adsorbed peptide over-layers reveal that the peptide chains form an aggregate that does not evolve further into ordered patterns. Instead, we find that the inter-chain interactions are driven by hydrogen bonding and charge–charge interactions that are not sufficiently specific to support pattern formation. Overall, we suggest that the experimentally-observed over-layer pattern may be due to the drying of the sample, and may not be prevalent at the solvated interface. However, our simulations indicate sequence modifications of GrBP5 to promote over-layer ordering under aqueous conditions.
Introduction
The growth and organisation of hard biological tissues may be directed by the spontaneous ordering of soft materials at the hard–soft interface.1,2 Peptides adsorbed on solid surfaces may also be capable of delivering this type of soft pattern formation,3,4 and promise a range of biotechnology applications, including biosensing, drug-delivery, and tissue engineering.5 Graphene is an ideal substrate for exploring this phenomenon. This requires peptide sequences capable of both recognizing graphene6–8 and self-organizing into supramolecular structures at the aqueous interface. Initial efforts to determine the molecular-level factors that influence this pattern formation process are promising9–12 but presently lack the molecular-level information required to rationally guide their design.The dodecapeptide GrBP5 (IMVTESSDYSSY), herein referred to as GrBP5-WT, is a graphite binding peptide sequence.9–12 Above a critical surface concentration this sequence self-assembles at graphite/graphene interfaces.9–12 This is attributed to the presence of three proposed sub-domains; the N-terminal hydrophobic subdomain SD1 (IMV-), the hydrophilic central subdomain SD2 (-TESSD-), and the C-terminal subdomain SD3 (-YSSY) which is also hydrophilic and contains aromatic residues. This segmentation of the physio-chemical properties of the sequence may confer an amphiphilic character.
Using atomic force microscopy (AFM) on freeze-dried samples, So et al. investigated the structural evolution of the graphite-adsorbed GrBP5-WT at different surface coverages.9 The adsorbed peptide chains aggregated and, for a surface coverage of ∼60% or more, a self-ordered 2D nanostructure displaying six-fold symmetry was reported. A schematic overivew of the previously-proposed mechanism of structural evolution for the peptide over-layer is provided in Fig. 1. Briefly, So et al. proposed adsorption to the graphite/graphene interface to primarily initiate via SD3 due to π–π interactions between Tyr and the graphite/graphene substrate. Once adsorbed, SD3 was suggested to facilitate aggregation of the adsorbed peptide chains into a disordered assembly. The hydrophobic and hydrophilic sub-domains (SD1 and SD2 respectively) were suggested to subsequently direct the lateral assembly of aggregate into an ordered pattern on graphite. However, due to the challenges of in situ AFM measurements under aqueous conditions, So et al. characterized the structural evolution of the peptide over-layers using ex situ samples that were flash-frozen and then freeze-dried. Therefore, it is not known if these self-assembled soft-patterned interfaces are stable under aqueous conditions.
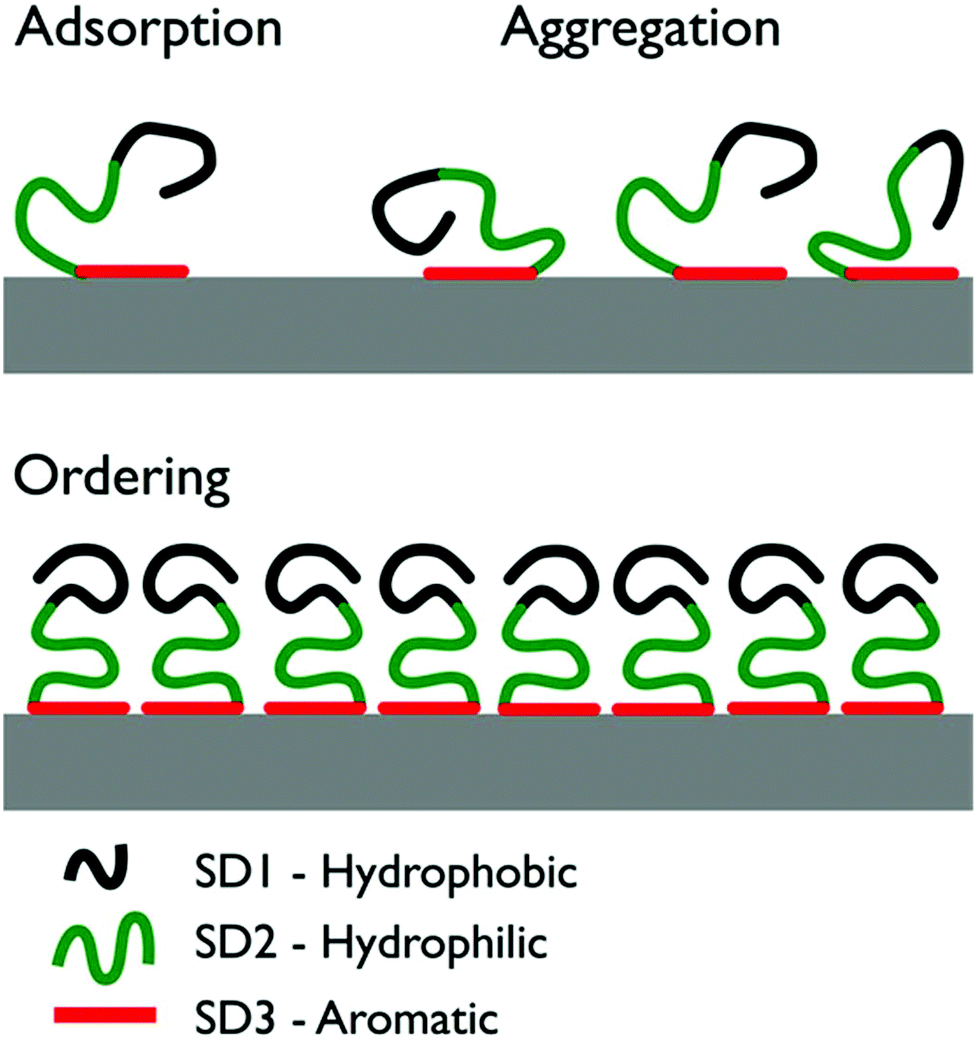 | ||
| Fig. 1 Illustration of the three stages of the previously-proposed surface-induced organization mechanism of GrBP5-WT.9 Initial surface contact proceeds via aromatic residues in SD3. As surface coverage increases, the peptide chains form a disordered surface-adsorbed aggregate. Ultimately, the coverage reaches a critical threshold and the chains self-organize into an ordered over-layer via inter-chain interactions of SD1. | ||
So et al. also approximated the binding affinity of the GrBP5 sequence by measuring the fractional surface coverage of the dried samples as a function of peptide concentration. While this is a reasonable first approximation that has been extensively used in past studies, currently it is acknowledged that factors other than the binding affinity of the peptide could affect this degree of surface coverage. Extrapolations of surface coverage to the peptide-surface binding affinity should therefore be viewed with caution. As part of a wider exploration of peptide-surface adsorption and assembly, So et al. also investigated the adsorption and assembly of a range of GrBP5 mutant sequences, using AFM.9
These mutant studies sought to elucidate the effect of changing a single sub-domain within the sequence (Table 1). In GrBP5-M1 (IMVTESSD![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) SS) the tyrosines in SD3 (Y9 and Y11) were mutated to alanine. In GrBP5-M2 (IMVTESSD
SS) the tyrosines in SD3 (Y9 and Y11) were mutated to alanine. In GrBP5-M2 (IMVTESSD![[W with combining low line]](https://www.rsc.org/images/entities/char_0057_0332.gif) SS) tyrosine was mutated to tryptophan. Mutations to SD1 probed the assumption of the amphiphilically-driven disorder-to-order mechanism. In GrBP5-M4 (
SS) tyrosine was mutated to tryptophan. Mutations to SD1 probed the assumption of the amphiphilically-driven disorder-to-order mechanism. In GrBP5-M4 (![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif)
![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif) TESSDYSSY), SD1 was transformed from hydrophobic to hydrophilic, eliminating the overall amphiphilic character or the peptide. In GrBP5-M5 (
TESSDYSSY), SD1 was transformed from hydrophobic to hydrophilic, eliminating the overall amphiphilic character or the peptide. In GrBP5-M5 (![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) TESSDYSSY), the mutated SD1 was designed to confer increased hydrophobicity compared to GrBP5-WT.
