
Pd-Catalyzed C–H aziridination of 3,3,5,5-tetrasubstituted piperazin-2-ones†
Thomas A.
Alanine
 *a,
Stephen
Stokes
a,
Craig A.
Roberts
b and
James. S.
Scott
a
*a,
Stephen
Stokes
a,
Craig A.
Roberts
b and
James. S.
Scott
a
aMedicinal Chemistry, Oncology, IMED Biotech Unit, AstraZeneca, Cambridge CB4 0FZ, United Kingdom. E-mail: t.alanine@sygnaturediscovery.com
bMedicinal Chemistry, Oncology, IMED Biotech Unit, AstraZeneca, Macclesfield SK10 4TG, United Kingdom
First published on 4th December 2017
Abstract
A palladium mediated C–H aziridination reaction of 3,3,5,5-substituted-piperazin-2-ones has been developed using phenyliodonium diacetate (PIDA) and succinic acid to give synthetically useful bicyclic aziridines, in moderate to good yields. Succinic acid was found to be key for selectively promoting C–N bond formation (aziridination) and suppressing competitive acetoxylation. Analysis of the reaction kinetics revealed the role of succinic acid in promoting an equilibrium between monomeric and dimeric palladium species in the rate determining step of the reaction. The aziridines can be ring-opened by nucleophiles under Lewis or Brønsted acidic conditions to give formal C–H functionalized products. The reaction conditions can be further manipulated to produce acetoxylated, diacetoxylated and even triacetoxylated materials through the use of acetic acid and increased oxidant stoichiometry.
Introduction
Cyclic amines are common scaffolds encountered in natural products (e.g., alkaloids) and man-made chemical structures. This class of molecules play an important part within medicinal chemistry as bioactive frameworks and side-chains.1 Hindered amines (where the cyclic nitrogen atom is flanked by quaternary carbons) are a significantly under-represented subset of cyclic amines.2 Reports in the literature have centered on their uses as non-nucleophilic, strong amide bases3a–c or stable nitroxide radicals.4a–e Limited by poor synthetic accessibility, these structures have seldom been investigated. However, improved synthetic routes and functionalizations around this core may reveal new uses in catalysis or medicinal chemistry. The ability to directly functionalize aliphatic C–H bonds opens up the opportunity to access complex hindered amine motifs. The field of palladium catalyzed C–H activation has matured over recent years with a variety of catalytic systems able to perform selective functionalizations on sp3 hybridized centers.5 Of particular interest to us was the discovery by Gaunt and co-workers that hindered aliphatic nitrogens are capable of directing palladium sp3 C–H activation β- to the amine.6a–d Given the prevalence of piperazine moieties in drug scaffolds,7a–d we were eager to investigate this approach to the functionalization of related 3,3,5,5-tetrasubstitued piperazin-2-ones, which has not previously been reported (Scheme 1). In particular, the ability to perform sp3 C–H functionalization of these systems would provide expedient access to asymmetrically-substituted cyclic amines, valuable building blocks for medicinal chemistry, with an additional vector for substitution.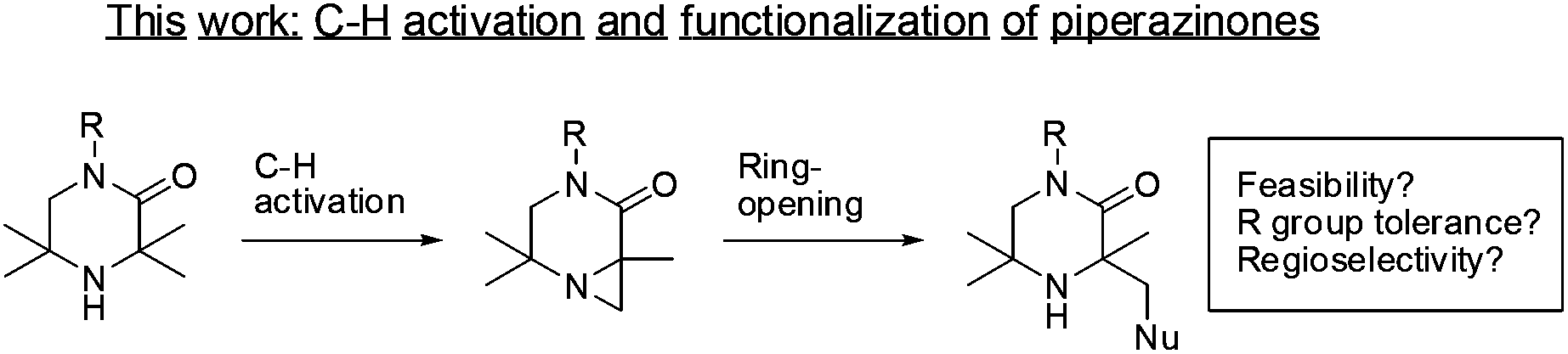 | ||
| Scheme 1 Proposed aliphatic C–H aziridination of piperazinones. | ||
We anticipated that 3,3,5,5-tetrasubstitued piperazin-2-ones might undergo similar reactive pathways to those reported for the morpholinone analogs.6a However, we had concerns that the presence of a more Lewis-basic carbonyl moiety or substitution on the lactam may not be tolerated, as this could present potentially reactive C–H bonds on the amide substituents, leading to competitive C–H insertion pathways.
