
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles†
Andrea
Longatti
a,
Christina
Schindler
 a,
Andie
Collinson
a,
Lesley
Jenkinson
a,
Carl
Matthews
a,
Laura
Fitzpatrick
a,
Margaret
Blundy
b,
Ralph
Minter
a,
Tristan
Vaughan
a,
Michael
Shaw
cd and
Natalie
Tigue
*a
a,
Andie
Collinson
a,
Lesley
Jenkinson
a,
Carl
Matthews
a,
Laura
Fitzpatrick
a,
Margaret
Blundy
b,
Ralph
Minter
a,
Tristan
Vaughan
a,
Michael
Shaw
cd and
Natalie
Tigue
*a
aDepartment of Antibody Discovery and Protein Engineering, MedImmune Ltd., Granta Park, Cambridge, CB21 6GH, UK. E-mail: tiguen@medimmune.com
bCore Tissue Culture, MedImmune Ltd., Granta Park, Cambridge, CB21 6GH, UK
cNational Physical Laboratory, Teddington, TW11 0LW, UK
dDepartment of Computer Science, University College London, London, WC1 6BT, UK
First published on 16th July 2018
Abstract
Exosomes are extracellular vesicles that mediate cell-to-cell communication by transferring biological cargo, such as DNA, RNA and proteins. Through genetic engineering of exosome-producing cells or manipulation of purified exosomes, it is possible to load exosomes with therapeutic molecules and target them to specific cells via the display of targeting moieties on their surface. This provides an opportunity to exploit a naturally-occurring biological process for therapeutic purposes. In this study, we explored the potential of single chain variable fragments (scFv) as targeting domains to achieve delivery of exosomes to cells expressing a cognate antigen. We generated exosomes targeting the Her2 receptor and, by varying the affinity of the scFvs and the Her2 expression level on recipient cells, we determined that both a high-affinity anti-Her2-scFv (KD ≤ 1 nM) and cells expressing a high level (≥106 copies per cell) of Her2 were optimally required to enable selective uptake. We also demonstrate that targeting exosomes to cells via a specific cell surface receptor can alter their intracellular trafficking route, providing opportunities to influence the efficiency of delivery and fate of intracellular cargo. These experiments provide solid data to support the wider application of exosomes displaying antibody fragments as vehicles for the targeted delivery of therapeutic molecules.
Introduction
Exosomes are small (50–100 nm in diameter) extracellular vesicles (EVs) that are released by cells and transmit information to other cells via delivery of biologically active cargo.1 Exosomes are formed by the invagination of the endosomal membrane leading to the formation of![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ulti
ulti![[v with combining low line]](https://www.rsc.org/images/entities/char_0076_0332.gif) esicular
esicular ![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) odies (MVBs). During this process cytosolic material is incorporated into the lumen of exosomes, which are subsequently expelled into the extracellular space upon fusion of MVBs with the plasma membrane.2 Once released, exosomes can be taken up by cells in an autocrine, paracrine or endocrine manner.3,4 Several routes by which exosomes are taken up by target cells have been described and include direct membrane fusion or endocytosis via clathrin-mediated endocytosis, macropinocytosis, and caveolae-dependent uptake.5–8 Except for membrane fusion, where the exosome content is released into the cytoplasm directly, whole exosomes enter the recipient cell, whereby cargo release is mediated by fusion of the exosome membrane with the endosome membrane.5,9,10
odies (MVBs). During this process cytosolic material is incorporated into the lumen of exosomes, which are subsequently expelled into the extracellular space upon fusion of MVBs with the plasma membrane.2 Once released, exosomes can be taken up by cells in an autocrine, paracrine or endocrine manner.3,4 Several routes by which exosomes are taken up by target cells have been described and include direct membrane fusion or endocytosis via clathrin-mediated endocytosis, macropinocytosis, and caveolae-dependent uptake.5–8 Except for membrane fusion, where the exosome content is released into the cytoplasm directly, whole exosomes enter the recipient cell, whereby cargo release is mediated by fusion of the exosome membrane with the endosome membrane.5,9,10
The capacity of exosomes to transfer biologically active molecules such as RNA, DNA and protein has been extensively studied1,11,12 and strategies to exploit this phenomenon to develop exosomes loaded with therapeutically relevant molecules are now emerging. For example, exosomes can be loaded with protein or RNA cargo by genetic modification of the producer cells, which leads to its incorporation during MVB biogenesis,12 or post-purification by electroporation, detergent permeabilisation, hypotonic dialysis, sonication, transfection or extrusion.13–19 Such “ex vivo” loading strategies have been primarily adopted to encapsulate small molecules, with the observation that the encapsulated cargo demonstrates superior efficacy compared to the free drug, both in vivo and in vitro.19–21
To further exploit the potential of exosomes as therapeutic nanovesicles, a means by which to target exosomes to enable delivery to the desired recipient cell will be important. Targeting mechanisms have been explored previously by displaying naturally occurring peptides or receptor ligands on the surface of exosomes,22 however, such approaches have limitations for universal application. Antibodies or antibody fragments, such as single chain variable fragment (scFv) domains, could provide a versatile alternative for the targeting of exosomes to specific cells because they can be raised to virtually any antigen and they are exquisitely specific. The affinity of an scFv can also be tuned to the desired level, which may be crucial in balancing targeting and exosomal cargo release kinetics.
Most studies to date have incorporated targeting moieties onto the surface of exosomes via genetic modification of cells to encode a fusion to a transmembrane domain that localises to exosomal membranes e.g. Lamp2b.23 In this study we employed a similar strategy; we modified HEK293 cells to express scFv domains with varying affinities for the cell surface receptor ERBB2 (Her2), fused to the C1C2 domain of lactadherin, which peripherally associates with the outer exosome membrane by interaction with phosphatidylserine24 and is highly enriched in exosome fractions.25 We demonstrate that anti-Her2 scFvs can be displayed on the surface of exosomes and confirm that these exosomes can bind to recombinant Her2. In uptake studies, we observed that incorporation of the C1C2 domain at the surface of the exosomes negatively impacts their ability to be taken up by cells, whereas exosomes displaying scFvs with a high affinity for Her2 results in selective uptake into cells expressing a high level of Her2. Furthermore, we show that, by targeting exosomes to specific cell surface antigens via the C1C2 domain, it is possible to alter the routes by which the exosomes are trafficked. Our data suggest that antibody-mediated targeting of exosomes has the potential to be broadly applicable for the development of targeted exosome therapeutics.
Results
Exosome uptake by recipient cells
To study the internalisation of HEK293-derived exosomes (hereafter referred to as exosomes) into recipient cells we established flow cytometry and microscopy-based uptake assays using purified, carboxyfluorescein succinimidyl ester (CFSE) labelled exosomes (CFSE-exosomes). CFSE labels exosomal protein cargo as well as transmembrane proteins via the action of esterases present in the exosome lumen, allowing the detection of intact exosomes, released cargo, and exosomal membrane proteins in recipient cells after uptake.When HEK293 cells were incubated with increasing concentrations of CFSE-exosomes we observed that association and uptake of the labelled exosomes into cells, as assessed by flow cytometry, is robust at low concentrations (0.5 × 1011 particles per ml) and is dose-dependent, with no saturation reached even at 3 × 1011 particles per ml during a 4 h incubation (Fig. 1A and ESI Fig. S2A†). Subsequently a time course was performed with 3 × 1011 particles per ml, and exosome association and uptake reached steady-state between 4 and 18 h continuous incubation, before a reduced signal was observed (Fig. 1B), likely due to degradation of the CFSE dye in lysosomes. For all subsequent experiments an incubation period of 4 h was used to achieve robust exosome uptake.