TESSDYSSY), the mutated SD1 was designed to confer increased hydrophobicity compared to GrBP5-WT.
| Peptide | Sequence | Coverage | Aggregation | Over-layer |
|---|---|---|---|---|
| GrBP5-WT | IMVTESSDYSSY | High | Strong | Ordered at ∼60% coverage |
| GrBP5-M1 | IMVTESSDSS |
Little/none | N/A | N/A |
| GrBP5-M2 | IMVTESSDSS |
Moderate | Low | Porous, ordered at ∼100% coverage |
| GrBP5-M4 | ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) TESSDYSSY TESSDYSSY |
High | Strong | Porous and amorphous |
| GrBP5-M5 | TESSDYSSY |
High | Strong | Ordered at ∼60% coverage |
So et al. reported that mutations to SD3 (GrBP5-M1 and GrBP5-M2) affected the degree of both surface coverage and peptide aggregation, while GrBP5-M1 (with no aromatic residues) yielded only minimal coverage. For GrBP5-M2 the peptide adsorbed with a lower surface coverage than GrBP5-WT, with less evidence of aggregation. In this case a more porous over-layer formed, with an ordered phase present only at ∼100% coverage. Mutations to SD1, (GrBP5-M4 and GrBP5-M5), yielded comparable coverages relative to GrBP5-WT. For GrBP5-M4, the peptide aggregated on the surface but the resulting over-layer was porous and disordered. In contrast, GrBP5-M5 was reported to form ordered over-layers akin to GrBP5-WT. However, these findings may be an artefact of the drying process, and in situ AFM measurements13–15 are yet to be reported for these interfaces. Alternatively, molecular simulations can complement these experimental approaches, providing molecular-level insights into the intrepretation of these experimental data.
Molecular simulations can predict and elucidate the structures of both the individual peptide adsorbed conformations as well as the overall structuring of the resulting peptide over-layer adsorbed at the aqueous graphene interface.5,16 However, many (but not all) previous MD studies, notably at the atomistic level, investigated the adsorption of a single isolated peptide chain and not a multi-peptide surface-adsorbed over-layer. Study of several co-adsorbed peptide chains is an acute challenge, which requires that both inter-peptide and peptide-surface interactions are captured adequately.
One strategy to address this challenge is the use of coarse-grained (CG) models, facilitating access to longer time- and length-scales.17–20 However, the subtle molecular-scale details that dictate the balance between inter-peptide and peptide-surface interactions relevant to surface-mediated self-assembly might not be reliably recovered using CG simulations. Atomistic simulations of multi-chain peptide adsorption have been previously reported,21–29 but in many (not all) instances featured either short peptides and/or low surface coverages, or lacked sufficient conformational sampling.
Previous atomistic simulations of peptides at the aqueous graphene interface indicated close contact between the aromatic residues and the graphene surface.17,30–37 Moreover, several studies suggest that many graphene binding sequences are intrinsically disordered.33,34,36 Intrinsically-disordered peptides (IDPs) typically do not possess a well-defined secondary structure, or set of secondary structures, and usually feature a complex conformational ensemble. Therefore, without a targeted approach to conformational sampling, the resulting simulation for IDPs might be biased, potentially leading to misleading conclusions.
Recently Penna et al. reported the use of a large number (100+) of standard MD simulations to investigate the initial stage of adsorption of a single chain of GrBP5-WT at the aqueous graphite interface, using non-polarizable force-fields (FFs).36 These authors proposed a staged adsorption mechanism, and concluded that SD1 was influential for all stages of the adsorption process, while SD3 dominated the latter stage. These findings are in broad agreement with the hypothesis proposed by So et al., but these predictions of the time-dependent adsorption mechanism have yet to be directly verified by experimental evidence. Moreover, neither the adsorbed structure of the mutant peptide sequences, nor the interaction of several adsorbed peptide chains was reported.
Here, we modelled the peptide/graphene interface in water, using a polarisable graphene model, in conjunction with Replica Exchange with Solute Tempering (REST) molecular dynamics (MD) simulations38,39 to predict the conformational ensemble of a single chain of five different peptide sequences adsorbed at the aqueous graphene interface; GrBP5-WT and four mutant (M1, M2, M4 and M5) sequences. We also performed REST-MD simulations of multi-chain over-layers of GrBP5-WT at two different surface coverages (50% and 70%).