As N-aryl piperazines are a common motif in bioactive scaffolds,8 we selected model substrate 1a for our studies. 1a was subjected to Gaunt's oxidative phenyliodonium diacetate mediated aziridination conditions (Scheme 2), developed for the aziridination of morpholinones.6a After 2 hours, the reaction showed disappearance of starting material to furnish a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of aziridine 2a and acetoxylation product 3 respectively, presumably derived from in situ ring-opening of the aziridine.6a Encouraged by this result, we examined the effect of addition of 20 equivalents of acid to the reaction, which has been reported to accelerate the reaction rate (Scheme 2).6b
:1 mixture of aziridine 2a and acetoxylation product 3 respectively, presumably derived from in situ ring-opening of the aziridine.6a Encouraged by this result, we examined the effect of addition of 20 equivalents of acid to the reaction, which has been reported to accelerate the reaction rate (Scheme 2).6b
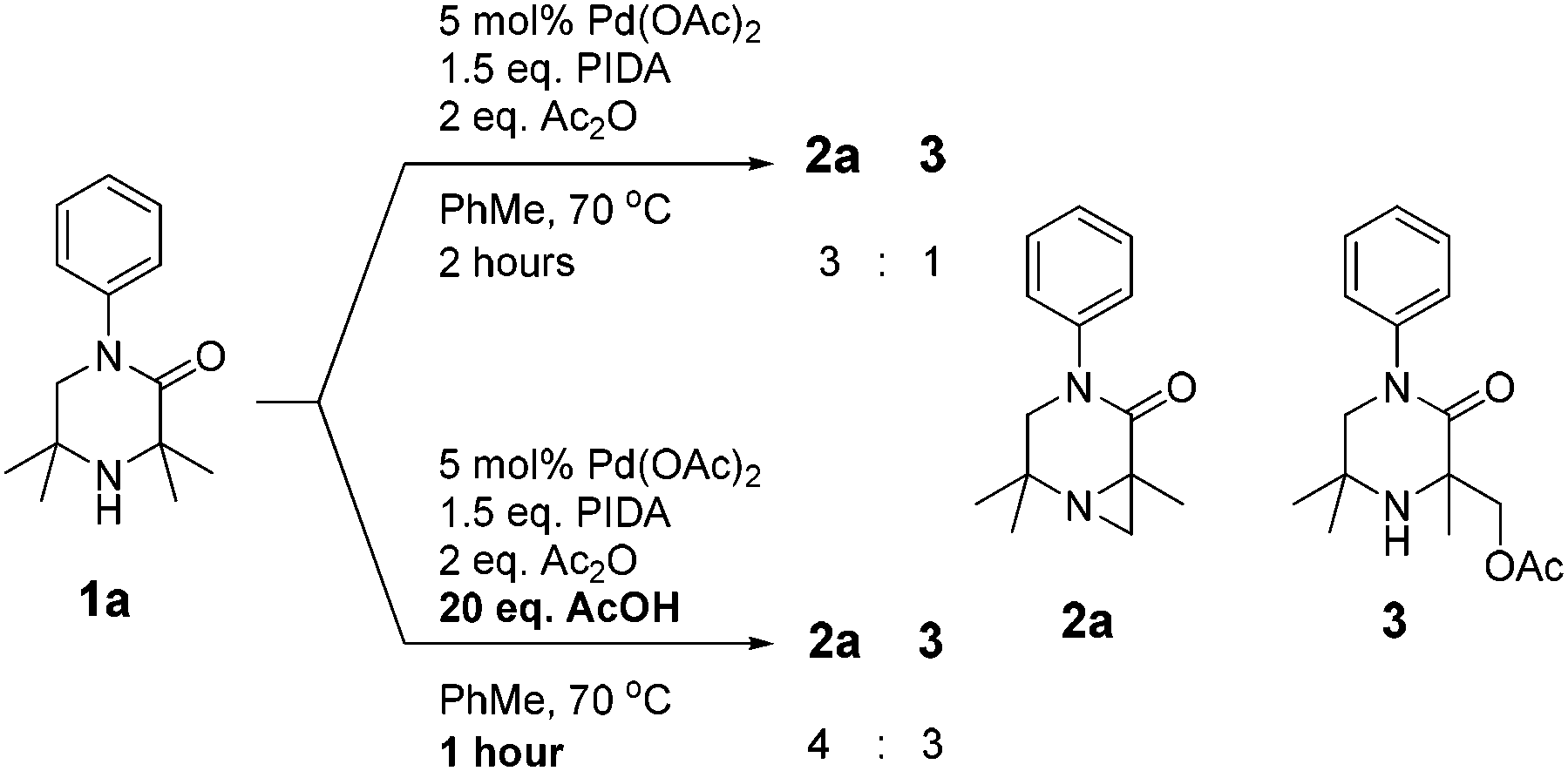 | ||
| Scheme 2 Initial investigations on the aziridination of 1a. | ||