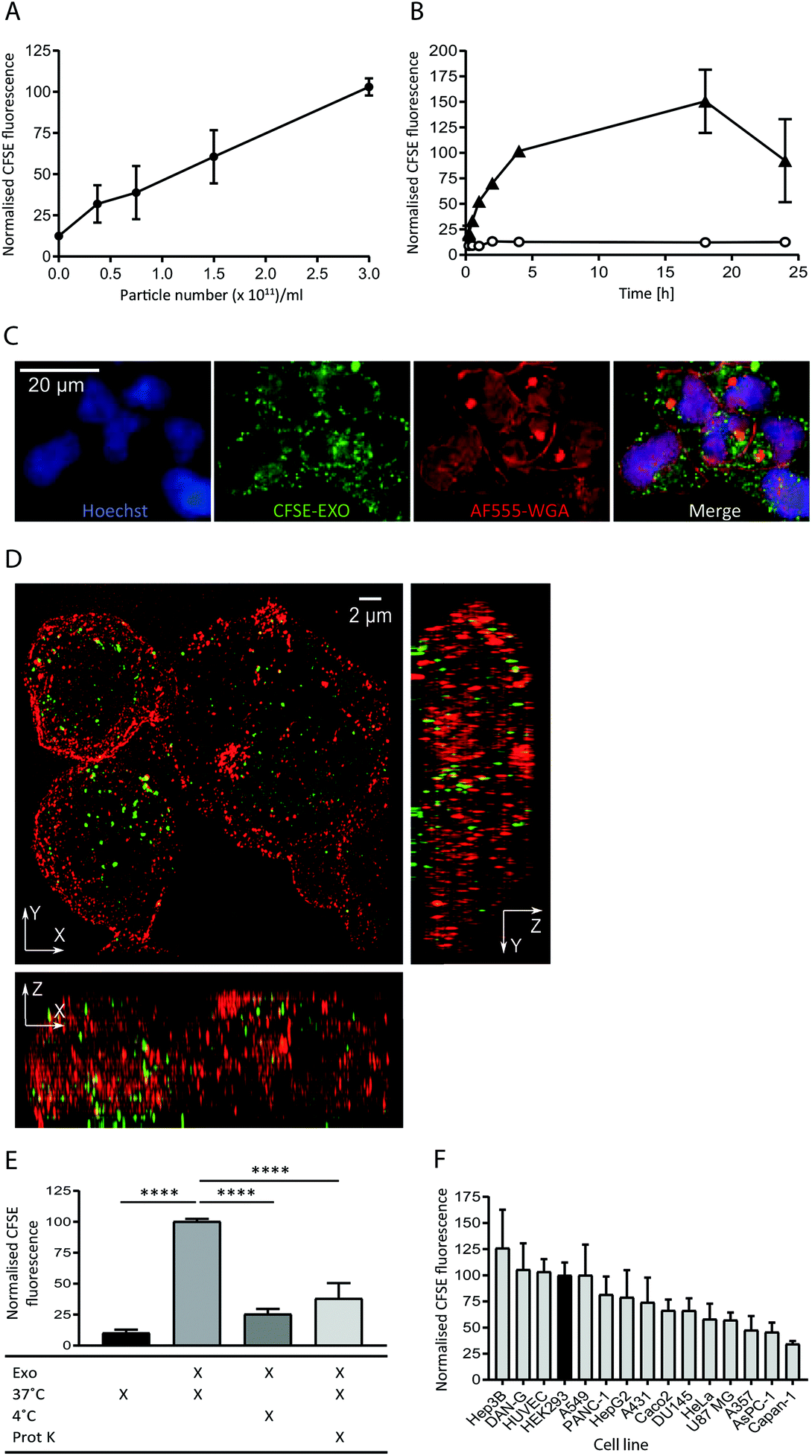 | ||
| Fig. 1 Exosomes derived from HEK293 cells are taken up efficiently by HEK293 cells in a time and dose dependent active manner. (A) HEK293 cells were incubated with increasing amounts of purified CFSE-exosomes, or with PBS, in duplicate for 4 h and recipient cell fluorescence was assessed by flow cytometry. Average mean fluorescence intensity (MFI) values were then calculated and normalised to the MFI value achieved at 3 × 1011 particles per ml. Error bars represent the standard error of the mean (SEM) for three independent experiments. Histograms can be found in ESI Fig. S2A.† (B) HEK293 cells were incubated for increasing amounts of time with 3 × 1011 particles per ml CFSE-exosomes (triangles), or with PBS (open circles) in duplicate and assessed by flow cytometry. MFI values were then calculated and normalised to the MFI obtained at 4 h incubation. Error bars represent the SEM for three independent experiments except for the 15, 30 and 60 minutes timepoints, which were only analysed once. (C) HEK293 cells were incubated with 3 × 1011 particles per ml CFSE-labelled exosomes (green) for 4 h and stained with Alexa Fluor-555 Wheat Germ Agglutinin (WGA; plasma membrane; red) and Hoechst (nucleus; blue) post-fixation, and visualised using confocal microscopy. (D) HEK293 cells were incubated with 3 × 1011 particles per ml CFSE-exosomes (green) for 4 h, then fixed (but not permeabilised) and stained with anti-CD81 to outline the plasma, followed by anti-mouse Alexa Fluor-647, and visualised using structured illumination super-resolution microscopy (SIM). The maximum intensity projection of each axis is shown. (E) HEK293 cells were incubated with CFSE-labelled exosomes or PBS, treated in the presence or absence of proteinase K, on ice or at 37 °C for 4 h, in duplicate. Uptake was assessed by flow cytometry, and the MFI values were normalised to the 37 °C values (second column). Statistical analyses were performed using one-way ANOVA with multiple comparisons, comparing to exosome uptake at 37 °C in the absence of proteinase K (column 2). Error bars represent the SEM of three 3 independent experiments. (F) Fifteen immortalised adherent cell lines were tested for their ability to take up HEK293-derived exosomes. Cells were incubated with 3 × 1011 particles per ml CFSE-labelled exosomes in duplicate for 4h and subsequently labelled with CellMask Deep Red to normalise for cell volume. CFSE-derived MFI values were then calculated and normalised to the CellMask Deep Red signal for each cell line. The values were subsequently normalised to the HEK recipient cell value and plotted. Error bars represent the SEM for three independent experiments. | ||
The intracellular localisation of CFSE-exosomes was confirmed using confocal microscopy where a CFSE-derived signal could be observed in the intracellular space following staining of the plasma membrane with labelled wheat germ agglutinin (Fig. 1C). The intracellular location of CFSE-exosomes was also visualised by super-resolution structured illumination microscopy (SIM) images, which showed an abundance of CFSE-labelled vesicles in the cell lumen (Fig. 1D and ESI Movies S1A and B†).
To confirm that, in the flow cytometry-based assay, we observe active exosome uptake as has been described previously5–7,26 HEK293 cells were incubated with CFSE-exosomes at 37 °C, or on ice for 4 h, and the fluorescence of the recipient cells was monitored by flow cytometry (Fig. 1E). As a control, PBS that had been incubated with an appropriate volume of FBS and CFSE and purified in the same manner as the exosome preps, was used. When the cells were placed on ice, where endocytosis and intracellular transport is inhibited, CFSE-exosome uptake was reduced to 25% of that observed at 37 °C, compared to 10% for the CFSE-only control, suggesting that the majority of CFSE-exosome transfer observed at 37 °C represents active cellular uptake. Consequently, and for simplicity, we have used the term “uptake” when describing subsequent flow cytometry experiments. Furthermore, when exosomes were pre-treated with proteinase K, which removes proteins exposed on the surface of exosomes, a 62% reduction in uptake was observed, indicating that protein(s) on the exosome membrane play a key role in mediating exosome binding and internalisation.
Next, we compared the exosome uptake efficiency of fifteen immortalised adherent cell lines, to identify a cell line that demonstrates efficient exosome uptake for use in subsequent experiments (Fig. 1F). The panel of cell lines was incubated for 4 h with CFSE-exosomes and analysed by flow cytometry. In parallel, CellTracker Deep Red staining was used to determine total cell volume. By normalising the CFSE-derived signal to the CellTracker Deep Red signal, we eliminated bias arising from differences in recipient cell volume, enabling comparisons to made between cell lines. We observed differences in uptake efficiency between the individual cell lines, with the most efficient cell line, Hep3B, demonstrating a 3-fold higher signal compared to the least efficient cell line, Capan-1. Interestingly, none of the cell lines in the panel demonstrated significantly increased exosome uptake compared to HEK293 cells; thus, HEK293 cells were used for all subsequent exosome uptake experiments.
Generation and validation of Her2-targeted exosomes
To assess whether antibody fragments are able to target exosomes to a specific cell receptor we chose to target Her2 as several anti-Her2 scFvs have been well characterised as tools for nanoparticle delivery.27 In particular, we utilised three different anti-Her2 scFvs that differ significantly in their affinities, primarily due to markedly different Koff rates: B1D2 which has the highest affinity for Her2 at 15 pM, ML39 which is 1 nM, and G98A which is 317 nM.27–29A transgene was constructed to encode one of the anti-Her2 scFvs or an irrelevant scFv, fused at its C-terminus to a myc tag followed by the C1C2 domain of lactadherin (Fig. 2A), which associates with phosphatidylserine, a lipid that has been shown to be enriched in exosomes.24 This fusion protein is preceded by a signal peptide (SP) to direct the fusion protein into the secretory pathway.30 These scFv-C1C2-encoding sequences were incorporated into JumpIn T-REx HEK293 cells to generate isogenic stable cell lines where the transgene is inserted at a single and predefined genomic locus, thereby reducing any positional effects on expression levels between the different cell lines. We validated the exosome-producing cell lines by performing flow cytometry detecting surface-exposed scFv via the myc tag, and demonstrated that each isogenic cell line exhibits a uniform cell population and surface scFv-expression level (ESI Fig. S2B†). We then isolated exosomes from all four cell lines and demonstrated via immunoblotting that fusion with the C1C2 domain enabled the scFv to partition into an exosome preparation as demonstrated by the presence of the exosomal markers CD63 and Alix, and absence of the endoplasmic reticulum contaminant calnexin (Fig. 2B, ESI Fig. S1B†). This immunoblotting experiment also confirmed that the different scFv-targeted exosomes were produced to an equivalent level and purity, suggesting that expression of the scFv-C1C2 fusion protein did not affect key exosome properties. To calculate the number of scFv molecules present in our exosome samples, we performed an immunoblotting-based quantification of the G98A-scFv exosomes compared to a purified myc-tagged scFv. Based on our calculations we estimate an average of approximately 1100–1400 scFv molecules per exosome to be present in our preparations (ESI Fig. S3†).
 | ||
| Fig. 2 Non-targeted and anti-Her2-scFv-exosomes can be generated via genetic manipulation of HEK293 cells. (A) Schematic illustration of the fusion protein expressed in HEK293 cells to produce Her2-targeted exosomes. SP, signal peptide; scFv, single chain Fragment variable; C1C2, C1C2-domain of lactadherin. (B) Immunoblot analysis of cell lysates and exosome lysates from cells expressing different scFv-C1C2 fusion proteins. Myc represents the C1C2 scFv fusion protein, CD63 and Alix are exosomal markers, and Calnexin is an endoplasmic reticulum protein, used to assess exosome purity. Molecular mass is indicated on the left. (C) Binding of recombinant Her2-Fc to immobilised exosomes by ELISA; irrelevant scFv (open circles), G98A scFv (squares), ML39 scFv (triangles) and B1D2 (closed circles). Error bars represent the SEM of three independent experiments where samples were analysed in duplicate. | ||
To confirm that Her2-targeted exosomes could bind to recombinant Her2 we performed an ELISA assay, and demonstrated that the two higher affinity Her2 scFv displaying exosomes (B1D2 and ML39) could bind to Her2 in a titratable manner, whereas the low affinity (G98A) and irrelevant scFv-exosomes exhibited little or no binding, respectively (Fig. 2C).