Results
Single chain adsorption
GrBP5-WT was found to adsorb principally via the SD3 tyrosines, with SD1 and SD2 featuring only modest surface contact, (Fig. 2a), with Table S1 of the ESI† providing numerical contact values. These data were consistent with the hypothesis of So et al. in that the SD3 sub-domain was chiefly responsible for mediating GrBP5-WT adsorption at aqueous graphene.9–11 A representative snapshot of GrBP5-WT in the surface adsorbed state, (Fig. 2b), illustrates these residue-surface contact data, with clear indication of a π-stacking arrangement between Tyr and graphene. The sharpness of the vertical density profile for SD1 (Fig. 2c) also supports this, along with the distribution of ring tilt (Fig. S13 of the ESI†) which indicates the near-planar orientation of the rings with respect to the graphene surface. In contrast with our findings, the study of Penna et al., the residues with the greatest degree of contact for the final adsorbed conformation of GrBP5-WT were in SD3 and SD1.36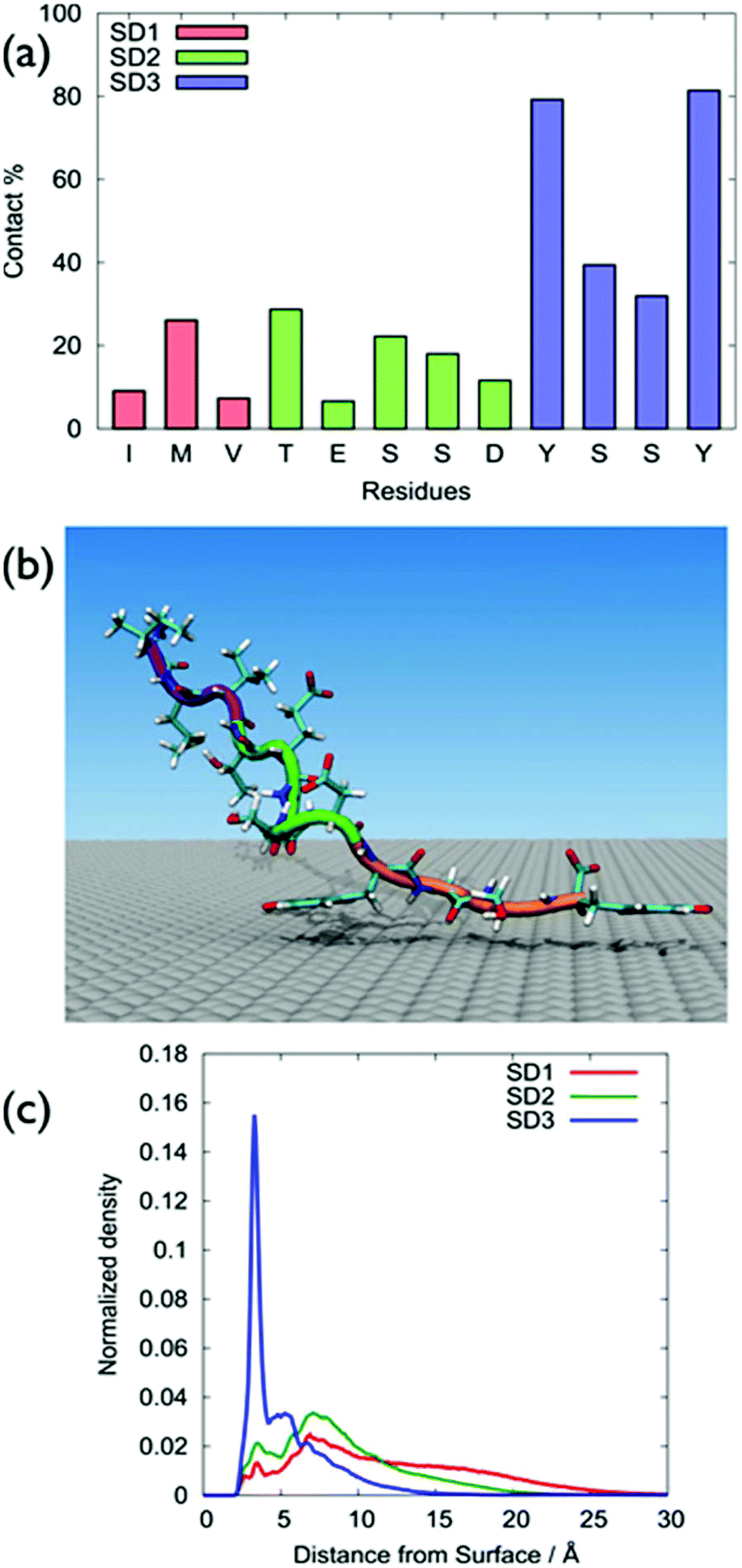 | ||
| Fig. 2 (a) Degree of surface-residue contact with the graphene surface for each residue in a single chain of GrBP5-WT, calculated from REST MD simulations. (b) Representative snapshot of the most populated cluster of a single chain of GrBP5-WT adsorbed at the aqueous interface, the backbone of SD1, SD2 and SD3 are coloured purple, green and orange, respectively water not shown for clarity. (c) Normalised atom number density profiles for the different subdomains of GrBP5-WT. | ||
The differences between our work and Penna et al.36 may arise from the different FFs used in the two studies (full details are provided in ‘Force-field comparison’ in the ESI†). π–π interactions are important in biological systems that contain aromatic residues.40 A study on the performance of the GRAPPA FF as regards π-stacking on graphene has been recently published for nucleic acids,41 and further discussion of GRAPPA and π-stacking is provided in the ESI† (‘Simulation details’ section). Another possible source of this discrepancy may be due to the different conformational sampling strategies used. We used REST-MD simulations, which can predict the equilibrium structural ensemble of the surface-adsorbed state, but cannot readily provide time-dependence information. Penna et al. used regular MD simulations to probe the time-dependence of GrBP5-WT attachment. However, even a large number of standard MD simulations may not be guaranteed to be sufficient to obtain the Boltzmann-weighted ensemble of adsorbed conformations.
All five sequences lacked appreciable secondary structure, and typically accessed hundreds of distinct conformations at room temperature (292–360 for the un-adsorbed peptides, 171–244 for the surface-adsorbed peptides; Tables S2 and S3 of the ESI†), as supported by our clustering analysis (Computational methods in the ESI†). Additionally, no single distinct structure was dominant (Tables S2 and S3 of the ESI†). Furthermore, none of these sequences showed any evidence of a well-defined secondary structure (Fig. S2 and S3 of the ESI†), with random coil character accounting for over 70% of the secondary structure (using the Dictionary of Secondary Structure of Proteins, DSSP,42 definitions). Further details can be found in the “Cluster and secondary structure analysis of peptide sequences” section of the ESI, Tables S2 and S3 and Fig. S1–S4.†
The surface-residue of the mutant sequences showed key differences with GrBP5-WT, Fig. 3 (numerical data in Table S1 of the ESI†). Of all the modifications of SD3, the Y → A mutations had the greatest impact on SD3-surface binding (Fig. 3c). This is consistent with the experimental observations of low surface coverage for GrBP5-M1. Moreover, the spatial extent of the impact of these mutations was found to be variable, depending on the location of the mutations. For instance, mutations in the SD1 region (e.g. GrBP5-M4/5) did not produce a significant non-localised influence on the relative degree of contact predicted for the SD3 residues (Fig. 3c). In contrast, mutations in SD3 influenced the degree of residue-surface contact in SD1, as detailed herein.