The acid was found to accelerate the reaction, leading to full consumption of starting material after 1 hour. Whilst the desired aziridine 2a remained the major product, an increase in the proportion of acetate 3 and diacetate 4 were observed.9 We sought thereafter to improve the product distribution towards 2a whilst retaining the effect of the acid on decreasing reaction time.
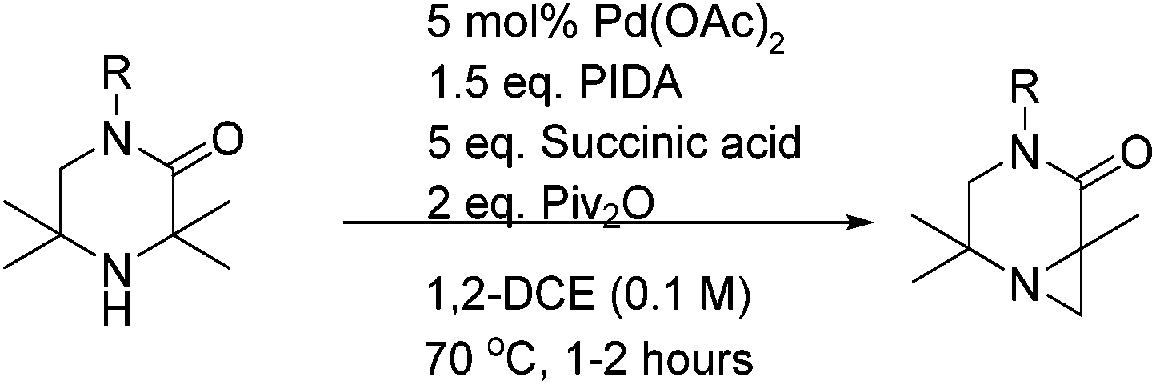
Further optimisation of the reaction was then performed (Table 1). A screen of palladium complexes and oxidants revealed that Pd(OAc)2 was the optimal source, and that crucially, the use of oxidants other than PIDA were unsuccessful.10
|
|
||||
|---|---|---|---|---|
| Entry | Solvent (0.1 M) | Anhydride | Acid | Assay yield of 2a (ratioa2a:3:4) |
| a Determined by UV integration of LC-MS traces after 1 h (unless indicated otherwise), 1H NMR assay yield obtained using CH2Br2 as internal standard. b Ratio obtained by 1H NMR. c Small amounts (∼10%) of the corresponding esters observed by 1H NMR, and the diester was detected by LC-MS. | ||||
| 1 | PhMe | Ac2O | None | 68% (2 h, 3:1:0) |
| 2 | PhMe | Ac2O | Acetic | 56% (4:3:1) |
| 3 | PhMe | Piv2O | Acetic | 57% (8:2:1) |
| 4 | PhMe | Piv2O | Pivalic | 71% (7:1:0)b |
| 5 | PhMe | Piv2O | Benzoic | 32%c (6:1:0) |
| 6 | PhMe | Piv2O | TFA | 0% |
| 7 | PhMe | Piv2O | 2-Toluic | 67%c (4:1:0) |
| 8 | PhMe | Piv2O | 2-Cl benzoic | <5% |
| 9 | PhMe | Piv2O | Oxalic | 0% |
| 10 | PhMe | Piv2O | Malonic | 0% |