Targeting of anti-Her2-scFv-exosomes to tumour cell lines
To test whether anti-Her2-scFvs displayed on the surface of exosomes can direct targeting to Her2-positive cell lines we quantified the uptake of purified recombinant exosomes in a panel of cancer cell lines with varying levels of Her2 expression. Prior to assessing exosome uptake, the level of surface Her2 expressed on each cell line was analysed by quantitative flow cytometry. Our analysis of HeLa (cervical), BT20 (breast), HCC1954 (breast), SK-BR-3 (breast) cell lines confirmed the previously reported Her2-expression status of these cells;31–34 HCC1954 and SK-BR-3 exhibit a high number of Her2 receptors per cell (3.6 × 106 and 4.2 × 106, respectively) and HeLa and BT-20 only display 1–2 × 105 receptors per cell, which is comparable to expression in HEK293 at just under 1 × 105 receptors per cell (Fig. 3A).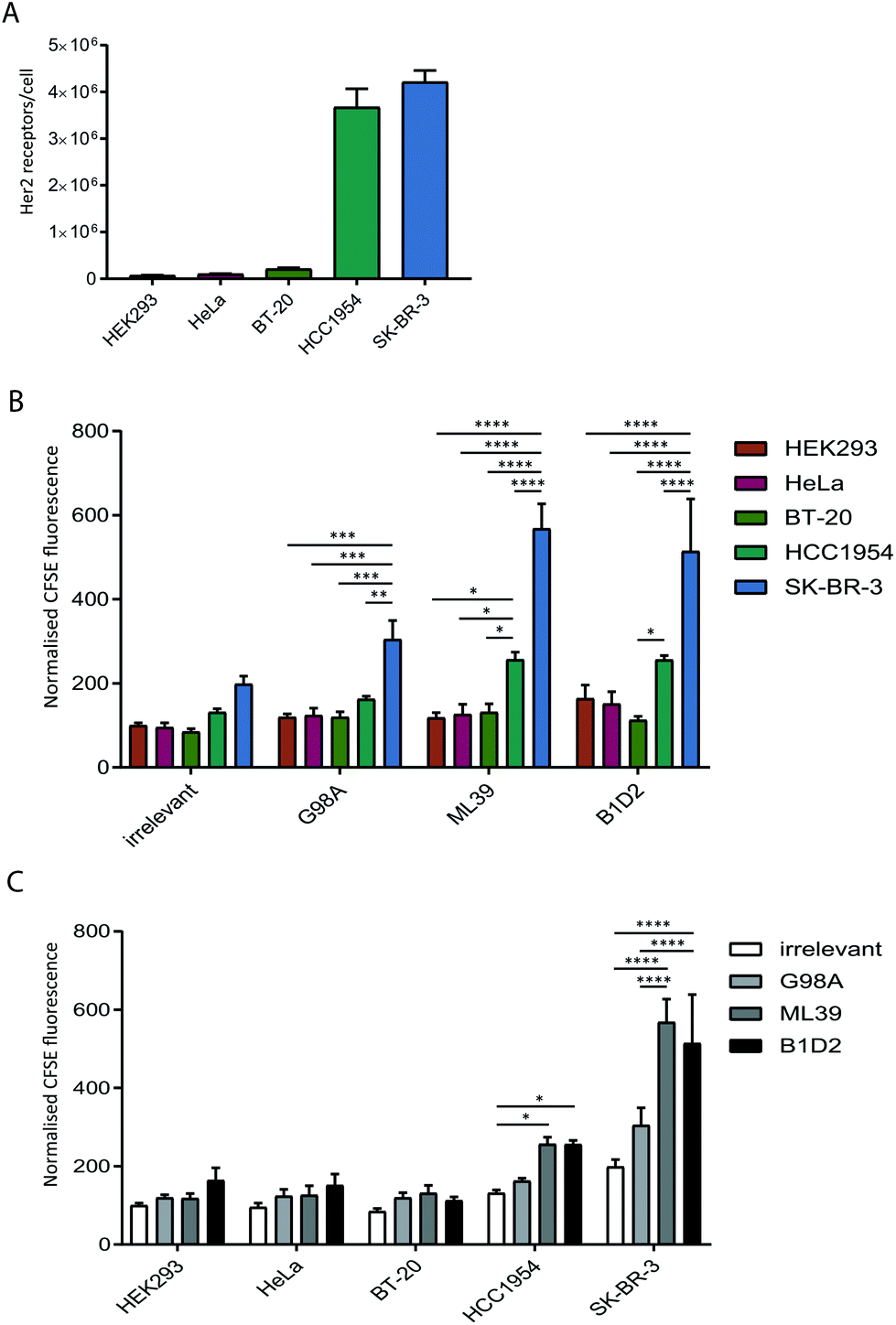 | ||
| Fig. 3 High affinity anti-Her2 targeted exosomes are required for efficient uptake into Her2 positive tumour cell lines. (A) The Her2 surface expression level on five cell lines was quantified by flow cytometry analysis using standardised beads. The receptor number was calculated by extrapolation to a standard curve. Error bars represent the SEM for three independent experiments. (B) and (C) The indicated cell lines were incubated with targeted (G98A, ML39 and B1D2) and irrelevant scFv CFSE-exosomes in duplicate for 4 h, and cells were analysed for CFSE-exosome uptake by flow cytometry. The MFI values from 3 independent experiments were calculated and normalised to the MFI of the HEK cells incubated with the irrelevant scFv exosomes. Statistical analysis to compare uptake between all groups was performed using two-way ANOVA with multiple comparisons; ****p < 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05; error bars indicate SEM. The data is depicted by comparing different exosomes (panel B) or different recipient cell lines (panel C). | ||
The uptake of scFv-exosomes into the cell line panel was tested by incubation of purified CFSE-labelled exosomes with cells for 4 h followed by flow cytometry analysis. When the uptake of different exosome preparations was compared between cell lines (Fig. 3B), there was a significantly higher signal observed for the SK-BR-3 cell line compared the other cell lines, for all the exosomes tested; this increase in uptake reached highly significant levels (2–3-fold increase) when Her2-targted exosomes were employed, reflective of the high Her2 level (4.2 × 106 receptors per cell) present on these cells. The HCC1954 cell line also demonstrated a somewhat increased propensity to take up non-targeted exosomes, however, despite displaying a similar level of Her2 on its surface (3.6 × 106 receptors per cell), uptake of Her-2 targeted exosomes was significantly lower than observed for the SK-BR-3 cell line.
By comparing the uptake observed for each cell line between the exosome preparations (Fig. 3C) we observed a universal increase in uptake for the targeted exosomes vs. the irrelevant control, which could reflect the presence of Her2 on the cell surface on all the cell lines tested. For HCC1954, and more strikingly for SK-BR-3, however, there was a statistically significant increase in signal when the two high affinity (ML39 and B1D2) scFv-bearing exosomes were used. Of note, there was no difference in uptake of ML39 and B1D2 exosomes into both HCC1954 and SK-BR-3 cells.
Targeting of anti-Her2-scFv-exosomes to isogenic Her2 expressing cells
We were able to show that Her2-targeting of exosomes to SK-BR-3 and HCC1954 cells can be achieved via high affinity scFv display, however the discrepancy between the signal obtained for the HCC1954 cells and its receptor number suggests inherent exosome uptake efficiencies or differences in Her2 trafficking between the two cell lines.To eliminate any cell line-specific differences in uptake, we created isogenic HEK293 cell lines with different Her2 surface levels. Recombinant human Her2 was stably introduced into HEK293 cells via lentiviral transduction and three clonal cell lines with either low (HEK-Her2-L), intermediate (HEK-Her2-M), or high (HEK-Her2-H) Her2 surface levels were isolated and the number of receptors per cell for each isogenic cell line were determined (Fig. 4A and ESI Fig. S2C†). The HEK-Her2-L cell line has a similar level of Her2 expression to HEK293 parental (1.4 × 105 receptors per cell), the HEK-Her2-M cell line has an intermediate expression level (7 × 105 receptors per cell) and the HEK-Her2-H cell line has expression levels similar to HCC1954 and SK-BR-3 cells (2.8 × 106 receptors per cell). We therefore concluded that these isogenic cell lines would be well suited to study Her-2 targeted exosome uptake.
 | ||
| Fig. 4 High Her2 expression is required for efficient targeted exosome uptake. (A) The absolute Her2 receptor number on the three clonal HEK293 cell lines HEK-Her2-L, HEK-Her2-M, and HEK-Her2-H, as well as parental HEK cells, was quantified by flow cytometry analysis using standardised beads, following staining with anti-Her2-FITC. The receptor number was calculated by extrapolation to a standard curve. Data shown is the average of 3 independent experiments; error bars indicate SEM. (B) and (C) Parental HEK293 and HEK-Her2 overexpressing cell lines were incubated with wild-type, non-targeted and targeted CFSE-exosomes in duplicate for 4 h and cells were analysed by flow cytometry. The MFI values were calculated for three independent experiments and normalised to the MFI of the HEK293 cells incubated with wild-type exosomes. Statistical analysis to compare uptake between all groups was performed using two-way ANOVA with multiple comparisons; ****p < 0.0001, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05; error bars indicate SEM. The data is depicted by comparing different exosomes (panel B) or different recipient cell lines (panel C). (D) Parental HEK293, and HEK-Her2 overexpressing cell lines were incubated with exosomes as described above, followed by staining with WGA-Alexa-fluor555 (red) and Hoechst (blue) post-fixation. Images were acquired on the Opera high-throughput confocal microscope. | ||
We investigated the uptake of both wild-type and recombinant (non-targeted and targeted) exosomes into the isogenic HEK293-Her2 cell lines. By comparing the uptake of different exosome preparations between cell lines, we observed a significant increase in uptake with ML39- and B1D2-bearing exosomes into HEK-Her2-H cells, but not into HEK parental or HEK-Her2-L or HEK-Her2-H, suggesting that a threshold of Her2 receptors per cell is required for efficient uptake (Fig. 4B). In agreement with the data obtained with the tumour cell lines (Fig. 3), there was no significant difference in the signal observed between the ML39 and B1D2 exosomes. When we compared the different cell lines, we could see a clear reduction (∼2-fold) in the signal between wild-type (non-modified) and C1C2-scFv bearing exosomes, suggesting that the expression of the C1C2-scFv fusion protein has an impact on binding of exosomes to HEK cells (Fig. 4C). Indeed, this reduction in uptake for C1C2-bearing exosomes was also observed in the tumour cell line panel (ESI Fig. S2E†), indicating that this phenomenon is not restricted to HEK cells. Interestingly, when ML39 and B1D2 exosomes were incubated with HEK-Her2-H cells, the signal obtained was higher than for the wild-type exosomes, whereas the irrelevant scFv-bearing exosome signal was two-fold lower.
We then assessed the contribution of association and uptake versus uptake alone for ML39 and B1D2 exosomes into the HEK-Her2-H cell line, and observed an increased proportion of CFSE signal associated with the recipient cells at 4 °C vs. 37 °C (albeit not significant, or of low significance, respectively), compared to unmodified or irrelevant scFv-bearing exosomes (ESI Fig. S2D†). This indicates that there is a higher proportion of exosomes bound to the cell surface when Her2-targeted exosomes are used.
Finally, the flow cytometry uptake data were verified by high-throughput confocal imaging of the HEK wt and HEK-Her2 cell lines incubated with non-targeted vs. targeted exosomes (Fig. 4D).