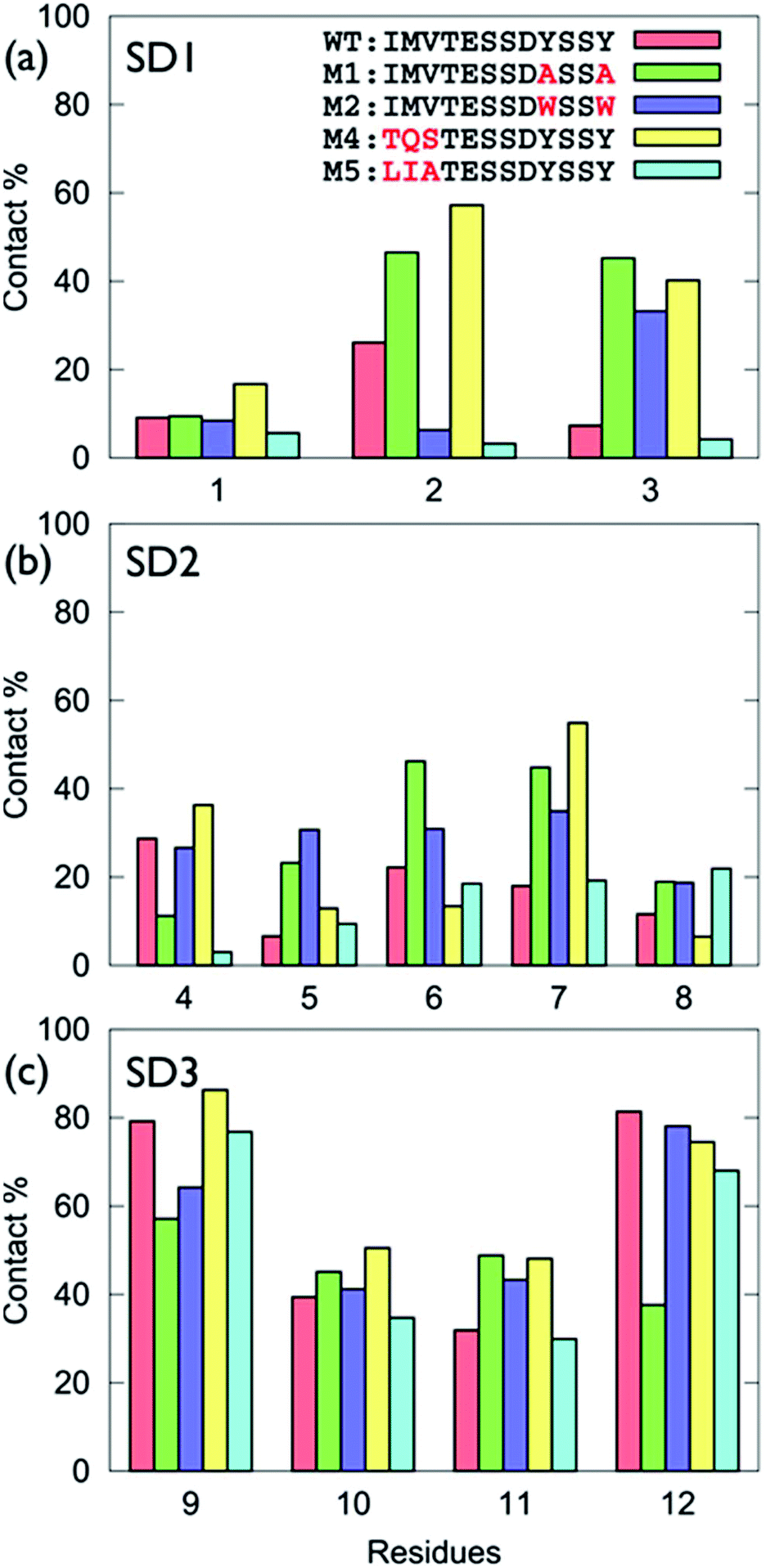 | ||
| Fig. 3 Degree of surface-residue contact for each residue with the graphene interface for the GrBP5 family of peptides, (a) sub-domain 1, (b) sub-domain 2, (c) sub-domain 3. | ||
The trends in residue-surface contact across SD1 showed a comparatively greater degree of variation (Fig. 3a), even for those sequences in which SD1 was conserved (i.e. for GrBP5-WT/M1/M2). Mutation of Tyr in SD3 for Ala (GrBP5-M1) enhanced surface contact in SD1 (Fig. 3a and Fig. S5 of the ESI†). We suggest that elimination of the aromatic residues in SD3 promoted a compensatory role for SD1 in maintaining overall peptide-surface contact.
The GrBP5-M4 mutant (replacing IMV- with TQS-), yielded enhanced contact via Gln. Previously-reported molecular simulations have indicated that amino acids with an amide-bearing side-chain, Gln and Asn (Q and N), absorbed strongly to aqueous graphene interfaces.31,34,43 The favourable interaction of Q2 resulted in GrBP5-M4/graphene contact that added SD1, along with SD3, to the surface contact region. In contrast, the SD1-surface contact predicted for GrBP5-M5, where IMV- was replaced LIA-, was diminished compared with GrBP5-WT. This is consistent with a GRAPPA-based previously-predicted ranking in adsorption strength34 as Met > Ala > Val ≈ Leu > Ile.
Desipte the fact that the SD2 motif was conserved across all five peptide sequences, mutations in both SD1 and SD3 were indirectly modified residue-surface contact in SD2 (Fig. S5 of the ESI†). Notably, in GrBP5-M4, the enhanced surface contact of Ser7 indicated a very different mode of adsorption for GrBP5-M4 compared with the other four sequences. The poor contact of Asp8 across all five sequences was again consistent with the weak free-energy of adsorption of the corresponding amino acid.34
On the basis of these data, the surface adsorption modes of the five sequences can be classified into three broad categories; representative snapshots of these are provided in Fig. 4. In the first category (GrBP5-WT, GrBP5-M2 and GrBP5-M5) surface contacts formed chiefly via SD3, with SD1 and SD2 playing a relatively lesser role. Within this category, the degree of interaction of SD2 was variable, following the trend GrBP5-M2 > GrBP5-WT > GrBP5-M5. In all three of these cases SD1 extended away from the surface (Fig. 2c and Fig. S5 of the ESI†), thus presenting an unbound chain segment for potential participation in surface-mediated inter-peptide interactions, akin to the schematic in Fig. 1. These three sequences were the only peptides in the set to show any signs of experimentally-observed ordering,9 albeit in the surface-dried state.
 | ||
| Fig. 4 Snapshots of representative structures of the mutant peptide sequences, (a) GrBP5-M1, (b) GrBP5-M2 (c) GrBP5-M4 and (d) GrBP5-M5, adsorbed at the aqueous graphene interface. Water not shown for clarity. The backbone of the peptides sequences is coloured by sub-domain: SD1, SD2 and SD3 are purple, green and orange respectively. | ||
The binding properties of GrBP5-M1 encompassed the second category, where the Y9A and Y12A mutants resulted in surface contact dominated by SD1 and SD2, while SD3 was extended away from the surface. The chief contributors to the peptide-surface contact were located in the N-terminal half of the sequence (M2, V3, S5, S6) (Fig. 4a and Fig. S5 of the ESI†). This appeared to compensate for the loss of SD3-graphene contact. Experimental studies of GrBP5-M1 supported a low surface coverage (which So et al. conflated with lower binding strength), with no observed pattern formation in the dried state. Our findings are also therefore consistent with the experimental hypothesis that SD1 (and possibly also SD2) may play a role in peptide aggregation at the interface.
GrBP5-M4 represented the third category, with strong contact points spaced along the length of the peptide chain, at positions 2, 3, 7, 9 and 12 (Fig. S5 of the ESI†). The adsorbed peptide adopted conformations were flat in profile on surface (Fig. 4c and Fig. S5 of the ESI†), lacking the unbound peptide segments of the other two adsorption-mode categories (where either SD1 or SD3 protruded from the surface). Previous experiments sought to conflate the high surface coverage of this sequence with strong binding strength, and moreover this sequence did not support pattern formation in the dried state. We remark here that while we also tried the more traditional structural analysis of the radius of gyration, which was, as expected, not very informative given the short length of these IDP-like peptide chains (section ‘Radius of gyration analysis’, ESI†).