| 11 | PhMe | Piv2O | Succinic | 77% (>19:1:0) |
| 12 | PhMe | Piv2O | Glutaric | 76% (>19:1:0) |
| 13 | PhMe | Piv2O | Phthalic | <5% |
| 14 | 1,2-DCE | Piv2O | Succinic | 80% (>19:1:0) |
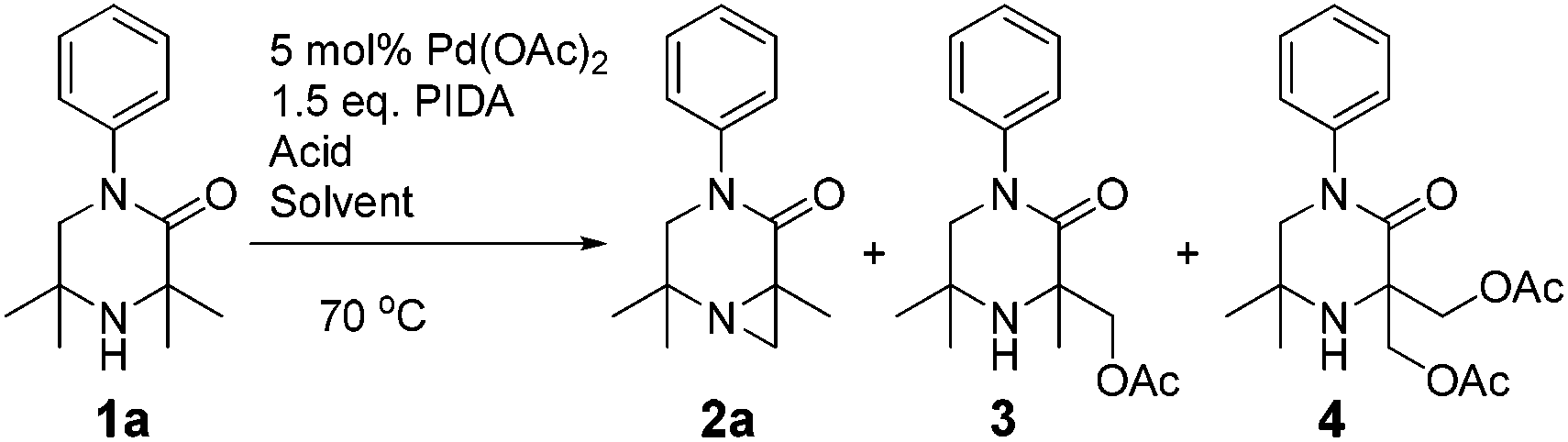
However, an improvement in selectivity towards 2a was observed by replacing acetic anhydride with pivalic anhydride with no impact on reaction time (entry 3). The acid additive was found to be key to product distribution. Low-pKa and hindered acids (entries 4–7) lengthened reaction time and/or encouraged ring-opening products. We investigated the use of 10 equivalents of di-carboxylic acids, as we speculated that the high concentration of carboxylate/carboxylic acid might lead to ring-opening of the desired aziridine under the reaction conditions. Flexible diacids of chain length four atoms and above (succinic and glutaric) successfully suppressed competitive C–O bond formation (entries 11 and 12). Oxalic, malonic and phthalic acid were ineffective (entries 9, 10 and 13). Selectivity was further improved on switching the solvent to 1,2-dichloroethane (1,2-DCE), and the stoichiometry of the acid could be reduced from 10 to 5 equivalents without hampering the reaction (entry 14). Using these optimised conditions, 2a was isolated in good yield (73%) on 1 g scale.