Intracellular trafficking of targeted, non-targeted and wild-type exosomes
To investigate whether modification of exosomes to incorporate scFv molecules on their surface influences their trafficking into Her2 over-expressing HEK293 cells we investigated their co-localisation with ligands that follow established endocytic and trafficking routes. Transferrin (Tfn) enters cells via clathrin-coated pits and is trafficked via sorting and recycling endosomes back to the plasma membrane. Dextran (Dex) is taken up via macropinocytosis and distributes to sorting/early and late endosomes, and lysosomes. Cholera toxin subunit B (CtxB) is taken up via caveolae, or lipid rafts, and transported in a retrograde fashion to the Golgi and endoplasmic reticulum (ER) via sorting endosomes.35 CFSE-labelled exosomes and either AlexaFluor647 labelled Tfn, Dex, or CtxB were co-incubated with HEK-Her2-H cells for 4 h, and analysed by high throughput confocal microscopy. Using images from >1500 cells for each ligand/exosome treatment, the Mander's Overlap Coefficient (MOC) was calculated, enabling comparisons to be made between co-localisation with the different ligands for each exosome preparation (Fig. 5A). For non-modified (wild-type) exosomes, the highest MOC value was obtained with CtxB. In contrast, for non-targeted irrelevant-scFv exosomes, a significantly higher MOC value was obtained for Dex, compared to Tfn or CtxB, suggesting that different pathways are adopted in the two exosome preparations. When targeted exosomes were used, an equivalent level of co-localisation with all ligands was observed indicating a shift towards clathrin- and caveolin-mediated uptake routes and/or downstream trafficking with targeted exosomes. To provide high resolution, qualitative visualisation of the co-localisation of CFSE-exosomes and ligands, we further employed SIM. Consistent with our confocal imaging analysis, examples were found where Tfn, Dex, and CtxB all co-localised with both targeted and non-targeted CFSE exosomes (Fig. 5B).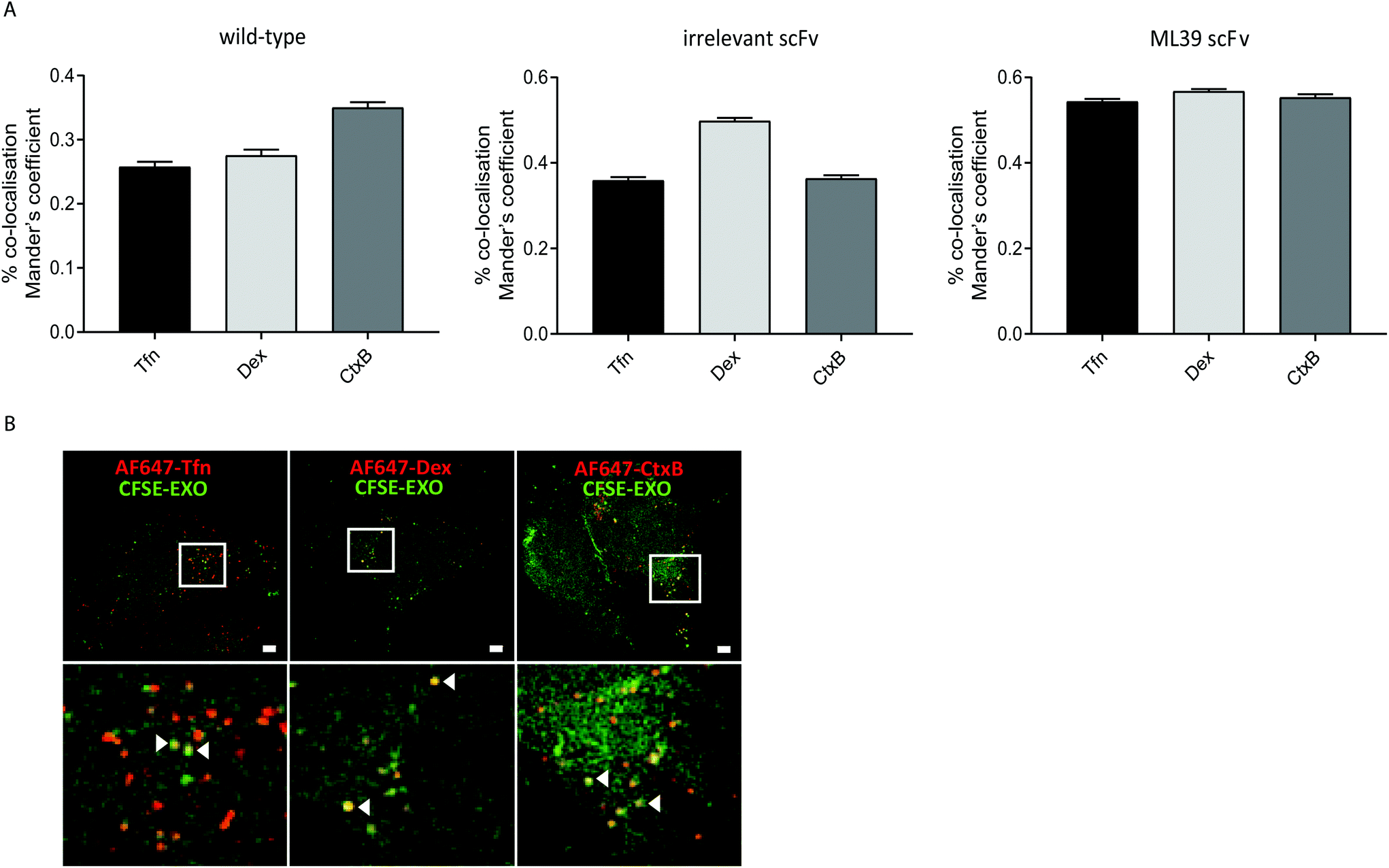 | ||
| Fig. 5 Targeted exosomes display a different co-localisation pattern than non-targeted exosomes. (A) Left panel: Quantification of co-localisation between CFSE-exosomes and different labelled ligands was determined via calculation of the mean percentage of exosomes co-localising with the indicated ligand (Mander's overlap coefficient) in the cytosol. Right panel: Percent increase in co-localisation of targeted (ML39) versus non-targeted (irrelevant) exosomes with each ligand. Data presented is from three independent experiments with at least 500 cells analysed per condition per experiment. Error bars indicate standard error of the mean. Statistical analysis was performed using one-way ANOVA to compare targeted exosomes to their untargeted counterparts (left panel) or to compare all three ligands (right panel); ****p < 0.0001. (B) Representative images acquired by SIM-super-resolution microscopy illustrating co-localisation of CFSE-labelled anti-Her2-scFv-ML39 exosomes (green) and AlexaFluor647-labelled ligands (shown in red) following incubation with HEK293-Her2-H cells for 4h. Scale bars indicate 2 μm; lower panel represents inset image. | ||
Discussion
Exploiting the intrinsic ability of exosomes to transfer biologically active cargo between cells for the delivery of therapeutic molecules is an attractive approach to the generation of better tolerated and more efficient nanomedicines.36 Targeting these exosomes to specific cell types represents a way to further expand the utility of these natural nanovesicles by improving the delivery of therapeutic cargo to diseased cells.In this study, we have assessed the feasibility of using scFvs as bespoke, specific targeting motifs for exosomes. We fused anti-Her2 scFvs to the C1C2 domain of lactadherin and evaluated the impact of different scFv affinities on the ability of the targeted exosomes to be taken up by a panel of tumour cell lines and recombinant Her2-expressing HEK cell lines. To monitor exosome uptake by recipient cells we first established a robust exosome uptake assay that could quantitate binding and uptake of CFSE-labelled exosomes, and used it to optimise exosome concentration, incubation time, and recipient cell type. We observed that exosome uptake was dose-dependent and efficient at concentrations of 0.5–3 × 1011 particles per ml, and reaches steady-state between 4–18 h of continuous incubation of the exosomes with recipient cells (Fig. 1A and B) similar to previous reports.5–7,26,37 We also showed that the majority of the signal measured was intracellular, representing active uptake and internalisation, rather than association with the cell surface alone (Fig. 1C–E).
A comparison of exosome uptake efficiencies across a panel of 15 cell lines identified that HEK293 cells are most efficient at taking up exosomes that have been derived from HEK293 cells, when compared to the other cell lines tested. Indeed, in a similar study using U-87 MG cell derived exosomes, none of the cell types tested (CHO K1, MEF, HUVEC, HeLa) were significantly more efficient at taking up exosomes than U-87 MG cells themselves.26 Although a comprehensive study is lacking, it is interesting to consider that this could be a common phenomenon that points towards a prominent role of exosomes in autocrine and paracrine signalling. In this analysis, we also found that, although all the cell lines demonstrated exosome uptake, some (e.g. Hep3B) are more efficient than others (e.g. Capan-1) (Fig. 1F). These results are in line with other studies that have observed differences in exosome uptake efficiency albeit using different exosome producing cells and different target cells.26,38–41 The ability of cells to take up non-targeted exosomes could reflect their overall endocytic activity or it could be influenced by the presence of different cell surface receptors on the individual cell lines, or a combination of both.
In this study, we generated Her2-targeted exosomes and analysed exosome uptake by both flow cytometry and microscopy-based analyses. First, we assessed the uptake of four different exosome preparations, all expressing an scFv fused to the C1C2 domain of lactadherin (an irrelevant scFv or scFvs of increasing affinity for Her2), into cells endogenously expressing different surface levels of the Her2 receptor. We observed robust uptake of the two high affinity scFv-bearing exosome preparations (ML39; 1 nM and B1D2; 15 pM) into SK-BR-3 cells, as expected based on the high level of Her2 present on these cells. Interestingly, however, HCC1954 cells did not take up targeted exosomes as well as SK-BR-3 cells (Fig. 3B and C) despite similar levels of surface Her2 expression (Fig. 3A); a finding that could be related to the presence of different cell surface receptors (e.g. ERBB3 (Her3) and their influence on Her2 internalisation in the two cell lines).42,43
To eliminate the contribution of such cell line-specific differences to targeted exosomal uptake we generated a panel of isogenic stable cell lines expressing distinct levels of Her2. We found that only in the HEK-Her2-H cell line, expressing 2.8 × 106 receptors per cell, was efficient uptake of the high affinity (ML39 and B1D2) anti-Her2-scFv exosomes observed (Fig. 4B and C); uptake into the HEK-Her2-M cell line (7.5 × 105 receptors per cell) was not achieved. Indeed, for both endogenous and recombinant Her2-expressing cell lines, Her2 levels at or above 2.8 × 106 receptor per cell (HCC1954, SK-BR-3 and HEK-Her2-H) were required to observe Her2 scFv-mediated uptake, and cell-specific differences were encountered. This requirement for high receptor number may limit the choice of therapeutic targets to those that are highly expressed on target cells, and these may not represent the most selectively expressed targets. On the other hand, this result could represent the relatively slow internalisation rate of Her2,44 and targeting a more rapidly internalising receptor (e.g. CD22) could overcome the requirement for high receptor levels and/or high affinity scFvs. In addition, the presence of the receptor at a certain level in the cell may not be sufficient to enable robust uptake, as endocytosis rates and expression of other cell surface receptors may play a role. Certainly, it will be interesting to understand whether exosome uptake can be improved by utilising different scFv-receptor pairings. It could be envisaged, for example, that by displaying scFvs to more than one target on the surface of an exosome, the efficiency and specificity of uptake could be improved. Indeed, it has been demonstrated for a Her2 biparatopic antibody, that the combination of two binding domains to different epitopes on the same receptor can drive rapid internalisation of an otherwise poorly internalising receptor.45
In the uptake experiments, we included non-modified exosomes and, intriguingly, we observed a significantly (∼2-fold) lower signal with C1C2-scFv exosomes, compared to these “wild-type” exosomes, for all the HEK cell lines tested (Fig. 4C). This decrease, also observed in the tumour cell line panel (ESI Fig. S2E†), could be a consequence of the C1C2-scFv domain on the surface of exosomes restricting the ubiquitous interaction of phosphatidylserine with recipient cells (e.g. via binding to TIM proteins on the cell surface), or the masking of other protein–protein interactions required for efficient exosome uptake. The presence of the C1C2 domain also appeared to influence the trafficking route of exosomes, such that the propensity for the exosome preparations to colocalise with ligands that prefer a particular trafficking route was altered (Fig. 5A); wild-type (non-modified) exosomes seem to colocalise with CtxB to a better extent than to Tfn and Dex, whereas those with the C1C2-irrelevant scFv on their surface are found more often co-localised with Dex, which may represent differences in how the exosomes engage with target cells, or downstream intracellular trafficking molecules. It should be emphasized that the colocalisation data presented here, represent the steady-state distribution of the selected ligands after several rounds of uptake, and recycling or degradation. Further longitudinal studies utilising pulse-chase experiments or real-time imaging would be needed to determine the exact uptake and intracellular trafficking routes of targeted and non-targeted exosomes. Nevertheless, the C1C2-based targeting approach appears to simultaneously inhibit ubiquitous uptake, and facilitate selective uptake via specific cell surface receptors, representing an opportunity to improve the therapeutic window of targeted exosome therapies.