In summary, our simulation data are consistent with the hypothesis proposed by So et al.,9–11 where the two common themes were the strong SD3-surface contact and the presence of SD1 as an unbound chain segment However, it is not clear if these experimentally observed ordered peptide over-layers were formed due to the drying process. We next investigated the viability of peptide assembly of GrBP5-WT at the aqueous graphene interface using advanced conformational sampling approaches.
Multi-chain adsorption of GrBP5-WT
From previously-reported experiments, the disorder-to-order transition was suggested to occur at ≈60% coverage.9–11 Therefore, two multi-chain systems were prepared and modelled using REST-MD simulations; one containing eight chains, and the second containing twelve chains, of GrBP5-WT. We established that these two systems corresponded with surface coverages of ∼50% and ∼70%, based on the calculation of the solvent accessible surface area (SASA) of the graphene sheet when covered with 8 and 12 peptide chains (‘Simulation details’, ESI†).We first quantified the degree of residue-surface contact for the over-layer systems and compared these with our single-chain data. Table S5 of the ESI† summarises the average residue-surface contact fraction for each residue in the multi-chain systems. For both coverages, the overall trends in residue surface contact reflected that of the single chain.
Despite the fact that the chains were initially positioned with a (relatively) even lateral distribution across the graphene interface, Fig. 5a, the over-layer morphologies quickly became dominated by arrangements of aggregated chains (Fig. 5b). The over-layers featured regions of high and low peptide density with an uneven lateral distribution, exposing patches of graphene directly to the solvent. However, the overall degree of surface coverage did not change significantly, from ∼50 (or ∼70%) during the simulation. Along the direction normal to the surface, the spatial distribution of the peptide chains in the over-layers (Fig. S6 of the ESI†) was effectively unchanged compared with the single chain data. Aggregation of the peptide chains was confirmed via calculations of the average number of inter-chain atomic pair contacts as a function of REST-MD simulation steps, as shown in Fig. 6a. We also calculated the average solvent accessible surface area (SASA) per chain of the peptide over-layer (i.e. the surface area of each surface-adsorbed chain in contact with water). This trend in peptide SASA values was also consistent with an aggregated state (Fig. 5b).
 | ||
| Fig. 5 Snapshots of the adsorbed over-layer of GrBP5-WT (50% surface coverage) (a) the initial geometry and (b) after 40 ns of REST-MD simulation. The purple surface indicates the solvent accessible surface area of the peptide over-layer. Water not shown for clarity. | ||
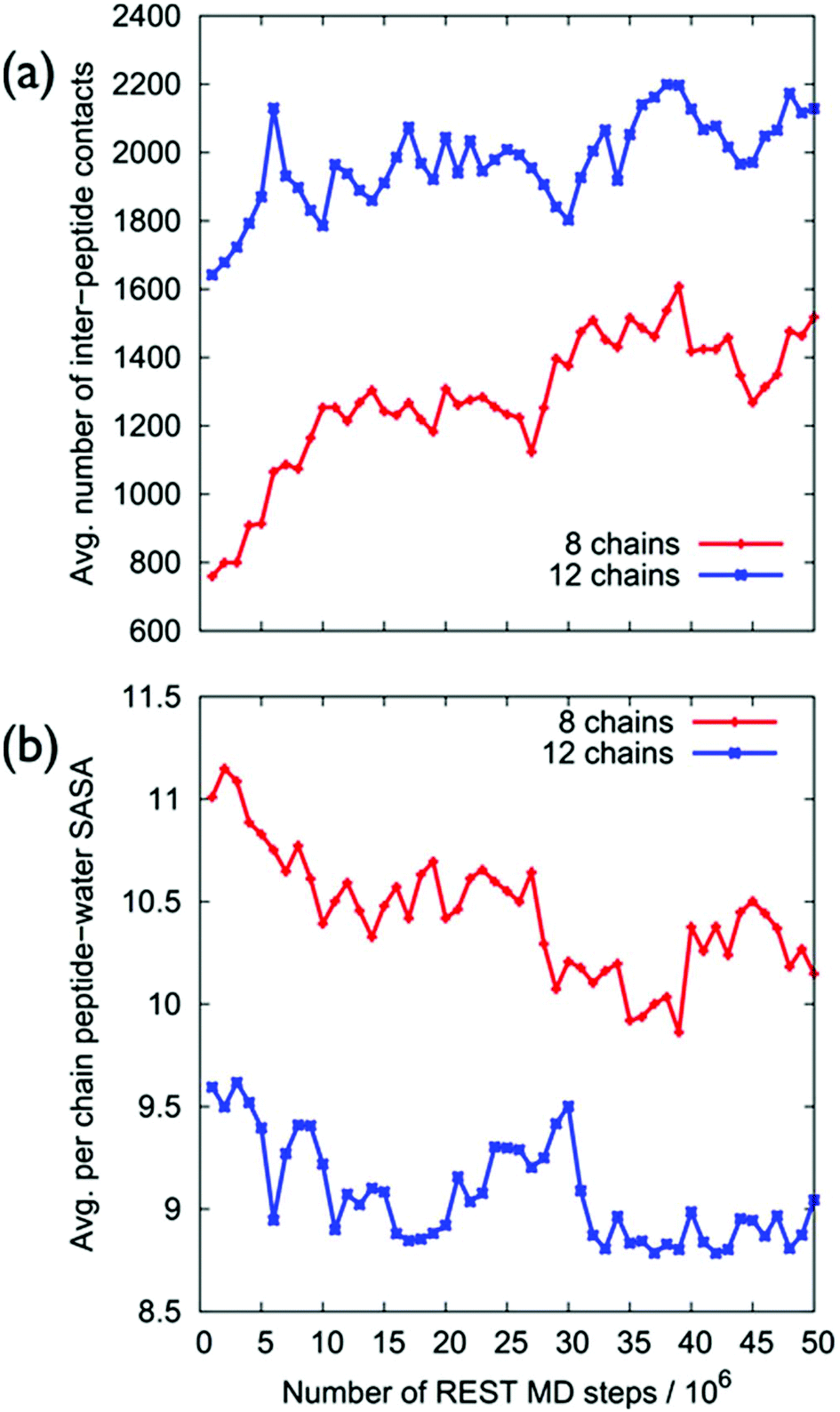 | ||
| Fig. 6 Aggregation of the peptide chains, (a) average number of inter-peptide atom contacts and (b) average per chain surface area of peptide exposed to water, as a function of REST-MD trajectory, for both 50 and 70% coverage, adsorbed at the aqueous graphene interface. | ||
Clustering analysis of each individual chain in the multi-chain over-layer (detailed in the ESI† section ‘Multi-chain clustering’) suggested that the change in coverage did not appreciably influence the overall trends in residue-surface contact. Up to this point, the findings from our multi-chain REST-MD simulations have broadly supported the hypothesis inferred from the experimental studies. However, as detailed herein, our evidence does not support the formation and/or stability of peptide over-layer ordering under aqueous conditions. Despite our extensive conformational sampling of the resulting over-layer structures, it was not apparent from visual inspection, nor by our analyses, that ordered morphologies were produced, or that ordering was actually incipient, from these aggregates.