We next sought to explore the role of succinic acid in the reaction. As no succinate adducts are observed, reducing the overall acetate concentration whilst retaining acid catalysis6b by succinate in the reaction mixture could lead to decreased ring-opening side reactions from acetate. We also hypothesised that the polydentate acid might allow for the formation of polynuclear palladium species to form, which are known to effect catalysis.11a–e A recent article by Chen and co-workers has reported a similar process where C–N bond reductive elimination is enforced through the use of diacid ligands from a palladium(IV) species, with supporting computational analysis.12 Gaunt and co-workers have studied the aziridination of morpholinones6b in detail and have demonstrated that the turn-over limiting step (TOLS) is C–H bond cleavage, and that acetic acid provides a rate enhancement through protonation of the amine, thus limiting the formation of palladium(bis)amine complexes.
We confirmed the TOLS is C–H bond cleavage for the reaction of 1a as a kinetic isotope effect was observed when we examined the reaction rate of a d6-analogue of 1a (KIE = 4.1). We next investigated the rate of reaction of 1a at different concentrations of palladium in the presence of succinic acid. Analysis of initial rates revealed a non-linear correlation with concentration of palladium. This gave a reaction order of 1.73 ± 0.15 for palladium, suggesting that under the reaction conditions an equilibrium between active mono- and bimetallic palladium species performing the catalysis exists. We believe the presence of the succinic acid allows for the formation of this bimetallic species as a bridging ligand.
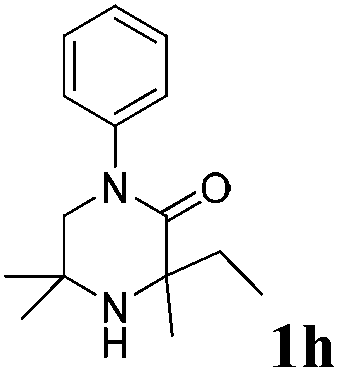
Next, a range of 3,3-substituted piperazinones were found to successfully undergo aziridination under the optimised conditions (Table 2).
|
|
||
|---|---|---|
| Entry | Substrate R = | Yield (isolated) |
| a 16 hours reaction time, with a significant amount (25%) of acetoxylated by-product observed. b 2.5 hours reaction time. c 5 hours reaction time with additional oxidant added after 2 hours. | ||
| 1 | C6H5 – 1a | 2a 73% (1 g scale) |
| 2 | (3-Br)C6H4 – 1b | 2b 52% |
| 3 | (4-OMe)C6H4 – 1c | 2c 72% |
| 4 | (2-Me)C6H4 – 1d | 2d 53% |
| 5 | C6H5CH2 – 1e | 2e 18%a |
| 6 | (2-CF3)C6H4CH2 – 1f | 2f 56% |
| 7 | (4-OMe)C6H4(CH2)2 – 1g | 2g 62%b |
| 8 |
|

|
| 9 |
|

|
| 10 |
|
|
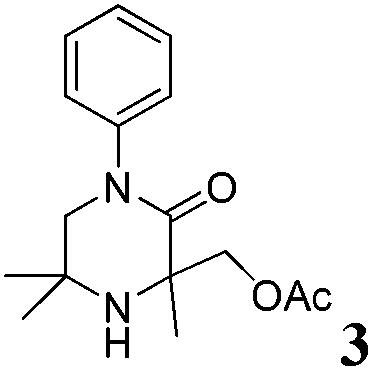
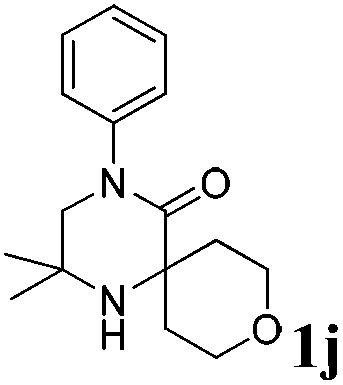
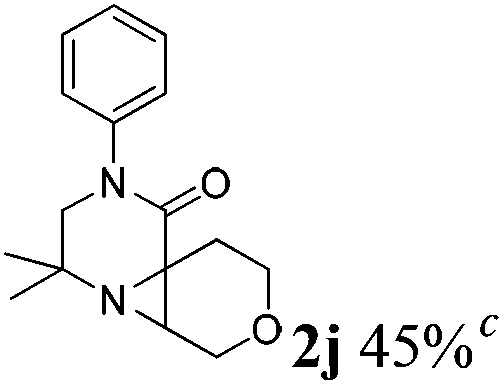
The reaction tolerates aryl groups on the amide nitrogen, with a range of substitutions (1a–d, entries 1–4), notably bromo (1b, entry 2) which provides a handle for further functionalisation.13 However, benzylic amides gave lower yields (entries 5 and 6), although trifluoromethyl-substituted benzylic amide 1f proceeded well.14 In contrast, phenethyl substitution was well tolerated. Moreover, primary and secondary alkyl substituents at the 3-position were reactive under these conditions. Unsymmetrical substrates 1h and 3 (entries 8 and 9) underwent regioselective aziridination on the less hindered methyl group. Tetrahydropyran 1j was also successful, albeit with an extended reaction time and with an additional charging of oxidant (entry 10).