Two approaches for the introduction of targeting ligands into exosomes have been described to date; one is based on the non-specific conjugation of ligands (e.g. nanobodies) to proteins on the surface of purified exosomes13 and the other, more commonly described method, involves the genetic engineering of exosome-producing cells to express fusion proteins that are displayed the exosome surface. The fusion partners that have been employed to generate targeted exosomes have included Lamp2b,13,23 PDGFR46 and a GPI anchor signal peptide,47 but their use has not been without limitations. For example, fusion with Lamp2b can lead to cleavage of peptide ligands48 or prevent fusion protein expression entirely (unpublished observations), and the use of a GPI anchor may not provide sufficient ligand density to enable exosome uptake.47 In this report, we used an alternative exosomal motif, the C1C2 domain of lactadherin, to generate targeted exosomes, similar to an approach described previously.24 The C1C2 domain binds to phosphatidylserine which is present on the surface of exosomes,49 and this binding is promoted by increased membrane curvature,49,50 making C1C2 a promising fusion partner for exosomal display. Indeed, characterisation of C1C2-scFv expressing cell lines and the exosomes produced from them, confirmed that the scFv molecules can be detected on the cell surface and are associated with the exosome fractions. In addition, we calculated an average of 1100–1400 scFv molecules per exosome, which compares favourably with the level of the F5 scFv on HER2 antibody-targeted liposomal doxorubicin (MM-302), where a density of 45 copies per liposome was achieved, and suggests that a significant proportion of the exosome surface is decorated with scFv molecules. In a similar exosome-targeting study whereby a GPI anchor-based method was employed to generate targeted exosomes, a value of 0.15–0.25 molecules per exosome was obtained using transmission electron microscopy.47 The number of exosome studies that have calculated this figure is limited, and the most appropriate method for quantification of surface-displayed molecules within exosome populations, or on individual exosomes has not been established, making such comparisons difficult. Nevertheless, fusion to the C1C2 domain appears to represent a very efficient method for the generation of exosomes with a high density of surface-displayed molecules.
Several ligands have been adopted for use as exosomal targeting moieties including peptides such as the GE11 peptide that binds to EGF-receptor,46 protein fragments such as the interleukin (IL)-3 fragment that binds to IL-3 receptor,51 viral proteins and peptides including gp350 of EBV that binds to CD21 on B cells, and a rabies virus glycoprotein peptide that binds to the acetylcholine receptor.23,52 Whilst these approaches provide valuable tools for targeting specific cells, we believe that antibodies or antibody fragments offer many advantages as targeting moieties. Firstly, antibodies can be selected against virtually any target protein or epitope using well-established technologies such as mouse immunisation or phage display.53,54 Secondly, a panel of antibodies to a chosen target will provide varying different affinities which can be optimised or tailored as required. Finally, antibodies demonstrate an exquisite specificity that can be exploited to generate exosomes that can be taken up by chosen cells of interest via binding to a specific cell surface protein. As antibodies require the production and association of two separate heavy and light chains, a single chain variable fragment (scFv), consisting of the variable heavy (VH) and variable light (VL) chains joined by a flexible linker, is often used in applications where only the binding function of the antibody is required. The potential of scFvs as highly specific and versatile exosome targeting motifs, however, has not been fully explored.
Interestingly, there was little difference between the uptake of B1D2 and ML39 exosomes into Her2 positive cells, suggesting that an affinity of 1 nM is sufficient to saturate the exosome uptake mechanism, and that increasing the affinity of the scFv cannot improve this further. This finding, together with the high scFv density on the exosome surface, could suggest that avidity of multiple binding entities has masked the impact of the higher affinity scFvs. Previous studies that have successfully achieved targeting13,23,46 have utilised peptide ligands with affinities for their targets in the same range (iRGD peptide; 17 nM,55 RVG peptide; 1.2 nM56 and GE11 peptide; 22 nM57), however, as the ligand densities were not calculated, and receptor internalisation rates will be different, it is difficult to compare these studies. It could be envisaged that different affinity and avidity constants could impact cargo trafficking downstream of cell surface receptor binding. It is known, for example, that antibodies binding with very high affinity to their target may not be able to dissociate within the endosome, leading to rapid lysosomal degradation.29,58 Extending this phenomenon, it could be envisaged that an internalised exosome that is tightly associated to its target may compromise the natural “back-fusion” process to release its cargo. This is highly relevant to the delivery of loaded exosomes as it would prevent efficient cytoplasmic delivery of the potential therapeutic cargo.
Although it has previously been demonstrated that exosomes can be taken up by target cells via macropinocytosis and clathrin- and caveolin-mediated endocytosis,5–8 the impact of arming exosomes with a targeting mechanism on their trafficking pathway has not yet been explored. In this study, we found that Her2-targeted exosomes showed equivalent co-localisation with Dextran, Tfn and CtxB at steady-state, whereas the non-targeted exosomes showed a preference for Dextran (Fig. 5A). Since it has been reported that Her2 can be taken up via caveolin-1 dependent endocytosis and, to a lesser extent, clathrin-mediated endocytosis,59–62 this difference likely represents anti-Her2-scFv-exosomes binding to Her2 and concomitant internalisation with the receptor. Thus, by directing exosomes to bind to a specific cell surface receptor, we have shown that it is possible to alter the trafficking and likely intracellular fate of the cargo. Targeting receptors that are known to traffic via specific pathways and with predefined kinetics could provide an opportunity to manipulate the fate of potential therapeutic cargo to ensure optimal delivery.
In this study, we have explored the use of scFvs as exosomal targeting motifs. Using Her2 and anti-Her2-scFvs as a model system we have shown that exosomes can be targeted to Her2-expressing cells by genetically engineering the exosome producer cells to incorporate a high-affinity anti-Her2-scFv onto the exosome surface. Targeting exosomes via a specific cell surface receptor may also provide a means to bias the uptake of exosomes towards a more efficient or “productive” intracellular route. If exosomes are to fulfil their promise as natural delivery vehicles for otherwise difficult to deliver therapeutic molecules many hurdles have yet to be overcome, but targeting therapeutic exosomes to a desired tissue will clearly be of high importance in any disease setting. The data presented here provide compelling evidence that scFvs can serve as broadly-applicable targeting motifs for exosomal delivery.
Materials and methods
DNA constructs
All sequences encoding transgenes were synthesized by GeneArt (Thermo Fisher Scientific) and codon-optimised for expression in human cells. Constructs to enable display of proteins on the surface of the exosome were designed to encode a validated leader sequence (MWWRLWWLLLLLLLLWPMVWA;30 followed by an scFv sequence) (irrelevant, G98A, ML39 or B1D228) flanked by NheI and BsrGI sites, a c-myc epitope tag (EQKLISEEDL), and the C1C2 domain of MFG-E8 (UniProt domain annotation Q08431). The DNA was then cloned into the JumpIn expression vector (pJTI-R4-CMV; Thermo Fisher Scientific) via XbaI and PmeI cloning sites to enable isogenic cell line generation. To enable the generation of recombinant Her2 cell lines, the coding sequence for human HER2 (UniProt P04626) preceded by a Kozak sequence (CACCATG) was synthesized by GeneArt, and cloned into a lentiviral vector (pCDH-CAG-MCS-IRES-puro, System Biosciences) via NheI-HF and EcoRI-HF. All constructs were confirmed by full-length sequencing.Cell lines
Cells were obtained from ECACC, ATCC or DSMZ and were maintained in the following media in 5% CO2 at 37 °C, unless otherwise stated. HEK293, HeLa, Caco-2, HepG2, Hep3B (MEM; 10% FBS; 1% NEAA); AsPC-1 (RPMI, 10% FBS, 1 mM sodium pyruvate); BT-20, U87 MG (MEM; 10% FBS; 1% sodium pyruvate; 1% NEAA); HCC1954, DAN G, DU-145 (RPMI 1640 (ATCC modification); 10% FBS); SK-BR-3 (McCoys 5A medium with L-glutamine; 10% FBS); HEK-scFv adherent (DMEM, 10% FBS, 0.1 mM NEAA, 5 μg ml−1 Blasticidin; 1 mg ml−1 G418); HEK-scFv suspension-adapted cells (FreeStyle293 medium (Thermo Fisher Scientific), 5 μg ml−1 Blasticidin, 1 mg ml−1 G418); HEK-Her2-low, HEK-Her2-medium, HEK-Her2-high (MEM, 10% FBS, 1% NEAA, 1 μg ml−1 Puromycin); Capan1 (IMDM, 20% FBS); HUVEC (EGM-2 Bullet kit Lonza); PANC1, A375, A431, A549 (DMEM, 10% FBS). All basal media and media additives were purchased from Thermo Fisher Scientific. Cells were passaged when they reached 80% confluency and cultured no longer than passage 15.Recombinant cell line generation and maintenance
To generate isogenic cell lines, expressing scFv-C1C2 fusion proteins, JumpIn HEK293 cells (Thermo Fisher Scientific) were transfected with the corresponding scFv vector and R4 integrase plasmid using Lipofectamine LTX reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. Transfected cells were selected by addition of 5 μg ml−1 Blasticidin and 1 mg ml−1 G418 to the normal culture medium. Surviving cells were expanded, analysed for myc expression by immunoblot and FACS analysis as described below. These cells were then adapted to growth in suspension in serum free FreeStyle medium (Thermo Fisher Scientific) supplemented with 5 μg ml−1 Blasticidin and 1 mg ml−1 G418, and cultured in a shaking incubator in 5% CO2 at 37 °C.To generate overexpressing Her2 cells, lentiviral particles were first generated. HEK293FT cells were co-transfected with lentiviral vector constructs and pPACKH1 packaging plasmid mix (System Biosciences) using Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer's instructions. 48 h post-transfection, culture supernatants containing lentiviral particles were harvested and used to infect HEK293 at 70–80% confluency in media containing 8 μg ml−1 polybrene (Sigma-Aldrich). Transduced cells were selected by addition of 1 μg ml−1 puromycin (Thermo Fisher Scientific) 48 h after transduction. Cells were maintained in selective media throughout. Clonal cell lines representing distinct levels of HER2 expression were isolated by limiting dilution and FACS sorting following cell surface staining using an anti-Her2 antibody (20 μl/1 × 106 cells; Clone Neu 24.7; BD Biosciences, #340553).