To this end, we next probed the structural ordering of the over-layers more systematically. For example, intuitively, we expect that an ordered multi-peptide layer morphology should feature a small number of distinct conformations. On the contrary, our simulations revealed an increase in conformational diversity as a function of increasing surface coverage, as supported by cross-comparisons of the two most populated clusters of each individual peptide chain in the over-layer for each coverage (Tables S8 and S9 of the ESI†).
Additionally, we would not expect to find a strong random coil character in the adsorbed peptide conformations of an ordered over-layer. The secondary structural characteristics of the chains were probed via calculations of the relative proportions of the different secondary structure motifs, averaged over all peptide chains in the over-layer, shown in Fig. S7 of the ESI.† These data further indicated a lack of secondary structural ordering for both coverages, where random coil character accounted for over 70% of the ensemble.
Despite our extensive conformational sampling of the multi-chain systems, it is possible that our simulations might not have captured the timescales on which structural ordering may eventually proceed. If this were true, we would still expect to observe key mechanistic details indicative of the progression from adsorption, to aggregation, and finally to ordering. To test for So et al.'s suggested ordering mechanism (via hydrophobic inter-chain interactions), we investigated different types of inter-peptide interactions (detailed below), but found little evidence to support this.
We first quantified the intra-chain and inter-chain hydrogen-bonding, which appear to be inconsistent with So et al.'s proposed mechanism. Our data indicate that the formation of hydrogen bonds was a significant factor in the peptide aggregation. The breakdown of the average number of inter-chain hydrogen bonds between each sub-domain pairing is shown in Fig. 7, while Tables S10 and S11 of the ESI† provide the relevant numerical details.
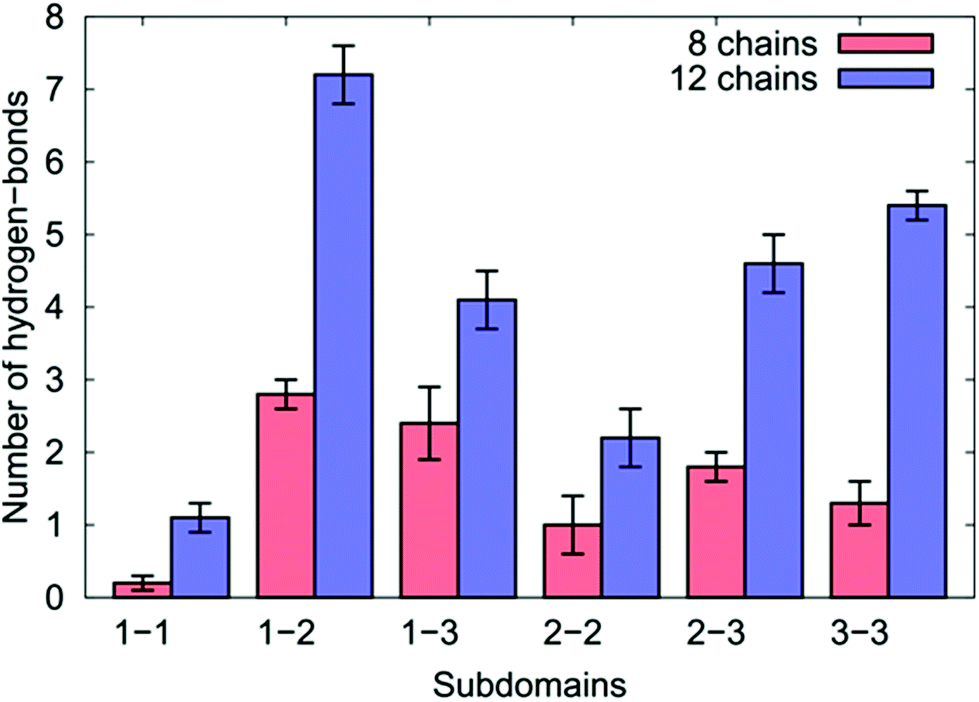 | ||
| Fig. 7 The number of inter-chain hydrogen bonds for the different sub-domains of the over-layers of GrBP5-WT adsorbed at the aqueous graphene interface. | ||
We found for both coverages that 29% of all the inter-chain hydrogen bonds formed were found between residues in SD1 and SD2, which comprised the single greatest contribution to the inter-chain hydrogen bonds of any pairing. This may seem surprising given that the side-chains in the hydrophobic residues in SD1 cannot participate in hydrogen bonding. This can be explained by the positively-charged N-terminus in SD1 interacting with the negatively-charged Asp/Glu residues in SD2 (accounting for more than 50% of the SD1–SD2 inter-chain hydrogen bonds).
In the case of the eight chain over-layer, the SD1–SD3 pairing was the second largest contributor to inter-chain hydrogen bonds, while the SD3–SD3 pairing was the second largest contributor for the 12-chain system, with a significant increase in the total number compared with the lower coverage. We also noted a concomitant decrease in the number of intra-chain hydrogen bonds per chain between SD2–SD3 and SD3–SD3 suggesting that as peptide coverage increased, residues in SD3 that previously supported intra-peptide hydrogen bonds were diverted into SD3–SD3 inter-chain hydrogen bonds. As the coverage increased, we also observed that residues in SD2 were diverted from intra-peptide hydrogen bonds with SD3 into forming SD2–SD2 intra-peptide hydrogen bonds.
Hydrophobic–hydrophobic inter-chain interactions, mediated via SD1, are a key aspect of the currently-proposed ordering mechanism. However, we did not find evidence to support this hypothesis, as detailed below. To characterise these interactions, we calculated the inter-chain radial distribution functions, g(r), related to specific sites (Table S12†) in the residues of SD1, for the twelve chain over-layer. As detailed in the ESI† section ‘Hydrophobic inter-chain contact’, there was only insubstantial inter-chain contact between the hydrophobic residues of SD1. In summary, our inter-chain interaction analyses do not support a mechanism driven by hydrophobic–hydrophobic contacts in SD1 of GrBP5-WT.
Our hydrogen-bonding analysis of the GrBP5-WT over-layers suggested that the Y9W/Y12W mutation (GrBP5-M2) might disrupt the SD3–SD3 inter-chain interactions in our GrBP5-WT multi-chain simulations. Such disruption might account for the experimentally-reported weaker aggregation of GrBP5-M2. Furthermore, the similarity in the experimentally determined over-layer morphologies of GrBP5-WT and GrBP5-M5 is consistent with our predicted single-chain surface-residue contact behaviour for these sequences. Our single-chain evidence suggests that the mutation of SD1 in GrBP5-M5 might not adversely affect the inter-chain interactions (SD1–SD2 and SD3–SD3) noted in the simulations of GrBP5-WT over-layers. In contrast, the enhanced surface contact of SD1 with the graphene interface for GrBP5-M4 could conceivably lead to the C-termini of the peptide chains competing with E5/D8 for interaction with the N-termini. In addition, the strong residue-surface contact of Q2 and S3 also suggest that an enhancement SD1–SD3 hydrogen bonding might be possible, which may explain the more porous over-layer observed experimentally for GrBP5-M4.