To further emphasise the utility of the C–H activation strategy, we sought to open the aziridine substrates by reaction with nucleophiles. Aziridine 2a smoothly reacted with a range of nucleophiles under Brønsted or Lewis acidic conditions,15 giving piperazinones 5a–e (Scheme 3).
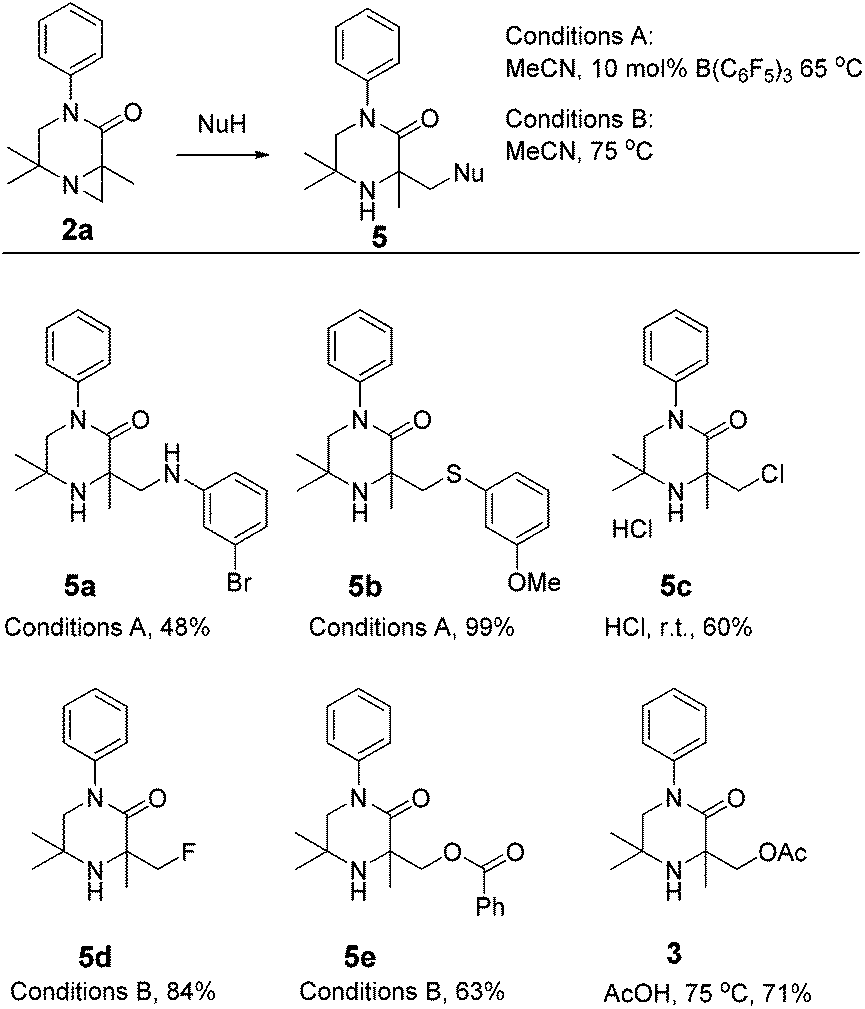 | ||
| Scheme 3 Nucleophilic addition to aziridine 2a. | ||
It was also possible to run sequential C–H activation processes to furnish a di- (4) and tri-acetate product16 (6) by conducting the reaction in acetic acid, with higher loadings of oxidant for longer time periods (Scheme 4).