Exosome isolation, quality control and CFSE-labelling
Suspension adapted HEK293 and HEK293-scFv cells were cultured in their respective medium and passaged three days prior to exosome isolation. Cell culture supernatant was harvested by spinning at 2000g for 30 min to remove cells and debris. The supernatants were further clarified by filtration through 0.2 μm filters. The resulting clarified conditioned medium, containing exosomes, was concentrated using tangential flow filtration (TFF) (similar to Heinemann et al.63) with a 300 kDa membrane (PALL) to 10% of the original volume. Buffer exchange to PBS was carried out by diluting the filtration retentate to its original volume followed by re-concentration. This step was carried out twice resulting in 99% buffer exchange. Further concentration of the TFF retentate to approximately 1/500 of the original volume of conditioned medium was performed using spin filters with a 300 kDa molecular weight cut-off (Sartorius). All procedures were performed at 4 °C. The concentration of exosomes as well as their mean size and size distribution was determined by nanoparticle tracking on a NS300 system (Malvern) using the NTA3.0 software, with the same instrument and software settings used for all samples. Settings were as follows: 3 movies of 60 seconds duration at room temperature were acquired for each sample. The threshold was kept constant (at 6) for all measurements. NTA3.0 software was used to yield average particle size and count and mode sizes of 90–120 nm in diameter were obtained for exosome samples (ESI Fig. S1A†). Exosome purity, as indicated by a particles per μg protein value, was determined64 by dividing the particle count by total protein concentration as measured using a Pierce micro BCA protein assay kit (Thermo Fisher Scientific). A value of ≥2 × 109 particles per μg total protein was consistently achieved, indicative of “low purity” preparations as specified by Webber et al., compared to the gold-standard yet laborious methods involving sucrose cushions.For labelling of exosomes, preparations were adjusted to 1e12 ml−1 and labelled with 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE; Sigma-Aldrich) at a final concentration of 5 μM in PBS by incubating at 37 °C for 30 minutes followed by purification through three successive Zeba Spin Desalting Columns (7K MWCO, Thermo Fisher Scientific) to remove free CFSE dye. A “PBS control” sample that contained CFSE, PBS and a volume of FBS equivalent to the protein concentration in the exosome preparation was included, to control for protein-bound CFSE that was captured during the purification steps. To determine the effect of removal of exosomal surface proteins on cellular uptake of CFSE labelled exosomes, CFSE labelled exosomes were treated with Proteinase K (Thermo Fisher Scientific) for 15 min at room temperature. Proteinase K action was quenched by addition of an excess of 20 mg ml−1 purified bovine serum albumin (New England Biolabs).
Immunoblotting
Cell lysates were prepared as described in Théry et al., 2006,65 using RIPA buffer supplemented with 1% Protease Inhibitor Cocktail (Sigma Aldrich) and protein concentration was determined by micro BCA assay. Samples containing 10 μg total protein were denatured and resolved on NuPAGE 4–12% Bis–Tris Protein Gels (Thermo Fisher Scientific) alongside a molecular mass marker (LI-COR Biosciences). Protein was transferred onto nitrocellulose membranes (Thermo Fisher Scientific) and membranes were blocked for 1 hour at RT using blocking buffer (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DPBS and Odyssey blocking buffer (LI-COR Biosciences)). Membranes were probed overnight at 4 °C with antibodies to myc (1:1000; Clone 9E10; Invitrogen #13-2500), Alix (1:1500; Abcam, #ab88388), CD63 (1:1000; Abcam, #ab59479) and Calnexin (1:2000; Abcam, # Ab22595), washed and incubated with donkey anti-rabbit IgG IRDye 680 (1:10000; LI-COR Biosciences, #926-32221) and donkey anti-mouse IgG IRDye 800CW (1:10000; LI-COR Biosciences, #926-32212). Immunoblots were washed as above and visualised on the Odyssey CLx Infrared Imaging System.
:1 DPBS and Odyssey blocking buffer (LI-COR Biosciences)). Membranes were probed overnight at 4 °C with antibodies to myc (1:1000; Clone 9E10; Invitrogen #13-2500), Alix (1:1500; Abcam, #ab88388), CD63 (1:1000; Abcam, #ab59479) and Calnexin (1:2000; Abcam, # Ab22595), washed and incubated with donkey anti-rabbit IgG IRDye 680 (1:10000; LI-COR Biosciences, #926-32221) and donkey anti-mouse IgG IRDye 800CW (1:10000; LI-COR Biosciences, #926-32212). Immunoblots were washed as above and visualised on the Odyssey CLx Infrared Imaging System.
Quantification of scFv molecules per exosome
Quantification of the number of scFv molecules present on the exosome preparations was performed by detecting the myc tag as a surrogate for the scFv. Four-fold serial dilutions of a purified myc-tagged scFv of known concentration were processed alongside four-fold serial dilutions of the G98A-scFv exosome preparation, and immunoblotting was performed to detect the myc tag as described above (ESI Fig. S3A†). The intensity of the relevant myc-positive bands was quantitated using the Odyssey Image Studio software, and a linear regression analysis for the purified myc protein was performed in Prism (GraphPad) (ESI Fig. S3B†). Interpolated values (in pmoles) for the exosome serial dilutions were then obtained and the number of myc molecules was calculated by multiplying the number of moles by Avogadro's constant (6.02214086 × 1023 mol−1). The values from the two most dilute samples, that correlated well, were chosen to define the number of myc molecules, and therefore, scFv molecules, per exosome (ESI Fig. S3C†).Flow cytometry-based exosome uptake assay
Recipient cells were seeded in 96 well collagen coated plates (GreinerBioOne) in appropriate medium for 2 days at a density of 20000 cells per well. For the assay, cells were washed once with Freestyle medium, then 50 μl per well Freestyle medium was added followed by 50 μl per well CFSE-labelled exosomes at 1 × 1012 particles per ml. This is equivalent to approximately 1 × 106 particles per cell assuming the cells have undergone a single population doubling before the assay. For ligand co-localisation studies 50 μl per well Freestyle medium containing 50 μg ml−1 Tfn-AF647, 200 μg ml−1 Dex-AF647 or 0.5 μg ml−1 CtxB (all Thermo Fisher Scientific) was added followed by 50 μl per well CFSE-labelled exosomes at 1 × 1012 particles per ml. After incubation at 37 °C 5% CO2 cells were washed twice with PBS and processed for flow cytometry analysis. For the ice control, immediately after the CFSE-labelled exosome addition plates were placed on ice for 4 h after which cells were washed twice with PBS and processed for flow cytometry analysis. For flow cytometry, cells were dissociated with accutase and washed 3 times in PBS supplemented with 1% BSA. For antibody staining, cells were incubated with anti-Her2-FITC (20μl/1 × 106 cells; Clone Neu 24.7; BD Biosciences, #340553) or anti-myc-FITC (20μl/1 × 106 cells; Clone 9E10, Sigma-Aldrich, SAB4700448) for 30 minutes at 4 °C in the dark and washed 3 times in PBS supplemented with 1% BSA. 5 μg ml−1 DAPI (Sigma-Aldrich) was added to the last wash to assess cell viability. Flow cytometry was performed immediately using an LSR Fortessa cell analyser (BD Biosciences). Data was analysed using the FlowJo version 10 software (TreeStar). For comparison between samples the geometric mean of the median fluorescence intensity (MFI) was determined. At least 10000 cells were analysed for each sample. Cell debris was excluded by analysis of forward and side scatter area. Cells were gated on single cell populations using forward/side scatter height vs. width and dead cells were excluded by gating on DAPI-negative cells. When different cell lines were compared by flow cytometry, cells were labelled with CellMask Deep Red (Thermo Fisher Scientific) to normalise cell mass after the 4 h incubation with exosomes. Unless otherwise indicated a final concentration of approximately 5 × 1011 particles per ml was used for uptake experiments. Each independent experiment was performed with duplicate samples.
Quantitative flow cytometry
To quantitate the absolute number of Her2 receptors on the surface of the cell lines, cells were dissociated using accutase (Thermo Fisher Scientific), counted by Trypan blue exclusion and then stained with FITC-conjugated antibodies to Her2 (20μl/1 × 106 cells; Clone Neu 24.7; BD Biosciences, #340553) or a corresponding isotype control (20μl/1 × 106 cells; Mouse IgG1-FITC; Clone MOPC-21; BD Biosciences, #556649) for 30 min at 4 °C in the dark according to manufacturer's instructions. QSC microspheres were prepared and stained with anti-Her2-FITC, according to the manufacturer's instructions (Bangs Laboratories Inc.). DAPI staining was used as a viability stain as described above. Absolute receptor numbers were obtained by using the QSC lot specific calculation sheet.High-throughput microscopy
For microscopy cells were seeded into black 96-well Greiner μ-clear PDL coated plates (Greiner Bio-One #655946) at 20000 cells per well and treated as described above. Cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) in PBS and blocked with PBS containing 1% BSA. To label nuclei Hoechst33342 (Thermo Fisher Scientific) was added at 1 μg ml−1 for 2 min. To label the plasma membrane Alexa Fluor-555 conjugated wheat germ agglutinin (AF555-WGA, Thermo Fisher Scientific) was added in PBS/1%BSA at 5 μg ml−1 for 20 minutes followed by 3x washes with PBS/1%BSA. Fixed and stained cells were stored in PBS at 4 °C until acquisition. 2 images per well were acquired at 20x magnification on an ImageXpress Micro widefield high content microscope (Molecular Devices) or 10 images per well were acquired at 60× magnification on a confocal Opera High Content Screening System (PerkinElmer).