Discussion
Overall, our REST-MD simulations of the multi-chain GrBP5-WT system revealed that the peptide chains maintained close engagement with the aqueous graphene interface primarily via the SD3 region. While the chains aggregated and supported enhanced inter-peptide interactions, the resulting over-layers showed little sign of ordering. The proposed amphiphilic behaviour of the peptide chains was not apparent from our simulations. This discrepancy with the previously-proposed formation mechanism could be attributed to number of factors.First, we acknowledge that it is highly challenging to sample the conformations of the adsorbed multi-chain peptide over-layers thoroughly, despite the efficiency and effectiveness of the REST-MD simulation technique. While the effective time-scales captured in our REST-MD simulations span the tens of μs range,39 this time-scale may still be too short to capture the entire ordering process. That said, our simulations did not reveal indicative signatures of the onset of a disorder-to-order structural transition, such as the formation of incipient hydrophobic inter-peptide interactions, and/or a reduction in conformational diversity in the adsorbed peptide ensemble as a function of simulation progression. Moreover, we cannot rule out possible finite size effects, which we cannot explore further given the REST-MD overlayer simulations of our current size are already extremely resource-intensive.
Alternatively, it is possible that the experimentally observed over-layers of GrBP5-WT may not actually self-assemble into ordered structures under aqueous conditions. Recent experimental evidence reported by Sun et al.44 suggests that this is a plausible scenario. In this recent study, dried over-layers of GrBP5-WT on HOPG were soaked overnight in water and re-examined using ex situ AFM observations. These authors found the peptide over-layer did not remain ordered, while the coverage also decreased.
Overall, our REST-MD simulations are indicate strong inter-peptide interactions; even if GrBP5-WT was conclusively found to not form an ordered over-layer under aqueous conditions, our data might indicate how the GrBP5 sequence may be modified to achieve this. For example, enlargement of the hydrophobic SD1 sub-domain, particularly with additional aliphatic residues that are predicted to have low affinity for aqueous graphene (such as Ile, Leu, and Val), may both discourage SD1-surface interactions and provide enhanced opportunities for hydrophobically-mediated SD1–SD1 inter-peptide interactions. Also, the charge–charge inter-peptide interactions could be manipulated to augment those arising from the N-termini and E5/D5 e.g. via the judicious insertion of acidic and basic residues. In partnership with such modifications, a combination of in situ AFM experiments and advanced molecular simulations would enable the systematic advances needed to rationally design of graphene binding peptides with a reliable self-patterning capability at the interface.
Computational methodology
All simulations were performed using GROMACS version 5.0.45 The GRAPPA FF35 was used to describe interactions between the graphene surface and the peptides and water molecules described using the CHARMM22*46,47 parameters and the modified version of the TIP3P water48,49 model, respectively. The Replica Exchange with Solute Tempering (REST)38,39 MD simulation approach has been employed throughout (see previous studies for further details34,39,50). Here, we used 16 replicas distributed over an effective temperature window of 300–433 K. All simulations were performed in the canonical (NVT) ensemble at 300 K and a time-step of 1 fs. Production runs were performed for 20 × 106 and 50 × 106 MD steps, for the single chain and multi-chain systems respectively, with exchanges between replicas attempted every 1000 steps (1 ps).In all simulations, the peptide chains were modeled in zwitterionic form (i.e. with no capping groups on the termini), consistent with previous experiments. We conducted five surface-adsorbed single-chain simulations, each comprising one peptide chain (one of each sequence from Table 1), the graphene surface, liquid water, and counter-ions where necessary. The initial conformation of the peptide chain in each of the 16 replicas was different, covering a range of different secondary structure motifs. For the over-layer simulations of GrBP5-WT, the system comprised eight/twelve peptide chains adsorbed on a graphene sheet, liquid water, and sufficient Na+ counter-ions to ensure overall charge neutrality. The six most populated distinct conformations of GrBP5-WT in the single-chain adsorbed state, identified from the clustering analysis of single chain simulations, were used in the construction of the initial over-layer configurations. Full details of all aspects of these REST-MD simulations, including their analyses and tests for equilibration, are provided in the ESI.†
Conclusions
Advanced conformational sampling using REST-MD simulations, in combination with a polarisable force-field specifically tailored for aqueous biomolecule-graphitic interfaces, was used to predict the adsorption and surface-mediated self-organisation of a family of graphene-binding peptides based on the GrBP5 sequence. In terms of the adsorption of single peptide chains to the aqueous graphene interface, our findings were consistent with existing experimental data. The parent GrBP5-WT sequence adsorbed to chiefly via the aromatic residues in SD3, with the other two sub-domains supporting only weak interactions with the surface. Two of the mutant sequences, GrBP5-M2 and GrBP5-M5, adopted a similar adsorption mode. The Y9A/Y12A mutations in GrBP5-M1 yielded reduced contact with the surface. Mutation of the hydrophobic SD1 region with hydrophilic residues led to adsorption mediated via SD1 and SD3, which consequently yielded enhanced surface contact and a flatter adsorbed structure relative to the other sequences in this family. Multi-chain simulations GrBP5-WT at 50% and 70% surface coverage revealed that the surface contact was also chiefly mediated by SD3. At both coverages, the peptide chains aggregated in an uneven spatial distribution, exposing patches of bare graphene to liquid water. However, neither a long-range ordering of the over-layer, nor an incipient disorder-to-order transition, was evident. In contrast with previous hypotheses, we found the aggregation of the peptide chains was primarily driven by hydrogen-bonding and charge–charge interactions between SD1 and SD2, and not hydrophobic inter-peptide contacts between SD1 and SD1. Our findings indicate modifications of the GrBP5 sequence to improve the self-organisation capability of this peptide, which may induce and stabilise the lateral ordering of multi-chain peptide over-layers on graphene under aqueous conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Air Force Office for Scientific Research (Grant #FA9550-12-1-0226). T. R. W. thanks veski for an Innovation Fellowship. The computational resources for this work were provided by the Victorian Life Sciences Computation Initiative (VLSCI).References
- F. Nudelman, B. A. Gotliv, L. Addadi and S. Weiner, J. Struct. Biol., 2006, 153, 176–187 CrossRef CAS PubMed
.
- F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582–6596 CrossRef CAS PubMed
- A. Care, P. L. Bergquist and A. Sunna, Trends Biotechnol., 2015, 33, 259–268 CrossRef CAS PubMed
- F. De Leo, A. Magistrato and D. Bonifazi, Chem. Soc. Rev., 2015, 44, 6916–6953 RSC
- M. R. Knecht and T. R. Walsh, Chem. Rev., 2017, 117, 12641–12704.