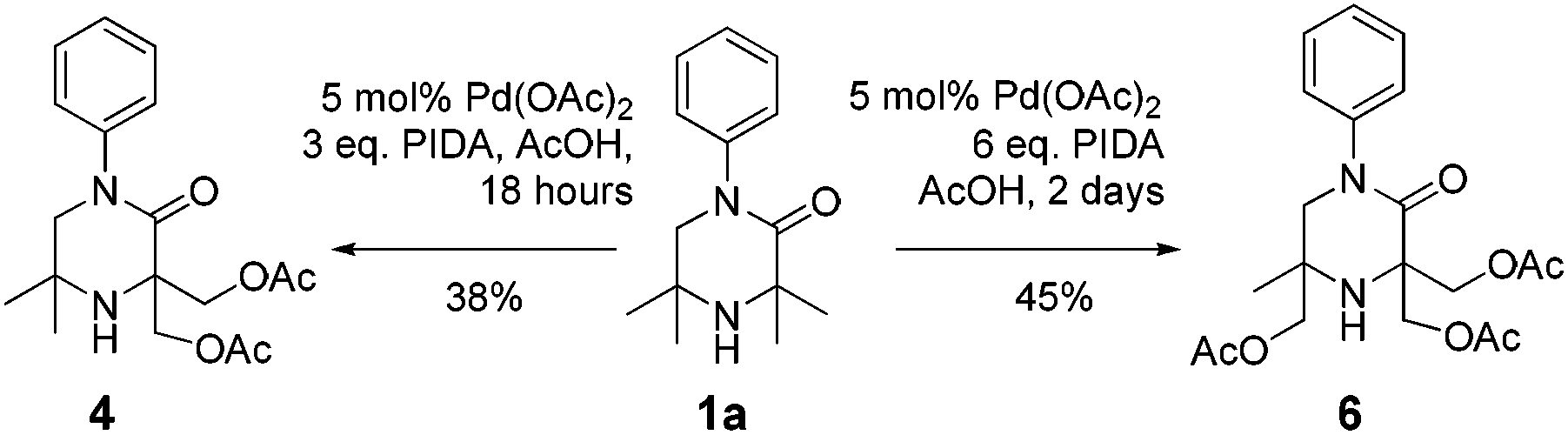 | ||
| Scheme 4 Poly-acetoxylation of 1a. | ||
We postulate that acetic acid opens the aziridine in situ, which allows for a second aziridination process to occur. In the case of the triacetate, which was isolated after a long reaction time, the third C–H functionalisation process likely does not proceed via an aziridination process but rather through a direct C–H acetoxylation process.17 This sequence allows for solvent-controlled aziridination or acetoxylation reaction pathways, depending on the solvent and oxidant loading.
Conclusions
Through optimisation of the reaction conditions, we have demonstrated that 3,3,5,5-tetrasubstituted piperazin-2-ones can undergo efficient nitrogen-directed palladium-catalysed aziridination. The selective formation of versatile aziridines through oxidative C–N bond formation was achieved using PIDA as the oxidant and succinic acid as the additive. Both primary and secondary alkyl C–H bonds can successfully be functionalised. The transformation tolerates aryl and phenethyl substituents on the lactam nitrogen, as well as remote functionality such as halogen, alkyl, and alkoxy. Benzylic substituents are less well tolerated. Analysis of the reaction kinetics with succinic acid revealed an almost second-order (1.73 ± 0.15) dependency of the reaction rate on palladium, indicative of an equilibrium between active monomeric and dimeric palladium species, mediated by succinic acid. Divergent aziridination and acetoxylation are possible by controlling the nature of the acid additive in the reaction. Furthermore, the aziridine products can further be derivatised through acid-mediated nucleophilic ring-opening with a range of halogen, N, O and S-nucleophiles. We anticipate that the use of diacids could be a useful additive to suppress competitive acetoxylation in similar C–H functionalisation processes, as well as a start point to introduce a chiral coordination sphere on the catalytic centers to develop enantioselective aziridination processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Oncology iMed, AstraZeneca for the post-doctoral funding and Professor Matthew Gaunt (University of Cambridge) for valuable discussions and insights. We also thank Paul Davey, Becky Burton and Joshua Hill (Oncology iMed, AstraZeneca) for purification and MS support and Eva Lenz (Oncology iMed, AstraZeneca) for NMR support.Notes and references
- For a recent review on piperazine moieties in drug discovery, see: V. R. Patel and S. Won Park, Mini-Rev. Med. Chem., 2013, 13, 1579–1601 CrossRef PubMed.
- A Scifinder substance search (April 2016) reveals >4000000 hits for the piperazine scaffold, >180000 hits for 2,2-disubstituted piperazines and only ∼1600 hits for 2,2,6,6-tetrasubstituted piperazines.