Co-localisation analysis was performed as described above but AlexaFluor-647 labelled ligands were added together with exosomes (50 μl at 1 × 1012 particles per ml with 50 μl fresh freestyle medium as above) for 4h at 37 °C (50 μg ml−1 transferrin-AlexaFluor-647, 200 μg ml−1 Dextran-AlexaFluor-647; or 0.5 μg ml−1 CtxB-AlexaFluor-647) (Thermo Fisher Scientific) after which cells were processed as described above for high-throughput confocal microscopy analysis.
Co-localisation was quantified using the Columbus software. Briefly, using the inbuilt software, nuclei were counted based on Hoechst labelling, cytosol was outlined based on wheat germ agglutinin labelling to define the region of interest, and co-localisation in the cytoplasm of each cell was quantified by calculating the Mander's Overlap Coefficients using an unbiased approach based on whole field and local median background subtraction in each image. Three independent experiments were performed where >500 images were analysed for each treatment, in each experiment.
Super-resolution microscopy
For super-resolution imaging cells were mounted in chambered coverslips (Lab-TEK II, Nunc) and images were captured using a custom-built structured illumination microscope.66 Sinusoidal excitation patterns were generated using a spatial light modulator (SLM) configured to display a series of binary phase gratings. A spatially filtered image of the SLM, illuminated with collimated laser light, was projected through the rear illumination port of an inverted microscope body (IX71, Olympus) and into the focal plane of the microscope objective lens (UPLSAPO 60×/1.3, Olympus). Sample images were acquired using a scientific CMOS camera (ORCA-Flash4.0, Hamamatsu Photonics), with the global exposure period of the camera's rolling shutter synchronised to the pattern displayed on the SLM. Each super resolution image was reconstructed from a sequence of nine raw images of the sample acquired under excitation with different sinusoidal irradiance patterns: three pattern orientations each separated by 60° and three pattern phases, separated by 2π/3, per orientation. Images were reconstructed as described previously,67 with out of focus light suppressed by multiplication of the zero and first order Fourier space passbands by Gaussian and complimentary inverted Gaussian functions. The lateral offset between different colour channels was measured using references images of 100 nm diameter TetraSpeck beads (Thermo Fisher Scientific), captured using the same illumination wavelengths and emission filters as the sample. The resulting geometric transformation required to maximise the overlap between these reference images was then applied to correct the corresponding images of fluorescent ligands and exosomes.To visualise the intracellular location of exosomes non-permeabilised cells were labelled for 20 minutes with mouse anti-CD81 (clone JS-81, BD Biosciences, #555675), followed by 20 minutes of anti-mouse Alexa Fluor-647 (Thermo Fisher Scientific; both in PBS with 1% BSA with 3× PBS/BSA washes in between). Orthogonal (xy, xz and yz) maximum intensity projection were created from reconstructed focal series of images (z-stacks) to show the 3D position of the exosomes relative to the plasma membrane. Further 3D visualisations of the plasma membrane and exosomes were generated from reconstructed z-stacks using the ImageJ plugin ClearVolume.68
ELISA
Exosomes were coated at a concentration of 8 × 108 exosomes per well onto 96-well Nunc MaxiSorp plates (Thermo Fisher Scientific) overnight at 4 °C. After incubation, plates were washed 3 times with DPBS and blocked for 1 h in 3% BSA in DPBS. Plates were then incubated for 2 h at room temperature with recombinant Human Her2-Fc (ErbB2/Fc Chimera; Sino Biological Inc.) at concentrations indicated in block solution. After washing 3 times with DPBS, bound exosomes were detected with anti-Human IgG (Fc specific)-Peroxidase antibody produced in goat (Sigma-Aldrich) for 1 h at room temperature at a final concentration of 2 μg ml−1 in block. After a further wash, the ELISA signal was detected using SuperSignal ELISA Pico Substrate (Thermo Fisher Scientific), according to the manufacturer's instructions and measured using an UltraSensitive Luminescence protocol on a 2104 EnVision Multilabel Reader (PerkinElmer).Statistical analysis
All data is presented as mean ± standard error of the mean (SEM) from n = 3 independent experiments, using duplicate samples, unless otherwise indicated. Where appropriate normalization was performed to compare independent experiments. Statistical analysis was performed in Prism (GraphPad) using one- or two-way ANOVA, as appropriate with multiple comparisons.Conflicts of interest
AL, CS, AC, LJ, CM, LF, MB, RM, TV and NT are current or former salaried employees of MedImmune Ltd (a member of the AstraZeneca Group) and CS, AC, LJ, CM, MB, RM, TV and NT have/had stock or stock options in AstraZeneca, the parent company of MedImmune.Acknowledgements
Miguel Carvalho for providing the C1C2 cloning vectors, Janette Dillon for cell line generation guidance, James Dodgson for writing the co-localisation algorithm and providing training and guidance, Core Tissue culture facility for maintenance of cell cultures, DNA chemistry facility for oligonucleotide synthesis and DNA sequencing.References
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nat. Cell Biol., 2007, 9(6), 654–659 CrossRef PubMed.
- N. P. Hessvik and A. Llorente, Current knowledge on exosome biogenesis and release, Cell. Mol. Life Sci., 2017, 75(2), 193–208 CrossRef PubMed.
- H. G. Zhang and W. E. Grizzle, Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions, Am. J. Pathol., 2014, 184(1), 28–41 CrossRef PubMed.
- A. V. Vlassov, S. Magdaleno, R. Setterquist and R. Conrad, Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials, Biochim. Biophys. Acta, 2012, 1820(7), 940–948 CrossRef PubMed.
- C. Escrevente, S. Keller, P. Altevogt and J. Costa, Interaction and uptake of exosomes by ovarian cancer cells, BMC Cancer, 2011, 11, 108 CrossRef PubMed.
- T. Tian, Y. L. Zhu, F. H. Hu, Y. Y. Wang, N. P. Huang and Z. D. Xiao, Dynamics of exosome internalization and trafficking, J. Cell. Physiol., 2013, 228(7), 1487–1495 CrossRef PubMed.
- L. A. Mulcahy, R. C. Pink and D. R. Carter, Routes and mechanisms of extracellular vesicle uptake, J. Extracell. Vesicles, 2014, 3(1), 24641 CrossRef PubMed.
- D. Fitzner, M. Schnaars, D. van Rossum, G. Krishnamoorthy, P. Dibaj and M. Bakhti, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis, J. Cell Sci., 2011, 124(Pt 3), 447–458 CrossRef PubMed.
- A. E. Morelli, A. T. Larregina, W. J. Shufesky, M. L. Sullivan, D. B. Stolz and G. D. Papworth, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells, Blood, 2004, 104(10), 3257–3266 CrossRef PubMed.
- A. Montecalvo, A. T. Larregina, W. J. Shufesky, D. B. Stolz, M. L. Sullivan and J. M. Karlsson, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes, Blood, 2012, 119(3), 756–766 CrossRef PubMed.
- A. Lo Cicero, P. D. Stahl and G. Raposo, Extracellular vesicles shuffling intercellular messages: for good or for bad, Curr. Opin. Cell Biol., 2015, 35, 69–77 CrossRef PubMed.
- N. Yim, S. W. Ryu, K. Choi, K. R. Lee, S. Lee and H. Choi, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module, Nat. Commun., 2016, 7, 12277 CrossRef PubMed.
- Y. Tian, S. Li, J. Song, T. Ji, M. Zhu and G. J. Anderson, et al., A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy, Biomaterials, 2014, 35(7), 2383–2390 CrossRef PubMed.
- T. N. Lamichhane, R. S. Raiker and S. M. Jay, Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery, Mol. Pharmaceutics, 2015, 12(10), 3650–3657 CrossRef PubMed.
- D. S. Sutaria, M. Badawi, M. A. Phelps and T. D. Schmittgen, Achieving the Promise of Therapeutic Extracellular Vesicles: The Devil is in Details of Therapeutic Loading, Pharm. Res., 2017, 1–14 Search PubMed.
- M. J. Haney, Y. Zhao, E. B. Harrison, V. Mahajan, S. Ahmed and Z. He, et al., Specific transfection of inflamed brain by macrophages: a new therapeutic strategy for neurodegenerative diseases, PLoS One, 2013, 8(4), e61852 CrossRef PubMed.
- S. El-Andaloussi, Y. Lee, S. Lakhal-Littleton, J. Li, Y. Seow and C. Gardiner, et al. Exosome-mediated delivery of siRNA in vitro and in vivo, Nat. Protoc., 2012, 7(12), 2112–2126 CrossRef PubMed.
- J. Wahlgren, T. D. L. Karlson, P. Glader, E. Telemo and H. Valadi, Activated Human T Cells Secrete Exosomes That Participate in IL-2 Mediated Immune Response Signaling, PLoS One, 2012, 7(11), e49723 CrossRef PubMed.
- G. Fuhrmann, A. Serio, M. Mazo, R. Nair and M. M. Stevens, Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins, J. Controlled Release, 2015, 205, 35–44 CrossRef PubMed.
- M. S. Kim, M. J. Haney, Y. Zhao, V. Mahajan, I. Deygen and N. L. Klyachko, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells, Nanomedicine, 2016, 12(3), 655–664 CrossRef PubMed.
- F. Aqil, H. Kausar, A. K. Agrawal, J. Jeyabalan, A.-H. Kyakulaga and R. Munagala, et al. Exosomal formulation enhances therapeutic response of celastrol against lung cancer, Exp. Mol. Pathol., 2016, 101(1), 12–21 CrossRef PubMed.
- K. B. Johnsen, J. M. Gudbergsson, M. N. Skov, L. Pilgaard, T. Moos and M. Duroux, A comprehensive overview of exosomes as drug delivery vehicles—Endogenous nanocarriers for targeted cancer therapy, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1846(1), 75–87 CrossRef PubMed.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes, Nat. Biotechnol., 2011, 29(4), 341–345 CrossRef PubMed.