- M. J. Pender, L. A. Sowards, J. D. Hartgerink, M. O. Stone and R. R. Naik, Nano Lett., 2006, 6, 40–44 CrossRef CAS PubMed
- S. N. Kim, Z. Kuang, J. M. Slocik, S. E. Jones, Y. Cui, B. L. Farmer, M. C. McAlpine and R. R. Naik, J. Am. Chem. Soc., 2011, 133, 14480–14483 CrossRef CAS PubMed
- M. Calvaresi and F. Zerbetto, Acc. Chem. Res., 2013, 46, 2454–2463 CrossRef CAS PubMed
- C. R. So, Y. Hayamizu, H. Yazici, C. Gresswell, D. Khatayevich, C. Tamerler and M. Sarikaya, ACS Nano, 2012, 6, 1648–1656 CrossRef CAS PubMed
- D. Khatayevich, C. R. So, Y. Hayamizu, C. Gresswell and M. Sarikaya, Langmuir, 2012, 28, 8589–8593 CrossRef CAS PubMed
- D. Khatayevich, T. Page, C. Gresswell, Y. Hayamizu, W. Grady and M. Sarikaya, Small, 2014, 10, 1505–1513 CrossRef CAS PubMed
- Y. Hayamizu, C. R. So, S. Dag, T. S. Page, D. Starkebaum and M. Sarikaya, Sci. Rep., 2016, 6, 33778 CrossRef CAS PubMed
- C. R. So, J. Liu, K. P. Fears, D. H. Leary, J. P. Golden and K. J. Wahl, ACS Nano, 2015, 9, 5782–5791 CrossRef CAS PubMed
- C.-L. Chen, R. N. Zuckermann and J. J. DeYoreo, ACS Nano, 2016, 10, 5314–5320 CrossRef CAS PubMed
- F. Jiao, Y. Chen, H. Jin, P. He, C.-L. Chen and J. J. De Yoreo, Adv. Funct. Mater., 2016, 26, 8960–8967 CrossRef CAS
- T. R. Walsh, Acc. Chem. Res., 2017, 50, 1617–1624 CrossRef CAS PubMed
- R. B. Pandey, Z. Kuang, B. L. Farmer, S. S. Kim and R. R. Naik, Soft Matter, 2012, 8, 9101–9109 RSC
- P. Gemünden and H. Behringer, J. Chem. Phys., 2013, 138, 024904 CrossRef PubMed
- K. L. Osborne, M. Bachmann and B. Strodel, Proteins, 2013, 81, 1141–1155 CrossRef CAS PubMed
- M. Möddel, W. Janke and M. Bachmann, Phys. Rev. Lett., 2014, 112, 148303 CrossRef PubMed
- N. M. Bedford, Z. E. Hughes, Z. Tang, Y. Li, B. D. Briggs, Y. Ren, M. T. Swihart, V. G. Petkov, R. R. Naik, M. R. Knecht and T. R. Walsh, J. Am. Chem. Soc., 2016, 138, 540–548 CrossRef CAS PubMed
- G. Zuo, S.-G. Kang, P. Xiu, Y. Zhao and R. Zhou, Small, 2012, 9, 1546–1556 CrossRef PubMed
- M. Hoefling, S. Monti, S. Corni and K.-E. Gottschalk, PLoS One, 2011, 6, e20925 CAS
- S. Monti, V. Carravetta, C. Li and H. Ågren, J. Phys. Chem. C, 2014, 118, 3610–3619 CAS
- N. M. Bedford, H. Ramezani-Dakhel, J. M. Slocik, B. D. Briggs, Y. Ren, A. I. Frenkel, V. Petkov, H. Heinz, R. R. Naik and M. R. Knecht, ACS Nano, 2015, 9, 5082–5092 CrossRef CAS PubMed
- H. Ramezani-Dakhel, L. Ruan, Y. Huang and H. Heinz, Adv. Funct. Mater., 2015, 25, 1374–1384 CrossRef CAS
- S. Monti, V. Carravetta and H. Ågren, Nanoscale, 2016, 8, 12929–12938 RSC
- S.-G. Kang, T. Huynh, Z. Xia, Y. Zhang, H. Fang, G. Wei and R. Zhou, J. Am. Chem. Soc., 2013, 135, 3150–3157 CrossRef CAS PubMed
- E. J. Wallace, R. S. G. D'Rozario, B. M. Sanchez and M. S. P. Sansom, Nanoscale, 2010, 2, 967–975 RSC
- B. Akdim, R. Pachter, S. S. Kim, R. R. Naik, T. R. Walsh, S. Trohalaki, G. Hong, Z. Kuang and B. L. Farmer, ACS Appl. Mater. Interfaces, 2013, 5, 7470–7477 CAS
- A. N. Camden, S. A. Barr and R. J. Berry, J. Phys. Chem. B, 2013, 117, 10691–10697 CrossRef CAS PubMed
- N. Dragneva, W. B. Floriano, D. Stauffer, R. C. Mawhinney, G. Fanchini and O. Rubel, J. Chem. Phys., 2013, 139, 174711 CrossRef CAS PubMed
- N. Todorova, A. J. Makarucha, N. D. M. Hine, A. A. Mostofi and I. Yarovsky, PLoS Comput. Biol., 2013, 9, e1003360 Search PubMed
- Z. E. Hughes and T. R. Walsh, J. Mater. Chem. B, 2015, 3, 3211–3221 RSC
- Z. E. Hughes, S. M. Tomásio and T. R. Walsh, Nanoscale, 2014, 6, 5438–5448 RSC
- M. J. Penna, M. Mijajlovic, C. Tamerler and M. J. Biggs, Soft Matter, 2015, 11, 5192–5203 RSC
- C. M. Welch, A. N. Camden, S. A. Barr, G. M. Leuty, G. S. Kedziora and R. J. Berry, J. Chem. Phys., 2015, 143, 045104 CrossRef PubMed
- T. Terakawa, T. Kameda and S. Takada, J. Comput. Chem., 2010, 32, 1228–1234 CrossRef PubMed
- L. B. Wright and T. R. Walsh, Phys. Chem. Chem. Phys., 2013, 15, 4715–4726 RSC
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed
- Z. E. Hughes, G. Wei, K. L. M. Drew, L. Colombi Ciacchi and T. R. Walsh, Langmuir, 2017, 33, 10193–10204 CrossRef CAS PubMed
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed
- X. Zou, S. Wei, J. Jasensky, M. Xiao, Q. Wang, I. Brooks, L. Charles and Z. Chen, J. Am. Chem. Soc., 2017, 139, 1928–1936 CrossRef CAS PubMed
- L. Sun, T. Narimatsu, S. Tsuchiya, T. Tanaka, P. Li and Y. Hayamizu, RSC Adv., 2016, 6, 96889–96897 RSC
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed
- S. Piana, K. Lindorff-Larsen and D. E. Shaw, Biophys. J., 2011, 100, L47 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- E. Neria, S. Fischer and M. Karplus, J. Chem. Phys., 1996, 105, 1902–1921 CrossRef CAS
- Z. Tang, J. P. Palafox-Hernandez, W.-C. Law, Z. E. Hughes, M. T. Swihart, P. N. Prasad, M. R. Knecht and T. R. Walsh, ACS Nano, 2013, 7, 9632–9646 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Methodology details, residue contact data, clustering data, secondary structure analyses, density profiles, conformational comparisons, hydrogen-bonding data, radial distribution functions, and force-field comparisons. See DOI: 10.1039/c7nr06441a |
| This journal is © The Royal Society of Chemistry 2018 |