- (a) R. A. Olofson and C. M. Dougherty, J. Am. Chem. Soc., 1973, 95, 582–584 CrossRef CAS; (b) C. Krinninger, Spec. Chem. Mag., 2010, 30, 20–21 CAS; (c) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef CAS PubMed.
- (a) Z. Zhou and L. Liu, Curr. Org. Chem., 2014, 18, 459–474 CrossRef CAS; (b) R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS; (c) K.-U. Schoning, Chim. Oggi, 2010, 28, 18–20 CAS; (d) K.-U. Schoening, Chim. Oggi, 2010, 28, 12–13 CAS; (e) E. Yoshida, Polymers, 2012, 4, 1125–1156 CrossRef.
- For selected recent reviews, see: (a) N. Dastbaravardeh, M. Christakakou, M. Haider and M. Schnuerch, Synthesis, 2014, 46, 1421–1439 CrossRef; (b) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC; (c) R. Giri, S. Thapa and A. Kafle, Adv. Synth. Catal., 2014, 356, 1395–1411 CrossRef CAS.
- (a) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed; (b) A. P. Smalley and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 10632–10641 CrossRef CAS PubMed; (c) J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS PubMed; (d) C. He and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 15840–15844 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Oh, H.-J. Ha, D. Chi and H. Lee, Curr. Med. Chem., 2001, 8, 999–1034 CrossRef CAS PubMed; (b) P. Kowalski, T. Kowalska, M. Mokrosz, A. Bojarski and S. Charakchieva-Minol, Molecules, 2001, 6, 784–795 CrossRef CAS; (c) Y. Zhou, J. T. Manka, A. L. Rodriguez, C. D. Weaver, E. L. Days, P. N. Vinson, S. Jadhav, E. J. Hermann, C. K. Jones, P. J. Conn, C. W. Lindsley and S. R. Stauffer, ACS Med. Chem. Lett., 2010, 1, 433–438 CrossRef CAS PubMed; (d) V. Soskic, V. Sukalovic and S. Kostic-Rajacic, Mini-Rev. Med. Chem., 2015, 15, 988–1001 CrossRef CAS PubMed.
- >100 marketed drugs contain a piperazine moiety. See DrugBank 5.0: D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2017, gkx1037, DOI:10.1093/nar/gkx1037.
- Small amounts of 2i were also detected.
- The use of di-(pivaloyloxy)iodobenzene led to the detection of some 2a, albeit in low amounts.
- Evidence for polynuclear species may be inferred from reported X-ray structures of cyclopalladated morpholinones in ref. 6a and computational data in ref. 6b, as well as precedent from: (a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed; (b) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234–11241 CrossRef CAS PubMed; (c) F. A. Cotton, J. Gu, C. A. Murillo and D. J. Timmons, J. Am. Chem. Soc., 1998, 120, 13280–13281 CrossRef CAS; (d) A. R. Dick, J. W. Kampf and M. S. Sanford, Organometallics, 2005, 24, 482–485 CrossRef CAS; (e) K. Muñiz, Angew. Chem., Int. Ed., 2009, 48, 9412–9423 CrossRef PubMed.
- G. He, G. Lu, Z. Guo, P. Liu and G. Chen, Nat. Chem., 2016, 8, 1131–1136 CrossRef CAS.
- We also investigated the aziridination of a 3-pyridyl analogue of 1a however this gave erratic results variable with the reaction scale.
- In the case of benzylic substrates, aldehyde signals were observed by 1H NMR in the crude reactions, suggesting these substrates undergo competitive benzylic oxidation processes. A 2-chlorobenzyl substituent and a 2-thiophenyl failed to give satisfactory amounts of aziridine due to competitive oxidative debenzylation.
- I. D. G. Watson and A. K. Yudin, J. Org. Chem., 2003, 68, 5160–5167 CrossRef CAS PubMed.
- The reaction also produced higher-order acetoxylation products (tetra- and penta-acetoxylation) in trace amounts, detectable by mass spectrometry.
- See ref. 6a; acetoxylation (not aziridination) was observed in cases where there was no carbonyl moiety in the substrate. We presume a similar process occurs on 6 on the methyl groups distal to the carbonyl.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob02486j |
| This journal is © The Royal Society of Chemistry 2018 |