- A. Delcayre, A. Estelles, J. Sperinde, T. Roulon, P. Paz and B. Aguilar, et al. Exosome Display technology: applications to the development of new diagnostics and therapeutics, Blood Cells, Mol., Dis., 2005, 35(2), 158–168 CrossRef PubMed.
- D. K. Kim, J. Lee, S. R. Kim, D. S. Choi, Y. J. Yoon and J. H. Kim, et al. EVpedia: a community web portal for extracellular vesicles research, Bioinformatics, 2015, 31(6), 933–939 CrossRef PubMed.
- K. J. Svensson, H. C. Christianson, A. Wittrup, E. Bourseau-Guilmain, E. Lindqvist and L. M. Svensson, et al. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1, J. Biol. Chem., 2013, 288(24), 17713–17724 CrossRef PubMed.
- S. M. Sadat, S. Saeidnia, A. J. Nazarali and A. Haddadi, Nano-pharmaceutical formulations for targeted drug delivery against HER2 in breast cancer, Curr. Cancer Drug Targets, 2015, 15(1), 71–86 CrossRef PubMed.
- R. Schier, A. McCall, G. P. Adams, K. W. Marshall, H. Merritt and M. Yim, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site, J. Mol. Biol., 1996, 263(4), 551–567 CrossRef PubMed.
- S. I. Rudnick, J. Lou, C. C. Shaller, Y. Tang, A. J. Klein-Szanto and L. M. Weiner, et al. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors, Cancer Res., 2011, 71(6), 2250–2259 CrossRef PubMed.
- S. Barash, W. Wang and Y. Shi, Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression, Biochem. Biophys. Res. Commun., 2002, 294(4), 835–842 CrossRef PubMed.
- B. S. Hendriks, S. G. Klinz, J. G. Reynolds, C. W. Espelin, D. F. Gaddy and T. J. Wickham, Impact of tumor HER2/ERBB2 expression level on HER2-targeted liposomal doxorubicin-mediated drug delivery: multiple low-affinity interactions lead to a threshold effect, Mol. Cancer Ther., 2013, 12(9), 1816–1828 CrossRef PubMed.
- T. A. Jarvinen, M. Tanner, V. Rantanen, M. Barlund, A. Borg and S. Grenman, et al. Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer, Am. J. Pathol., 2000, 156(3), 839–847 CrossRef PubMed.
- J. M. Nam, K. H. Jeon, H. Kwon, E. Lee, K. Y. Jun and Y. B. Jin, et al. Dithiiranylmethyloxy azaxanthone shows potent anti-tumor activity via suppression of HER2 expression and HER2-mediated signals in HER2-overexpressing breast cancer cells, Eur. J. Pharm. Sci., 2013, 50(2), 181–190 CrossRef PubMed.
- K. Subik, J. F. Lee, L. Baxter, T. Strzepek, D. Costello and P. Crowley, et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines, Breast Cancer, 2010, 4, 35–41 Search PubMed.
- S. V. Barysch, R. Jahn and S. O. Rizzoli, A fluorescence-based in vitro assay for investigating early endosome dynamics, Nat. Protocols, 2010, 5(6), 1127–1137 Search PubMed.
- D. Ha, N. Yang and V. Nadithe, Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges, Acta Pharm. Sin. B, 2016, 6(4), 287–296 CrossRef PubMed.
- W. Heusermann, J. Hean, D. Trojer, E. Steib, S. von Bueren and A. Graff-Meyer, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER, J. Cell Biol., 2016, 213(2), 173–184 CrossRef PubMed.
- D. Feng, W. L. Zhao, Y. Y. Ye, X. C. Bai, R. Q. Liu and L. F. Chang, et al. Cellular internalization of exosomes occurs through phagocytosis, Traffic, 2010, 11(5), 675–687 CrossRef PubMed.
- M. Mittelbrunn, C. Gutiérrez-Vázquez, C. Villarroya-Beltri, S. González, F. Sánchez-Cabo and M.Á González, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells, Nat. Commun., 2011, 2, 282 CrossRef PubMed.
- T. J. Smyth, J. S. Redzic, M. W. Graner and T. J. Anchordoquy, Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro, Biochim. Biophys. Acta, Biomembr., 2014, 1838(11), 2954–2965 CrossRef PubMed.
- M. Chiba, M. Kimura and S. Asari, Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma HepG2 and lung cancer A549 cell lines, Oncol. Rep., 2012, 28(5), 1551–1558 CrossRef PubMed.
- F. Henjes, C. Bender, S. Von Der Heyde, L. Braun, H. A. Mannsperger and C. Schmidt, et al. Strong EGFR signaling in cell line models of ERBB2-amplified breast cancer attenuates response towards ERBB2-targeting drugs, Oncogenesis, 2012, 1(7), e16 CrossRef PubMed.
- Ö. Sahin, H. Fröhlich, C. Löbke, U. Korf, S. Burmester and M. Majety, et al. Modeling ERBB receptor-regulated G1/S transition to find novel targets for de novo trastuzumab resistance, BMC Syst. Biol., 2009, 3, 1 CrossRef PubMed.
- A. Sorkin and L. K. Goh, Endocytosis and intracellular trafficking of ErbBs, Exp. Cell Res., 2008, 314(17), 3093–3106 CrossRef PubMed.
- J. Y. Li, S. R. Perry, V. Muniz-Medina, X. Wang, L. K. Wetzel and M. C. Rebelatto, et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy, Cancer Cell, 2016, 29(1), 117–129 CrossRef PubMed.
- S. Ohno, M. Takanashi, K. Sudo, S. Ueda, A. Ishikawa and N. Matsuyama, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells, Mol. Ther., 2013, 21(1), 185–191 CrossRef PubMed.
- S. A. Kooijmans, C. G. Aleza, S. R. Roffler, W. W. van Solinge, P. Vader and R. M. Schiffelers, Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting, J. Extracell. Vesicles, 2016, 5(1), 31053 CrossRef PubMed.
- M. E. Hung and J. N. Leonard, Stabilization of exosome-targeting peptides via engineered glycosylation, J. Biol. Chem., 2015, 290(13), 8166–8172 CrossRef PubMed.
- J. Shi, C. W. Heegaard, J. T. Rasmussen and G. E. Gilbert, Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature, Biochim. Biophys. Acta, 2004, 1667(1), 82–90 CrossRef PubMed.
- D. E. Otzen, K. Blans, H. Wang, G. E. Gilbert and J. T. Rasmussen, Lactadherin binds to phosphatidylserine-containing vesicles in a two-step mechanism sensitive to vesicle size and composition, Biochim. Biophys. Acta, Biomembr., 2012, 1818(4), 1019–1027 CrossRef PubMed.
- D. Bellavia, S. Raimondo, G. Calabrese, S. Forte, M. Cristaldi and A. Patinella, et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth, Theranostics, 2017, 7(5), 1333–1345 CrossRef PubMed.
- R. Ruiss, S. Jochum, R. Mocikat, W. Hammerschmidt and R. Zeidler, EBV-gp350 confers B-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant B cells–a new option for the treatment of B-CLL, PLoS One, 2011, 6(10), e25294 CrossRef PubMed.
- N. Lonberg, Human monoclonal antibodies from transgenic mice, Therapeutic Antibodies: Springer, 2008, pp. 69–97 Search PubMed.
- A. R. Bradbury, S. Sidhu, S. Dübel and J. McCafferty, Beyond natural antibodies: the power of in vitro display technologies, Nat. Biotechnol., 2011, 29(3), 245–254 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy and O. M. Girard, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors, Cancer Cell, 2009, 16(6), 510–520 CrossRef PubMed.
- M. Oswald, S. Geissler and A. Goepferich, Targeting the Central Nervous System (CNS): A Review of Rabies Virus-Targeting Strategies, Mol. Pharmaceutics, 2017, 14(7), 2177–2196 CrossRef PubMed.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao and J. Li, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics, FASEB J., 2005, 19(14), 1978–1985 CrossRef PubMed.
- N. Bien-Ly, Y. J. Yu, D. Bumbaca, J. Elstrott, C. A. Boswell and Y. Zhang, et al., Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants, J. Exp. Med., 2014, 211(2), 233–244 CrossRef PubMed.
- C. D. Austin, A. M. De Maziere, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot and M. X. Sliwkowski, et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin, Mol. Biol. Cell, 2004, 15(12), 5268–5282 CrossRef PubMed.
- K. Roepstorff, L. Grøvdal, M. Grandal, M. Lerdrup and B. van Deurs, Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer, Histochem. Cell Biol., 2008, 129(5), 563–578 CrossRef PubMed.
- S. C. Sekhar, T. Kasai, A. Satoh, T. Shigehiro, A. Mizutani and H. Murakami, et al. Identification of caveolin-1 as a potential causative factor in the generation of trastuzumab resistance in breast cancer cells, J. Cancer, 2013, 4(5), 391–401 CrossRef PubMed.
- Y. C. Chung, J. F. Kuo, W. C. Wei, K. J. Chang and W. T. Chao, Caveolin-1 Dependent Endocytosis Enhances the Chemosensitivity of HER-2 Positive Breast Cancer Cells to Trastuzumab Emtansine (T-DM1), PLoS One, 2015, 10(7), e0133072 CrossRef PubMed.
- M. L. Heinemann, M. Ilmer, L. P. Silva, D. H. Hawke, A. Recio and M. A. Vorontsova, et al. Benchtop isolation and characterization of functional exosomes by sequential filtration, J. Chromatogr., A, 2014, 1371, 125–135 CrossRef PubMed.
- J. Webber and A. Clayton, How pure are your vesicles?, J. Extracell. Vesicles, 2013, 2(1), 19861 CrossRef PubMed.
- C. Théry, S. Amigorena, G. Raposo and A. Clayton, Isolation and characterization of exosomes from cell culture supernatants and biological fluids, Curr. Protoc. Cell Biol., 2006, ch. 3, Unit 3.22 Search PubMed.
- M. Shaw, L. Zajiczek and K. O'Holleran, High speed structured illumination microscopy in optically thick samples, Methods, 2015, 88, 11–19 CrossRef PubMed.
- K. O'Holleran and M. Shaw, Optimized approaches for optical sectioning and resolution enhancement in 2D structured illumination microscopy, Biomed. Opt. Express, 2014, 5(8), 2580–2590 CrossRef PubMed.
- L. A. Royer, M. Weigert, U. Gunther, N. Maghelli, F. Jug and I. F. Sbalzarini, et al. ClearVolume: open-source live 3D visualization for light-sheet microscopy, Nat. Methods, 2015, 12(6), 480–481 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr03970d |
| This journal is © The Royal Society of Chemistry 2018 |